DOI:10.32604/biocell.2021.014602
www.techscience.com/journal/biocell
| Biocell DOI:10.32604/biocell.2021.014602 | www.techscience.com/journal/biocell |
| Article |
Loss of the ubiquitin-conjugating enzyme Rad6B disturbs mitochondrial function and cellular homeostasis in mouse skin
Department of Anatomy and Histology, School of Basic Medical Sciences, Lanzhou University, Lanzhou, 730000, China
*Address correspondence to: Yanfeng Song, songyanfeng@lzu.edu.cn; Degui Wang, wangdegui@lzu.edu.cn
#These authors contributed equally to this work
Received: 12 October 2020; Accepted: 21 December 2020
Abstract: Various factors can induce cell degeneration by altering the phenotype and metabolism of cells. Mitochondria play an essential role in cellular homeostasis and function, rendering aging processes highly associated with mitochondrial function and status. Herein, we describe an aging-prone phenotype of murine skin cells caused by depletion of Rad6B (Ube2b), an E2 ubiquitin-conjugating enzyme. In this study, using Masson’s trichrome, we showed that loss of Rad6B causes physiological structure changes in mouse skin with age. In addition, a combination of western blotting experiments, transmission electron microscopy and employment of immunofluorescence staining revealed that depletion of Rad6B was characterized by an abnormal mitochondrial metabolic profile, including decreased mitochondrial biosynthesis and mitochondrial complex activities, increased ROS production and suppressed mitophagy resulting in metabolic disorders. Taken together, these results highlight an important relationship between Rad6B and skin senescence, suggesting that Rad6B plays an indispensable role in maintaining a healthy mitochondrial network and intracellular homeostasis.
Keywords: Rad6B; Skin; Mitochondria; Respiratory chain; Senescence
Abbreviations
| PGC-1α: | peroxisome proliferator-activated receptor-γ co-activator 1α |
| Nrf2: | nuclear factor (erythroid-derived 2)-like 2 protein |
| MFN1: | mitofusin 1 |
| MFN2: | mitofusin 2 |
| PVDF: | polyvinylidene difluoride |
| SDS-PAGE: | sodium dodecyl sulfate polyacrylamide gel electrophoresis |
| ATP: | adenosine triphosphate |
| OXPHOS: | oxidative phosphorylation |
| ROS: | reactive oxygen species |
| DAPI, 4’: | 6-diamidino-2-phenylindole |
| CCK-8: | Cell Counting Kit-8 |
| PMSF: | phenylmethylsulfonyl fluoride |
| PBS: | phosphate-buffered saline |
| FBS: | fetal bovine serum |
| RTCA: | Real-Time Cellular Analysis |
| RIPA: | radioimmunoprecipitation assay |
| BCA: | bicinchoninic acid |
| DCFH-DA, 2’: | 7’-dichlorodihydrofluorescein diacetate |
| 2-DG: | 2-deoxy-D-glucose |
| FCCP: | carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone |
| OCR: | oxygen consumption rate |
| ECAR: | extracellular acidification rate |
| ETC: | electron transport chain |
| DRP1: | dynamin-relatedprotein 1 |
| TFAM: | mitochondrial transcription factor A |
| LC3: | microtubule-associated protein 1 |
The skin, which is the self-regulating protective barrier organ. Its structure consists of epidermis, dermis, subcutaneous fat with adnexal structures, and its critical function is to maintain homeostasis of the body and ultimately organism survival (Slominski et al., 2012; Slominski et al., 2018). Various signaling pathways affect skin degeneration, and the regulators involved influence the metabolic and phenotypic changes that accompany the senescence status. Skin aging is affected by internal and external factors, which ultimately leads to cumulative changes in skin structure, function, and appearance. It is a complex biological process (Bocheva et al., 2019). The fundamental mechanisms of skin aging are varied, with recent researches illustrating that multiple pathways, including those regulating mitochondrial function, changes in gene expression, defects in cellular DNA repair, decreased antioxidant defense, telomere attrition, and ubiquitin-regulated proteolysis, are collectively involved in this process (Birch-Machin and Bowman, 2016; Bocheva et al., 2019; Bourdens et al., 2019; Marcheggiani et al., 2019).
One characteristic of cell aging is mitochondrial degeneration and dysfunction. Mitochondria are pivotal cellular organelles that produce adenosine triphosphate (ATP) through oxidative phosphorylation (OXPHOS) in their inner membranes, proceeding in parallel to glycolysis in the cytoplasm. While undergoing fusion and fission, mitochondria remain stable metabolic networks to support essential functions and promote cellular homeostasis (Abate et al., 2019). Mitochondria are also the major sources of reactive oxygen species (ROS), assigning a dual character to them with regard to cellular environmental homeostasis, which has profound implications for the occurrence of various diseases, such as neurodegenerative disorders, senescence, and cancer (Yan et al., 2013; Rimessi et al., 2016). It is reported that the function of skin barrier construction directly or indirectly depends on mitochondria, and mitochondria play an important role in different signal pathways and processes, for example, cell proliferation, hair follicle development, and epidermal differentiation. Furthermore, it is demonstrated that ubiquitous deficiency of mtDNA in mice, which leads to wrinkles and visual hair loss, as well as increased inflammatory responses and a number of dysfunctional hair follicles. In addition, mitochondrial dysfunction is considered to be a driving force of potential age-related human diseases (Slominski et al., 2017; Singh et al., 2018).
The ubiquitin-proteasome system is composed of three categories of enzymes that act in sequential order: Ubiquitin-activating (E1), ubiquitin-conjugating (E2), and ubiquitin ligase (E3) enzymes. Ubiquitin can be covalently attached to proteins through an enzymatic cascade by E1-3, consequently regulating protein activity or degradation through monoubiquitination or polyubiquitination (polyUb) pathways. Previous studies have confirmed that the ubiquitin-proteasome system is involved in the regulation of mitochondrial fission and fusion in response to oxidative stress and mitochondrial remodeling through the conjunction of E2 and E3 enzymes, with damaged mitochondria being separated from the mitochondrial network to prevent further cell degeneration (Norris et al., 2015). Studies have revealed that ubiquitination and autophagy systems cooperate to degrade damaged and dysfunctional mitochondria with polyUb chains via a specialized type of autophagy named mitophagy (Dikic, 2017; McWilliams and Muqit, 2017; Harper et al., 2018). A recent study reported that cells depleted of the ubiquitin-conjugating enzyme UBE2E3 tended to senescence in the absence of DNA damage and exhibited a distinct senescence-associated secretory phenotype due to the disruption of the mitochondrial network (Plafker et al., 2018).
Rad6B belongs to a group of E2 enzymes that is well known to participate in protein degradation. Humans possess two Rad6 genes, UBE2A and UBE2B, which are also known to encode Rad6A and Rad6B proteins, respectively, which exert largely overlapping functions (Game and Chernikova, 2009). However, the effects of Rad6B deficiency on mitochondria and the cellular events elicited are still unknown. In this study, we found that skin senescence caused by depletion of Rad6B with age is characterized by defective mitochondrial metabolic profiles as well as mitochondrial dysfunction and subsequent metabolic disorders. Our data provide evidence that Rad6B is essential for mitochondrial function and for maintaining normal tissue metabolism.
We used male wild-type (WT) and Rad6B knockout (KO) mice throughout the experiments and were obtained from COMM (MGI: 102944). The embryonic stem cells of Rad6B knockout C57BL/6 were generated from the knockout mouse project (KOMP) library (UC Davis), embryonic stem cells were implanted into the uterus of mice to breed transgenic mice (Guo et al., 2018; Guo et al., 2019). These two mice strains were divided into 1-, 3-, and 12-month-old groups. All animals were housed at the Lanzhou University Animal Center. The mice were kept in standard animal facilities and given free access to food and water, and efforts were made to alleviate the suffering of the mice.
Murine skin fibroblasts were isolated from dorsal skin tissues, and the 0.5 × 1 cm full-thickness dorsal skin tissues were obtained. Briefly, skin tissues were steeped with 75% alcohol for about 30 s and subsequently washed three times in phosphate-buffered saline (PBS) with antibiotics (penicillin and streptomycin). Afterwards, the skin tissues were cut into small pieces and incubated with Type I collagenase (BIOSHARP) at 37°C for 2–3 h. After being centrifuged and washed repeatedly, the skin fibroblasts isolated were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS) in an incubator at 37°C with 5% CO2.
Adenosine triphosphate (ATP) contents of mouse skin tissues were detected using a commercial ATP Content Assay Kit (Solarbio, BC0300). Briefly, skin tissues (0.1 g) were lysed with 1 mL extraction buffer and centrifuged at 8000 × g for 10 min, the supernatant was collected, and 500 mL of chloroform were added, after which the samples were centrifuged at 10000 × g for 3 min. The absorbance of the supernatant was measured immediately using the Automatic Enzyme Marker System (TECAN, SPARK, 1710002409) at 340 nm. Relative ATP contents were normalized against the control samples.
For this, the dorsal skins of the mice were fixed in 4% buffered formalin, embedded in paraffin, and sectioned into 5 μm thick slices. After dewaxing, the sections were stained with hematoxylin and eosin (H&E). For immunohistochemistry analyses, the paraffin sections were incubated with anti-TFAM (Abcam, ab272885), anti-PGC-1α (Proteintech, 66369-1-AP) primary antibodies, subsequently with biotin-conjugated, affinity-purified goat anti-rabbit IgG (H&L) and HRP-conjugated streptavidin at 37°C, with 3,3-diaminobenzidine serving as the chromogenic substrate. For Masson’s trichrome staining, the sections were used sequentially with hematoxylin and ferric oxide, ponceau, glacial acetic acid, phosphomolybdic acid, and aniline blue, and then the sections were mounted with neutral gum.
Skin tissues were ground and lysed with radioimmunoprecipitation assay (RIPA) lysis buffer (Solarbio, 20180929) containing 1% phenylmethylsulfonyl fluoride (PMSF, Solarbio, 2877K). After being centrifuged, the supernatant was collected, and the protein concentration was determined using the bicinchoninic acid (BCA) Protein Assay Kit (Solarbio, PC0020). Afterwards, the samples were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto polyvinylidene fluoride (PVDF) membranes. The membranes were incubated with primary antibodies for 8–16 h at 4°C and incubated with appropriate peroxidase-conjugated secondary antibodies for 1.5 h at 28°C, and the signals were visualized by enhanced chemiluminescence. The following primary antibodies were used in this study at the dilutions indicated: anti-Rad6B (Proteintech, 10733-1-AP, 1:700), anti-PGC-1α (Proteintech, 66369-1-AP, 1:1000), anti-Nrf2 (Immunoway, YT3189, 1:2000), anti-TFAM (Abcam, ab272885, 1:800), anti-Actin (Proteintech, 20536-1-AP, 1:5000), anti-ND1 (Proteintech, 19703-1-AP, 1:1000), anti-UQCRC2 (Proteintech, 14742-1-AP, 1:1000), anti-SDHB (Proteintech, 10620-1-AP, 1:20000), anti-COX4 (Proteintech, 11242-1-AP, 1:10000), anti-ATP5A (Proteintech, 14676-1-AP, 1:1000), anti-Mfn1 (Proteintech, 13798-1-AP, 1:700), anti-Mfn2 (Proteintech, 12186-1-AP, 1:4000), anti-Drp1 (Proteintech, 12957-1-AP, 1:1500), anti-p16 (Abcam, ab51243, 1:4000), anti-p21 (Abcam, ab109199, 1:1000), anti-P62 (Proteintech, 18420-1-AP, 1:2000), anti-LC3 (Proteintech, 14600-1-AP, 1:1000).
Measurements of oxygen consumption and extracellular acidification rates (OCR and ECAR, respectively) of primary skin fibroblasts
For OCR measurements, primary mouse skin fibroblasts were seeded on Seahorse cell culture plates at 2 × 104 cells/well and incubated for 10–16 h. Before the examination, the medium was exchanged for 500 µL of assay medium (pH 7.4), which was composed of 10 mM glucose, 1 mM pyruvate, and 2 mM glutamine in XF Base Medium. Inhibitors of electron transport chain (ETC) complexes were injected into different ports of the Seahorse cartridge, including oligomycin A (oligo, 1 µM), carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP, 1 µM), antimycin A (AA, 0.5 µM), rotenone (rot, 0.5 mM), after which the corresponding OCR values were measured with the XF24 Seahorse Biosciences Extracellular Flux Analyzer (Seahorse Bioscience, 102238-100). Five replicates were analyzed for each experimental group.
For ECAR measurements, primary skin fibroblasts were plated on Seahorse cell culture plates at 2 × 104 cells/well and incubated as described above. Experiments were performed in assay medium (pH 7.4) with 1 mM glutamine. Additional reagents required, i.e., glucose (10 mM), oligo (1 µM), and 2-deoxyglucose (2-DG, 50 mM), were injected into different ports of the Seahorse cartridge, after which the ECAR values were measured. Five replicates were performed for each experimental group.
Mitochondrial membrane potential and ROS measurements in primary skin fibroblasts
To evaluate intracellular ROS levels, primary cells were incubated with 10 μM 2’,7’-dichlorodihydrofluorescein diacetate (DCFH-DA, Solarbio, CA1410) for 20 min at 37°C, subsequently washed three times with serum-free medium, and imaged using an Olympus fluorescence microscope (BX53).
For mitochondrial membrane potential, primary skin fibroblasts were loaded with MitoTracker Red (Solarbio, M9940) in serum-free medium for 15 min at 37°C in the dark, washed three times with PBS, and counterstained with DAPI before being imaged by laser confocal microscopy (Leica, TCS-SP8).
Immunofluorescence staining of primary skin fibroblasts
Primary skin fibroblasts were fixed in 4% paraformaldehyde, subsequently washed twice with PBS, and permeabilized using 0.5% Triton X-100. After being blocked with 3% bovine serum albumin in PBS, the cells were incubated with the primary antibodies against Vimentin (Proteintech, 10366-1-AP, 1:2000), COX4, and ND1 at 4°C for 8–16 h. For detection, goat anti-rabbit IgG-FITC (Abcam, ab6717) and anti-rabbit IgG-Cy3 (Abcam, ab6939) secondary antibodies were used according to standard immunostaining procedures. The nuclei were counterstained with 4’,6-diamidino-2-phenylindole (DAPI).
Cell viability was measured using the Cell Counting Kit-8 (CCK-8, Dojindo). To that end, primary fibroblasts were plated at a density of 3 × 103 cells/well on 96-well plates and were incubated at 37°C for 24, 48, and 72 h, respectively, after which 100 µL of the assay reagent were added per well, and the plates were incubated for another 3 h. Absorbance was recorded at 450 nm using enzymatic activity as a marker, and relative cell viability was calculated.
Analysis of cell proliferation
The Real-Time Cellular Analysis System (ACEA Biosciences, xCELLigence RTCA S16) was used for measuring cell proliferation. After having determined the background impedance value of the assay plate (E-Plate), primary skin fibroblasts (3 × 103 cells/well) were added, and the assay plate was placed on the test bench and incubated for 72 h. Growth curves were then recorded and analyzed.
Beta-galactosidase (SA-β-gal) staining
Primary skin fibroblasts were seeded on 24-well plates at 2 × 104 cells/well and incubated for 48 h. Afterwards, the cells were prepared using the commercial Senescence β-gal Staining Kit (Beyotime) according to the manufacturer’s instruction and imaged using an Olympus fluorescence microscope.
Transmission electron microscopy
Primary skin fibroblast clusters were fixed with 2.5% glutaraldehyde in 0.1 M PBS (pH 7.4), washed three times with PBS, and stained with 1% osmium tetroxide. Subsequently, the samples were dehydrated and embedded, and ultrathin sections (70 nm thick) were obtained using an ultramicrotome. Then, the sections were stained with 2% uranyl acetate and lead citrate and viewed under a transmission electron microscope (TEM, HITACHI HT7700).
Total DNA was isolated from skin tissues using Wizard® SV Genomic DNA Purification System (Promega), and 1 µL DNA solution was added to PCR mixture for amplification. Subsequently, the 1% agarose gel was prepared, heated, and mixed, then poured into the gel mold to solidify for 30 min. Electrophoresis with 6 µL amplification product was added into each well (constant voltage 100 V, 40 min). After electrophoresis, the agarose gel was imaged with a multifunctional molecular imaging analysis system (Azure Biosystems, C500), and the genotype was identified. The primer sequences were used as following: Rad6B (5’- CCCCTGTGTATGTGTTATACCATCC-3’, 5’-TGAATTCAGGACTCAGGGATGCCTG-3’).
The data were analyzed using SPSS17.0 software. Figures were generated using Origin 8.5 software. Student’s t-test was used to assess differences between two groups. A P-value < 0.05 was considered statistically significant.
Rad6B KO induces phenotypic differences in the skin with age
The results of the genotyping of WT and Rad6B KO C57 mice are shown in Suppl. Fig. S1. H&E staining of the skin showed that its thickness was reduced in KO groups, especially in 1-month-old KO mice compared with the WT group (Figs. 1A–1B). Quantitative histomorphometry revealed that the number of hair follicles in anagen were increased in 1-month-old WT groups, and there was evidence of a raised number of hair follicles in telogen in 1-month-old KO groups compared with other groups. Moreover, the number of hair follicles in the catagen was enhanced in 3-, and 12-month-old groups, especially in the 12-month-old KO group (Figs. 1C–1E). Masson’s trichrome staining showed that the collagen fiber content was also decreased in KO groups but significantly reduced in the 12-month-old KO group (Figs. 1F–1G). Taken together, the phenotypic skin-related differences between WT and KO mice indicated that Rad6B KO might induce alterations in skin physiological structure with age.
Rad6B KO affects mitochondrial biogenesis of mouse skin cells
Mice with this genotype are known to feature decreased protein degradation and DNA damage repair capacities. In this study, the metabolism states of cells in the skin were determined. Immunohistochemical staining of samples from each group showed that cells positive for proliferator-activated receptor-γ co-activator 1α (PGC-1α), a regulator of mitochondrial synthesis, were less frequently detected in the skin of both 1- and 12-month-old KO mice, but there was no striking difference in the 3-month-old group (Figs. 2A, 2B). The number of mitochondrial transcription factor A (TFAM) positive cells in the skin was also decreased in both 1- and 12-month-old KO mice (Figs. 2C, 2D). Consistently, the expression of PGC-1α and TFAM proteins in skin tissues was decreased, especially in the 1- and 12-month-old KO groups (Figs. 2E–2G). Nrf2 is involved in the regulation of oxidative stress in the case of mitochondrial dysfunction (Sabouny et al., 2017; Hu et al., 2018). In this study, the expression of nuclear factor (erythroid-derived 2)-like 2 protein (Nrf2) was down-regulated in the KO group (Figs. 2E and 2H), which might have been indicative of different levels of ROS in WT and KO groups. These results suggested that Rad6B KO interfered with the metabolism of cells resident in mouse skin, and this phenotype was particularly prominent in both the 1- and 12-month-old groups.
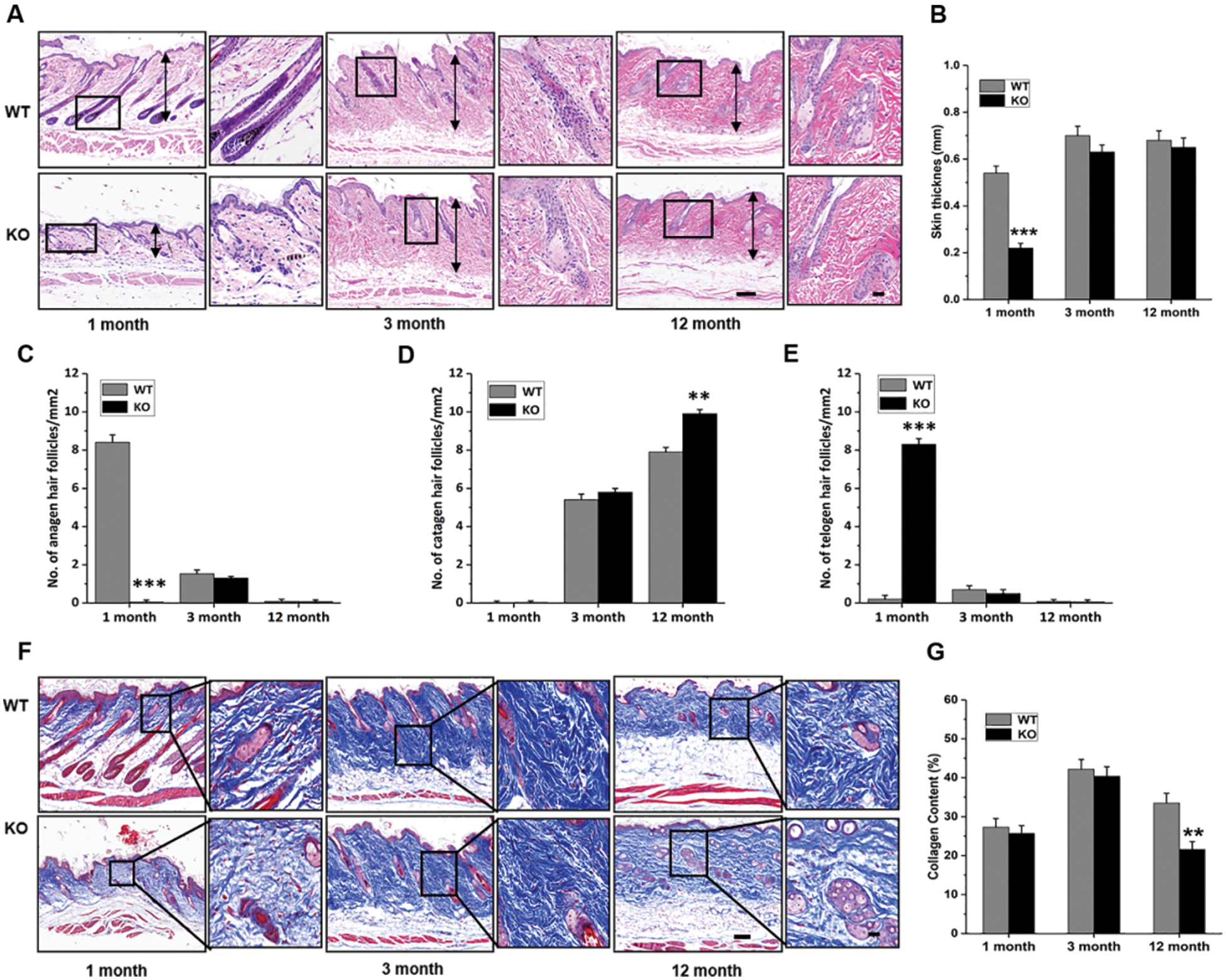
Figure 1: Rad6B knockout (KO) induces phenotypic differences in the skin.
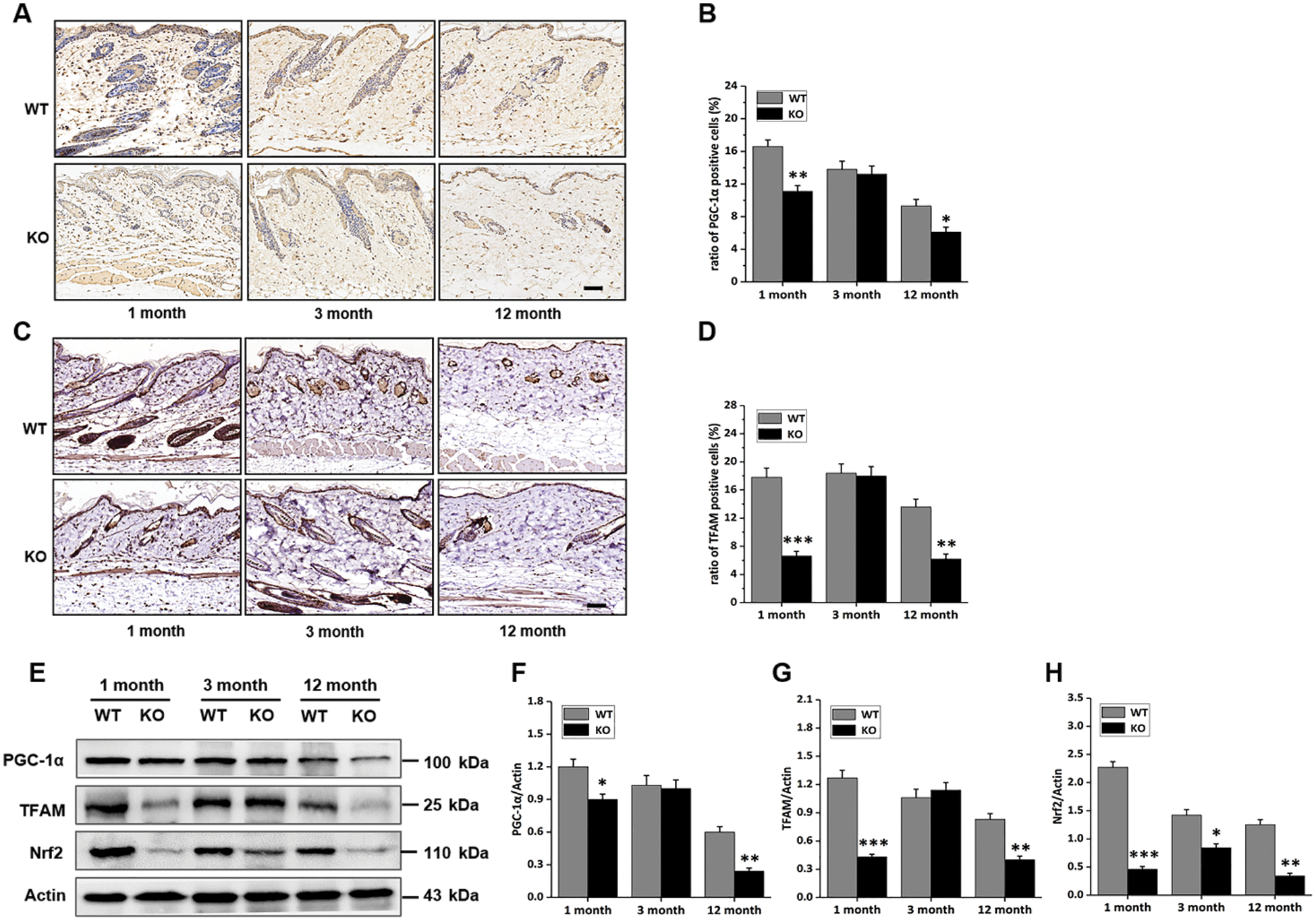
Figure 2: Rad6B knockout (KO) affects the mitochondrial biogenesis of mouse skin cells.
Changes in mitochondrial complex activities and dynamics in mouse skin following Rad6B loss
The cells of the skin are highly proliferative relative to those in other organs; therefore, the mitochondria are presumed to be highly active and of prime importance. To investigate whether mitochondrial function was changed due to Rad6B KO, the expression of five mitochondrial complex proteins was analyzed. As shown in Figs. 3A, 3B and 3E, loss of Rad6B caused evident alternations in ND1 and COX4 expression profiles, which correspond to complexes I and IV, respectively. Protein expression analyses revealed that ND1 was markedly increased in the 1-month-old KO group but decreased in the 12-month-old KO group. Interestingly, COX4 expression was depressed in mice of the 1-month-old KO group and enhanced in those of the 12-month-old KO group. However, expression of SDHB, URCRC2, and ATP5A, subunits of complexes II, III, and V, respectively, was unchanged in each group (Figs. 3C, 3D and 3F). Subsequently, we examined the ATP content of skin tissues in each group, and the results showed that the ATP content was increased in 1-month-old KO mice but diminished significantly in 12-month-old KO mice compared with WT mice (Fig. 3G). Evidence has emerged that mitochondrial fusion and fission play important roles in cell metabolism and growth (Abate et al., 2019). Therefore, in the present study, we further investigated mitochondrial fusion (mitofusin 1 [MFN1] and mitofusin 2 [MFN2]) and fission (dynamin-related protein 1 [DRP1]) proteins in mouse skins. As shown in Figs. 3H–3K, expression of MFN1, MFN2, and DRP1 was down-regulated in the 12-month-old KO group compared with the WT group, but no differences were observed between other age groups. Hence, we speculated that the whole mitochondrial mass and status in the skin were degenerating with age in the case of Rad6B KO mice. Taken together, these data suggested that Rad6B KO reduced the activity of mitochondrial complexes I and IV, thus impairing mitochondrial function. Moreover, mitochondrial dynamics were declining with old age.
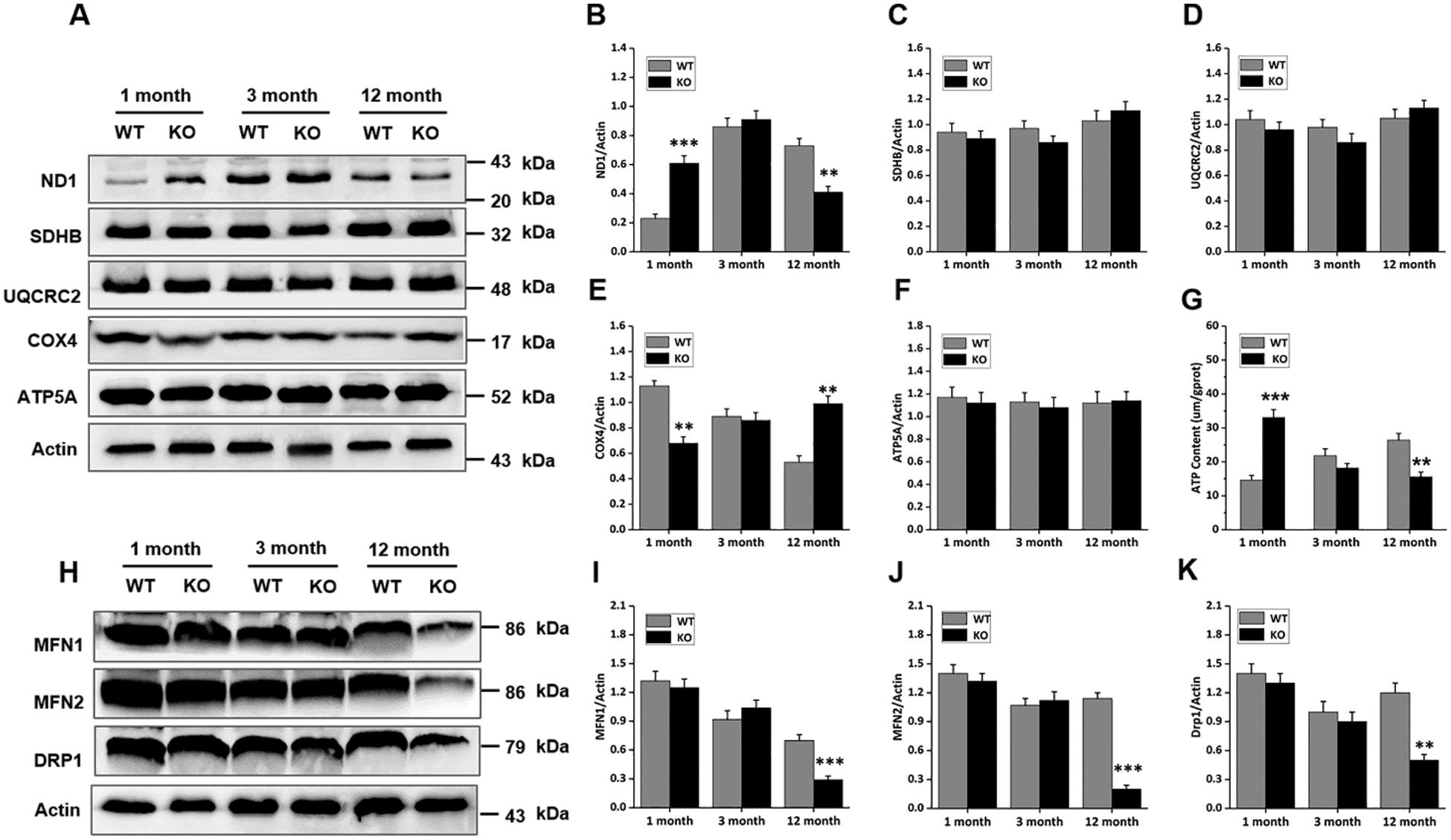
Figure 3: Changes in mitochondrial complex activities and dynamics in mouse skin following Rad6B loss.
Abnormal mitochondrial function and cell performance in primary skin fibroblasts lacking Rad6B
We extracted primary fibroblasts from mouse skins for the following experiments. First, the mitochondrial membrane potential of cells from each group was analyzed with MitoTracker Red (Figs. 4A, 4B), and the results revealed that the fluorescence intensity was markedly decreased in the 12-month-old KO group. As oxidative stress plays an important role in aging, we then clarified whether differences in ROS levels were related to Rad6B KO and mitochondrial complex dysfunction. As shown in Figs. 4C–4D, compared with WT cells, the ROS level was significantly increased in the KO groups. In addition, electron microscopic analysis showed that there was no striking difference in mitochondrial morphology in the 1- and 3-month-old groups; however, diminished mitochondrial cristae, elevated swollen and degenerated mitochondria were observed in cells of the 12-month-old KO group (Fig. 4E). To evaluate the viability and proliferation ability of the cells, CCK-8 and RTCA assays were performed, respectively. Rad6B KO resulted in a significant decrease in cell viability and proliferation ability over time (Figs. 4F–4I), and the impact on cell proliferation was particularly pronounced in the 12-month-old KO group. These results suggested that loss of Rad6B led to the dysfunction and destruction of the mitochondrial network in primary skin fibroblasts.
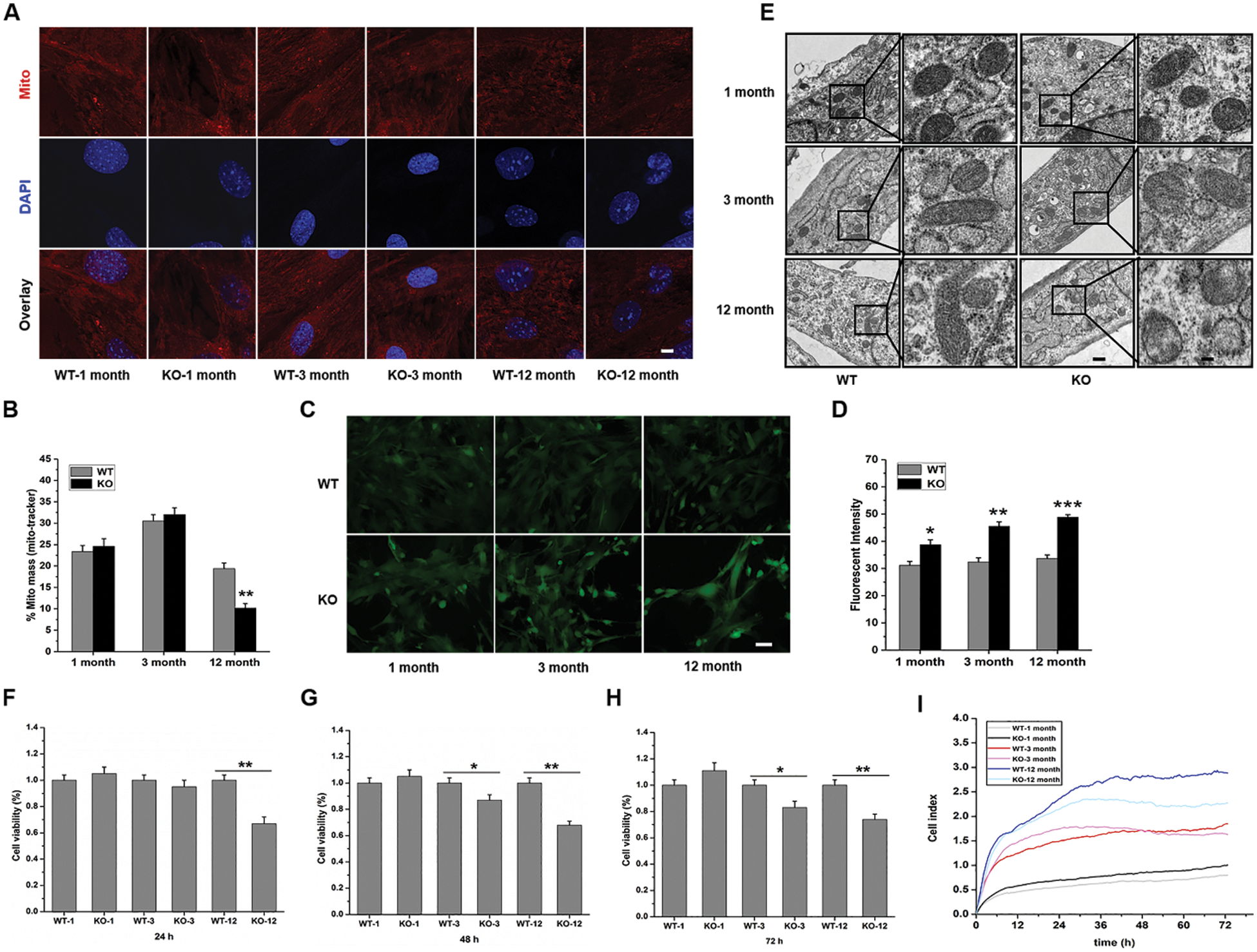
Figure 4: Rad6B knockout (KO) results in abnormal mitochondrial membrane potential and structure, reactive oxygen species (ROS) level production, and reduced cell viability and proliferation of primary skin fibroblasts.
Rad6B KO causes cell senescence
In fact, it has been shown that autophagy is a hallmark of the senescence process, and the mitophagy markers P62 and LC3-II/LC3-I also seem to be changed in the senescence process (Moreira et al., 2017; Bakula and Scheibye-Knudsen, 2020). Based on this, we decided to investigate the mitophagy proteins in primary skin fibroblasts. As shown in Figs. 5A–5C, the rate between microtubule-associated protein 1 light chain 3 (LC3)-II and LC3-I was significantly highest in the 12-month-old KO group. Consistently, expression of the Sequestosome 1 (P62) was also increased dramatically in the 12-month-old KO group compared had increased significantly in primary skin fibroblasts of the 12-month-old KO group (Figs. 5D and 5E). In line with this, p16 and p21 expression increased with age with the WT groups. To test whether Rad6B KO was associated with accelerated aging, we evaluated senescence-associated SA-β-gal activity, as well as the expression of p16 and p21 proteins, which are known biomarkers of aging, for each group. SA-β-gal activity and was hence highest in the 12-month-old KO group (Figs. 5F–5H). These data indicated that Rad6B KO caused the elimination of damaged mitochondria through mitophagy was disturbed, leading to accumulation of damaged mitochondria and accelerated aging.
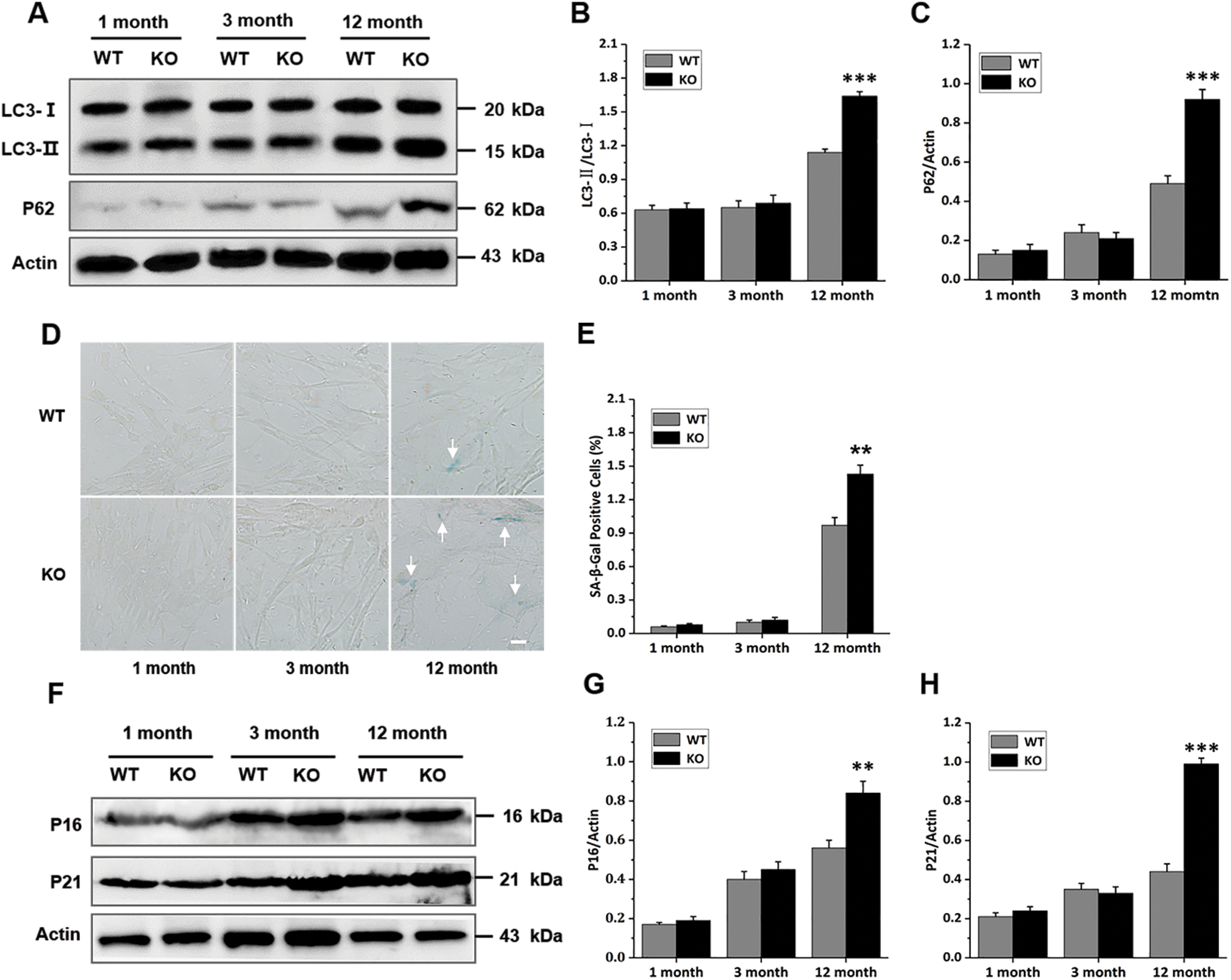
Figure 5: Rad6B knockout (KO) leads to accelerated cell aging.
Energy metabolism changes in primary skin fibroblasts with Rad6B KO
In a previous study, the ATP content was changed in skin tissues from Rad6B KO mice (Fig. 3G), allowing us to speculate that OXPHOS in mitochondria of skin cells was affected by Rad6B KO. Moreover, considering that glycolysis was probably coupled with this cellular response, the capacity of OXPHOS and glycolysis of primary skin fibroblasts was examined using the Seahorse XF24 Extracellular Flux Analyzer. As shown in Fig. 6, the mitochondrial OCR was elevated in the 1-month-old KO group, and the ECAR was reduced (Figs. 6A and 6D). Dramatically decreased sOCR and increased EACR values were observed in the 12-month-old KO group compared with the WT group (Figs. 6C and 6F). It was considered that glycolysis might have been redeployed to compensate for the reduction in mitochondrial OXPHOS in the case of Rad6B KO. The OCR and ECAR values measured in the 3-month-old groups showed no differences (Figs. 6B and 6E). Together, these results clearly indicated that Rad6B KO caused a switch from OXPHOS to glycolysis with age, which was probably realized by reprogramming the metabolic network, thus compensating the lack of ATP production in old primary skin fibroblasts featuring a KO of Rad6B.
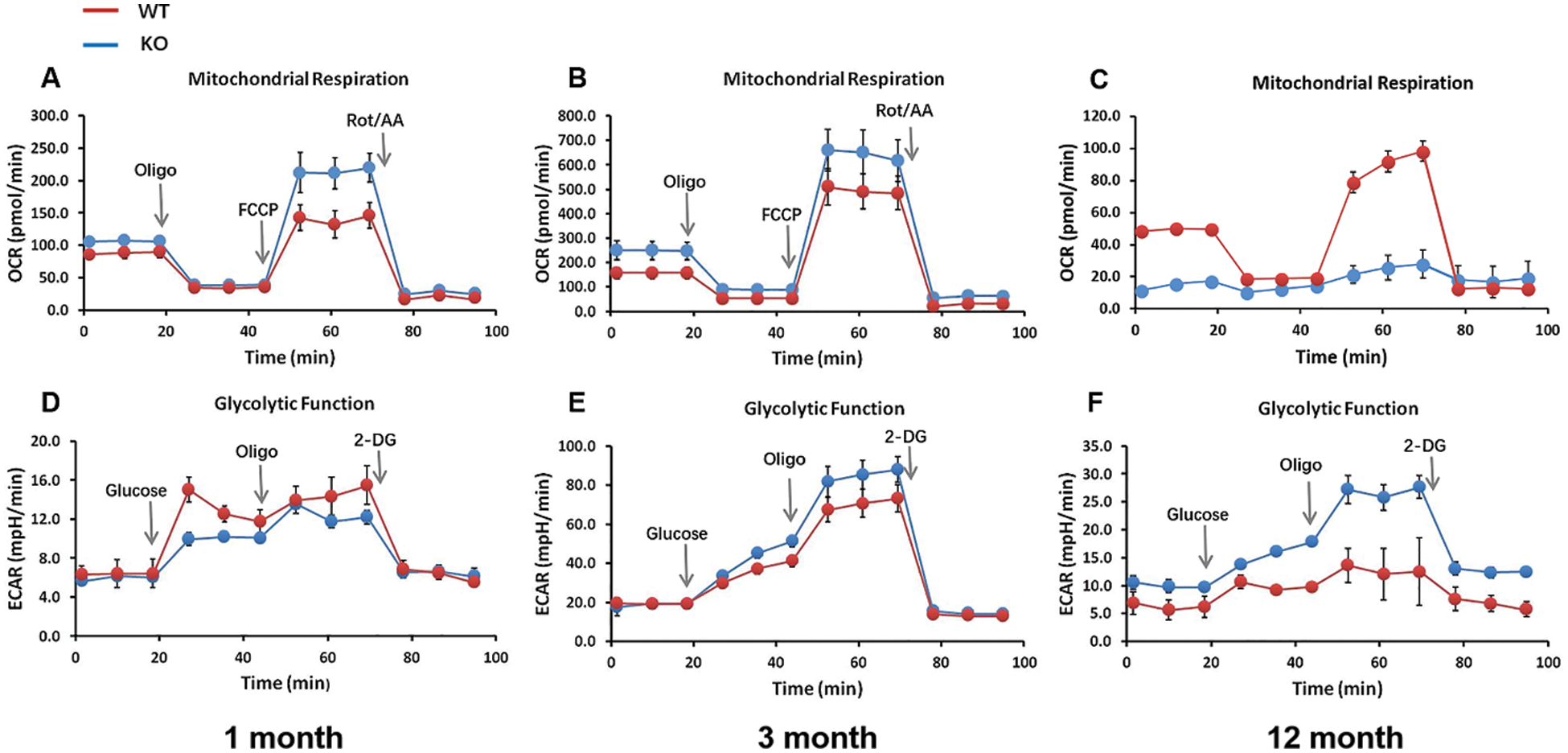
Figure 6: Changes in energy metabolism upon Rad6B knockout (KO) in primary skin fibroblasts.
In this study, an aging-prone phenotype was induced in mice by depletion of Rad6B. As a ubiquitin-conjugating enzyme, Rad6B could associate with the corresponding E3 protein to maintain the ubiquitination status of substrates. Efforts to illustrate the cellular functions of Rad6B have reported that it is required for cell repair and proliferation (Gerard et al., 2011; Liu et al., 2013). In tumor cells, over-expression of Rad6B leads to chromosomal instability, including multi-nucleation, abnormal mitosis, aneuploidy, and transformation (Shekhar et al., 2002). Blocking Rad6B by small molecule inhibitors induces mitochondrial dysfunction and PARP-1 hyper-activation in breast cancer cells, eventually leading to cell death (Sanders et al., 2013; Haynes et al., 2016). In addition, the homologous gene Rad6A has been confirmed to play an important role in mitigating oxidative stress, triggering mitophagy, and maintaining mitochondrial integrity. Accordingly, mutations of Rad6A cause mitochondrial depolarization and neuronal dysfunction in vivo (Haddad et al., 2013). Our previous research showed that KO of Rad6B in mice led to sperm degeneration and sterile males (Guo et al., 2018), and one explanation for this phenotype might be that spermatocytes lacking Rad6B had defective cell proliferation and underwent early senescence.
The histological features of aging skin were reduced of dermal thickness, reduced of adnexal structures and the function, thinning of subcutaneous fat, and reduced number of cutaneous microvessels and nerve endings, etc. (Zouboulis and Makrantonaki, 2011). And proper hair follicle morphogenesis in the skin tissue mainly depends on a functional mitochondrial (Kloepper et al., 2015). We observed that the number of hair follicles in the anagen in 1-month-old KO groups were a sharp decrease and a remarkable increase in the telogen (Figs. 1C and 1E). It showed that the hair follicles morphogenesis was severely retarded in 1-month-old KO mice, probably due to their mitochondrial biosynthesis was reduced. The number of hair follicles increased in the catagen in 12-month-old KO groups, and the lower end of the hair follicle retracted to the epithelium and the hair shaft atrophied significantly compared with the WT group (Fig. 1D), which possibly indicated the decline in mitochondrial function or the number of functional mitochondria. Because the specific transcription factors of PGC-1α and TFAM, which participate in the regulation of mitochondrial biosynthesis and oxidative stress (Wenz, 2013), were significantly decreased in the 1- and 12-month-old KO groups in this study (Fig. 2). Previous studies have indicated that decreased mitochondrial mass or function is often accompanied by depressed levels of PGC-1α and TFAM, which are commonly up-regulated with age and in neurodegenerative diseases, cancers, and some other diseases (Zhu et al., 2013; Gureev et al., 2019). We considered that Rad6B KO induced mitochondrial synthesis dysfunction with age, which contributed to the disordered metabolism. There are many E3 ubiquitin ligase enzymes, and the corresponding substrates could be covalently combined with Rad6B, but most of them are still unknown. The weakened capacity of the ubiquitin-proteasome system induced by Rad6B depletion resulted in aberrant substrate expression. However, these substrates might be direct regulators of mitochondrial functions or key pathways upstream of mitochondrial synthesis, which are still needed to be explored.
Mitochondria, which are considered intracellular powerhouses, are essential organelles whose major function is to produce ATP via OXPHOS in order to maintain basic physiological functions within cells (Willems et al., 2015). In eukaryotic cells, the respiratory chain exists in the inner membrane of the mitochondria, which performs a series of hydrogen and electron transfer reactions to produce ATP and ROS continually. We investigated the five complexes in the respiratory chain, with the results showing that Rad6B KO led to altered activities of the complexes I and IV in the 1- and 12-month-old groups (Figs. 3A, 3B and 3E). Consistently, ATP production in skin tissue of the KO group was elevated in the case of 1-month-old mice but decreased in 12-month-old mice (Fig. 3G), indicating that Rad6B KO induced mitochondrial respiratory chain dysfunction by altering the activities of complexes I and IV with age. Commonly, cells are able to adjust the imbalances of mitochondrial fission and fusion to ensure normal function and maintain intracellular homeostasis (Wenz, 2013). Furthermore, an aggravated cell status and mitochondrial dysfunction caused by affecting replication and transcription associated with mitochondrial synthesis and dynamics in both transgenic mice and patients (Niedernhofer et al., 2018). In our study, Rad6B KO caused remarkably reduced expression of the mitochondrial fusion and fission proteins MFN1, MFN2, and DRP1 in 12-month-old mice (Figs. 3H–3K). The results demonstrated that Rad6B KO caused abnormal mitochondrial dynamics, which led to mitochondrial dysfunction.
Subsequently, we extracted primary skin fibroblasts to elucidate the effect of Rad6B deficiency on skin cells. The results confirmed that Rad6B KO reduced cell viability and proliferation with age (Figs. 4F–4I). As byproducts of the mitochondrial respiratory chain, ROS exist as cell stabilization signals, but excessive ROS can induce serious oxidative stress damage to intracellular components and cause organelle dysfunction (Rimessi et al., 2016; Livingston et al., 2019; Oka et al., 2019). According to our results, Rad6B KO led to remarkably up-regulated intracellular ROS contents in all age groups analyzed (Figs. 4C–4D), which hinted at persistent damage in these cells, and clear degeneration of mitochondria at 12 months of age was verified by electron microscopic analysis (Fig. 4E). In addition, mitochondrial dysfunction and continuously elevated ROS levels are further believed to cause senescence in the cell (Hekimi et al., 2011; Wiley et al., 2016). Consistently, various reports on humans and different model organisms have suggested that impaired mitochondrial function might lead to aging and phenotypes of age-related diseases (Kauppila et al., 2017).
Some studies have shown that inhibiting mitophagy promotes senescence, and senescent cells show reduced levels of mitophagy(Wang et al., 2012; Kang et al., 2015). During autophagosome formation, LC3-I is processed into LC3-II, and LC3-II is degraded after autophagosome fusion with lysosomes. A low LC3-II /LC3-I ratio (LC3-II/LC3-I) indicates insufficient autophagosome formation, while a high LC3-II/LC3-I ratio demonstrates an insufficiency in autophagosome degradation or raised autophagosome formation. P62 is required for mitophagy, and inhibition of mitophagy increases p62 protein level (Sebori et al., 2018). LC3-II/LC3-I have been shown to increase in aged mice muscle in several studies(Sebastian et al., 2016). In our study, the LC3-II/LC3-I and P62 protein levels were significantly higher in the 12-month-old KO group than in the WT group (Figs. 5A–5C). These results demonstrated that mitophagy was suppressed in the 12-month-old KO mice, which resulted in a defective mitochondrial network that may lead to metabolic dysfunction in senescence. Our data also indicated that the senescence-associated phenotype had progressively developed in the 12-month-old KO group (Figs. 5D–5H), with SA-β-gal activity, as well as p16 and p21 expression is affected. The characterization of Rad6B KO-mediated senescence exhibited a senescence signature that might have been derived from mitochondrial dysfunction and degeneration. Based on this model, one explanation would be that KO of Rad6B alters mitochondrial homeostasis, including mitochondrial biosynthesis, complex activities, membrane potential, ROS production, and mitophagy.
Interestingly, senescent cells lacking mitochondria showed increased glycolysis. In some contexts, senescent cells must reprogram their metabolism to support their metabolic needs (Kaplon et al., 2013; Moreira et al., 2017). Indeed, metabolic flux analysis confirmed that Rad6B KO triggered a metabolic switch from OXPHOS to glycolysis in primary skin fibroblasts with age (Fig. 6), which we assume to be a metabolic reprogramming mechanism for maintaining intracellular energy supplies.
Research on human diseases has demonstrated that cells are highly dependent on mitochondrial function for their proliferation and survival. Thus, it is important to understand the mitochondrial status in ubiquitin-conjugating enzyme-deficient animals. In addition, more investigations need to be undertaken to explore the mechanisms by which Rad6B KO affects mitochondrial biosynthesis, as well as those underlying the effects on senescence. In conclusion, our work links disordered metabolism caused by Rad6B deficiency to mitochondrial dysfunction and cell senescence with age and reveals that Rad6B plays an indispensable role in maintaining a healthy mitochondrial network and intracellular homeostasis.
Acknowledgement: We would like to thank Editage (www.editage.com) for English language editing.
Availability of Data and Materials: ll data supporting the conclusions of this article are included in this manuscript.
Author Contributions: D. Wang and R. Shen designed the study. L. Yu and Y. Guo collated the data and carried out data analyses. L. Yu constructed the figures. D. Wang and Y. Song contributed to drafting the manuscript. Y. Guo and C. Li raised the animals. All authors have read and approved the final manuscript submitted.
Ethics Approval: All animals’ protocols were in accordance with the Ethics Committee for Laboratory Animals of Lanzhou University (JCYX-2017-0222).
Funding Statement: This work was funded by the National Natural Science Foundation of China (Grant Nos. 82071695 and 81772907 to DW and No. 82060535 to YT) and the Fundamental Research Funds for the Central Universities (Grant lzujbky-2017-k12 to DW).
Conflicts of Interest: The authors declare that they have no conflicts of interest.
Abate M, Festa A, Falco M, Lombardi A, Luce A, Grimald A, Zappavigna S, Sperlongano P, Irace C, Caraglia M, Misso G. (2019). Mitochondria as playmakers of apoptosis, autophagy and senescence. Seminars in Cell & Developmental Biology 98: 139–153. [Google Scholar]
Bakula D, Scheibye-Knudsen M. (2020). MitophAging: Mitophagy in aging and disease. Frontiers in Cell and Developmental Biology 8: 287. DOI 10.3389/fcell.2020.00239. [Google Scholar] [CrossRef]
Birch-Machin MA, Bowman A. (2016). Oxidative stress and ageing. British Journal of Dermatology 175: 26–29. DOI 10.1111/bjd.14906. [Google Scholar] [CrossRef]
Bocheva G, Slominski RM, Slominski AT. (2019). Neuroendocrine aspects of skin aging. International Journal of Molecular Sciences 20: 2798. DOI 10.3390/ijms20112798. [Google Scholar] [CrossRef]
Bourdens M, Jeanson Y, Taurand M, Juin N, Carriere A, Clement F, Casteilla L, Bulteau AL, Planat-Benard V. (2019). Short exposure to cold atmospheric plasma induces senescence in human skin fibroblasts and adipose mesenchymal stromal cells. Scientific Reports 9: 265sr6. DOI 10.1038/s41598-019-45191-2. [Google Scholar] [CrossRef]
Dikic I. (2017). Proteasomal and Autophagic Degradation Systems. Annual Review of Biochemistry 86: 193–224. DOI 10.1146/annurev-biochem-061516-044908. [Google Scholar] [CrossRef]
Game JC, Chernikova SB. (2009). The role of RAD6 in recombinational repair, checkpoints and meiosis via histone modification. DNA Repair 8: 470–482. DOI 10.1016/j.dnarep.2009.01.007. [Google Scholar] [CrossRef]
Gerard B, Tait L, Nangia-Makker P, Shekhar MP. (2014). Rad6B acts downstream of Wnt signaling to stabilize beta-catenin: implications for a novel Wnt/beta-catenin target. Journal of Molecular Signaling 6: 6. DOI 10.1186/1750-2187-6-6. [Google Scholar] [CrossRef]
Guo Y, Song Y, Guo Z, Hu M, Liu B, Duan H, Wang L, Yuan T, Wang D. (2018). Function of RAD6B and RNF8 in spermatogenesis. Cell Cycle 17: 162–173. DOI 10.1080/15384101.2017.1361066. [Google Scholar] [CrossRef]
Guo Z, Tian Y, Guo Y, Li B, Liu X, Xie K, Song Y, Wang D. (2019). RAD6B plays a critical role in neuronal DNA damage response to resist neurodegeneration. Frontiers in Cellular Neuroscience 13: 157. DOI 10.3389/fncel.2019.00392. [Google Scholar] [CrossRef]
Gureev AP, Shaforostova EA, Popov VN. (2019). Regulation of mitochondrial biogenesis as a way for active longevity: interaction between the Nrf2 and PGC-1α signaling pathways. Frontiers in Genetics 10: 64. DOI 10.3389/fgene.2019.00435. [Google Scholar] [CrossRef]
Haddad DM, Vilain S, Vos M, Esposito G, Matta S, Kalscheuer VM, Craessaerts K, Leyssen M, Nascimento RM, Vianna-Morgante AM, Strooper BD, Esch HV, Morais VA, Verstreken P. (2013). Mutations in the intellectual disability gene Ube2a cause neuronal dysfunction and impair parkin-dependent mitophagy. Molecular Cell 50: 831–843. DOI 10.1016/j.molcel.2013.04.012. [Google Scholar] [CrossRef]
Harper JW, Ordureau A, Heo JM. (2018). Building and decoding ubiquitin chains for mitophagy. Nature Reviews Molecular Cell Biology 19: 93–108. DOI 10.1038/nrm.2017.129. [Google Scholar] [CrossRef]
Haynes B, Zhang Y, Liu F, Li J, Petit S, Kothayer H, Bao X, Westwell AD, Mao G, Shekhar MPV. (2016). Gold nanoparticle conjugated Rad6 inhibitor induces cell death in triple negative breast cancer cells by inducing mitochondrial dysfunction and PARP-1 hyperactivation: Synthesis and characterization. Nanomedicine: Nanotechnology, Biology and Medicine 12: 745–757. DOI 10.1016/j.nano.2015.10.010. [Google Scholar] [CrossRef]
Hekimi S, Lapointe J, Wen Y. (2011). Taking a “good” look at free radicals in the aging process. Trends in Cell Biology 21: 569–576. DOI 10.1016/j.tcb.2011.06.008. [Google Scholar] [CrossRef]
Hu Q, Ren J, Li G, Wu J, Wu X, Wang G, Gu G, Ren H, Hong Z, Li J. (2018). The mitochondrially targeted antioxidant MitoQ protects the intestinal barrier by ameliorating mitochondrial DNA damage via the Nrf2/ARE signaling pathway. Cell Death & Disease 9: 1007. DOI 10.1038/s41419-018-0436-x. [Google Scholar] [CrossRef]
Kang C, Xu Q, Martin TD, Li MZ, Demaria M, Aron L, Lu T, Yankner BA, Campisi J, Elledge SJ. (2015). The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 349: 6225. DOI 10.1126/science.aaa5612. [Google Scholar] [CrossRef]
Kaplon J, Zheng L, Meissl K, Chaneton B, Selivanov VA, Mackay G, van der Burg SHV, Verdegaal EM, Cascante M, Shlomi T, Gottlieb E, Peeper DS. (2013). A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature 498: 109–112. DOI 10.1038/nature12154. [Google Scholar] [CrossRef]
Kauppila TES, Kauppila JHK, Larsson NG. (2017). Mammalian mitochondria and aging: An update. Cell Metabolism 25: 57–71. DOI 10.1016/j.cmet.2016.09.017. [Google Scholar] [CrossRef]
Kloepper JE, Baris OR, Reuter K, Kobayashi K, Weiland D, Vidali S, Tobin DJ, Niemann C, Wiesner RJ, Paus R. (2015). Mitochondrial function in murine skin epithelium is crucial for hair follicle morphogenesis and epithelial–mesenchymal interactions. Journal of Investigative Dermatology 135: 679–689. DOI 10.1038/jid.2014.475. [Google Scholar] [CrossRef]
Liu C, Wang D, Wu J, Keller J, Ma T, Yu X. (2013). RNF168 forms a functional complex with RAD6 during the DNA damage response. Journal of Cell Science 126: 2042–2051. DOI 10.1242/jcs.122945. [Google Scholar] [CrossRef]
Livingston MJ, Wang JH, Zhou JL, Wu GY, Ganley IG, Hill JA, Yin XM, Dong Z. (2019). Clearance of damaged mitochondria via mitophagy is important to the protective effect of ischemic preconditioning in kidneys. Autophagy 15: 2142–2162. DOI 10.1080/15548627.2019.1615822. [Google Scholar] [CrossRef]
Marcheggiani F, Cirilli I, Orlando P, Silvestri S, Vogelsang A, Knott A, Blatt T, Weise JM, Tiano L. (2019). Modulation of Coenzyme Q10 content and oxidative status in human dermal fibroblasts using HMG-CoA reductase inhibitor over a broad range of concentrations. From mitohormesis to mitochondrial dysfunction and accelerated aging. Aging 11: 2565–2582. [Google Scholar]
McWilliams TG, Muqit MM. (2017). PINK1 and Parkin: Emerging themes in mitochondrial homeostasis. Current Opinion in Cell Biology 45: 83–91. DOI 10.1016/j.ceb.2017.03.013. [Google Scholar] [CrossRef]
Moreira OC, Estebanez B, Martinez-Florez S, de Paz JA, Cuevas MJ, Gonzalez-Gallego J. (2017). Mitochondrial function and mitophagy in the elderly: Effects of exercise. Oxidative Medicine and Cellular Longevity 2017: 1–13. DOI 10.1155/2017/2012798. [Google Scholar] [CrossRef]
Niedernhofer LJ, Gurkar AU, Wang Y, Vijg J, Hoeijmakers JHJ, Robbins PD. (2018). Nuclear genomic instability and aging. Annual Review of Biochemistry 87: 295–322. DOI 10.1146/annurev-biochem-062917-012239. [Google Scholar] [CrossRef]
Norris KL, Hao R, Chen LF, Lai CH, Kapur M, Shaughnessy PJ, Chou D, Yan J, Taylor JP, Engelender S, West AE, Lim KL, Yao TP. (2015). Convergence of Parkin, PINK1, and α-Synuclein on stress-induced mitochondrial morphological remodeling. Journal of Biological Chemistry 290: 13862–13874. DOI 10.1074/jbc.M114.634063. [Google Scholar] [CrossRef]
Oka S, Hayashi M, Taguchi K, Hidaka M, Tsuzuki T, Sekiguchi M. (2019). ROS control in human iPS cells reveals early events in spontaneous carcinogenesis. Carcinogenesis 41: 36–43. [Google Scholar]
Plafker KS, Zyla K, Berry W, Plafker SM. (2018). Loss of the ubiquitin conjugating enzyme UBE2E3 induces cellular senescence. Redox Biology 17: 411–422. DOI 10.1016/j.redox.2018.05.008. [Google Scholar] [CrossRef]
Rimessi A, Previati M, Nigro F, Wieckowski MR, Pinton P. (2016). Mitochondrial reactive oxygen species and inflammation: Molecular mechanisms, diseases and promising therapies. International Journal of Biochemistry & Cell Biology 81: 281–293. DOI 10.1016/j.biocel.2016.06.015. [Google Scholar] [CrossRef]
Sabouny R, Fraunberger E, Geoffrion Mèle, Ng ACH, Baird SD, Screaton RA, Milne R, McBride HM, Shutt TE. (2017). The Keap12013;Nrf2 stress response pathway promotes mitochondrial hyperfusion through degradation of the mitochondrial fission protein drp1. Antioxidants & Redox Signaling 27: 1447–1459. DOI 10.1089/ars.2016.6855. [Google Scholar] [CrossRef]
Sanders MA, Brahemi G, Nangia-Makker P, Balan V, Morelli M, Kothayer H, Westwell AD, Shekhar MPV. (2013). Novel inhibitors of Rad6 ubiquitin conjugating enzyme: design, synthesis, identification, and functional characterization. Molecular Cancer Therapeutics 12: 373–383. DOI 10.1158/1535-7163.MCT-12-0793. [Google Scholar] [CrossRef]
Sebastian D, Sorianello E, Segales J, Irazoki A, Ruiz-Bonilla V, Sala D, Planet E, Berenguer-Llergo A, Munoz JP, Sanchez-Feutrie M, Plana N, Hernandez-Alvarez MI, Serrano AL, Palacin M, Zorzano A. (2016). Mfn2 deficiency links age-related sarcopenia and impaired autophagy to activation of an adaptive mitophagy pathway. EMBO Journal 35: 1677–1693. DOI 10.15252/embj.201593084. [Google Scholar] [CrossRef]
Sebori R, Kuno A, Hosoda R, Hayashi T, Horio Y. (2018). Resveratrol decreases oxidative stress by restoring mitophagy and improves the pathophysiology of dystrophin-deficient mdx mice. Oxidative Medicine and Cellular Longevity 2018: 1–13. DOI 10.1155/2018/9179270. [Google Scholar] [CrossRef]
Shekhar MP, Lyakhovich A, Visscher DW, Heng H, Kondrat N. (2002). Rad6 overexpression induces multinucleation, centrosome amplification, abnormal mitosis, aneuploidy, and transformation. Cancer Research 62: 2115–2124. [Google Scholar]
Singh B, Schoeb TR, Bajpai P, Slominski A, Singh KK. (2018). Reversing wrinkled skin and hair loss in mice by restoring mitochondrial function. Cell Death & Disease 9: 6. DOI 10.1038/s41419-018-0765-9. [Google Scholar] [CrossRef]
Slominski AT, Zmijewski MA, Plonka PM, Szaflarski JP, Paus R. (2018). How UV light touches the brain and endocrine system through skin, and why. Endocrinology 159: 1992–2007. DOI 10.1210/en.2017-03230. [Google Scholar] [CrossRef]
Slominski AT, Zmijewski MA, Semak I, Kim TK, Janjetovic Z, Slominski RM, Zmijewski JW. (2017). Melatonin, mitochondria, and the skin. Cellular and Molecular Life Sciences 74: 3913–3925. DOI 10.1007/s00018-017-2617-7. [Google Scholar] [CrossRef]
Slominski AT, Zmijewski MA, Skobowiat C, Zbytek B, Slominski RM, Steketee JD. (2012). Sensing the environment: regulation of local and global homeostasis by the skin's neuroendocrine system. Advances in Anatomy Embryology and Cell Biology 212: 1–115. [Google Scholar]
Wang Y, Wang XD, Lapi E, Sullivan A, Jia W, He YW, Ratnayaka I, Zhong S, Goldin RD, Goemans CG, Tolkovsky AM, Lu X. (2012). Autophagic activity dictates the cellular response to oncogenic RAS. Proceedings of the National Academy of Sciences of the United States of America 109: 13325–13330. DOI 10.1073/pnas.1120193109. [Google Scholar] [CrossRef]
Wenz T. (2013). Regulation of mitochondrial biogenesis and PGC-1alpha under cellular stress. Mitochondrion 13: 134–142. DOI 10.1016/j.mito.2013.01.006. [Google Scholar] [CrossRef]
Wiley CD, Velarde MC, Lecot P, Liu S, Sarnoski EA, Freund A, Shirakawa K, Lim HW, Davis SS, Ramanathan A, Gerencser AA, Verdin E, Campisi J. (2016). Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metabolism 23: 303–314. DOI 10.1016/j.cmet.2015.11.011. [Google Scholar] [CrossRef]
Willems PH, Rossignol R, Dieteren CE, Murphy MP, Koopman WJ. (2015). Redox homeostasis and mitochondrial dynamics. Cell Metabolism 22: 207–218. DOI 10.1016/j.cmet.2015.06.006. [Google Scholar] [CrossRef]
Yan MH, Wang X, Zhu X. (2013). Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease. Free Radical Biology and Medicine 62: 90–101. DOI 10.1016/j.freeradbiomed.2012.11.014. [Google Scholar] [CrossRef]
Zhu J, Wang KZ, Chu CT. (2014). After the banquet: Mitochondrial biogenesis, mitophagy, and cell survival. Autophagy 9: 1663–1676. DOI 10.4161/auto.24135. [Google Scholar] [CrossRef]
Zouboulis CC, Makrantonaki E. (2011). Clinical aspects and molecular diagnostics of skin aging. Clinics in Dermatology 29: 3–14. DOI 10.1016/j.clindermatol.2010.07.001. [Google Scholar] [CrossRef]
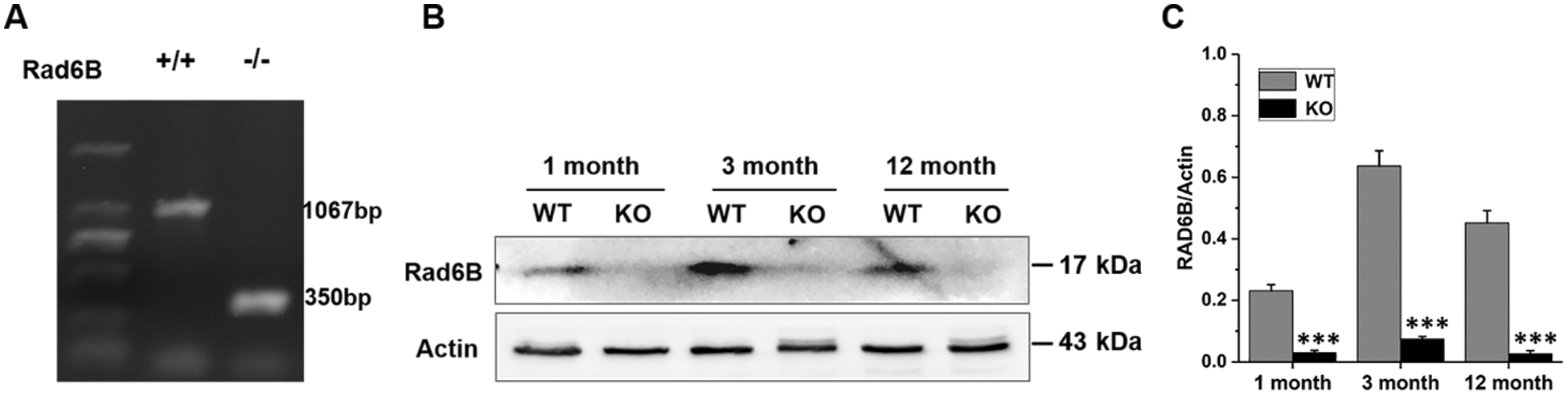
SUPPLEMENTARY FIGURE S1: Genotyping of wild-type (WT) and Rad6B knockout (KO) of C57BL/6 mice.
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |