DOI:10.32604/biocell.2021.014704
www.techscience.com/journal/biocell
| Biocell DOI:10.32604/biocell.2021.014704 | www.techscience.com/journal/biocell |
| Review |
A short review of the genes involved in the development and progression of colorectal cancer
Department of Life Sciences, School of Environment and Life Sciences, Independent University, Bangladesh (IUB), Dhaka, 1229, Bangladesh
*Address correspondence to: Rashed Noor, rashednoor@iub.edu.bd
Received: 22 October 2020; Accepted: 21 December 2020
Abstract: The extent and aggression of colorectal cancer is a worldwide public health threat. Extensive research has been conducted on the pre-requisites leading to this fatal cancer. An array of genes along with their mutations and the signal transduction pathways leading to the cellular transformation into the cancerous cells have been investigated. Based on the knowledge gained so far, present review shortly discussed the role of the major genes especially those are involved in instigating abnormalities in the cellular cycles, cellular proliferation and differentiation. A simple but novel molecular scheme of the colorectal cancer development has also been plotted.
Keywords: Colorectal cancer; Genes; Signal transduction pathways
Defects in genomic organization have been linked to the tumor progression have been suggested (defects in the mitotic spindle leading to the formation of malignant tumors) more than one hundred years ago; however, still the research on cancer genetics ongoing with the aim of finding the novel genes as well as the signal transduction pathways that are associated with the abnormal genetic constitution acquired as a result of mutation (Liu et al., 2020; Forootan et al., 2019; Boveri, 1914). Colorectal cancers (CRCs) have long been known to develop through a cascade of events (i.e., the adenoma-carcinoma sequence) starting with the transformation of normal colonic epithelium to an adenomatous intermediate and then ultimately adenocarcinoma (Pino and Chung, 2010; Morson, 1974). Familial adenomatous polyposis (FAP) along with the syndromes not related to polyposis; for example, the Lynch syndrome among the hereditary non-polyposis colorectal cancer (HNPCC), have been reported to be the most frequent cause for the development of the onset of hereditary colorectal cancer (HCRC), which is actually a group of syndromes, accounting up to 10% among all cases of CRCs including the hereditary polyposis colorectal cancer (HPCC), so far reported (Medina Pabón and Babiker, 2020; Valle et al., 2019). Among the HNPCC, the Lynch syndrome (LS) is the most common, whereby the flat polyps within the right colon may trigger CRC (Medina Pabón and Babiker, 2020). On the contrary, in the case of HPCC-FAP, hundreds or thousands of precancerous adenomatous polyps form that, in turn, instigate the CRC (Medina Pabón and Babiker, 2020).
Akin to the model of an adenoma probably induced by the DNA mutation due to environmental agent(s) or more specifically by the carcinogenic agents whose continuous impact resulting in the progression into a carcinoma (through a series of molecular signal transduction pathways, like the chromosomal instability pathway or CIN, the microsatellite instability pathway, MSI and the serrated adenoma pathway, etc.), the history of knowledge on (1) The inherited susceptibilities of the colorectal adenomatous polyps undergoing the malignant transformation (formation of cancerous cells) and (2) The progressive research on the long-term risk of colorectal cancer is widely known effectively starting nearly 45 years ago continuing till date (Beggs and Hodgson, 2008; Hill et al., 1978; Muto et al., 1975).
Indeed, the multistep genetic model of colorectal carcinogenesis has been reported thirty years ago, which is still the accepted model for solid tumors (Fearon and Vogelstein, 1990). According to the model, (1) The Adenomatous Polyposis Coli (APC) tumor suppressor gene is first inactivated, followed by the activation of mutations of the Kirsten rat sarcoma viral oncogene homolog or KRAS (belongs to a class of oncogenes which, upon mutation, have the potential to transform normal cells into cancerous ones); (2) and the succeeding malignant transformation is propelled by the superfluous mutations in the transforming growth factor-β (TGF-β) signaling pathway (involved in cellular proliferation, differentiation, apoptosis, extracellular matrix remodeling, angiogenesis, inflammation, etc.); (3) The phosphatidyl-inositol 3-kinase (PIK3) signaling pathway boosts a major signaling pivot downstream of the epidermal growth factor receptor and serves a key role in the pathogenesis of several cancers), and (4) finally, the TP53 signaling pathways influence the cell cycle arrest, apoptosis, and angiogenesis (Pino and Chung, 2010; Fearon and Vogelstein, 1990). The projection of the clinical concept about colorectal cancer (CRC) is mainly based on the fact that in relation to the size of the colon and rectum, tumors are likely to be of larger size and eventually may pose a higher cancer rate as measured by repeated colonoscopy, sigmoidoscopy and polypectomy (Atkin et al., 1992). Deciphering the current state of knowledge on the genomics of colorectal cancer including (1) The genetic changes in polyps undergoing the malignant transformation as well as the possible epigenomic changes in colorectal polyps; (2) Mechanism by which the key tumor suppressor gene APC (adenomatous polyposis coli, whose product, the APC protein, plays a grave role in several cellular processes) is mutated (leading to adenomatous polyposis, FAP or the attenuated FAP, i.e., AFAP) provoking colorectal carcinogenesis have long been a great interest for cancer research (Beggs and Hodgson, 2008). Besides, a study on the application of the gene expression profiles of cancer types/subtypes to detect the prognostic genes, as well their clinical implementations, is also going on (Liu et al., 2020). Based on the previous literature, the current review shortly described the associated genes (as well as the mutation points) and the relevant signal transduction pathways involved in the formation of colorectal cancer.
Genes Involved in the Development of Colorectal Cancer
CRCs have been reported to develop along with long time achieving successive accretion of genetic alterations oppressing (1) The regulators of cell proliferation, (2) The cell cycle mechanisms, (3) Apoptosis, and (4) The DNA repair system (Boland and Goel, 2010). The genome-wide sequencing projected around 80 mutated genes per colorectal tumor; however, nearly 15 among them were pondered to be the major instigators of tumorigenesis (Pino and Chung, 2010; Leary et al., 2008; Wood et al., 2007). The inherited colorectal cancer syndromes known as the familial adenomatous polyposis or FAP (as stated earlier), which is caused by germline mutations in the APC gene resulting in the loss of the second, normal allele of the tumor suppressor APC gene; and Lynch Syndrome (also known as the hereditary nonpolyposis colorectal cancer or HNPCC) are indeed easily detectable along with a well understandable sequence of genetic mutations which serve as the underlying predisposing factors to instigate colorectal cancer (Beggs and Hodgson, 2008). The other inherited variants like the MutY human homolog (MYH) gene (encoding a protein product of a base excision repair enzyme which is involved in the repair of oxidative DNA damage) whose mutation has been reported to result in the heritable predisposition to colon and stomach cancer as well as to form the polyposis and hyperplastic polyposis syndrome (Beggs and Hodgson, 2008; Sampson et al., 2005).
The commonest form of hereditary non-polyposis colorectal cancer is the Lynch syndrome (also known as Hereditary colorectal cancer syndrome), resulted due to the mutation of the DNA mismatch repair (MMR) enzymes (which recognize and repair the flawed insertion, deletion, and mis-incorporation of the DNA bases during replication and recombination); i.e., (1) The mutation of hMSH2 gene (also known as the caretaker gene which encodes the DNA MMR protein, MSH2), (2) Human MutL homolog 1 or the hMLH1 gene (whose mutation may lead to the missense codons which in turn trim down the adeptness of MMR), (3) hPMS1 gene (whose mutation may result in the hereditary nonpolyposis colorectal cancer), (4) hPMS2 gene (also encoding the components of the DNA-mismatch repair complex) and (5) The human mutS homolog 6 or the hMSH6 gene which is also known as p160 (encodes the DNA MMR protein Msh6; and is the homologue of the human G/T binding protein, GTBP), is actually the early onset of colorectal cancer derived from colorectal endometrial, ovarian, gastric and urinary tract carcinoma (Lynch et al., 1988).
The risk of colorectal carcinogenesis in the untreated FAP patients appears to be is 100%, while among the attenuated APC variant (i.e., AAPC due to mutations in the 3’ and 5’ end of the APC gene accompanied with the mutation in the alternatively spliced exon 9) although the risk remains the same, fewer polyps can be generated in the late stage of life (Beggs and Hodgson, 2008). Interestingly, the FAP-like and AAPC-like phenotypes with the absence of germline mutation in the APC gene have been noticed to carry bi-allelic mutations of the MYH gene by 25% (Venesio et al., 2004). In the 1st–2nd decade of life, the Peutz-Jegher Syndrome (PJS) can be expressed which is characterized by the multiple hamartomatous polyps in the gastrointestinal (GI) tract (Juvenile polyposis syndrome) due to the germline mutation of the tumor suppressor gene STK11, encoding a serine-threonine kinase which is known to regulate the cell polarity, i.e., the cellular proliferation; and acts as a tumor suppressor (Beggs and Hodgson, 2008).
Another tumor suppressor, known as the PTEN gene, encoding the phosphatase and tensin homolog, has been reported to control the cell growth and proliferation, i.e., the regulation of cell cycle; preventing cells from growing and dividing too rapidly, and to prevent angiogenesis (a precarious step in cancer progression); and hence the somatic mutations of PTEN are likely to be associated with the colorectal malignancies as observed through the GI polyps (Beggs and Hodgson, 2008; Chow and Baker, 2006).
In course of colorectal cancer among the children, several important molecular aspects have been discovered: (1) The Juvenile polyposis syndrome (JPS), involving the mutations in the SMAD2 gene (whose product is known to mediate the signal of the TGF-β, thereby regulating the multiple cellular processes including cell proliferation, apoptosis and differentiation), (2) Mutation is the SMAD4 gene (encoding SMAD4, which is the central mediator of TGF-β signaling pathway), (3) Mutation in the bone morphogenetic protein receptor type 1A (BMPR1A) gene, whose product serves as the ligand for the TGF-β superfamily, and has a significant role in cell differentiation; and (4) Finally, the mutation in the ENG genes (encoding a component of the TGF-β receptor complex); and it is also to be noted that these events have been reported throughout the GI tract in a patient with the family history of JPS with multiple polyps developed before 20 years of age (Sweet et al., 2005). Besides, the increased frequency of chromosomal 1p allelic loss as well as the somatic BRAF and k-RAS2 mutations may lead to Hyperplastic polyposis syndrome (Beggs and Hodgson, 2008).
Pathways Involved in the Development of Colorectal Cancer
Tumor progression involves multiple genetic events accompanied by genomic instability and mutations (Forootan et al., 2019). As mentioned in the beginning, and has been modeled within Fig. 1, several major signal transduction pathways somatic genetic pathways in leading to sporadic colorectal cancer have been identified including the (1) Chromosomal instability (CIN) pathway, (2) Involvement of the tumor suppressor APC, (3) The K-ras (involved in the epidermal growth factor receptor pathway, i.e., the EGFR signaling pathway which regulates growth, survival, proliferation, and cellular differentiation) mutations, (4) Mutation within another tumor suppressor as well as the cell cycle regulator p53, (5) Mutations in the TGF-β pathway, the microsatellite instability pathway (MSI) leading to the defective DNA mismatch repair, (6) The serrated adenoma pathway (whereby the serrated polyps have been noticed to replace the traditional adenoma as the precursor lesion to the CRC), (7) Mutation of BRAF gene (encoding the B-Raf protein which is known to be involved transmitting signals inside cells that are engaged in directing the cell growth), and (8) The CpG island (genome regions containing a huge number of CpG dinucleotide repeats) methylation, i.e., CpG island methylator phenotype (CIMP) in colorectal cancers (due to the unusual methylation of multiple CpG islands including those in several tumor suppressor genes) have been well reported so far (Pino and Chung, 2010; Beggs and Hodgson, 2008). CpG (cytosine preceding guanine) islands are the regions (rich in CpG dinucleotides) within the genome which are common in the promoter sites; and several CpG dinucleotides, which remain methylated in normal cells, have been found to be unmethylated in case of the colorectal cancer cases (Nazemalhosseini Mojarad et al., 2013). The principal trend in the development of tumor depends on the attainment of genomic instability through the distinct pathways in colorectal cancer pathogenesis, mainly the CIN pathway, the MSI pathway, and the CpG island methylator phenotype (CIMP) pathways of which the most cases have been reported to arise through the CIN pathway, characterized by the widespread imbalances in the chromosome number (aneuploidy) and the loss of heterozygosity (LOH) [3]. Multiple pathways have also been reported in the progression of a tumor with a noticeable fraction of MSI colorectal cancers with the chromosomal abnormalities as well as the CIMP phenotype, and CIN-positive tumors have been found to be associated with high levels of MSI; CIMP-positive tumors were linked to the chromosomal abnormalities (Pino and Chung, 2010; Sinicrope et al., 2006).
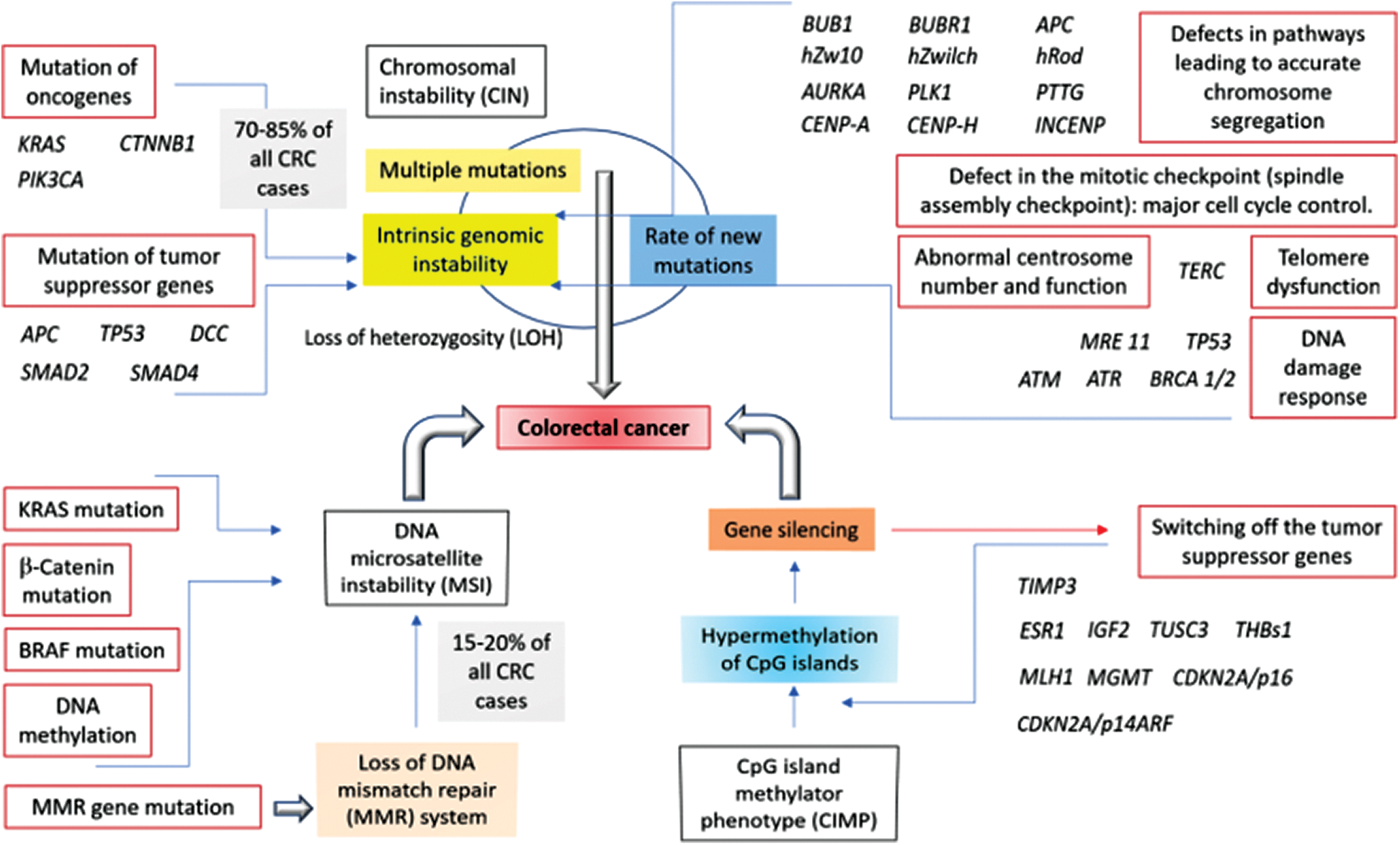
Figure 1: Involvement of the chromosomal instability (CIN), microsatellite instability (MSI), and CpG island methylator phenotype (CIMP) pathways in the development of colorectal cancer. The model consisting of the mutations or the overexpression of the relevant genes along with the associated pathways has been proposed on the basis of the published reports (Nazemalhosseini Mojarad et al., 2013; Boland and Goel, 2010; Pino and Chung, 2010).
Cross Talk between the Signal Transduction Pathways
A significant array of the molecular crosstalk has been reported within the signal transduction oncogenic pathways (Koveitypour et al., 2019). Significant interaction has been shown within the mitogen-activated protein kinase (MAPK) cascades downstream of the epidermal growth factor receptor (EGFR)/Ras signaling pathway (participating in the cellular processes including the cell growth and proliferation as well as in the survival of normal cells), PI3K/AKT pathway (required for cell growth and proliferation followed by cell differentiation and migration), the transforming growth factor-β (TGF-β) pathway (involved in cell proliferation and differentiation, cell migration; and the cellular apoptosis and adhesion), the Wnt/B-catenin signaling pathway (involved in cellular fate measurement, cell proliferation, cell migration and the asymmetric cell division), and the Notch pathway (necessary for cell differentiation, proliferation and apoptosis) in such ways whereby the Notch signaling regulated the Wnt pathway as well stimulated the EGFR pathway with a concomitant suppression of the TGF-β pathway (Jiang et al., 2019; Koveitypour et al., 2019; Ziouti et al., 2019).
As it is known from the previous literature, the development of colorectal cancer requires multiple genetic pathways along with the mutations in DNA mismatch repair genes, in APC and other genes, and due to the global genome hypermethylation. The present review incremented the existing knowledge on colorectal cancer-related genes and pathways through a simplified scheme which may further bring up an overall scenario of such cancer formation as well as the molecular mechanism. The present review did not discuss the specific pathological features either the diagnosis or the treatment aspects of the disease; however, the carcinogenesis with the concomitant procedure of tumor development has been presented for a better understanding of colorectal cancer genetics. The metabolic and biochemical networks between the genes and signal transduction pathways involved in colorectal cancer that have been simply elucidated in this review may aid to foster the research on the diagnostic and therapeutic methodologies for the efficient control and impediment of colorectal cancer.
Acknowledgement: Authors thank the groups whose literatures have been cited during writing this article.
Author Contribution: The authors confirm contribution to the paper as follows: Study conception and design: RN; data collection: AZ, SAIA; analysis and interpretation of results: ZN, RN; draft manuscript preparation: AZ. HJS, RN. All authors reviewed the results and approved the final version of the manuscript.
Funding Statement: The authors received no specific funding for this study file.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
Atkin WS, Morson BC, Cuzick J. (1992). Long-term risk of colorectal cancer after excision of rectosigmoid adenomas. New England Journal of Medicine 326: 658–662. DOI 10.1056/NEJM199203053261002. [Google Scholar] [CrossRef]
Beggs AD, Hodgson SV. (2008). The genomics of colorectal cancer: State of the art. Current Genomics 9: 1–10. DOI 10.2174/138920208783884865. [Google Scholar] [CrossRef]
Boland CR, Goel A. (2010). Microsatellite instability in colorectal cancer. Gastroenterology 138: 2073–2087.e3. DOI 10.1053/j.gastro.2009.12.064. [Google Scholar] [CrossRef]
Boveri T. (1914). Zur Frage der Entstehung Maligner Tumoren. Jena, Germany: Gustav Fisher, pp. 1–64. [Google Scholar]
Chow LM, Baker SJ. (2006). PTEN function in normal and neoplastic growth. Cancer Letters 241: 184–196. DOI 10.1016/j.canlet.2005.11.042. [Google Scholar] [CrossRef]
Fearon E, Vogelstein B. (1990). A genetic model for colorectal tumorigenesis. Cell 61: 759–767. DOI 10.1016/0092-8674(90)90186-I. [Google Scholar] [CrossRef]
Forootan F, Nasr Esfahani MH, Ghaedi K. (2019). Signaling pathways involved in colorectal cancer progression. Cell & Bioscience 2: 97. [Google Scholar]
Hill MJ, Morson BC, Bussey HJ. (1978). Aetiology of adenoma-carcinoma sequence in large bowel. Lancet 1: 245–247. DOI 10.1016/S0140-6736(78)90487-7. [Google Scholar] [CrossRef]
Jiang Z, Cao Q, Dai G, Wang J, Liu C, Lv L, Pan J. (2019). Celastrol inhibits colorectal cancer through TgF-β1/Smad signaling. OncoTargets & Therapy 12: 509–518. DOI 10.2147/OTT.S187817. [Google Scholar] [CrossRef]
Koveitypour Z, Panahi F, Vakilian M, Peymani M, Seyed Forootan F, Nasr Esfahani MH, Ghaedi K. (2019). Signaling pathways involved in colorectal cancer progression. Cell & Bioscience 2: 97. DOI 10.1186/s13578-019-0361-4. [Google Scholar] [CrossRef]
Leary R, Lin J, Cummins J, Boca S, Wood LD, Parsons DW, Jones S, Sjoblom T, Park BH, Parsons R, Willis J, Dawson D, Willson JKV, Nikolskaya T, Nikolsky Y, Kopelovich L, Papadopoulos N, Pennacchio LA, Wang TL, Markowitz SD, Parmigiani G, Kinzler KW, Vogelstein B, Velculescu VE. (2008). Integrated analysis of homozygous deletions, focal amplifications, and sequence alterations in breast and colorectal cancers. Proceedings of the National Academy of Sciences 105: 16224–16229. DOI 10.1073/pnas.0808041105. [Google Scholar] [CrossRef]
Liu H, Li H, Luo K, Sharma A, Sun X. (2020). Prognostic gene expression signature revealed the involvement of mutational pathways in cancer genome. Journal of Cancer 11: 4510–4520. DOI 10.7150/jca.40237. [Google Scholar] [CrossRef]
Lynch HT, Watson P, Lanspa SJ, Marcus J, Smyrk T, Fitzgibbons RJJr, Kriegler M, Lynch JF. (1988). Natural history of colorectal cancer in hereditary nonpolyposis colorectal cancer (Lynch syndromes I and II). Diseases of the Colon & Rectum 31: 439–444. DOI 10.1007/BF02552613. [Google Scholar] [CrossRef]
Morson B. (1974). The polyp-cancer sequence in the large bowel. Proceedings of the Royal Society of Medicine 67: 451–457. DOI 10.1177/00359157740676P115. [Google Scholar] [CrossRef]
Muto T, Bussey HJ, Morson BC. (1975). The evolution of cancer of the colon and rectum. Cancer 36: 2251–2270. DOI 10.1002/cncr.2820360944. [Google Scholar] [CrossRef]
Medina Pabón MA, Babiker HM. (2020). A review of hereditary colorectal cancers. in: StatPearls [Internet]. Treasure Island (FLStatPearls Publishing. https://www.ncbi.nlm.nih.gov/books/NBK538195/ [Google Scholar]
Nazemalhosseini Mojarad E, Kuppen PJ, Aghdaei HA, Zali MR. (2013). The CpG island methylator phenotype (CIMP) in colorectal cancer. Gastroenterology and Hepatology from Bed to Bench 6: 120–128. [Google Scholar]
Pino MS, Chung DC. (2010). The chromosomal instability pathway in colon cancer. Gastroenterology 138: 2059–2072. DOI 10.1053/j.gastro.2009.12.065. [Google Scholar] [CrossRef]
Sampson JR, Jones S, Dolwani S, Cheadle JP. (2005). MutYH (MYH) and colorectal cancer. Biochemical Society Transactions 33: 679–683. DOI 10.1042/BST0330679. [Google Scholar] [CrossRef]
Sinicrope F, Rego R, Halling K, Foster N, Sargent DJ, La Plant B, French AJ, Laurie JA, Goldberg RM, Thibodeau SN, Witzig TE. (2006). Prognostic impact of microsatellite instability and DNA ploidy in human colon carcinoma patients. Gastroenterology 131: 729–737. DOI 10.1053/j.gastro.2006.06.005. [Google Scholar] [CrossRef]
Sweet K, Willis J, Zhou XP, Gallione C Sawada T. (2005). Molecular classification of patients with unexplained hamartomatous and hyperplastic polyposis. JAMA 294: 2465–2473. DOI 10.1001/jama.294.19.2465. [Google Scholar] [CrossRef]
Valle L, Vilar E, Tavtigian SV, Stoffel EM. (2019). Genetic predisposition to colorectal cancer: Syndromes, genes, classification of genetic variants and implications for precision medicine. Journal of Pathology 247: 574–588. DOI 10.1002/path.5229. [Google Scholar] [CrossRef]
Venesio T, Molatore S, Cattaneo F, Arrigoni A, Risio M, Ranzani GN. (2004). High frequency of MYH gene mutations in a subset of patients with familial adenomatous polyposis. Gastroenterology 126: 1681–1685. DOI 10.1053/j.gastro.2004.02.022. [Google Scholar] [CrossRef]
Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, Silliman N, Szabo S, Dezso Z, Ustyanksky V, Nikolskaya T, Nikolsky Y, Karchin R, Wilson PA, Kaminker JS, Zhang Z, Croshaw R, Willis J, Dawson D, Shipitsin M, Willson JKV, Sukumar S, Polyak K, Park BH, Pethiyagoda CL, Pant PVK, Ballinger DG, Sparks AB, Hartigan J, Smith DR, Suh E, Papadopoulos N, Buckhaults P, Markowitz SD, Parmigiani G, Kinzler KW, Velculescu VE, Vogelstein B. (2007). The genomic landscapes of human breast and colorectal cancers. Science 318: 1108–1113. DOI 10.1126/science.1145720. [Google Scholar] [CrossRef]
Ziouti F, Ebert R, Rummler M, Krug M, Müller-Deubert S, Lüdemann M, Jakob F, Willie BM, Jundt F. (2019). NOTCH signaling is activated through mechanical strain in human bone marrow-derived mesenchymal stromal cells. Stem Cells International 2019: 5150634. DOI 10.1155/2019/5150634. [Google Scholar] [CrossRef]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |