DOI:10.32604/biocell.2021.015586
www.techscience.com/journal/biocell
| BIOCELL DOI:10.32604/biocell.2021.015586 | www.techscience.com/journal/biocell |
| Article |
Long intergenic noncoding RNAs differentially expressed in Staphylococcus aureus-induced inflammation in bovine mammary epithelial cells
1College of Animal Science and Technology, Yangzhou University, Yangzhou, 225009, China
2Joint International Research Laboratory of Agriculture and Agri-Product Safety, Ministry of Education, Yangzhou University, Yangzhou, 225009, China
3Hubei Blue Valley Microbial Technology Co., Ltd., Yichang, 44300, China
*Address correspondence to: Yongjiang Mao, cattle@yzu.edu.cn; Zhangping Yang, yzp@yzu.edu.cn.
Received: 29 December 2020; Accepted: 01 March 2021
#These authors contributed equally to this work
Abstract: Cow mastitis is the most common disease that affects the dairy farming industry and causes serious harm to dairy cows and humans, and Staphylococcus (S.) aureus is one of the main pathogens that cause mastitis in dairy cows. In this study, a mastitis model was established through the infection of bovine mammary epithelial cells (BMECs) with S. aureus (bacterial concentration of 1 × 109/mL), and these cells and a blank group (untreated) were analyzed by flow cytometry (10000 cells, 200 cells collected per second), hematoxylin and eosin (H&E) staining and immunohistochemistry. In addition, the lncRNAs (long non-coding RNAs) in the normal and S. aureus-infected BMEC group were screened by second-generation sequencing. Flow cytometry, H&E staining, and immunohistochemistry assays were performed to verify the successful construction of an S. aureus infection model in BMECs. A close relationship was found between the differential expression of lncRNAs and S. aureus mastitis. The total original sequencing reads were 627.13 M, and the average reads from each sample were approximately 104.52 M. After removing the unwanted reads, the total clean reads were 606.43 M, and the average reads from each sample were approximately 101.07 M. After S. aureus infection, 30 lncRNAs were differentially expressed, and these included 21 upregulated and nine down-regulated lncRNAs. This research will not only expand our understanding of the lncRNA map in dairy cows but also help us hypothesize the function of lncRNAs in the genome and identify novel molecular markers of mastitis.
Keywords: Mastitis; Staphylococcus aureus; Bovine mammary epithelial cells; lncRNA
Mastitis is a mammary gland disease associated with a high prevalence and high economic loss that not only affects the production and quality of quality but also endangers human health (De Vries et al., 2018; Fujimoto et al., 2020). Pathogenic microorganism infection is the main cause of cow mastitis, and Staphylococcus (S.) aureus is one of the most important pathogens causing mastitis in dairy cows (Monistero et al., 2020). S. aureus is sensitive to some antibiotics (amoxicillin and cephalosporin) but easily develops drug resistance (Bandyopadhyay et al., 2015; Hasanian et al., 2020). In addition, antibiotic residues in milk are associated with substantial food safety concerns (Tanhaeian et al., 2020; Zorraquino et al., 2011) related to human health (Pitkala et al., 2004). Therefore, the study of using nonantibiotics for the treatment of mastitis has become a focus of research regarding the prevention and treatment of mastitis in recent years. Animal disease models are mainly used in experimental physiology, pathology, and therapeutics (including new drug screening) (Freyssin et al., 2020). The development of mastitis is very complex, and the use of dairy cows as an experimental model to further explore the pathogenesis of this disease will result in the relatively slow development of a treatment for mastitis (Chen et al., 2018). Through indirect studies using a cell model, we can experimentally change factors that are impossible or difficult to manipulate under natural conditions, thus observe the experimental results of this manipulation more accurately and subsequently perform a comparative study with mastitis disease. This approach will be helpful for obtaining an understanding of the occurrence and development of mastitis using a more convenient and effective approach and for studying prevention and control measures. The purpose of this study was to construct a pathological model of BMECs infected with S. aureus and perform lncRNA screening using this S. aureus-infected model. A few studies have investigated lncRNAs in BMECs, but the analysis of lncRNAs in S. aureus models of BMECs has not been reported. Therefore, studying the expression profile of lncRNAs in normal and infected BMECs will be of great significance for identifying molecular markers of mastitis (Ozdemir and Altun, 2020; Wang et al., 2019b; Wang et al., 2020).
A number of studies have investigated lncRNAs. In 2012, Qu et al. identified 12614 intergenic lncRNAs and 9337 intragenic ncRNAs in expressed sequence tags (ESTs) in cattle (Qu and Adelson, 2012). Huang et al. (2012) predicted 449 unknown lncRNAs with lengths greater than 200 bp and multiple exons distributed in 405 intergenic regions in the EST database of cows (Huang et al., 2012). In 2013, Weikard et al. used next-generation sequencing technology to identify intergenic lncRNAs in cattle skin and found 4848 lncRNAs, most of which were intergenic lncRNAs. In 2014, Weikard et al. used the same technology to identify intergenic lncRNAs in cattle muscle and found 584 intergenic lncRNAs (Weikard et al., 2013). Koufariotis et al. identified lncRNAs in 18 tissues of cattle and compared these with those of humans and mice (Koufariotis et al., 2015). Cow mastitis is a very complex disease due to its multiple disease-causing factors. S. aureus-induced mastitis, which is one of the main research interests of our lab, is a complicated and regulated process involving lncRNA regulation (Ma et al., 2019a; Wang et al., 2019b; Wang et al., 2020).
Previous studies have shown that lncRNAs (>200 nt) are involved in the regulation of gene expression during the disease process (Li et al., 2021; Ma et al., 2019b). Although these were originally considered transcripts in the “junk DNA” region of the genome, lncRNAs are becoming increasingly known as key regulators in various life fields, such as the biological processes of development, differentiation, and disease (Sebastian-Delacruz et al., 2021). Studies have shown that lncRNAs regulate the physiology of cows at multiple levels, but the research on the roles of lncRNAs in the regulation of cow pathology is very limited. Our objectives were (1) to screen lncRNAs, (2) to identify intergenic lncRNAs that are differentially expressed in S. aureus-induced inflammation in BMECs compared with noninfected cells, and (3) to hypothesize the function of lncRNAs in the genome and identify novel molecular markers of mastitis. This study will not only improve our understanding of the function of the mammary gland and the molecular mechanism of mastitis at the molecular level of RNA but also provide new methods and ideas for the prevention and treatment of mastitis.
The mammary gland tissue was collected via an operation. Three dairy cows at the peak of lactation were selected for mammary gland biopsy (Chen et al., 2019b). After washing with phosphate-buffered saline (PBS), fat and connective tissues were removed. BMECs were separated using the tissue block method, purified by differential digestion, and cryopreserved after subculturing (Chen et al., 2019b). The laboratory has established culture technology for BMECs (Chen et al., 2019a). The basic medium used for primary mammary epithelial cells of dairy cattle is composed of Dulbecco’s modified Eagle’s medium (DMEM)/F12 (Sigma, China) and 10% fetal bovine serum (Sigma, China) supplemented with a variety of cytokines (5 μg/mL bovine insulin, 10 kU/L penicillin/streptomycin). The thawed mammary epithelial cells were cultured at 37°C and 5% CO2, and the medium was changed once every 48 h. The BMECs were digested with 0.25% trypsin for passage, and the growth of the cells was observed using an inverted microscope (LEICA, DMI3000B) (Chen et al., 2019b).
Construction of the S. aureus model
S. aureus was cultured and thawed one day before use. Single colonies were selected, placed on LB liquid culture medium, and incubated on a shaker for 4 h, which resulted in a bacterial concentration of 1 × 109/mL. One milliliter of suspension was centrifuged at 10000 rpm for 10 min. The supernatant was resuspended in the same amount of DMEM (Sigma, China). The cells were treated at a multiplicity of infection (MOI) of 100:1 for 3 h, the supernatant was aspirated, and the cells were washed with PBS (Chen et al., 2018).
Detection of apoptosis by flow cytometry
Annexin V (5 μL) and PI (10 μL) were added to the system, and the cells were incubated in the dark at room temperature for 20 min. Annexin V-FITC/PI is the most commonly used reagent for the detection of cell apoptosis. These two types of fluorescein can be excited by a laser at 488 nm and then observed in the first (FITC) and third (ECD) channels. The entire cell population can be analyzed to determine the proportion of apoptotic cells (Chen et al., 2019c). Cell apoptosis was detected using a computer (Beckman, USA).
The treated cells were dewaxed with xylene for 10 min, and the xylene was then washed off with 100% ethanol. The cells were subsequently hydrated with 95%, 85%, and 75% ethanol for 5 min. Hematoxylin staining was performed for 3 min, and the cells were then washed with water for 2 min. The sections were subsequently differentiated with 1% hydrochloric acid alcohol for 2 s and rinsed with water for 15 min. In the experiment, a drop of neutral gum was added to the center of the paraffin section, and the section was sealed with a cover glass. The sections were observed, and images were taken under a microscope (Dmi4000b inverted fluorescence microscope, Leica, DM2000 LED Germany) (Ohtomo et al., 2021; Wei et al., 2020).
The sections were dewaxed, hydrated, and subjected to two 10-min washes with xylene. The slides were then incubated with 100%, 95%, 85%, and 75% ethanol for 5–10 min and soaked in distilled water for 5 min. For antigen retrieval, the sections were incubated in citrate buffer (pH 6.0) and heated in a microwave at high heat for 8 min. The cells were then subjected to three 3 min-washes with 1× PBS (pH 7.2–7.6) and then incubated with 3% H2O2 at room temperature for 10 min to inactivate endogenous peroxidases. The slides were then incubated with the following primary antibodies: tumor necrosis factor-alpha (TNF-α; Invitrogen, Product #14-7321-81) and interleukin 4 (IL-4; Invitrogen, Product #PA5-25165). The secondary antibodies were then added, and the slides were incubated at 37°C for 1.5 h and subjected to three 5-min washes with PBS. After the runoff was transparent, the slides were subjected to two 10-min incubations with xylene (Wang et al., 2019). Each slide was sealed with neutral gum and observed under a microscope (Chen et al., 2020; Zhang et al., 2021). Sequencing was performed by Shanghai Jingzhun Biomedical Co., Ltd., Shanghai, China.
Construction and sequencing of the sequencing library
Total RNA (ribonucleic acid) was extracted using a kit according to the instructions provided. rRNA was removed using the Ribo-Zero Gold rRNA Removal Kit (Sigma, USA). After obtaining RNA samples of sufficient quality, the library was constructed, and the steps included single-chain synthesis, the addition of a 3’ terminal plus-strand poly(A) and plus-strand splice site, PCR enrichment, and any other steps needed to complete the construction of the sequencing library (Xing et al., 2020). Quantitative analysis of the library (using a qubit machine) and Illumina HiSeq 3000 sequencing were then performed (Andric et al., 2021; Wang et al., 2020).
Genome alignment and assembly of sequencing data
The clean data obtained in the previous step were compared to the reference genome (Bos taurus UMD3.1) using TopHat2 software. The reads used in the comparison were assembled using Cufflinks, and all the assembled transcripts were combined into a single transcript using Cuffcompare (Wang et al., 2019b).
Differential expression analysis of lncRNAs
To identifying real and reliable lncRNAs with a low false-positive rate, lncRNAs need to be screened using strict conditions. The coding-noncoding index (CNCI), coding potential calculator (CPC), PfamScan (Pfam), and predictor of long noncoding RNAs and messenger RNAs, based on an improved k-mer scheme (PLEK), were used to predict the sequences of lncRNAs. The overlapping results from the predictions obtained using the four software programs were defined as lncRNAs. The differential expression of lncRNAs between normal BMECs and S. aureus-infected BMECs was determined using the Cuffdiff program, which uses the number of fragments per thousand base length for a protein-coding gene per million fragments (FPKM) (Hou et al., 2020). FPKM is the most commonly used method to estimate the expression level of protein-coding genes and considers both the sequencing depth and the length of the protein-coding genes.
Detection of lncRNA expression
PrimeScript from Baobio Company (Dalian, China) was used with a Gamma RT Reagent kit with gDNA Eraser for cDNA synthesis in this study. The synthesized cDNA was diluted 1:100 and stored at −80°C. The lncRNAs were then amplified and detected using SYBR® Premix Ex Taq™ II and specific reverse transcripts. Each real-time fluorescent quantitative PCR was included in three replicate wells. The expression of other genes was quantified based on that of 18S rRNA, which served as the internal reference gene. The fold change in the target gene relative to the average value of the control group was determined using the 2−ΔCt method (Suppl. Tab. S1) (Ma et al., 2019a).
SPSS 18.0 (SPSS Inc., Chicago, IL, USA) software was used for the statistical analyses, and continuous variables are represented as the means ± standard deviations (Chen et al., 2019b). One-way ANOVA was used to analyze the variance and significance of the differences, and P < 0.01 and P < 0.05 were considered to indicate a significant difference. The graphs were generated using GraphPad Prism V5.0.
Construction of the S. aureus mastitis model (in vivo)
To verify the effect of S. aureus on inducing apoptosis in BMECs, we successfully constructed a model of S. aureus infection in BMECs. As demonstrated by flow cytometry, the apoptosis rate of mammary epithelial cells infected with S. aureus was significantly higher than that of the control cells. The proportion of early-apoptotic cells increased from 1.90% to 22.13%, and that of late-apoptosis cells increased from 3.24% to 16.13% (Fig. 1). We also observed the cell state of the different treatment groups. As shown by HE-staining, the cells in the control group were in good condition, whereas the nuclei of the cells in the S. aureus-infected group were significantly hyperchromatic (Fig. 2). The estimation of the protein expression levels of TNF-α and IL-4 based on staining showed that large amounts of TNF-α (Fig. 3) and IL-4 (Fig. 4) proteins were expressed in the S. aureus-infected cells. These experiments showed that the model of S. aureus mastitis was successfully constructed.
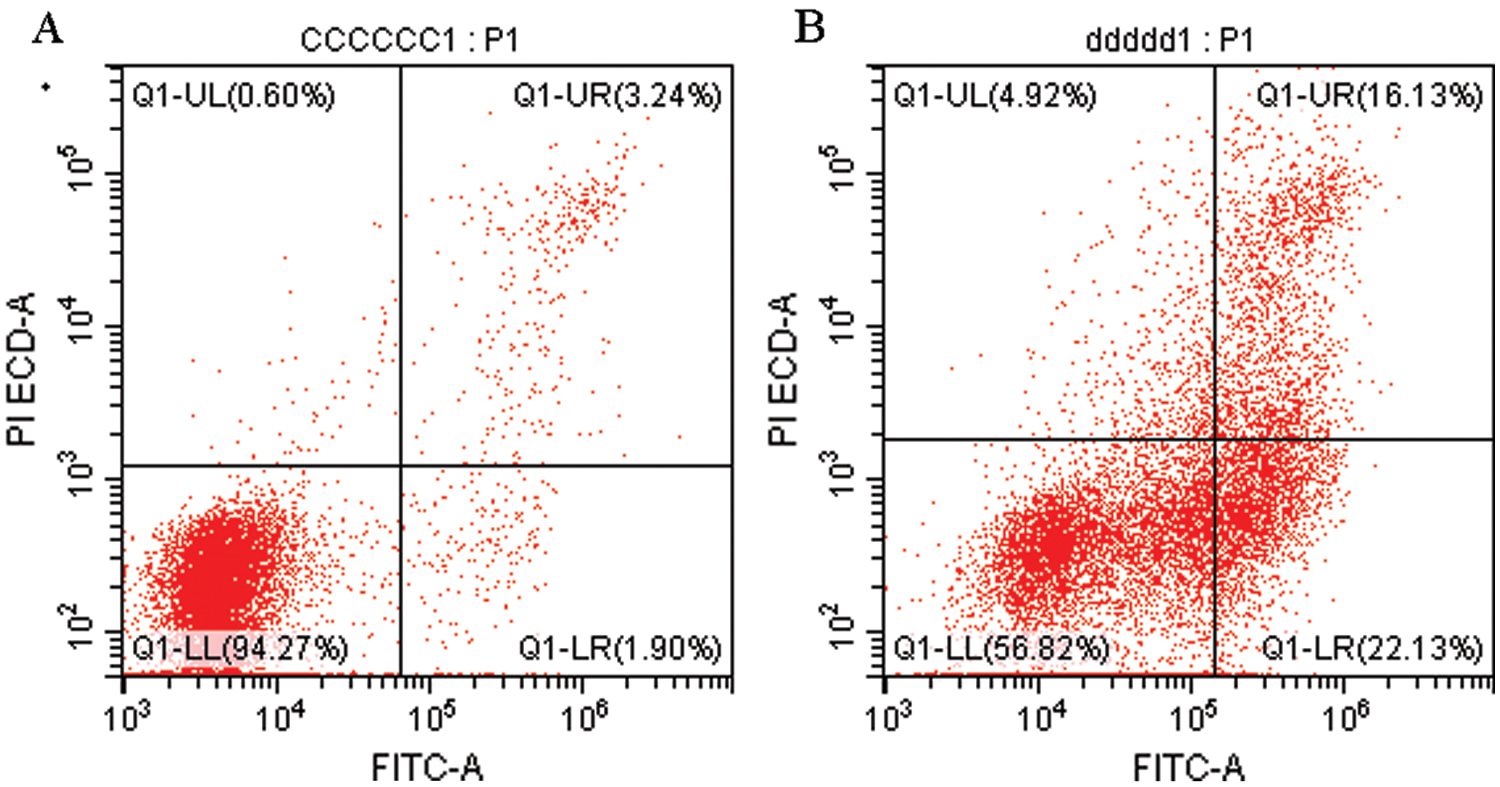
Figure 1: Flow cytometry detection of apoptotic BMECs.
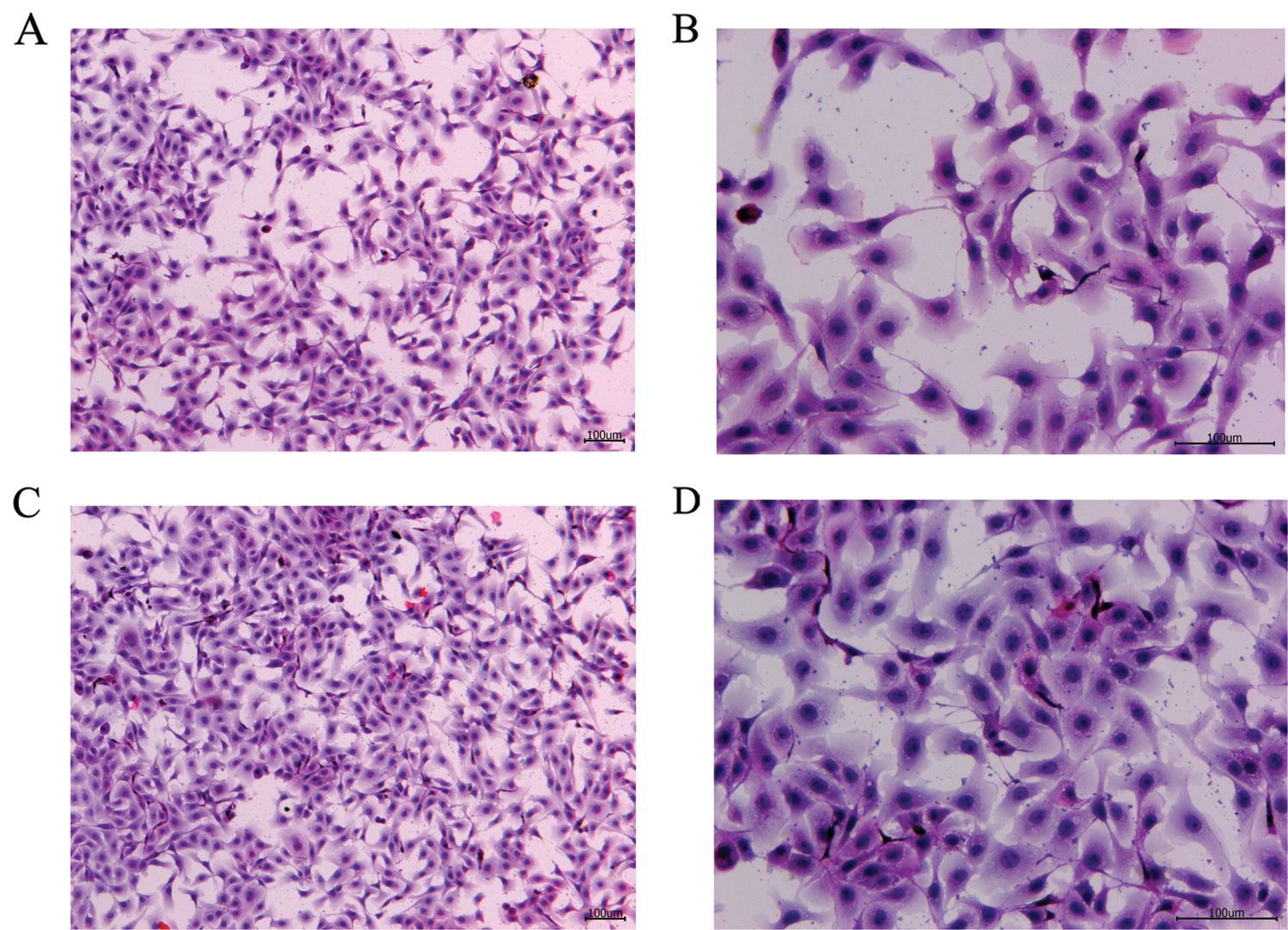
Figure 2: H&E staining of the blank control and S. aureus-treated groups.
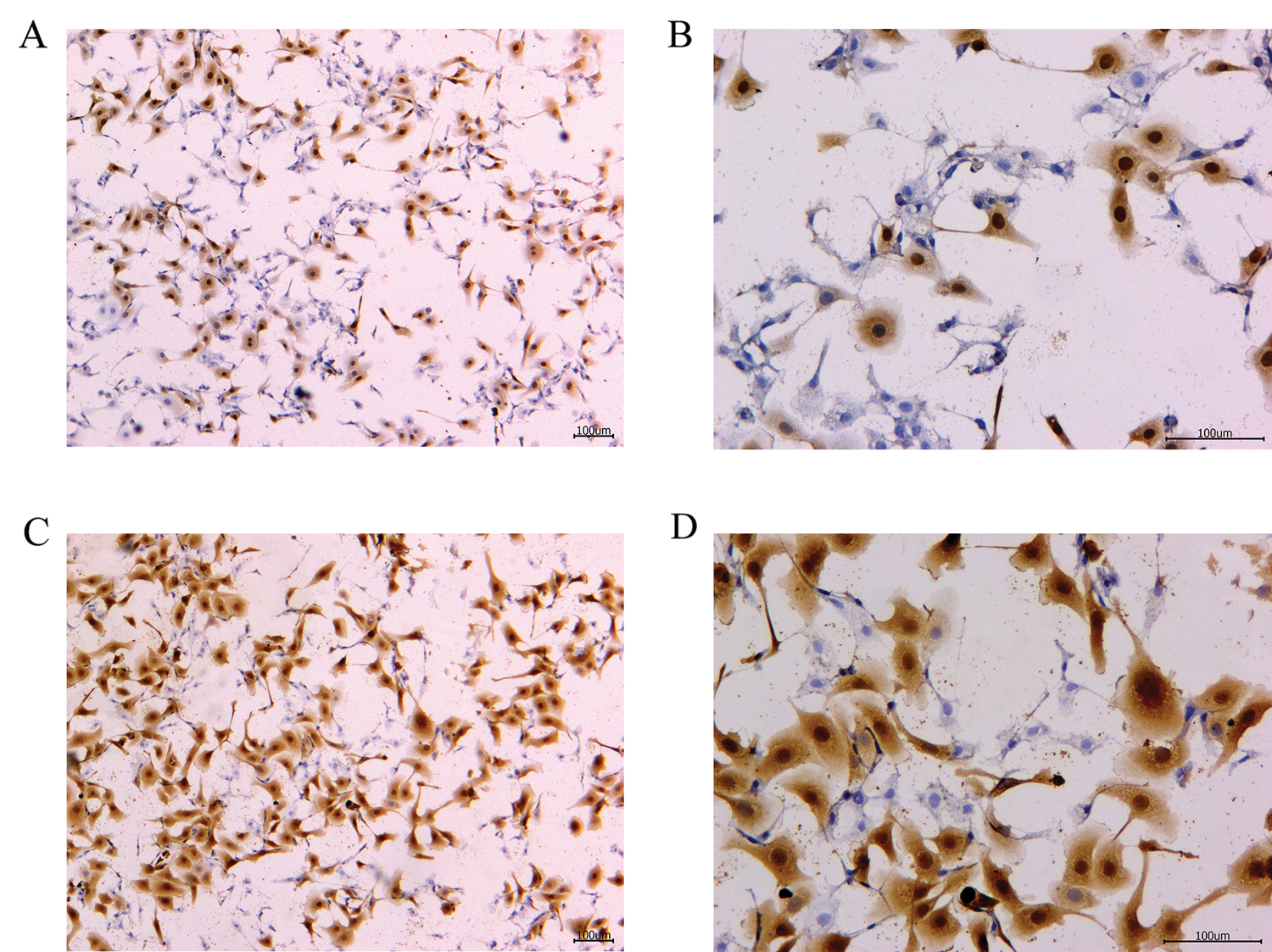
Figure 3: TNF-α protein immunohistochemistry of the blank control and S. aureus-treated groups.
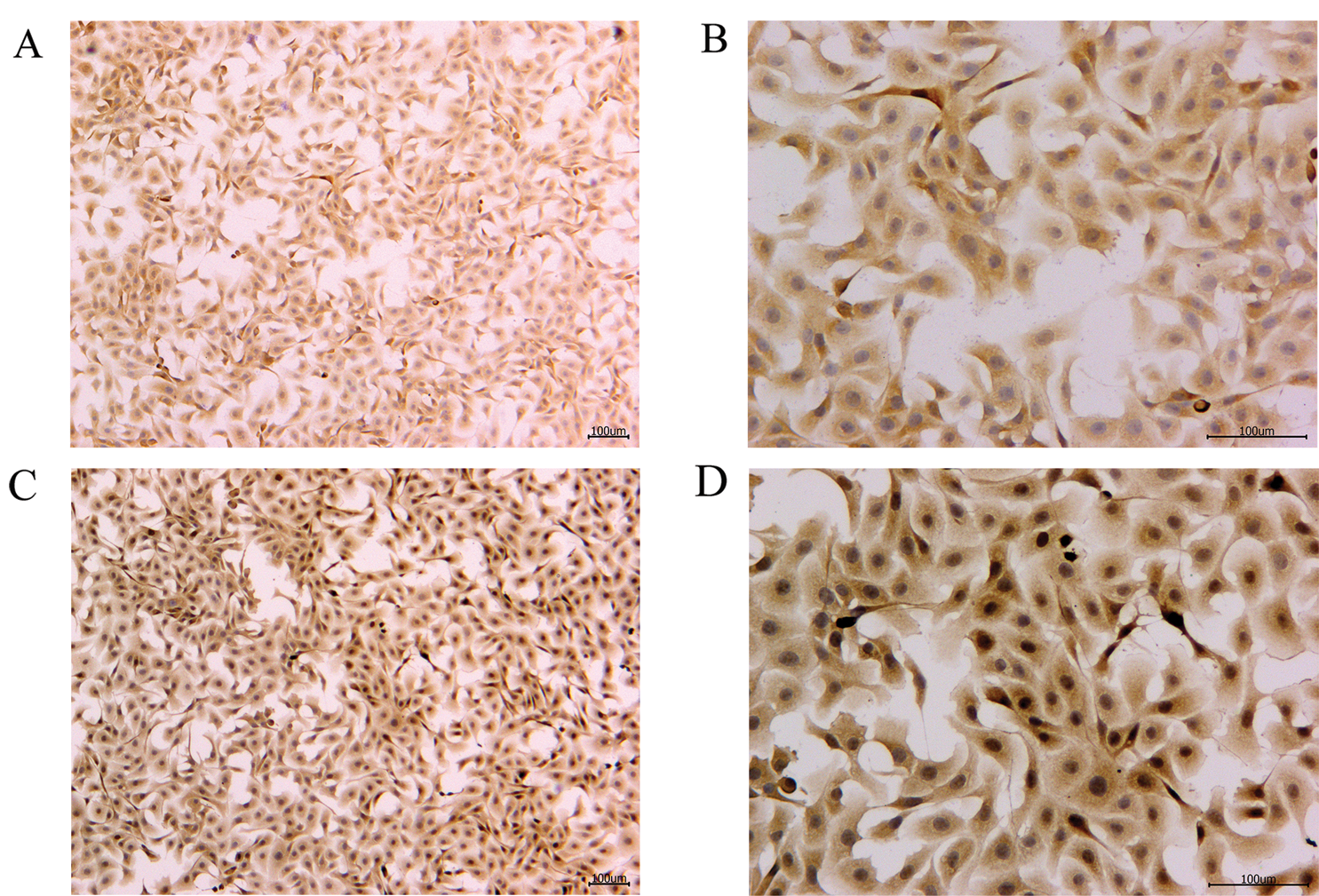
Figure 4: IL-4 protein immunohistochemistry of the blank control and S. aureus-treated groups.
Analysis of the sequencing quality
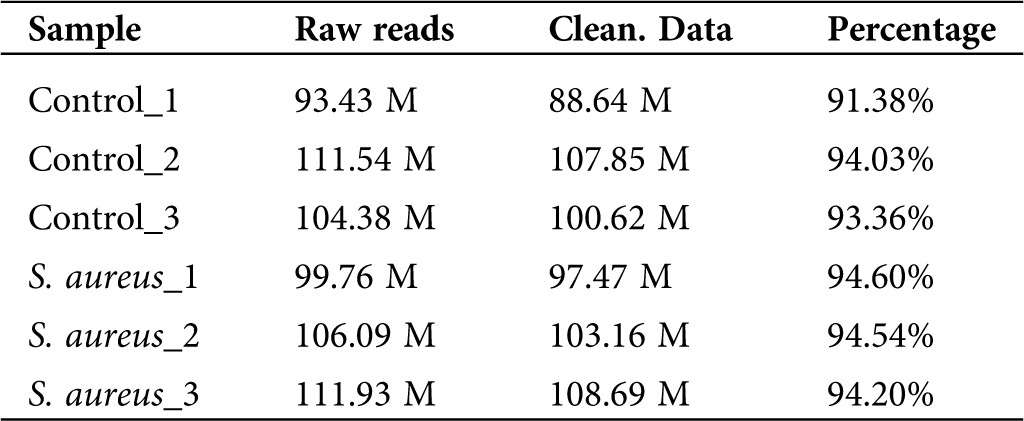
To determine the differential expression of lncRNAs in normal and S. aureus-infected BMECs, we used the two-terminal 150-bp sequencing mode of the HiSeq 3000 sequencing platform developed by Illumina and analyzed six samples, namely, three samples from the uninfected group (Control_1, Control_2, and Control_3) and three S. aureus-infected samples (S. aureus 1, S. aureus 2, and S. aureus 3). The total raw reads obtained were 627.13 M, and the average reads from each sample were approximately 104.52 M. The original sequencing data were processed, and sequences containing adapter reads, N, or rRNA and showing an abnormal length or low quality were filtered out (Tab. 1). The total clean data reads were 606.43 M, and the average reads from each sample were approximately 101.07 M. The high-quality clean data accounted for 91.38% to 94.54% of the original sequence, which indicated a good sequencing quality and that most of the sequencing data were of sufficient quality.
Table 1: Sequencing data quality
Genome alignment and assembly of sequencing data
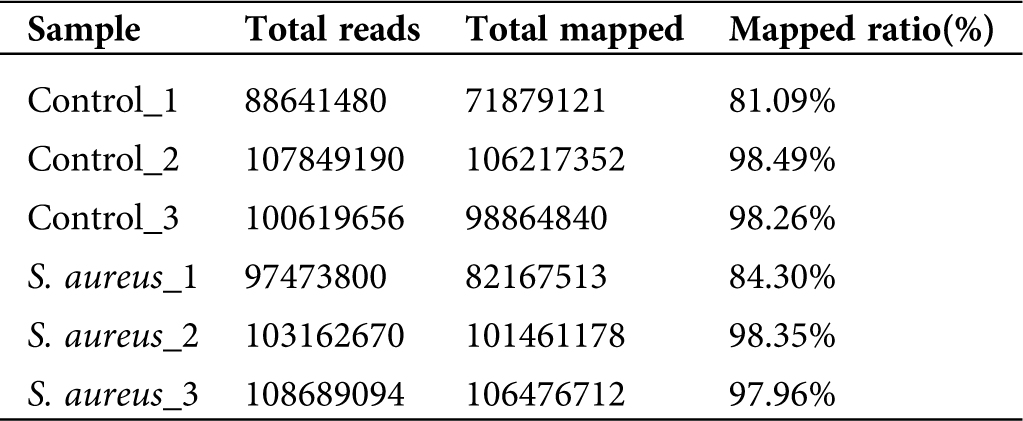
A comparison of the clean data with the bovine reference genome UMD3.1 using TopHat2 software yielded a comparison rate ranging from 81.09% to 98.49% (Tab. 2). The results showed that most of the sequencing results were bovine sequences, which also revealed the absence of RNA pollution from other species in the data obtained in the previous study. The alignment sequence was assembled using Cufflinks software for the next step of the lncRNA analysis.
Table 2: Alignment analysis of clean read with bovine genomics
Prediction and analysis of lncRNAs
For the identification of real and reliable lncRNAs with low false-positive rates, strict conditions need to be used for the screening of lncRNAs. The CPC, PLEK, CNCI, and Pfam software programs predicted 1657 noncoding transcripts, 1267 noncoding transcripts, 1638 coding transcripts, and 2380 noncoding transcripts, respectively. Based on a comprehensive analysis of the predicted results from the four software packages, the overlapping lncRNAs in the results from the four software packages were defined as the final lncRNAs. The results are shown in Fig. 5, and 1029 lncRNAs were ultimately identified.
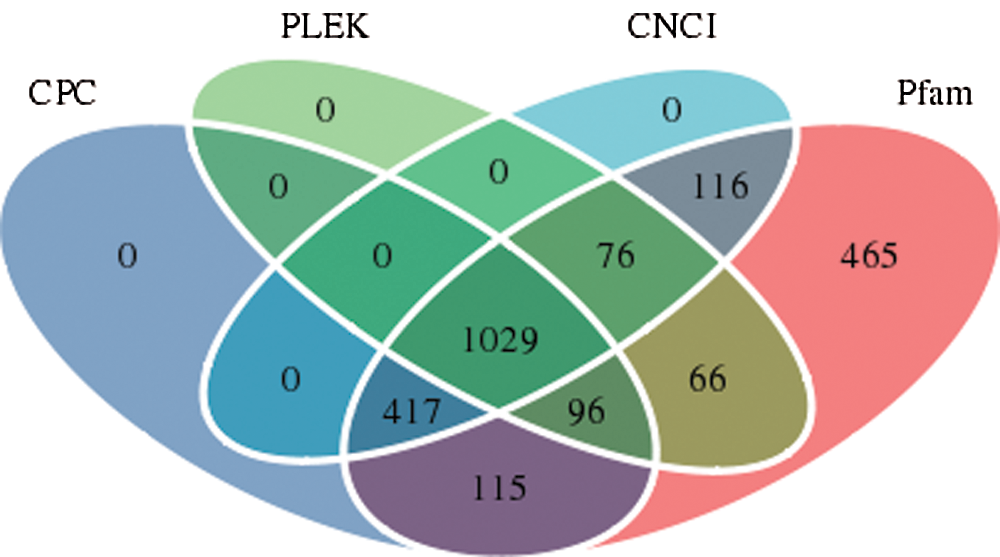
Figure 5: Number of lncRNAs identified by each or a combination of four software programs.
Analysis of lncRNA expression levels
A box and whisker plot uses five data statistics: minimum, first quartile (25%), median (50%), third quartile (75%), and maximum. This plot can also roughly indicate whether the data exhibit symmetry, the degree of distribution of the data, and other information. As shown in Fig. 6, the symmetry of the test sample was relatively good.
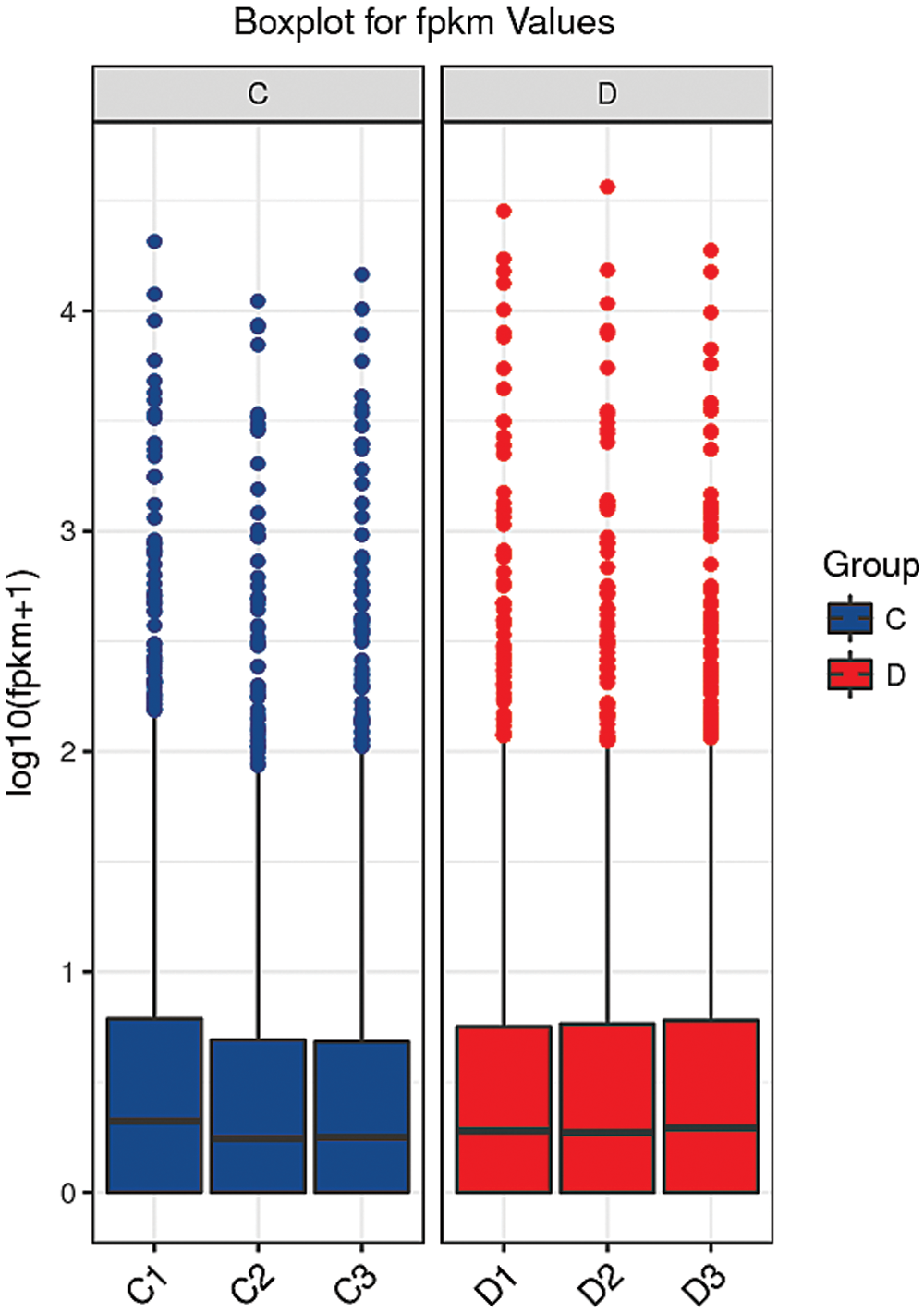
Figure 6: Box diagram of the blank control and S. aureus-treated groups.
The abscissa shows the sample name, and the ordinate shows the log10(FPKM+1) values. The box graph for each region shows five statistics (maximum, upper quartile, median, lower quartile and minimum from top to bottom). C: blank control group. D: S. aureus-treated group.
Differential expression analysis of lncRNAs
The comparison of normal mammary epithelial cells and S. aureus-infected mammary epithelial cells identified 30 differentially expressed lncRNAs. Among them, TCONS_00001500 showed 3.79-fold upregulation, and TCONS_00027546 exhibited 2.47-fold upregulation, whereas the expression of TCONS_00001464 was downregulated 2.88-fold (Fig. 7, Suppl. Tab. S2).
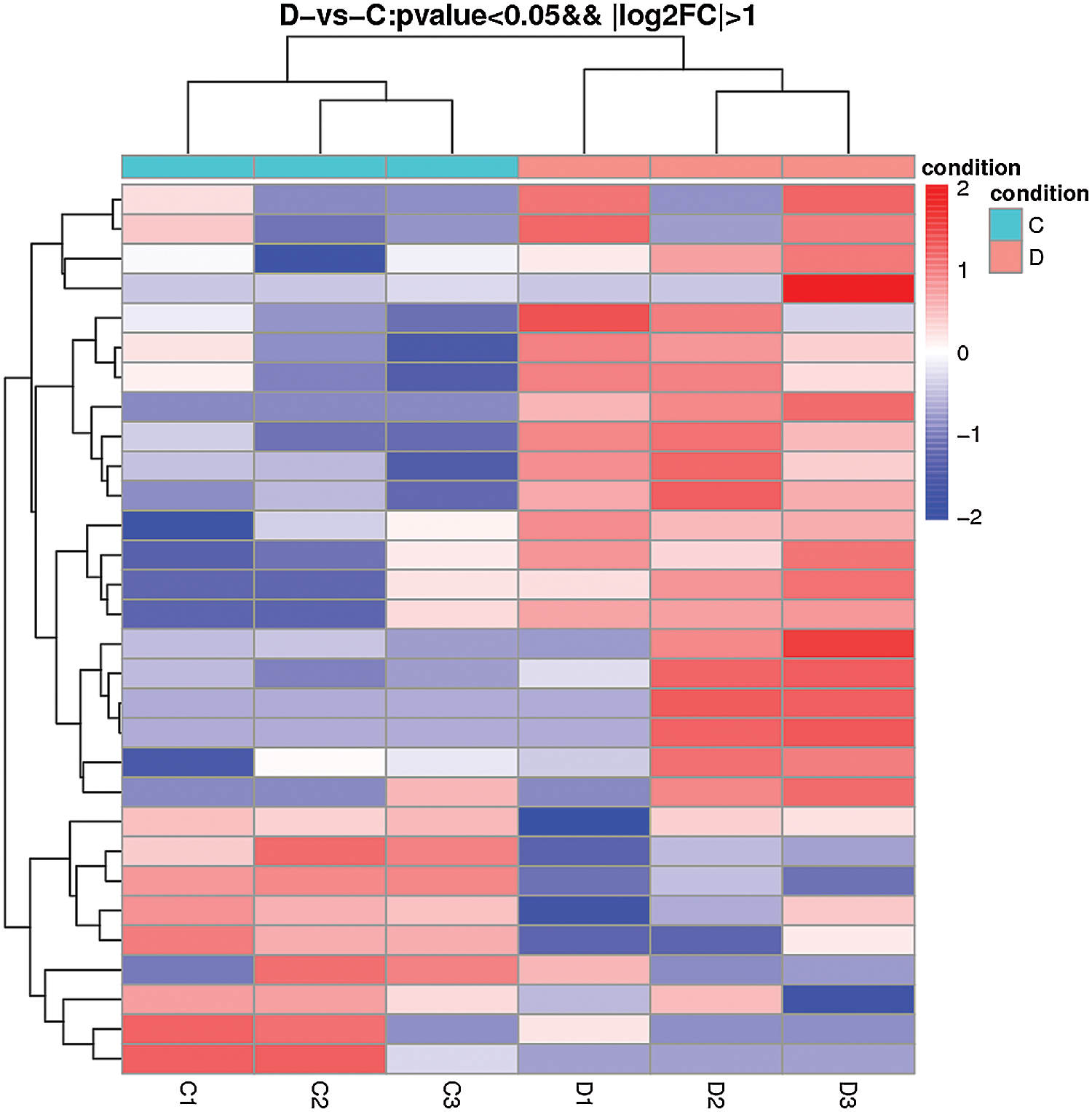
Figure 7: Heatmap of differentially expressed lncRNAs obtained from the comparison of the blank control and S. aureus-treated groups.
Analysis of the GO enrichment of differentially expressed lncRNAs
After obtaining the differentially expressed lncRNAs, we performed an enrichment analysis according to the GO functional annotations of the lncRNA source genes, analyzed the genes adjacent to the differentially expressed lncRNAs, and described their functions (combined with the GO annotation results). The method used for GO functional enrichment analysis was based on using all lncRNAs as the background list and the list of differentially expressed lncRNAs as the candidate list, screened from the background list. The hypergeometric distribution test was used to calculate the P-value of a representative GO function in the list of differential lncRNAs, and the P-value was corrected using the Benjamin-Hochberg procedure for multiple tests to obtain the false discovery rate (FDR). The GO annotation results indicate that the differentially expressed lncRNAs might participate in biological attention, biological regulation, cell killing, cell component organization or biogenesis, cell process, development process, establishment of localization, and immune system process, am ong other functions (Fig. 8, Suppl. Tab. S3).

Figure 8: GO functional annotation of potential target genes of differentially expressed lncRNAs identified from the comparison of the blank control S. aureus-treated groups.
Analysis of the KEGG pathway enrichment of the differentially expressed lncRNAs
KEGG is the main public database used for pathway analyses, and in this study, this database was used to analyze the pathways enriched in the differentially expressed lncRNAs based on KEGG annotation results. In addition, the hypergeometric distribution test was used to calculate the significance of the enrichment of the differentially expressed lncRNAs in each pathway entry. The calculated results will return a P-value indicating the degree of enrichment, and a small P-value indicates that the differentially expressed lncRNAs were enriched in the pathway. The results from the KEGG pathway analysis indicate that the differentially expressed lncRNAs might participate in aging, the circulatory system, development, the digestive system, the endocrine system, environmental adaptation, the exception system, and the immune system, among other pathways (Fig. 9, Suppl. Tab. S4).
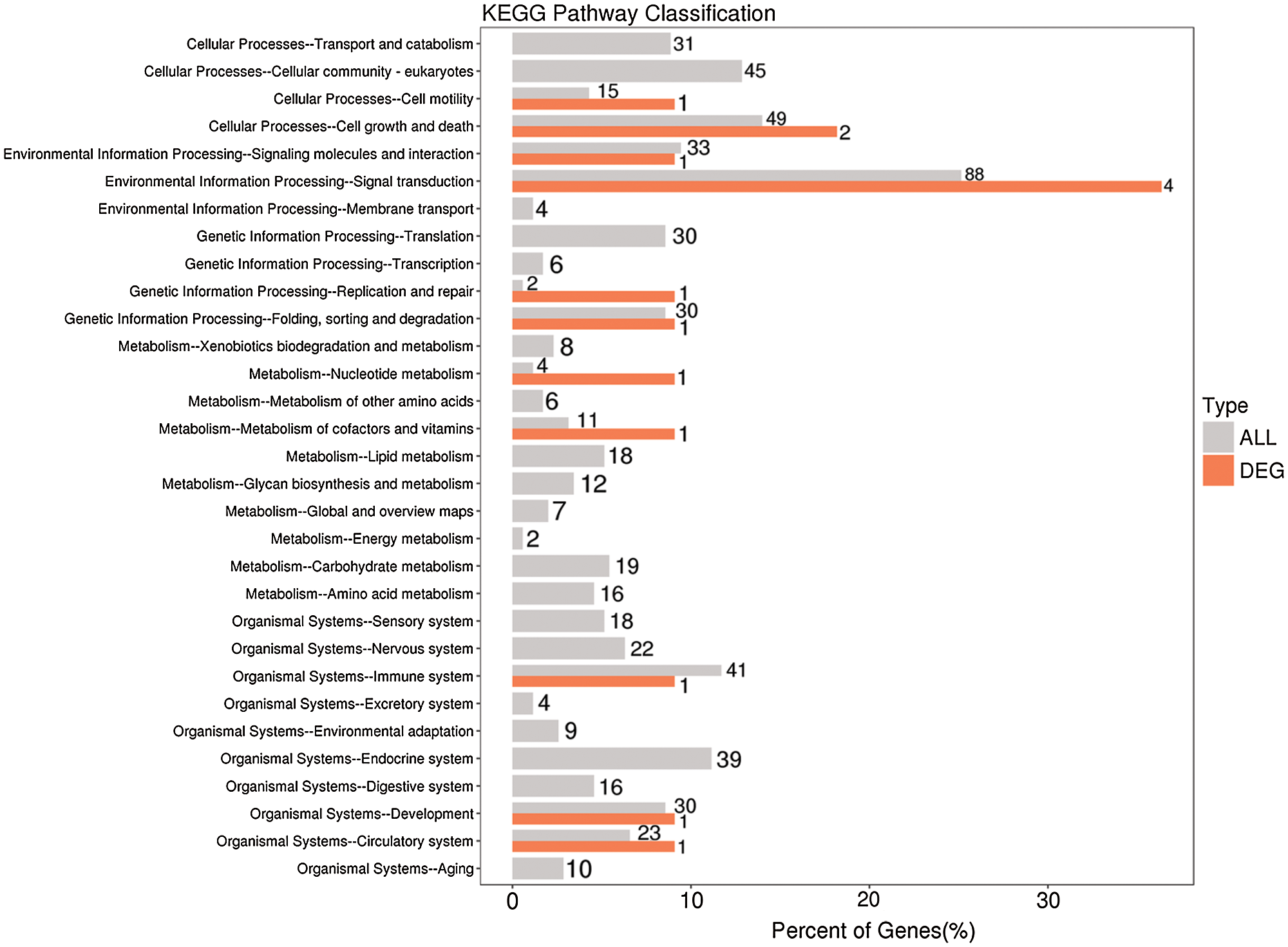
Figure 9: KEGG functional annotation of potential target genes of differentially expressed lncRNAs identified from the comparison of the blank control and S. aureus-treated groups.
RT-PCR verification of differentially expressed lncRNAs
Four different lncRNAs, namely, TCONS_00001500 (Fig. 10A), TCONS_00027546 (Fig. 10B), TCONS_00001464 (Fig. 10C) and TCONS_00007608 (Fig. 10D), were further analyzed by fluorescent quantitative PCR. As shown in Fig. 10, their expression trends were consistent with the results obtained by second-generation sequencing. For example, the second-generation sequencing of TCONS_00001500 showed that it was upregulated 3.79-fold in the infected cells, whereas the quantitative fluorescence analysis revealed that it was upregulated 2.25-fold. The second-generation sequencing of TCONS_00027546 revealed 2.47-fold upregulation in the infected cells, and quantitative fluorescence analysis showed that it was upregulated 2.49-fold.
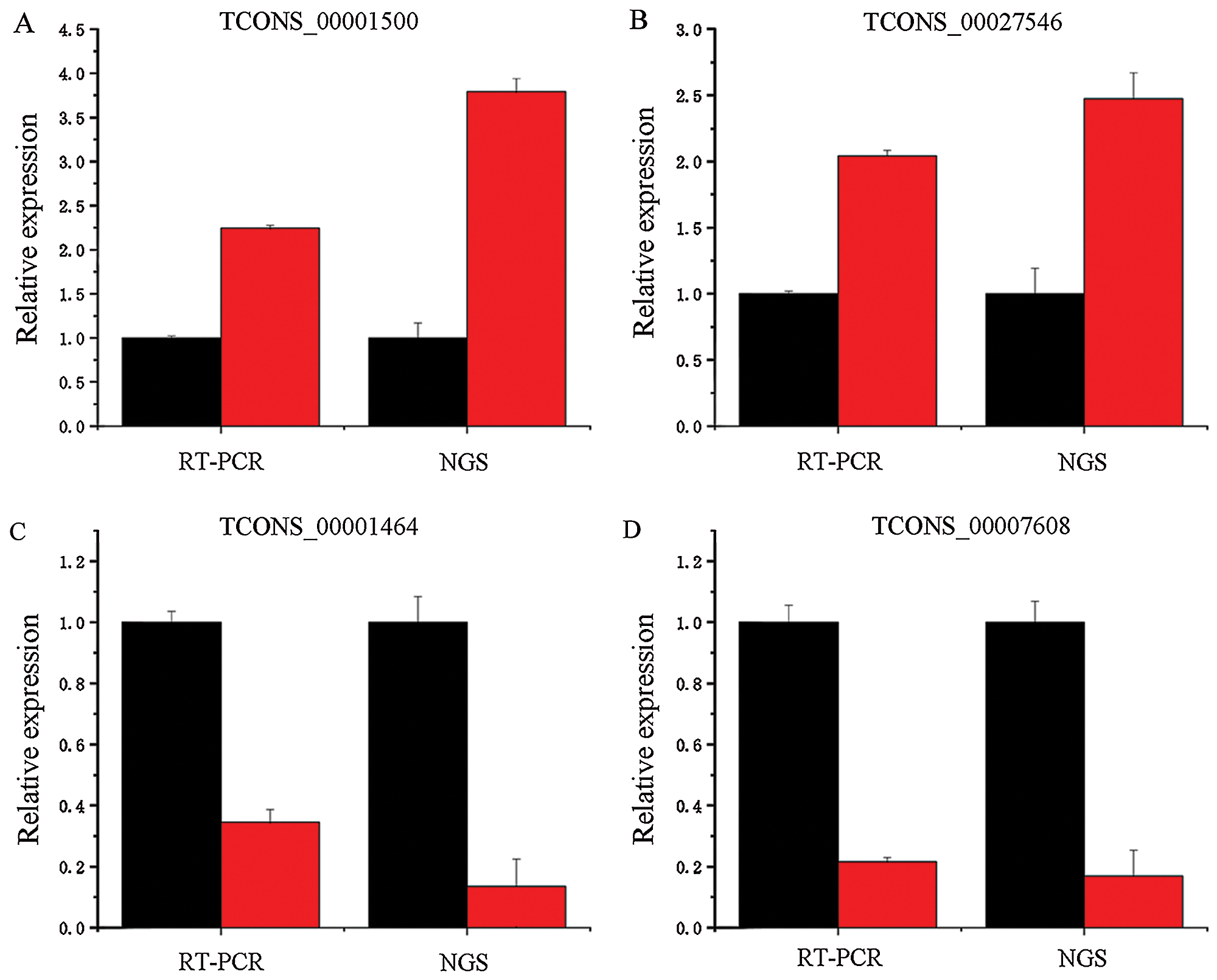
Figure 10: Fold changes in the expression of the differentially expressed lncRNAs obtained by RT-PCR and next-generation sequencing (NGS).
In 1976, the study conducted by Chandler and colleagues aimed to observe experimental mastitis in mice with reference to summer mastitis in cattle and successfully constructed the first mouse model of mastitis induced by S. aureus (Chandler et al., 1976). The structure of the mammary gland of mice is similar to that of cows, and mice can yield more samples and are more economical (Greenhalgh and Turos, 2009; Lepak and Andes, 2016). However, mice are small in size, and unwanted damage to their mammary gland can easily be caused during surgery. Some experiments have established acute mouse mastitis models induced by S. aureus and Streptococcus lactis (Mordmuang et al., 2019). In these models, the number of mast cells increases gradually, and the cytoplasm is filled with easily stained granules that are unevenly distributed. The degranulation state is also substantial and even exhibits depletion of granules, and a certain degree of mast cell infiltration can be detected in the acini. In addition, the expression of TNF-α, IFN-γ, and IL-4 is increased at the early stage of inflammation. A mouse model of mastitis induced by S. aureus has been successfully constructed (Zhang et al., 2010). According to the number of leukocytes and HE-staining, the two indexes were consistent with clinical manifestations. The analyses have shown that the injection of 50 μl of S. aureus (1.2 × 105 CFUs/mL) into a mouse can induce the typical symptoms of mastitis. qRT-PCR and Western blot analyses have shown that with increases in the S. aureus concentration used for infection, the expression of IL-2 in mammary gland tissue first increases and then decreases, whereas the expression of IL-4 first decreases and then increases (Komura et al., 2005). In this study, flow cytometry was used to detect the apoptosis of mammary epithelial cells treated with S. aureus. In addition, the infection upregulated the expression levels of the TNF-α and IL-4 genes, as demonstrated by immunohistochemistry. This finding is consistent with previous findings, which showed that IL-4 expression is increased at the early stage of inflammation (Zhang et al., 2010). It has been suggested that the immune response can be effectively promoted, which will eventually lead to the occurrence of S. aureus mastitis. A model of S. aureus mastitis has been successfully constructed, and this work provides a scientific theoretical basis for further exploration of the pathogenesis of mammalian mastitis in the future and reliable technical support for further research. We also expanded the knowledge regarding S. aureus-induced mastitis related to the processes of lncRNA regulation. The lncRNA profile can be used as a network biomarker to predict the response to mastitis and to help further understand the molecular basis of different responses. The use of S. aureus models of dairy cow mammary epithelial cells for the study of lncRNAs has not been reported. The lncRNAs in the normal and S. aureus-infected BMEC groups were screened by second-generation sequencing. These findings will not only expand our understanding of the lncRNA profile in dairy cows but also help us hypothesize the functions of lncRNAs in the genome and identify novel molecular markers of mastitis.
With the continuous development of genomic technology, researchers have obtained a more in-depth understanding of many biological transcripts, and these findings have changed the theories regarding the roles of RNAs in gene regulation (Liu et al., 2020). The use of these new technologies has shown that the genomes of mammals and other organisms can produce thousands of lncRNAs without protein-coding ability (Hitachi et al., 2020). According to their distance from protein-coding genes, lncRNAs can be divided into the following categories: noncoding RNAs (lncRNAs) between genes, which do not overlap with any other genes (Zhao et al., 2019); lncRNAs of introns, which are located in the intron region of a gene; lncRNAs derived from bidirectional promoters, whose transcriptional direction is opposite to the protein-coding direction and which are located within 1 kb of the promoter; enhancer lncRNAs, which are usually less than 2 kb away and are transcribed from the enhancer region of the genome (Nowosad et al., 2019); sense lncRNAs, which start from the sense chain encoded by the protein, can partially overlap with the intron region and partially or completely overlap with the exon; and antisense lncRNAs, which are transcribed from another strand of DNA and overlap with exons, introns, or both of protein-coding genes (Deng et al., 2019). lncRNAs play a variety of functions in chromosome remodeling, transcription, posttranscriptional regulation, development, cancer, and inflammation. The development of high-throughput sequencing technology and chip technology has allowed the identification of some lncRNAs, and an increasing number of studies have been conducted on these lncRNAs (Graindorge et al., 2019). Cui et al. (2014) identified lncRNAs that show differential expression after lipopolysaccharide (LPS) stimulation. In their study, the most obviously upregulated lncRNA was lnc-IL17R, which overlapped with the 3’ noncoding region of the human IL17R gene. The researchers then transfected lnc-IL17R into cells and found that this transfection reduced the LPS-induced inflammatory response (Cui et al., 2014). Another lncRNA, Ptprj-as1, is expressed in macrophages, and high expression of this lncRNA can be immediately induced by pro-inflammatory factors, such as LPS (Dave et al., 2013). Rapicavoli et al. (2013) studied the expression of lncRNAs induced by TNF-α and identified hundreds of differentially expressed lncRNAs, including 54 pseudogenes. When TNF-α activates the NF-κB signaling pathway, the lncRNA Lethe is selectively induced by binding to NF-κB transcription factors and preventing its binding to DNA (Rapicavoli et al., 2013). These studies indicate that lncRNAs play an important role in the inflammatory response. However, similar studies of S. aureus infection in BMECs have not been performed. Therefore, the study of lncRNAs in S. aureus mastitis can increase our understanding of lncRNA functions, provide guidance for the prevention and treatment of mastitis, and identify markers for screening mastitis. In this study, we compared the expression of lncRNAs between normal and S. aureus-infected mammary epithelial cells and identified 30 differentially expressed lncRNAs. These differentially expressed lncRNAs are related to the invasion of mammary epithelial cells by S. aureus, which might be due to communication between cells, the defense mechanism initiated by cells, or related pathways involved in the inflammatory response, but these findings need further study. These 30 differentially expressed lncRNAs can also be used as markers of mastitis and as a reference standard for the early detection of mastitis in the future.
Authors’ Contributions: Jingpeng Zhou and Xiaoguang Ji incepted the idea, Yuhao Wang performed the necessary search and drafted the first version, Xiaolong Wang reviewed the work in progress, Yongjiang Mao recommended changes in the concept to shape the final draft, Zhangping Yang proofread and edited the final version.
Availability of Data and Materials: All data generated or analyzed during this study are included in this published article (and its supplementary information files).
Supplementary Material: The supplementary material is available online at DOI: 10.32604/biocell.2021.015586.
Ethical Statement: The animal use protocol used in this study was approved by the Institutional Animal Care and Use Committee of the College of Animal Science and Technology, Yangzhou University, Yang Zhou, China (NSFC2020-DKXY-30, March 28, 2020).
Funding Statement: This research was supported by the National Natural Science Foundation of China (Grant Nos. 31872324, 31802035, and 31601915), the Natural Science Foundation of the Jiangsu Higher Education Institutions of China (18KJA230003), Six Talent Peaks Project in Jiangsu Province (NY-093), the China Postdoctoral Science Foundation (Grant Nos. 2017M621841 and 2019T120472).
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
Andric V, Nevers A, Hazra D, Auxilien S, Menant A, Graille M, Palancade B, Rougemaille M (2021). A scaffold lncRNA shapes the mitosis to meiosis switch. Nature Communications 12: 770. DOI 10.1038/s41467-021-21032-7. [Google Scholar] [CrossRef]
Bandyopadhyay S, Samanta I, Bhattacharyya D, Nanda PK, Kar D, Chowdhury J, Dandapat P, Das AK, Batul N, Mondal B, Dutta TK, Das G, Das BC, Naskar S, Bandyopadhyay UK, Das SC, Bandyopadhyay S (2015). Co-infection of methicillin-resistant Staphylococcus epidermidis, methicillin-resistant Staphylococcus aureus and extended spectrum beta-lactamase producing Escherichia coli in bovine mastitis--three cases reported from India. Veterinary Quarterly 35: 56–61. DOI 10.1080/01652176.2014.984365. [Google Scholar] [CrossRef]
Cui H, Xie N, Tan Z, Banerjee S, Thannickal VJ, Abraham E, Liu G (2014). The human long noncoding RNA lnc-IL7R regulates the inflammatory response. European Journal of Immunology 44: 2085–2095. DOI 10.1002/eji.201344126. [Google Scholar] [CrossRef]
Chandler RL, Anger HS, Smith K (1976). Observations on experimental mastitis in mice with reference to summer mastitis in cattle. Journal of Comparative Pathology 86: 319–327. DOI 10.1016/0021-9975(76)90056-6. [Google Scholar] [CrossRef]
Chen MM, Zeng GP, Li J, Fu JH, Long YY et al. (2020). High infiltration of CD20(+) B lymphocytes in extranodal natural killer/T-cell lymphoma is associated with better prognosis. British Journal of Haematology 191: e116–e120. [Google Scholar]
Chen Z, Chu S, Wang X, Fan Y, Zhan T, Arbab AAI, Li M, Zhang H, Mao Y, Loor JJ, Yang Z (2019a). MicroRNA-106b regulates milk fat metabolism via ATP Binding Cassette Subfamily A Member 1 (ABCA1) in bovine mammary epithelial cells. Journal of Agricultural and Food Chemistry 67: 3981–3990. DOI 10.1021/acs.jafc.9b00622. [Google Scholar] [CrossRef]
Chen Z, Chu S, Wang X, Sun Y, Xu T, Mao Y, Loor JJ, Yang Z (2019b). MiR-16a regulates milk fat metabolism by targeting large tumor suppressor kinase 1 (LATS1) in bovine mammary epithelial cells. Journal of Agricultural and Food Chemistry 67: 11167–11178. DOI 10.1021/acs.jafc.9b04883. [Google Scholar] [CrossRef]
Chen Z, Xia H, Shen H, Xu X, Arbab I, Li M, Zhang H, Mao Y, Yang Z (2018). Pathological features of Staphylococcus aureus induced mastitis in dairy cows and isobaric-tags-for-relative-and-absolute-quantitation proteomic analyses. Journal of Agricultural and Food Chemistry 66: 3880–3890. DOI 10.1021/acs.jafc.7b05461. [Google Scholar] [CrossRef]
Chen Z, Xu X, Tan T, Chen D, Liang H, Sun K, Li M, Zhang H, Mao Y, Yang Z (2019c). MicroRNA-145 regulates immune cytokines via targeting FSCN1 in Staphylococcus aureus-induced mastitis in dairy cows. Reproduction in Domestic Animals 54: 882–891. DOI 10.1111/rda.13438. [Google Scholar] [CrossRef]
Dave RK, Dinger ME, Andrew M, Askarian-Amiri M, Hume DA, Kellie S, Thomas PG (2013). Regulated expression of PTPRJ/CD148 and an antisense long noncoding RNA in macrophages by proinflammatory stimuli. PLoS One 8: e68306. DOI 10.1371/journal.pone.0068306. [Google Scholar] [CrossRef]
De Vries SPW, Hadjirin NF, Lay EM, Zadoks RN, Peacock SJ, Parkhill J, Grant AJ, McDougall S, Holmes MA (2018). Streptococcus bovimastitidis sp. nov., isolated from a dairy cow with mastitis. International Journal of Systematic and Evolutionary Microbiology 68: 21–27. DOI 10.1099/ijsem.0.002321. [Google Scholar] [CrossRef]
Deng RY, Hong T, Li CY, Shi CL, Liu C, Jiang FY, Li J, Fan XM, Feng SB, Wang YF (2019). Long non-coding RNA zinc finger antisense 1 expression associates with increased disease risk, elevated disease severity and higher inflammatory cytokines levels in patients with lumbar disc degeneration. Medicine 98: e18465. DOI 10.1097/MD.0000000000018465. [Google Scholar] [CrossRef]
Freyssin A, Page G, Fauconneau B, Rioux Bilan A (2020). Natural stilbenes effects in animal models of Alzheimer’s disease. Neural Regeneration Research 15: 843–849. DOI 10.4103/1673-5374.268970. [Google Scholar] [CrossRef]
Fujimoto Y, Ito H, Higuchi H, Ohno H, Makita K (2020). A case-control study of herd- and cow-level risk factors associated with an outbreak of Mycoplasma mastitis in Nemuro. Japan Preventive Veterinary Medicine 177: 104946. DOI 10.1016/j.prevetmed.2020.104946. [Google Scholar] [CrossRef]
Graindorge A, Pinheiro I, Nawrocka A, Mallory AC, Tsvetkov P, Gil N, Carolis C, Buchholz F, Ulitsky I, Heard E, Taipale M, Shkumatava A (2019). In-cell identification and measurement of RNA-protein interactions. Nature Communications 10: 5317. DOI 10.1038/s41467-019-13235-w. [Google Scholar] [CrossRef]
Greenhalgh K, Turos E (2009). In vivo studies of polyacrylate nanoparticle emulsions for topical and systemic applications. Nanomedicine 5: 46–54. DOI 10.1016/j.nano.2008.07.004. [Google Scholar] [CrossRef]
Hasanian S, Amirmozafari N, Sheikh AF, Mehregan I (2020). Molecular typing of methicillin and vancomycin-resistant Staphylococcus aureus isolated from clinical specimens by doublelocus sequence typing (DLST) method. Biocell 44:411–419. DOI 10.32604/biocell.2020.08976. [Google Scholar] [CrossRef]
Hitachi K, Nakatani M, Funasaki S, Hijikata I, Maekawa M, Honda M, Tsuchida K (2020). Expression levels of long non-coding RNAs change in models of altered muscle activity and muscle mass. International Journal of Molecular Sciences 21: 1628. DOI 10.3390/ijms21051628. [Google Scholar] [CrossRef]
Hou X, Tang D, Zheng F, Ou M, Xu Y et al. (2020). Expression profiling of immune cells in systemic lupus erythematosus by single-cell RNA sequencing. Biocell 44:559–582. DOI 10.32604/biocell.2020.011022. [Google Scholar] [CrossRef]
Huang W, Long N, Khatib H (2012). Genome-wide identification and initial characterization of bovine long non-coding RNAs from EST data. Animal Genetics 43: 674–682. DOI 10.1111/j.1365-2052.2012.02325.x. [Google Scholar] [CrossRef]
Komura K, Hasegawa M, Hamaguchi Y, Yukami T, Nagai M, Yachie A, Sato S, Takehara K (2005). Drug-induced hypersensitivity syndrome associated with human herpesvirus 6 and cytomegalovirus reactivation. Journal of Dermatology 32: 976–981. DOI 10.1111/j.1346-8138.2005.tb00885.x. [Google Scholar] [CrossRef]
Koufariotis LT, Chen YP, Chamberlain A, Vander Jagt C, Hayes BJ (2015). A catalogue of novel bovine long noncoding RNA across 18 tissues. PLoS One 10: e0141225. DOI 10.1371/journal.pone.0141225. [Google Scholar] [CrossRef]
Lepak AJ, Andes DR (2016). In vivo pharmacodynamic target assessment of delafloxacin against Staphylococcus aureus, Streptococcus pneumoniae, and Klebsiella pneumoniae in a murine lung infection model. Antimicrobial Agents and Chemotherapy 60: 4764–4769. DOI 10.1128/AAC.00647-16. [Google Scholar] [CrossRef]
Li C, Wei B, Zhao J (2021). Competing endogenous RNA network analysis explores the key lncRNAs, miRNAs, and mRNAs in type 1 diabetes. BMC Medical Genomics 14: 35. DOI 10.1186/s12920-021-00877-3. [Google Scholar] [CrossRef]
Liu W, Gan CY, Wang W, Liao LD, Li CQ, Xu LY, Li EM (2020). Identification of lncRNA-associated differential subnetworks in oesophageal squamous cell carcinoma by differential co-expression analysis. Journal of Cellular and Molecular Medicine 24: 4804–4818. DOI 10.1111/jcmm.15159. [Google Scholar] [CrossRef]
Ma M, Pei Y, Wang X, Feng J, Zhang Y, Gao MQ (2019a). LncRNA XIST mediates bovine mammary epithelial cell inflammatory response via NF-kappaB/NLRP3 inflammasome pathway. Cell Proliferation 52: e12525. DOI 10.1111/cpr.12525. [Google Scholar] [CrossRef]
Ma T, Wang F, Wang X (2019b). LncRNA LINC01772 promotes metastasis and EMT process in cervical cancer by sponging miR-3611 to relieve ZEB1. Biocell 43:191–198. DOI 10.32604/biocell.2019.06989. [Google Scholar] [CrossRef]
Monistero V, Barberio A, Biscarini F, Cremonesi P, Castiglioni B, Graber HU, Bottini E, Ceballos-Marquez A, Kroemker V, Petzer IM, Pollera C, Santisteban C, Veiga Dos Santos M, Bronzo V, Piccinini R, Re G, Cocchi M, Moroni P (2020). Different distribution of antimicrobial resistance genes and virulence profiles of Staphylococcus aureus strains isolated from clinical mastitis in six countries. Journal of Dairy Science 103: 3431–3446. DOI 10.3168/jds.2019-17141. [Google Scholar] [CrossRef]
Mordmuang A, Brouillette E, Voravuthikunchai SP, Malouin F (2019). Evaluation of a Rhodomyrtus tomentosa ethanolic extract for its therapeutic potential on Staphylococcus aureus infections using in vitro and in vivo models of mastitis. Veterinary Research 50: 49. DOI 10.1186/s13567-019-0664-9. [Google Scholar] [CrossRef]
Nowosad K, Hordyjewska-Kowalczyk E, Tylzanowski P (2019). Mutations in gene regulatory elements linked to human limb malformations. Journal of Medical Genetics 57: 361–370. DOI 10.1136/jmedgenet-2019-106369. [Google Scholar] [CrossRef]
Ohtomo S, Otsubo H, Arai H, Shimoda Y, Homma Y, Tominaga T (2021). Hyperperfusion in the thalamus on arterial spin labelling indicates non-convulsive status epilepticus. Brain Communications 3: fcaa223. DOI 10.1093/braincomms/fcaa223. [Google Scholar] [CrossRef]
Ozdemir S, Altun S (2020). Genome-wide analysis of mRNAs and lncRNAs in Mycoplasma bovis infected and non-infected bovine mammary gland tissues. Molecular and Cellular Probes 50: 101512. DOI 10.1016/j.mcp.2020.101512. [Google Scholar] [CrossRef]
Pitkala A, Haveri M, Pyorala S, Myllys V, Honkanen-Buzalski T (2004). Bovine mastitis in Finland 2001--prevalence, distribution of bacteria, and antimicrobial resistance. Journal of Dairy Science 87: 2433–2441. DOI 10.3168/jds.S0022-0302(04)73366-4. [Google Scholar] [CrossRef]
Qu Z, Adelson DL (2012). Bovine ncRNAs are abundant, primarily intergenic, conserved and associated with regulatory genes. PLoS One 7: e42638. DOI 10.1371/journal.pone.0042638. [Google Scholar] [CrossRef]
Rapicavoli NA, Qu K, Zhang JJ, Mikhail M, Laberge RM, Chang HY (2013). A mammalian pseudogene lncRNA at the interface of inflammation and anti-inflammatory therapeutics. eLife 2: e00762. [Google Scholar]
Sebastian-Delacruz M, Gonzalez-Moro I, Olazagoitia-Garmendia A, Castellanos-Rubio A, Santin I (2021). The role of lncRNAs in gene expression regulation through mRNA stabilization. Noncoding RNA 7. DOI 10.3390/ncrna7010003. [Google Scholar] [CrossRef]
Tanhaeian A, Mirzaii M, Pirkhezranian Z, Sekhavati MH (2020). Generation of an engineered food-grade Lactococcus lactis strain for production of an antimicrobial peptide: in vitro and in silico evaluation. BMC Biotechnology 20: 19. DOI 10.1186/s12896-020-00612-3. [Google Scholar] [CrossRef]
Wang H, Wang K, Guo J, Wen T (2019a). Gene expression profile of Sox1, Sox2, p53, Bax and Nestin in neural stem cells and adult mouse brain tissues. Biocell 43:59–64. DOI 10.32604/biocell.2019.05731. [Google Scholar] [CrossRef]
Wang H, Wang X, Li X, Wang Q, Qing S, Zhang Y, Gao M (2019b). A novel long non-coding RNA regulates the immune response in MAC-T cells and contributes to bovine mastitis. FEBS Journal 286: 1780–1795. DOI 10.1111/febs.14783. [Google Scholar] [CrossRef]
Wang X, Wang H, Zhang R, Li D, Gao MQ (2020). LRRC75A antisense lncRNA1 knockout attenuates inflammatory responses of bovine mammary epithelial cells. International Journal of Biological Sciences 16: 251–263. DOI 10.7150/ijbs.38214. [Google Scholar] [CrossRef]
Wei Z, Nie G, Yang F, Pi S, Wang C, Cao H, Guo X, Liu P, Li G, Hu G, Zhang C (2020). Inhibition of ROS/NLRP3/Caspase-1 mediated pyroptosis attenuates cadmium-induced apoptosis in duck renal tubular epithelial cells. Environmental Pollution 273: 115919. DOI 10.1016/j.envpol.2020.115919. [Google Scholar] [CrossRef]
Weikard R, Hadlich F, Kuehn C (2013). Identification of novel transcripts and noncoding RNAs in bovine skin by deep next generation sequencing. BMC Genomics 14: 789. DOI 10.1186/1471-2164-14-789. [Google Scholar] [CrossRef]
Xing J, Liu H, Jiang W, Wang L (2020). LncRNA-encoded peptide: Functions and predicting methods. Frontiers in Oncology 10: 622294. DOI 10.3389/fonc.2020.622294. [Google Scholar] [CrossRef]
Zhang HY, Zhu FF, Zhu YJ, Hu YJ, Chen X (2021). Effects of IL-18 on the proliferation and steroidogenesis of bovine theca cells: Possible roles in the pathogenesis of polycystic ovary syndrome. Journal of Cellular and Molecular Medicine 25: 1128–1139. DOI 10.1111/jcmm.16179. [Google Scholar] [CrossRef]
Zhang P, Wang J, Shi Y (2010). Structure and mechanism of the S component of a bacterial ECF transporter. Nature 468: 717–720. DOI 10.1038/nature09488. [Google Scholar] [CrossRef]
Zhao K, Wang M, Kang H, Wu A (2019). A prognostic five long-noncoding RNA signature for patients with rectal cancer. Journal of Cellular Biochemistry 121: 3854–3860. DOI 10.1002/jcb.29549. [Google Scholar] [CrossRef]
Zorraquino MA, Althaus RL, Roca M, Molina MP (2011). Heat treatment effects on the antimicrobial activity of macrolide and lincosamide antibiotics in milk. Journal of Food Protection 74: 311–315. DOI 10.4315/0362-028X.JFP-10-297. [Google Scholar] [CrossRef]
Appendix:
Supplementary files
Supplementary Table S1: Detection of lncRNA expression
Supplementary Table S2: Differential expression analysis of lncRNAs
Supplementary Table S3: Analysis of the GO enrichment of differentially expressed lncRNAs
Supplementary Table S4: Analysis of the KEGG pathway enrichment of the differentially expressed lncRNAs
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |