2021 45(1): 79-87
DOI:10.32604/biocell.2021.013583
www.techscience.com/journal/biocell
| BIOCELL 2021 45(1): 79-87 DOI:10.32604/biocell.2021.013583 | www.techscience.com/journal/biocell |
DNA methylation and alternative splicing modulate FBXW11 gene expression in Holstein bull testis and are correlated with sperm quality
1Dairy Cattle Research Center, Shandong Academy of Agricultural Sciences, Jinan, 250100, China
2College of Life Sciences and Food Engineering, Hebei University of Engineering, Handan, 056021, China
*Address correspondence to: Jinming Huang, huangjinm@sina.com
Received: 12 August 2020; Accepted: 25 September 2020
#These authors contributed equally to this study
Abstract: F-box and WD-40 domain protein 11 (FBXW11) is an important component of the E3 ubiquitin-ligase enzyme that plays a key role in the ubiquitin-dependent regulation of spermatogenesis. In our previous research, the mRNA expression of FBXW11 in bull sperm with high motility is significantly higher than that with low motility. In the present study, the protein expression levels of FBXW11 in bull testicular tissues with low-performance sperm quality groups were significantly higher than those in normal performance groups. The immunohistochemistry result demonstrated that FBXW11 protein was located in the periphery of Leydig cells and seminiferous tubules. Three splice variants of the FBXW11 gene, namely, FBXW11-tv1, FBXW11-tv2, and FBXW11-tv3, were identified in testicular tissues. The splicing patterns of the three variants are exon skipping. The transcript FBXW11-tv2 expressions were the highest in each sample. The low-performance groups displayed higher FBXW11-tv1 and FBXW11-tv2 transcript expressions than the normal performance groups. Two CpG islands were located within the 5’ UTR and exon 1-2 region of the FBXW11 gene. Bisulfite sequencing PCR results demonstrated that the methylation levels of 11 methylation sites in the CpG island 2 from −99 to −43 in the normal performance groups were significantly lower than those in the low-performance groups. Pearson correlation analysis suggested that the CpG island 2 methylation level was negatively correlated with sperm motility and the transcript FBXW11-tv2 expression level. Our data revealed that alternative splicing and DNA methylation jointly regulated FBXW11 gene expression and were correlated with sperm quality traits during spermatogenesis in Holsteins.
Keywords: FBXW11; Alternative splicing; DNA methylation; Bull; Sperm quality traits
Spermatogenesis is an extremely complex and precisely regulated process. Each stage of the spermatogenesis process is accompanied by complex regulatory mechanisms, including hormone regulation and protein ubiquitination (Baarends et al., 2000; Sofikitis et al., 2008). The ubiquitination of proteins occurs in numerous processes needed for the progression of spermatozoa (Manku et al., 2012). Many proteins are degraded by ubiquitination during spermatogenesis (Liu et al., 2005). The systemic tissue reports in rats revealed that the testis had the highest rate of ubiquitination, that is, almost fourfold higher than the others (Rajapurohitam et al., 2002).
The ubiquitin-proteasome system is essential in spermatogenesis and mainly consists of E1 ubiquitin-activating enzyme, E2 ubiquitin-conjugating enzyme, and E3 ubiquitin-ligase enzyme. The selectivity and specificity of E3 ubiquitin ligase enzyme for substrate proteins are important in maintaining the stability of spermatogenesis (Sutovsky, 2003). E3 ubiquitin ligase enzyme is mainly composed of five proteins, namely, Skp1, Ring-finger, Cullin, Rbxl, E3 ubiquitin ligase enzyme is mainly composed of five proteins, namely, Skp1, Ring-finger, Cullin, Rbxl, and F-box proteins (Zheng et al., 2002). F-box and WD-40 domain protein 11 (FBXW11) (also called β-TrCP2) is a member of the F-box protein family and a key component of the E3 ubiquitin ligase enzyme. FBXW11 can recognize and ubiquitinate specific phosphorylated substrates and regulate the NF-κB signaling pathway, the Wnt signaling pathway, and cell cycle and invasion, thereby affecting cell growth, differentiation, apoptosis, and tumor occurrence (Nakayama et al., 2003). FBXW11 is essential for spermatogenesis in mice, and FBXW11 gene conditional knockdown disrupts testicular organization (Kanarek et al., 2010; Morohoshi et al., 2019). In our previous research, we identified differentially expressed genes in semen from bulls with high and low sperm motility by using RNA-seq; FBXW11 is one of the differentially expressed genes (Wang et al., 2019). The expression level of the FBXW11 gene in sperm with high sperm motility is significantly higher than that with low sperm motility, thereby indicating the FBXW11 gene has a potential effect on bull sperm motility.
Alternative splicing is a vital mechanism in regulating gene expression in eukaryotes (Kornblihtt et al., 2013). Approximately 74% of multiexon genes are alternatively spliced (Johnson et al., 2003). Alternative splicing is predominant in the testis and plays an important role in spermatogenesis (Elliott and Grellscheid, 2006). Few studies have examined alternative splicing in bovine testes and sperm and reported that such splicing of reproduction-related genes is one of the influencing factors of sperm motility during bull spermatogenesis (Guo et al., 2014; Zhang et al., 2014). DNA methylation is also an important mechanism for controlling gene expression. DNA methylation is involved in spermatogenesis (Kropp et al., 2017). Abnormal DNA methylation in genes is associated with human defective sperm quality (Trasler, 2009).
The bovine FBXW11 gene is a multiexon gene that consists of 14 exons and 13 introns. We assumed that the expression of the bovine FBXW11 gene may be regulated by alternative splicing and DNA methylation. Therefore, we analyzed the expression and localization of the FBXW11 protein in bull testicular tissues by Western blotting (WB) analysis and immunohistochemistry (IHC). We also identified the splice variants and determined the core promoter region and CpG islands methylation pattern of the FBXW11 gene in testicular tissues of Holstein bulls with normal and low-performance sperm quality.
This study was carried out according to the Regulations for the Administration of Affairs Concerning Experimental Animals published by the Ministry of Science and Technology, China in 2004 and approved by the Animal Care and Use Committee from the Dairy Cattle Research Center, Shandong Academy of Agricultural Sciences, Shandong, China.
Testicular tissue samples were collected from eight healthy adult Chinese Holstein bulls (3–4 years old) at a commercial slaughterhouse in Jinan, Shandong Province, China. Based on the sperm motility traits of these bulls, the samples were classified into normal sperm motility (H1, H2, H3, and H4) and low sperm motility group (L1, L2, L3, and L4) (Tab. 1). The fresh sperm motility assays were performed by the Computer-Assisted Semen Analysis (CASA) system. Various sperm motility parameters were analyzed, including average path velocity (VAP), straight-line velocity (VSL), and curvilinear velocity (VCL). Assessment of sperm motility was performed using criteria: rapid progressive along a linear track. Small pieces of testicular tissues were taken and soaked with a 4% paraformaldehyde fixing solution for IHC analysis. The remaining testicular tissues were immediately frozen in liquid nitrogen.
Table 1: Sperm quality traits of eight healthy adult Chinese Holstein bull samples (normal performance [H1, H2, H3, and H4] and low-performance [L1, L2, L3, and L4] groups)
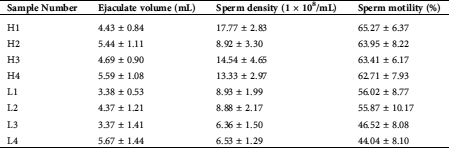
Hematoxylin–eosin staining, Western blotting analysis, and immunohistochemistry
The hematoxylin–eosin staining (HE-staining) of testicular tissues was performed to observe the intact structures and physiological states of the tissues. Western blotting and immunohistochemistry techniques were applied to examine the expression and localization of the FBXW11 protein in testicular tissues. The total protein of the testicular tissues was extracted using the RIPA lysis buffer. The testicular tissues and samples were embedded in paraffin and sectioned for HE-staining and IHC. FBXW11 antibodies were purchased from Proteintech Group. The immunogen of the antibody sequence and the bovine FBXW11 reference amino acid sequence was aligned to analyze the applicability of the antibody. WB and IHC procedures were performed according to a previously described protocol (Guo et al., 2014). Alpha Ease FC 4.0 software was used to quantify the band density in WB analysis. Relative protein levels were quantified using Alpha Ease FC 4.0 software after normalization with β-actin.
Identification of splice variants
Total RNA was extracted from eight testicular tissues using the RNAsimple Total RNA Kit (Tiangen). RNA quality was monitored using visualization bands in 1.5% agarose gels after electrophoresis. The RNA samples were reverse transcribed into cDNA. The RT-PCR reaction conditions are as follows: 5 min at 94°C, followed by 35 cycles of 94°C for 30 s, 58°C for 30 s, and 72°C for 1 min, with a final extension at 72°C for 5 min.
To determine the potential splice variant of bovine FBXW11, we designed two primer pairs (i.e., AS1 and AS2, Tab. S1) to amplify the exon 1–6 region sequence and three primer pairs (i.e., T2, T3, and T4, Tab. S1) to amplify exon 7 to the 3’ UTR region according to the bovine FBXW11 full-length reference mRNA sequence. The PCR products were purified, cloned into the pMD-19T vector by using T4 DNA ligase and transformed into Trans5α. Individual positive clones were picked and sequenced. The sequence alignments were performed using DNAMAN v5.2.2 software to identify splice variants. The three transcripts, that is, FBXW-tv1, FBXW-tv2, and FBXW-tv3 were identified in testicular tissues. The protein sequence of the transcripts FBXW-tv1, FBXW-tv2, and FBXW-tv3 were predicted using ExPASy software (https://web.expasy.org/translate/). The presence and location of the signal peptide cleavage sites in amino acid sequences from three FBXW transcripts were predicted using SignalP 4.0 (Petersen et al., 2011).
Relative mRNA expression analysis
RT-PCR experiments using the primer pairs QS1, QS2, and QS3 (Tab. S1) were performed to determine the expression of the three transcripts in testicular samples from four normal performance and four low-performance bulls. qPCR experiments were conducted to detect the relative expression of the transcripts FBXW-tv1, FBXW-tv2, and FBXW-tv3 among different samples. The qPCR procedure was based on the method from our previous report (Ju et al., 2015). The housekeeping gene β-actin was used as a reference to normalize data. 2−ΔΔCt was used to calculate the data.
FBXW11 promoter plasmid construction, cell transfection, and luciferase reporter assay
To analyze whether DNA methylation is involved in the regulation of FBXW11 gene expression, we used the MethPrimer 2.0 software (http://www.urogene.org/ methprimer/) (the software settings: Pick primers for bisulfite sequencing PCR and use CpG island prediction for primer selection) to predict and analyze the CpG island located within 5’ UTR and the exon of FBXW11, for which two CpG islands were found. To determine the position of the FBXW11 gene core promoter and ascertain whether the CpG island is located in the core promoter region, we designed three pair primers (i.e., P1, P2, and P3, Tab. S1) to amplify the 2000 bp sequence upstream of the FBXW11 gene transcription initiation site (TSS) segmentally. The amplified PCR products of the three fragments were purified, recovered, and cloned into the pGL3 vector. Then, the resulting plasmids were extracted.
The murine Leydig tumor cell line (MLTC-1) was cultured in RPMI-1640 medium with 10% fetal bovine serum containing 10 mg/L of penicillin and streptomycin. The sub-cultured cells were digested, centrifuged, and added to the RPMI-1640 medium without penicillin and streptomycin. The cell suspension was dispensed into a 24-well plate, and cell density was observed under a microscope. The preferred cell density per well was 5 × 104/mL. When the cells were adherent, 80–85%, transfection was performed with pGL3-FBXW11 constructs and an empty vector. After transfection for 48 h, the original culture medium was discarded, and the cells were washed with PBS. Dual-luciferase activity assays were conducted using the Dual-Luciferase® Reporter Assay System kit according to our previous report (Pan et al., 2013).
Bisulfate sequencing PCR (BSP) was used to analyze the methylation levels of the CpG island in eight testicular samples. Four pairs of specific primers (MY1-1, MY1-2, MY2-1, and MY2-2; Tab. S1) were designed to amplify CpG island fragments by nested PCR and touchdown PCR. BSP sequencing results were analyzed according to a previously described protocol (Guo et al., 2014).
The SPASS statistics V17.0 software was used to perform the statistical test on the relative protein expression level of FBXW11 between the low-performance sample group and the normal-performance sample group.
FBXW11 protein expression and localization in testicular tissues
The HE staining technique was used to detect the physiological states and pathological conditions of the bull testicular samples. The results showed that samples had a normal physiological state with no lesions and could be used in the experiment (Fig. 1A). Then, the FBXW11 protein expression and localization in testicular samples were analyzed by WB and IHC. WB analysis results showed that the expression levels of the FBXW11 protein in low-performance sample groups (0.4550 ± 0.058) were significantly higher than those in the normal performance sample groups (0.2675 ± 0.046) by SPASS statistics V17.0 software (p = 0.045, Fig. 1B). IHC results demonstrated that the FBXW11 proteins were located in the periphery of the Leydig cells and seminiferous tubules in testicular samples (Fig. 1C).
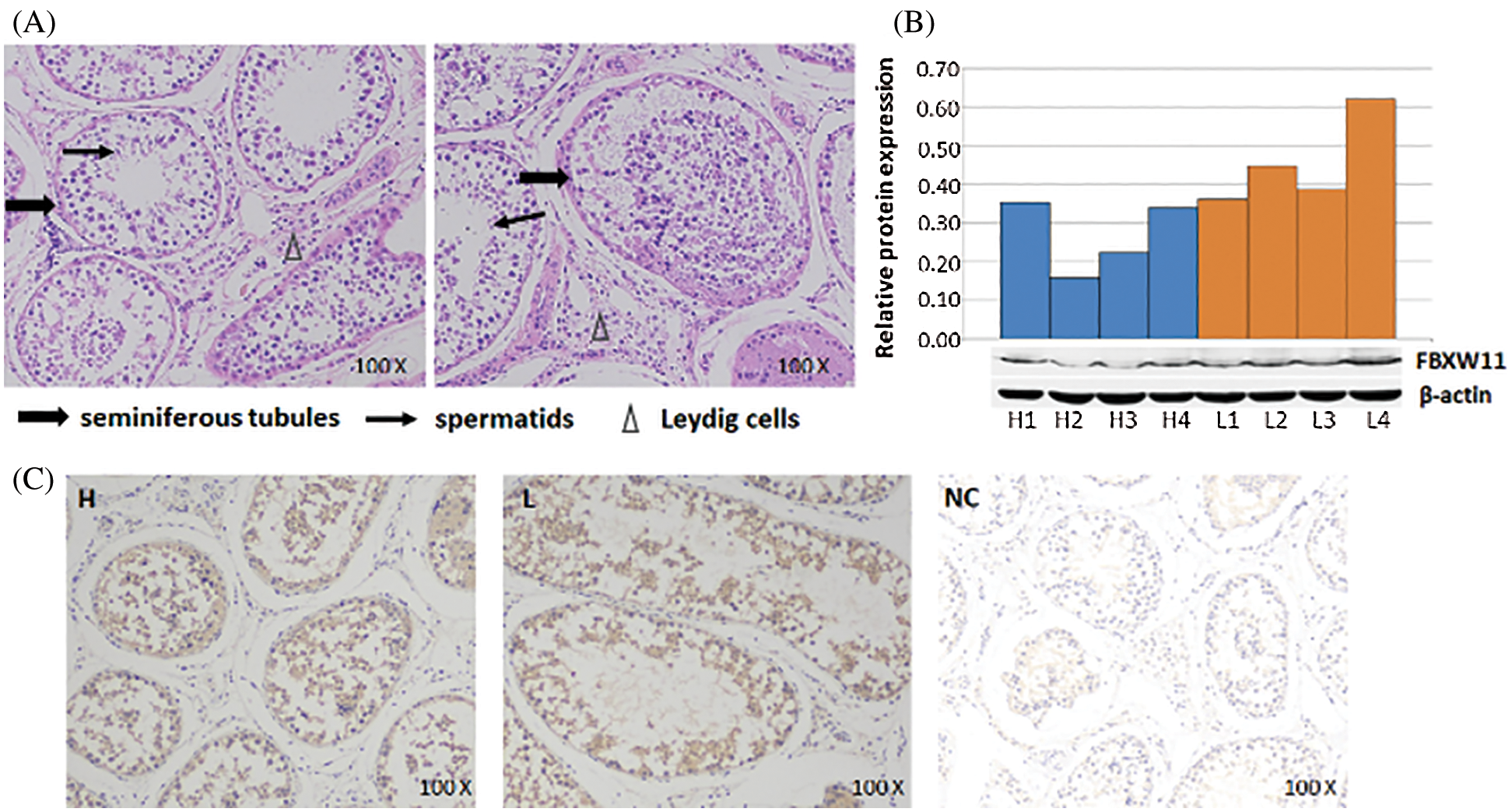
Figure 1: FBXW11 protein expression and localization in testicular tissues.
FBXW11 gene alternative splicing identification
To identify potential splice variants of bovine FBXW11, we designed five primer pairs to amplify the full-length coding region. Three PCR product bands were observed in agarose gel electrophoresis. Through clone sequencing and alignment analysis, three transcripts were identified, as follows: FBXW11-tv1 (XM_003587500.5, lacked exons 3–4), FBXW11-tv2 (XM_003587498.5, lacked exons 2–4), and FBXW11-tv3 (XM_003587499.5, lacked exons 2–5, Fig. 2A). The patterns of the three splice variants were exon skipping.
The FBXW11 gene contains three protein domains, as follows: F-box-like domain, D domain of β-TrCP, and WD40 domain. Comparison of the amino acid sequences encoded by the three splice variants revealed that the three protein domains remained intact in three transcripts after exon skipping, but the N-terminal sequences were different. The transcript FBXW11-tv2 was 19 aa less than transcript FBXW11-tv1, and transcript FBXW11-tv3 was 21 aa less than transcript FBXW11-tv1 (Fig. S1).
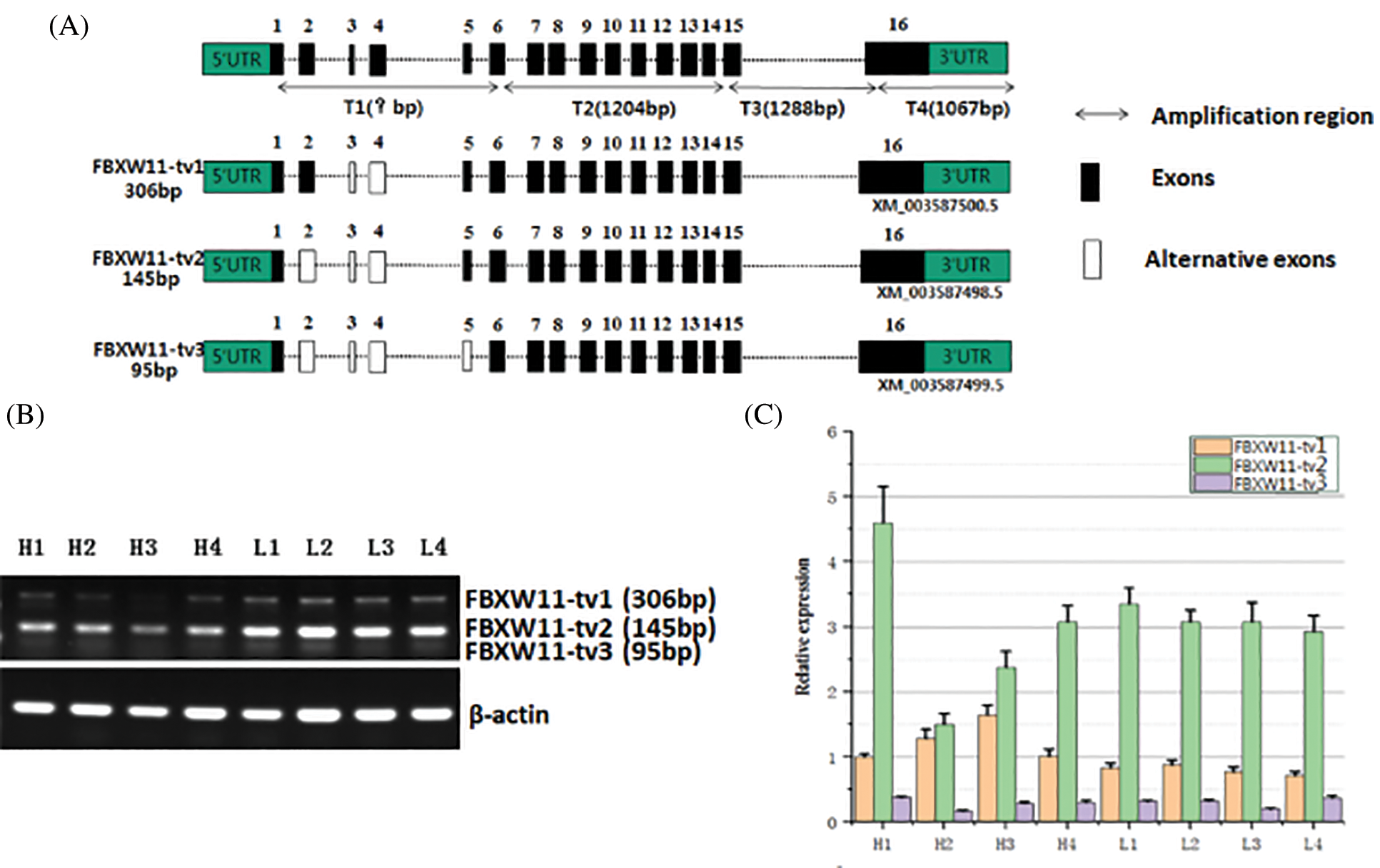
Figure 2: Identification and expression analysis of FBXW11 gene splicing variants.
FBXW11 gene mRNA expression analysis
The expression levels of the three transcripts from the testicular samples were analyzed by RT-PCR and qPCR. The RT-PCR results showed that transcript FBXW11-tv2 expression levels were highest for all samples, followed by transcripts FBXW11-tv1 and FBXW11-tv3 (Fig. 2B). The transcript FBXW11-tv2 was the primary transcript of the FBXW11 gene. The low-performance group testicular tissues displayed higher FBXW11-tv1 and FBXW11-tv2 transcript expression than the normal performance group testicular tissues (p < 0.05). The qPCR results were consistent with those of RT-PCR (Fig. 2C). We also analyzed the ratio change between FBXW11-tv1 and FBXW11-tv2 (Fig. S2). The result suggested that the ratio of FBXW11-tv1/FBXW11-tv2 in the normal performance group is higher than that in the low-performance group.
Cloning and luciferase activity analysis of FBXW11 gene promoter
To investigate whether DNA methylation regulates FBXW11 gene expression, we predicted that the CpG island was located within 5’ UTR and the exon 1–2 region of the FBXW11 gene and found two CpG islands. CpG island 1 was located at upstream −761 bp to −430 bp of the gene TSS. CpG island 2 included the entire exon 1 and the 140 bp at the junction of exon 1 and intron 1 (Fig. 3A).
To determine whether the two CpG islands are located in the core promoter region of the FBXW11 gene, we generated the truncated constructs (P1, P2, and P3) by the gradual deletion of nucleotides from the 5’ terminals and cloned into the pGL3 vector. These constructs were transiently transfected into MLTC-1 cells. The luciferase activity was measured to determine the minimum sequence required for FBXW11 transcription. Dual-luciferase reporter gene assays revealed that the construct P2 from g. −912 to g. +36 promoter activity was significantly higher than that of construct P1 from g. −1918 to g. +36, P3 from g. −415 to g. +36, and the control pGL3 vector (p < 0.05) (Fig. 3B). These results indicated that the region between −912 to −415 was the core promoter region of the bovine FBXW11 gene, and CpG island 1 was located in the core promoter region.
Figure 3: Luciferase activity analysis of FBXW11 gene promoter and CpG island methylation level analysis.
FBXW11 gene CpG island methylation level analysis
BSP sequencing was used to analyze the methylation levels of the FBXW11 gene in eight testicular samples. The results of the CpG island 1 indicated no difference in the methylation levels between the low-performance and normal performance groups. Thus, we continued to analyze the methylation level of CpG island 2. The methylation levels of 11 methylation sites in CpG island 2, from −99 to −43, were significantly different in the two group samples (p < 0.05), that is, the methylation levels of the normal performance groups were lower than those of the low-performance groups. The 11 methylation sites were located at the FBXW11 gene −99, −92, −86, −77, −71, −68, −65, −56, −52, −47, and −43, respectively. The methylation rates of the normal performance groups were H1, 35.0%; H2, 38.9%; H3, 53.2%; and H4, 46.7%. The methylation rates of the low-performance groups were L1, 74.0%; L2, 70.1%; L3, 68.8%; and L4, 70.1% (Fig. 3C). The methylation rates of the samples for the normal performance group were 43.45% on average, and those of samples for the low-performance group were 70.75% on average.
Pearson correlation analysis was performed on the methylation level of the two CpG islands and the bull sperm motility. No correlation was found between the CpG island 1 methylation level and sperm motility (Pearson coefficient r = 0.01), while the CpG island 2 methylation level was negatively correlated with sperm motility (r = −0.80, Fig. 3D). The correlation between the methylation level of CpG island 2 and the expression level of FBXW11-tv2 (the primary transcript of the FBXW11 gene) was also examined. The CpG island 2 methylation level was not significant correlated with transcript FBXW11-tv2 expression level (r = −0.28, p = 0.495).
The spermatogonial stem cells induce testicular maturation during spermatogenesis, which relies on the remodeling of protein complexes and the rapid transformation of proteins in developing germ cells and spermatogenic epithelial supporting cells (Sassone-Corsi, 2002). Therefore, protein ubiquitination is important in spermatogenesis. E3 ubiquitin-ligase enzyme recognizes the target protein and links ubiquitin tags during protein ubiquitination (Deshaies and Joazeiro, 2009). The FBXW11 gene is a component of the E3 ubiquitin-ligase enzyme that plays a major role in the regulation of the process from spermatogenesis to sperm maturation and is associated with cell apoptosis during spermatogenesis (Richburg et al., 2014; Nakagawa et al., 2017). Our research group also found that the FBXW11 gene is significantly differentially expressed between bull semen samples of high and low sperm motility groups. Therefore, we hypothesized that the bovine FBXW11 gene may have a potential role in spermatogenesis in bulls.
Western blotting analysis of the FBXW11 protein in testicular tissues showed that the low-performance group had significantly higher FBXW11 protein expression than those of the normal-performance group. IHC experiments also revealed that the FBXW11 proteins were located in the periphery of the Leydig cells and seminiferous tubules in testicular samples. The seminiferous tubule is the site of spermatogenesis and maturation, and the Leydig cells provide energy and enzymes to assist sperm production and promote sperm maturation (Holstein et al., 2003). Therefore, expression and localization analysis indicated that the FBXW11 gene may have an effect on testicular development.
Alternative splicing is an important mechanism in regulating gene expression. In the present study, three different splicing variants of the FBXW11 gene were found in the testicular tissues. The signal peptide and secondary structure of the different transcripts of the FBXW11 gene were predicted with software. The signal peptide is a short peptide chain located at the N-terminal guiding protein trans-membrane transfer. The difference in the signal peptides caused by N-terminal truncation has an effect on protein function (Yu et al., 2000). We predicted that the presence and location of signal peptide cleavage sites of the FBXW11 gene different transcripts. The result showed that no signal peptide was found at the N-terminal of the three transcripts. However, alternative splicing resulted in differences in the N-terminus of the three transcripts. The N-terminal of the FBXW11 protein is responsible for the interaction with the SKP1 protein, and the C-terminal recognizes the specific substrate protein (Gupta-Rossi et al., 2001). In humans, FBXW11 has many variants, and the differences among these variants involve the changes in their N-terminal sequences. The elimination of the N-terminal F-box domain renders the FBXW11 unable to interact with the SKP1 protein, thereby affecting sperm motility (Ohsaki et al., 2008).
The expression of the three splicing variants was analyzed. qPCR analysis showed that the FBXW11-tv1 and FBXW11-tv2 expression levels significantly differed between the normal- and low-performance groups. Most splicing transitions did not completely switch mRNA transcripts but instead changed the expression ratios of different transcripts. Significant changes in the ratios between DIS3-tv3 and DIS3-tv1 can indicate hematological cancers (Robinson et al., 2018). The addition of cisplatin D to CD4+ and CD8+ T cells changed the expression ratio of HTERT splicing transcripts and consequently inhibited telomerase activity (Zhdanov et al., 2017). Therefore, we analyzed the change in expression ratio between FBXW11-tv1 and FBXW11-tv2. The result suggested that the FBXW11-tv1/FBXW11-tv2 ratio in the normal performance group was higher than that in the low-performance group. The change in FBXW11-tv1and FBXW11-tv2 ratio may affect sperm quality traits.
DNA methylation is an important mechanism for controlling gene expression. For example, aberrant sperm DNA methylation pattern affects male fertility status (Aston et al., 2015). In the present study, two CpG islands in the 5’ UTR and exon 1–2 region of the FBXW11 gene. The luciferase activity assays indicated that CpG island 1 was located in the core promoter region of the FBXW11 gene. However, the difference in the methylation levels of CpG island 1 between the low-performance and normal-performance groups was insignificant. The CpG island 1 methylation level was not correlated with sperm motility. Thus, we examined the methylation level of CpG island 2. The CpG island 2 methylation level in the testicular tissues of the normal performance group was lower than that of the low-performance group. Pearson correlation analysis demonstrated that the CpG island 2 methylation levels were negatively correlated with sperm motility, which suggested that CpG island 2 DNA methylation may have an influence on bull sperm motility.
In summary, we determined the splice variants and CpG islands methylation pattern of the FBXW11 gene in testicular tissues from normal- and low-performance sperm quality groups, thereby indicating that alternative splicing and DNA methylation may modulate FBXW11 gene expression in Holstein bull testes and affect sperm quality.
Availability of Data and Materials: Data supporting this article are details in this manuscript.
Funding Statement: This project was supported by the Major Project of National Transgene in China (2018ZX08007001–002), the National Natural Science Foundation of China (31671286, 31672397 and 31771374), Shandong Provincial Natural Science Foundation for Distinguished Young Scholars of China (JQ201709), the Program of National Cow Industrial Technology System of China (CARS-37), and the Shandong Provincial Key Research and Development Program of China (2017GNC10120).
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
Aston KI, Uren PJ, Jenkins TG, Horsager A, Cairns BR, Smith AD, Carrell DT. (2015). Aberrant sperm DNA methylation predicts male fertility status and embryo quality. Fertility and Sterility 104: 1388–1397.e5. DOI 10.1016/j.fertnstert.2015.08.019. [Google Scholar] [CrossRef]
Baarends WM, van der Laan R, Grootegoed JA. (2000). Specific aspects of the ubiquitin system in spermatogenesis. Journal of Endocrinological Investigation 23: 597–604. DOI 10.1007/BF03343782. [Google Scholar] [CrossRef]
Deshaies RJ, Joazeiro CA. (2009). RING domain E3 ubiquitin ligases. Annual Review of Biochemistry 78: 399–434. DOI 10.1146/annurev.biochem.78.101807.093809. [Google Scholar] [CrossRef]
Elliott DJ, Grellscheid SN. (2006). Alternative RNA splicing regulation in the testis. Reproduction 132: 811–819. DOI 10.1530/REP-06-0147. [Google Scholar] [CrossRef]
Guo F, Yang B, Ju ZH, Wang XG, Qi C, Zhang Y, Wang CF, Liu HD, Feng MY, Chen Y, Xu YX, Zhong JF, Huang JM. (2014). Alternative splicing, promoter methylation, and functional SNPs of sperm flagella 2 gene in testis and mature spermatozoa of Holstein bulls. Reproduction 147: 241–252. DOI 10.1530/REP-13-0343. [Google Scholar] [CrossRef]
Gupta-Rossi N, Le Bail O, Gonen H, Brou C, Logeat F, Six E, Ciechanover A, Israël A. (2001). Functional interaction between SEL-10, an F-box protein, and the nuclear form of activated Notch1 receptor. Journal of Biological Chemistry 276: 34371–34378. DOI 10.1074/jbc.M101343200. [Google Scholar] [CrossRef]
Holstein AF, Schulze W, Davidoff M. (2003). Understanding spermatogenesis is a prerequisite for treatment. Reproductive Biology and Endocrinology 1: 107. DOI 10.1186/1477-7827-1-107. [Google Scholar] [CrossRef]
Johnson JM, Castle J, Garrett-Engele P, Kan Z, Loerch PM, Armour CD, Santos R, Schadt EE, Stoughton R, Shoemaker DD. (2003). Genome-wide survey of human alternative pre-mRNA splicing with exon junction microarrays. Science 302: 2141–2144. DOI 10.1126/science.1090100. [Google Scholar] [CrossRef]
Ju Z, Wang C, Wang X, Yang C, Sun Y, Jiang Q, Wang F, Li MJ, Zhong JF, Huang J. (2015). Role of an SNP in alternative splicing of bovine NCF4 and mastitis susceptibility. PLoS One 10: e0143705. DOI 10.1371/journal.pone.0143705. [Google Scholar] [CrossRef]
Kanarek N, Horwitz E, Mayan I, Leshets M, Cojocaru G, Davis M, Tsuberi B, Pikarsky E, Pagano M, Ben-Neriah Y. (2010). Spermatogenesis rescue in a mouse deficient for the ubiquitin ligase SCFβ-TrCP by single substrate depletion. Genes & Development 24: 470–477. DOI 10.1101/gad.551610. [Google Scholar] [CrossRef]
Kornblihtt AR, Schor IE, Alló M, Dujardin G, Petrillo E, Muñoz MJ. (2013). Alternative splicing: A pivotal step between eukaryotic transcription and translation. Nature Reviews Molecular Cell Biology 14: 153–165. DOI 10.1038/nrm3525. [Google Scholar] [CrossRef]
Kropp J, Carrillo JA, Namous H, Daniels A, Salih SM, Song J, Khatib H. (2017). Male fertility status is associated with DNA methylation signatures in sperm and transcritomic profiles of bovine preimplantation embryos. BMC Genomics 18: 734. DOI 10.1186/s12864-017-3673-y. [Google Scholar] [CrossRef]
Liu Z, Oughtred R, Wing SS. (2005). Characterization of E3Histone, a novel testis ubiquitin protein ligase which ubiquitinates histones. Molecular and Cellular Biology 25: 2819–2831. DOI 10.1128/MCB.25.7.2819-2831.2005. [Google Scholar] [CrossRef]
Manku G, Wing SS, Culty M. (2012). Expression of the ubiquitin proteasome system in neonatal rat gonocytes and spermatogonia: role in gonocyte differentiation. Biology of Reproduction 87: 44. DOI 10.1093/biolreprod/87.s1.44. [Google Scholar] [CrossRef]
Morohoshi A, Nakagawa T, Nakano S, Nagasawa Y, Nakayama K. (2019). The ubiquitin ligase subunit β-TrCP in Sertoli cells is essential for spermatogenesis in mice. Developmental Biology 445: 178–188. DOI 10.1016/j.ydbio.2018.10.023. [Google Scholar] [CrossRef]
Nakagawa T, Zhang T, Kushi R, Nakano S, Endo T, Nakagawa M, Yanagihara N, Zarkower D, Nakayama K. (2017). Regulation of mitosis-meiosis transition by the ubiquitin ligase β-TrCP in male germ cells. Development 144: 4137–4147. DOI 10.1242/dev.158485. [Google Scholar] [CrossRef]
Nakayama K, Hatakeyama S, Maruyama S, Kikuchi A, Onoé K, Good RA, Nakayama KI. (2011). Impaired degradation of inhibitory subunit of NF-κB (IκB) and β-catenin as a result of targeted disruption of the β-TrCP1 gene. Proceedings of the National Academy of Sciences of the United States of America 100: 8752–8757. DOI 10.1073/pnas.1133216100. [Google Scholar] [CrossRef]
Ohsaki K, Oishi K, Kozono Y, Nakayama K, Nakayama KI, Ishida N. (2008). The role of β-TrCP1 and β-TrCP2 in circadian rhythm generation by mediating degradation of clock protein PER2. Journal of Biochemistry 144: 609–618. DOI 10.1093/jb/mvn112. [Google Scholar] [CrossRef]
Pan Q, Ju Z, Huang J, Zhang Y, Qi C, Gao Q, Zhou L, Li QL, Wang LL, Zhong JF, Liu M, Wang C. (2013). PLCz functional haplotypes modulating promoter transcriptional activity are associated with semen quality traits in Chinese Holstein bulls. PLoS One 8: e58795. DOI 10.1371/journal.pone.0058795. [Google Scholar] [CrossRef]
Petersen TN, Brunak S, Heijne G, Nielsen H. (2011). SignalP 4.0: discriminating signal peptides from transmembrane regions. Nature Methods 8: 785–786. DOI 10.1038/nmeth.1701. [Google Scholar] [CrossRef]
Rajapurohitam V, Bedard N, Wing SS. (2002). Control of ubiquitination of proteins in rat tissues by ubiquitin conjugating enzymes and isopeptidases. American Journal of Physiology-Endocrinology and Metabolism 282: E739–E745. DOI 10.1152/ajpendo.00511.2001. [Google Scholar] [CrossRef]
Richburg JH, Myers JL, Bratton SB. (2014). The role of E3 ligases in the ubiquitin-dependent regulation of spermatogenesis. Seminars in Cell & Developmental Biology 30: 27–35. DOI 10.1016/j.semcdb.2014.03.001. [Google Scholar] [CrossRef]
Robinson SR, Viegas SC, Matos RG, Domingues S, Bedir M, Stewart HJS, Chevassut TJ, Oliver AW, Arraiano CM, Newbury SF. (2018). DIS3 isoforms vary in their endoribonuclease activity and are differentially expressed within haematological cancers. Biochemical Journal 475: 2091–2105. DOI 10.1042/BCJ20170962. [Google Scholar] [CrossRef]
Sassone-Corsi P. (2002). Unique chromatin remodeling and transcriptional regulation in spermatogenesis. Science 296: 2176–2178. DOI 10.1126/science.1070963. [Google Scholar] [CrossRef]
Sofikitis N, Giotitsas N, Tsounapi P, Baltogiannis D, Giannakis D, Pardalidis N. (2008). Hormonal regulation of spermatogenesis and spermiogenesis. Journal of Steroid Biochemistry and Molecular Biology 109: 323–330. DOI 10.1016/j.jsbmb.2008.03.004. [Google Scholar] [CrossRef]
Sutovsky P. (2003). Ubiquitin-dependent proteolysis in mammalian spermatogenesis, fertilization, and sperm quality control: killing three birds with one stone. Microscopy Research and Technique 61: 88–102. DOI 10.1002/jemt.10319. [Google Scholar] [CrossRef]
Trasler JM. (2009). Epigenetics in spermatogenesis. Molecular and Cellular Endocrinology 306: 33–36. DOI 10.1016/j.mce.2008.12.018. [Google Scholar] [CrossRef]
Wang X, Yang C, Guo F, Zhang Y, Ju Z, Jiang Q, Zhao XM, Liu Y, Zhao H, Wang JP, Sun Y, Wang CF, Zhu HB, Huang J. (2019). Integrated analysis of mRNAs and long noncoding RNAs in the semen from Holstein bulls with high and low sperm motility. Scientific Reports 9: 127. DOI 10.1038/s41598-018-38462-x. [Google Scholar] [CrossRef]
Yu G, Nishimura M, Arawaka S, Levitan D, Zhang L, Tandon A, Song YQ, Rogaeva E, Chen F, Kawarai T, Supala A, Levesque L, Yu H, Yang DS, Holmes E, Milman P, Liang Y, Zhang DM, Xu DH, Sato C, Rogaev E, Smith M, Janus C, Zhang Y, Aebersold R, Farrer LS, Sorbi S, Bruni A, Fraser P, St George-Hyslop P. (2000). Nicastrin modulates presenilin-mediated notch/glp-1 signal transduction and betaAPP processing. Nature 407: 48–54. DOI 10.1038/35024009. [Google Scholar] [CrossRef]
Zhang X, Wang C, Zhang Y, Ju Z, Qi C, Wang X, Huang J, Zhang S, Li J, Zhong J, Shi F. (2014). Association between an alternative promoter polymorphism and sperm deformity rate is due to modulation of the expression of KATNAL1 transcripts in Chinese Holstein bulls. Animal Genetics 45: 641–651. DOI 10.1111/age.12182. [Google Scholar] [CrossRef]
Zhdanov DD, Vasina DA, Orlova VS, Orlova EV, Grishin DV, Gladilina YA, Pokrovskaya MV, Aleksandrova SS, Sokolov NN. (2017). Induction of apoptotic endonuclease EndoG with DNA-damaging agents initiates alternative splicing of telomerase catalytic subunit hTERT and inhibition of telomerase activity hTERT in human CD4+ and CD8+ T-lymphocytes. Biomeditsinskaya Khimiya 63: 296–305. DOI 10.18097/PBMC20176304296. [Google Scholar] [CrossRef]
Zheng N, Schulman BA, Song L, Miller JJ, Jeffrey PD, Wang P, Chu C, Koepp DM, Elledge SJ, Pagano M, Conaway RC, Conaway JW, Harper JW, Pavletich NP. (2002). Structure of the Cul1–Rbx1–Skp1–F boxSkp2 SCF ubiquitin ligase complex. Nature 416: 703–709. DOI 10.1038/416703a. [Google Scholar] [CrossRef]
Supplement Materials

Figure S1: Software prediction results for the FBXW11 gene splice variants. The amino acid sequence comparison results of three splice variants revealed that the N-terminal sequences of FBXW11-tv2 and FBXW11-tv3 were 19 aa and 21 aa less than that of FBXW11-tv1, respectively.
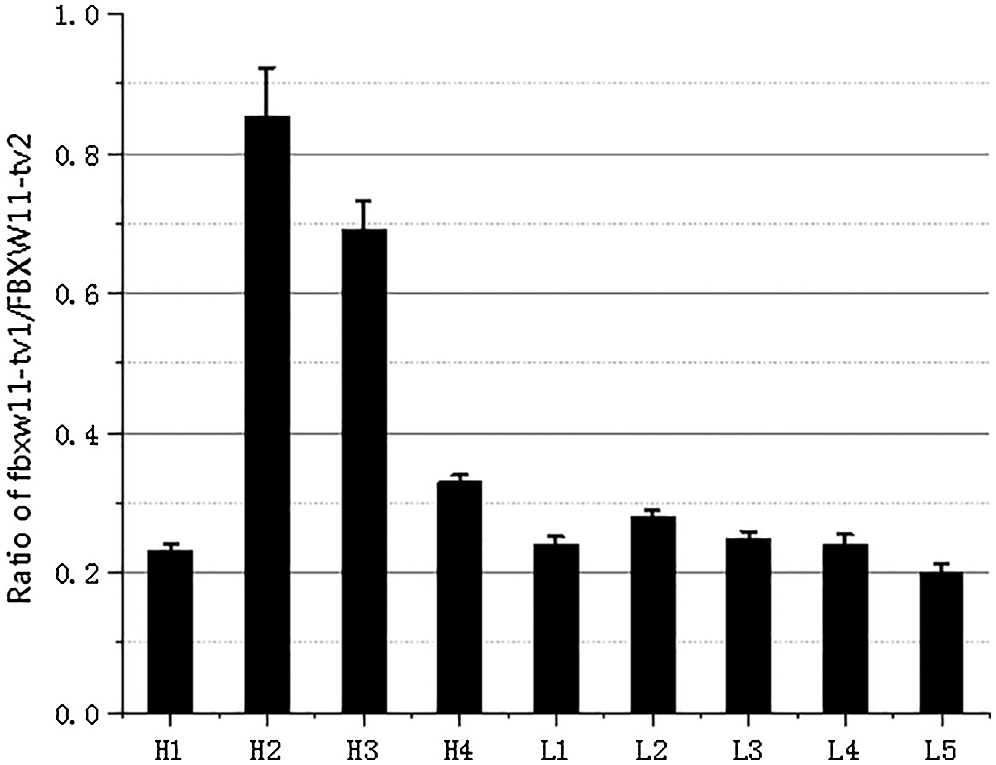
Figure S2: The transcripts FBXW11-tv1/FBXW11-tv2 expression ratio.
Table S1: Primer information on bovine FBXW11 gene

 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |