DOI:10.32604/biocell.2021.013993
www.techscience.com/journal/biocell
| Biocell DOI:10.32604/biocell.2021.013993 | www.techscience.com/journal/biocell |
| Review |
Mechanisms adopted by cancer cells to escape apoptosis–A review
Department of Basic Sciences, Deanship of Preparatory Year & Supporting Studies, Imam Abdulrahman Bin Faisal University, Dammam, 34212, Saudi Arabia
*Address correspondence to: Sayequa Dandoti, SSDandoti@iau.edu.sa
Received: 28 August 2020; Accepted: 03 November 2020
Abstract: Inactivation of apoptosis is the prime phenomenon in cancer development and cancer treatments. Mutations in the apoptotic pathway not only exert resistance to apoptosis and provide a survival advantage to cancer cells but also confer resistance to cancer therapies. Escaping apoptosis is the “hallmark” of cancer cells. Cancer cells can withstand many apoptotic stimuli, such as DNA damage, unfavorable environments, and cytotoxic therapies. Substantial research has been carried out and is in good progress on the various mechanisms adopted by cancer cells to evade apoptosis. This article reviews the apoptosis escape mechanisms by cancer cells, viz. apoptotic gene alterations (in few essential and accessory apoptotic genes), post-translational modifications (phosphorylation and ubiquitination of apoptotic proteins), metabolic alterations, mitochondrial alterations, immunity escape, epigenetics, cancer cell dormancy, cancer clonal theory, and reversibility of apoptosis. The review reveals that there is a wide scope for further research to address the various challenges in realizing successful cancer therapies that involve reversing the apoptotic resistance and/or inducing apoptosis in tumor cells.
Keywords: Apoptosis; Caspase; Apoptotic Genes; APAF-1; Death receptor; Cancer metabolism; Oncogene; miRNA
Cancer is one of the leading causes of death. As research in the therapeutic approach of cancer is developing, cancer is also developing its resistance towards various therapeutic agents. The most prevalent factor providing resistance to cancer cells is the escape from the default cell death program— ‘apoptosis’. In order to survive, cancer cells override many barriers that would cause apoptosis. Apoptosis is a highly regulated cell death process that occurs in multicellular organisms. It plays important role in the organism’s life from embryogenesis to aging, and in many diseases, such as cancer, neurodegenerative diseases, ischemic injury, AIDS (Acquired Immunodeficiency Syndrome), and autoimmune diseases. It is a homeostatic mechanism where organisms eliminate unnecessary cells from the body in the course of development, mop out infected or damaged cells from the system, and maintain the level by replacing these cells with the new ones. In response to the apoptotic stimuli, a complex cascade of events causes changes such as chromatin condensation, membrane blebbing, cell shrinkage, nuclear fragmentation, and DNA fragmentation, eventually leading to the cell demise without eliciting an immune response (Sjöström and Bergh, 2001). The process is carried out by cysteine aspartate proteases recognized as caspases. Many human diseases occur due to either the death of cells that should live or the presence of cells that should die. Hence, realizing the fact that apoptosis has the ability to restrict abnormal growth of tissue, the research interest on apoptosis regulation has suddenly emerged. By extensive research on diseases showing a characteristic decrease in apoptotic rate and on how the diseased cells escape apoptosis, we may be able to use these escape mechanisms to treat the diseases characterized by an increased rate of apoptosis.
The mechanisms of apoptosis evasion by cancer cells are of central importance in drug development as many cancer therapies intend to initiate cell death. Many researchers have elaborated the diverse aspects of cancer and apoptosis, such as apoptosis-inducing drug resistance due to apoptotic gene modifications and targeting these modifications to develop new therapies (Lowe and Lin, 2000; Schmitt, 2003; Mashima and Tsuruo, 2005; Fulda, 2010; Wong, 2011; Kaleigh and Kurokawa, 2013; Dasgupta et al., 2017; Aleksakhina et al., 2019; Jan and Chaudhry, 2019), cancer cell interaction with immune system and immune escape mechanisms (Messerschmidt et al., 2017; Mohme et al., 2017; Rodriguez, 2017; Leone et al., 2018; Steven and Seliger, 2018), cancer cell and their microenvironment (Gao et al., 2017; Sun et al., 2019), epigenetics (Yan et al., 2017; Hervouet et al., 2013), and anti-tumor drugs (Yan et al., 2017; Rodriguez et al., 2013; Cesari et al., 2014; Leone et al., 2018). However, the present article provides a comprehensive review of the various apoptosis-escape mechanisms reported so far, such as genetic alterations, post-translational modifications, metabolic alterations, mitochondrial alterations, immune system escape, epigenetics, cancer stem cells (CSCs), cancer cell dormancy, cancer clonal theory, circulating and disseminated tumor cells (CTCs and DTCs), and reversibility of apoptosis. After sequentially covering these sub-topics, the article highlights the challenges and future research directions to realize these mechanisms in cancer therapies and ends with concluding remarks.
Pathways to initiate apoptosis
The two most common pathways to initiate apoptosis are denoted as intrinsic and extrinsic (Sjöström and Bergh, 2001; Kessel, 2015). The intrinsic pathway can be termed as auto-activation as the cell itself senses the stimuli and undergoes apoptosis, while the extrinsic pathway is paracrine-activation, where the cell receives signals from neighboring cells and extracellular space; both pathways work by the activation of caspases (Elmore, 2007).
In the extrinsic pathway or death receptor pathway, extracellular ligands activate the responsible receptors that are located in the cell membrane. According to Kaleigh and Kurokawa (2013), the engagement of extracellular ligands such as FAS (FS-7-Associated Surface antigen) and TNF (Tumor Necrosis Factor) with cell surface receptor leads to the formation of DISC (Death Inducing Signal Complex) and eventual activation of initiator caspase-8 and caspase-10. Caspase-8 has a dominant function in the extrinsic pathway. When the death ligand binds to its cognate receptor, the cell recruits receptor-specific adaptor proteins like FADD (Fas-Associated Death Domain), which forms DISC that further activates caspase-8. The activated caspase-8 either directly catalyzes the activation of execution caspase or acts on its substrate BID (BH-3 Interacting Domain death agonist) to form tBID (truncated BID) that leads to the mitochondrial release of cyt c (cytochrome complex). Studies have suggested the inexplicable dual role of caspase-8 as pro-apoptosis and pro-survival (Salvesen and Walsh, 2014).
The intrinsic pathway (also known as the mitochondrial pathway) is activated by diverse stimuli (extra- and intracellular) like oxidative stress, irradiation, and cytotoxic drug treatment. The pro-apoptotic BCL-2 (B-Cell Lymphoma 2) family members like BIM (Bcl-2 Interacting Mediator of cell death), BID (BH-3 Interacting Domain death agonist), BAD (Bcl-2 Associated Agonist of cell death), BAX (Bcl-2 Associated Protein X), and BAK (Bcl-2 Homologous Antagonist Killer) promote MOMP (Mitochondrial outer membrane permeabilization) due to which pro-apoptotic proteins (such as cyt c) are released from mitochondrial intermembrane into the cytoplasm, where cyt c forms “Apoptosome” by binding to adaptor protein APAF-1 (apoptotic protease activating factor) and then it binds to procaspase-9 (Schafer and Kornbluth, 2006). Apoptosome is a caspase-9-activating complex, which activates initiator caspase-9. The activated caspase-9 sequentially activates caspase-3 that initiates the proteolytic reactions. Along with cyt c, mitochondria release other polypeptides, including AIF (Apoptosis-Inducing Factor), Endonuclease G, SMAC (Second Mitochondrial Activator of Caspases) also known as DIABLO, and Serine proteases Omi (also known as HtrA2), from the intermembrane space (Elmore, 2007). AIF and endonuclease G cause DNA damage and condensation. And SMAC binds to XIAP (X-linked Inhibitor of Apoptosis Protein), which inhibits several caspases (caspase -3, -7, -9); this binding neutralizes XIAP and facilitates cyt c-induced caspase activation (Chipuk et al., 2006).
The granzyme pathway is another pathway to initiate apoptosis, where CTLs (Cytotoxic T Lymphocytes) kill the damaged/ infected cells by secreting perforin (Trapani and Smyth, 2002). Perforin is a transmembrane pore-forming molecule that forms a pore in the target cell membrane and then releases granules (containing Serine proteases, granzyme A & B) through these pores into the target cell cytoplasm. Other pathways inducing apoptosis might also exist, but the intrinsic and the extrinsic pathways have been studied widely and demonstrated in detail.
The execution phase/pathway is where the abovementioned pathways converge to the final phase that starts by activation of execution caspases (Fig. 1). The execution of several caspases (caspase -3, -6, -7) activates cytoplasmic endonuclease and protease, which degrades nuclear material and proteins, respectively, ultimately leading to morphological and biochemical changes (Slee and Adrain, 2001). Among the execution caspases, caspase-3 is the most frequently activated during apoptosis. It is activated in apoptotic cells by extrinsic, intrinsic, and granzyme pathways where the activation is catalyzed by caspase-8, caspase-9, and granzyme B, respectively. As the execution involves the destruction of cellular structures (like chromatin condensation, DNA fragmentation, cleavage of many key cellular proteins, and formation of apoptotic bodies), caspase-3 is also termed as executioner caspase. Studies reported that caspase-3 also has functions before the cell commits to death (Porter and Jänicke, 1999).
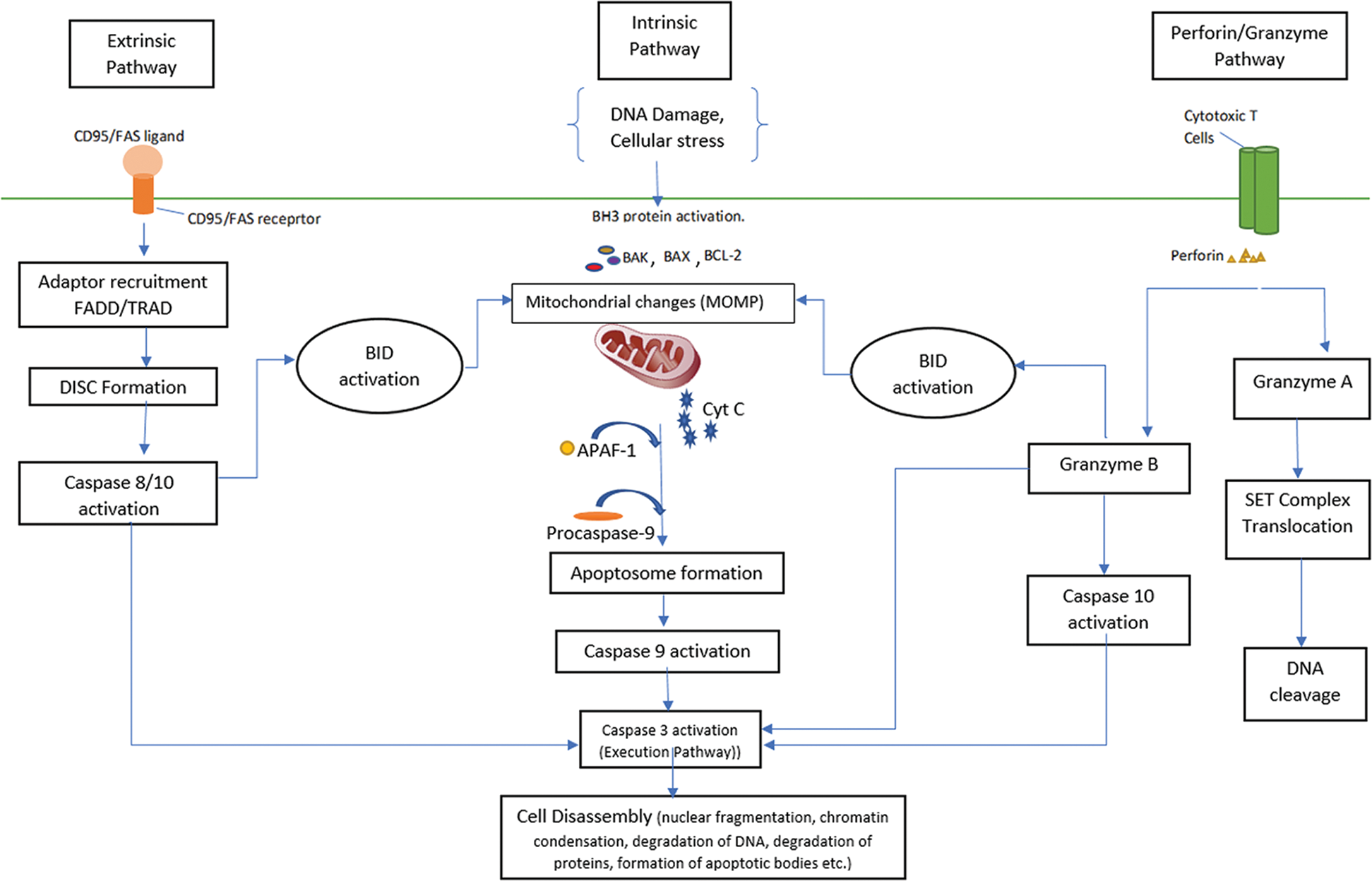
Figure 1: An overview of apoptotic pathway.
How cancer cells escape apoptosis
In cancer, some of the body’s cells divide irregularly without stopping and spread to other tissues in the body. Cancer occurs due to various genetic causes, and it is a complex process involving multiple steps in its development, such as mutation, tumor formation, and metastasis. Tissue homeostasis is affected not only by over-proliferation of the cells but also by decreased removal of cells, i.e., being a “defective cell,” cancer cells also need to undergo apoptosis. But during tumor development, these cells acquire few genetic alterations and adopt few mechanisms that cause evasion of apoptosis: one of the hallmarks of cancer. For decades, to find out the mechanisms in cancer cells to escape apoptosis is a topic of high interest to researchers.
The outcome of each apoptotic phase is regulated by the various factors involved in apoptosis, genes, and their interactive networks. These apoptotic regulatory genes are often dysregulated in cancer cells. These genomic abnormalities provide proliferative and survival advantages to the cancer cells.
One of the types of alterations cancer cells use to evade apoptosis is a shift in the balance between pro- and anti-apoptotic gene expression programs. Most tumor cells evade apoptosis by either increasing the expression of pro-survival genes (anti-apoptotic) or decreasing the expression of pro-apoptotic genes. These modulations in gene expressions can be achieved by transcriptional and translational alterations such as gene–deletion, –silencing, –copy number amplification, and transcription factors’ activation or inactivation, thereby affecting the expression of apoptotic regulators (Kumar and Cakouros, 2004). Also, mutations in genes that regulate apoptosis are commonly observed in cancer cells; this phenomenon eliminates the pro-apoptotic proteins and/or amplifies anti-apoptotic proteins, a key step in the progression to cancer. Somatic mutations and germline mutations of apoptosis-related genes were reported in human cancers (Pai et al., 1998; Park et al., 2001; Lee et al., 2004; Ghavami et al., 2009; Timofeev et al., 2019). Even the loss of anti-oncogenes (or tumor-suppressor gene) has been observed regulating the cell cycle by controlling growth and proliferation (Levine and Puzio-Ku, 2010). Mutation or inactivation of these genes results in reduction or loss of their function (leading to abnormal cell growth), which is believed to be a key factor in the development of several tumors. In human cancers, the mutation of the anticancer gene family member loses its original function. Tumor suppressor genes are inactivated by point mutations or deletion in both alleles of the gene (Wang et al., 2018). When tumor suppressor genes are inactivated, cell cycle control is lost, and the cell displays uncontrolled growth and division. The “loss of function” of multiple tumor suppressor genes is considered to be the main reason causing malignancy (Lam and Schmidt, 2012). Tumors also display resistance to receptor-induced cell death (Mohammad et al., 2015).
Genetic studies have identified many genes that function in apoptosis (Hoeppner et al., 2001), and few of the essential and secondary apoptotic genes altered in cancer cells are discussed in the following sub-sections.
BCL-2 (B-Cell Lymphoma-2) gene was originally found in B-cell follicular lymphoma; the pro-apoptotic and anti-apoptotic genes are members of this family (Fig. 2). In normal cells, the sensitivity of cells to apoptotic stimuli depends on the balance between pro-apoptotic and anti-apoptotic BCL-2 proteins. It has been reported that genetic inactivation of a BH3-only (BCL-2 Homology 3-only) protein leads to resistance to certain pro-apoptotic stimuli and also accelerates tumor formation in mice (Faqar-Uz-Zaman et al., 2018).
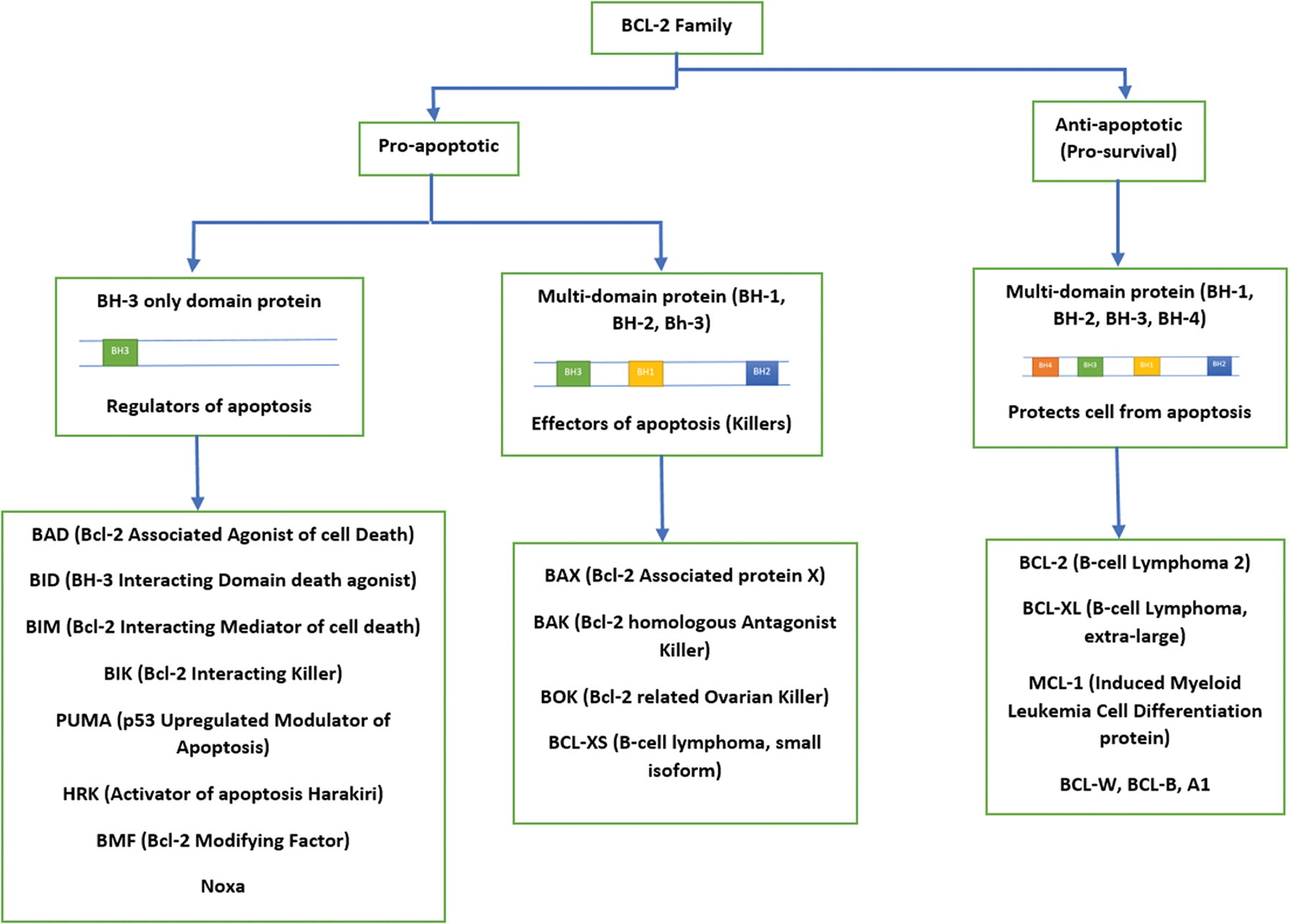
Figure 2: Family of BCL-2 genes.
Over-expression of anti-apoptotic BCL-2 was observed in cancer cells, e.g., amplification of BCL-XL in lung cancer and giant-cell tumor of bone (Adams and Cory, 2007); and amplification of anti-apoptotic MCL-1 in breast and lung cancers (Faqar-Uz-Zaman et al., 2018; Soderquist et al., 2018). In more than 80% of the bladder cancer tissues, anti-apoptotic BCL-2 protein was found positive, and its expression was correlated with the stage and grade of the tumor (Konstantinidou et al., 2002). Several cancer types such as lung-, breast-, prostate-, gastric-, renal-, pancreatic-, colorectal- and hepatocellular- cancer showed overexpression of members of BCL-2 family genes (Yip and Reed, 2008; Lessene et al., 2008). A study on transgenic mice reported collaboration of BCL-2 and/or BCL-XL expression with oncogene (Wang et al., 2003). Alternatively, the transcription factors like CREB (cAMP response element-binding protein) and STAT3 (signal transducer and activator of transcription 3), which activate genes in response to pro-survival stimuli, can increase the expression of MCL-1 and BCl-2 (Wang et al., 1999; Real et al., 2002). Hence, activation of CREB or STAT3 could promote the expression of anti-apoptotic genes in cancer cells.
Gene coding for pro-apoptotic protein BAX was found mutated in human hematological malignancies (Liu et al., 2013). In some lymphomas and leukemia, due to BAX frame-shift mutations, the BAX translation is terminated prematurely, due to which BAX loses its function, ultimately leading to apoptosis resistance (Brimmell et al., 1998). Deletion polymorphism of one BCL-2 family member, BIM, was reported in certain human populations and is significantly associated with innate resistance to TKI (Tyrosine Kinase Inhibitor) therapies (Kaleigh and Kurokawa, 2013). BIM transcription is induced in response to the withdrawal of growth factors (or other apoptotic stimuli). FOXO is one of the transcription factors involved in BIM expression (Jain et al., 2013); the FOXO activity is suppressed by pro-survival kinases, AKT, and ERK. Thus, cancer cells with high levels of AKT/ERK show suppressed BIM expression (Costa et al., 2007; Hübner et al., 2008).
Beroukhim et al. (2010) found two BCL-2 family genes deleted in cancer; a pro-apoptotic BH-3-only family gene PUMA (p53 Up-regulated Modulator of Apoptosis) and a pro-apoptotic multi-BH-domain BOK (Bcl-2 Related Ovarian Killer). PUMA is found to be most frequently deleted in cancer. PUMA is a crucial mediator of p53-dependent and p53-independent apoptosis; it is expressed in response to apoptotic stimuli and regulated by p53. On receiving death signals, PUMA binds to anti-apoptotic BCL-2 family member in mitochondria and relieves the inhibition on BAX and/or BAK, which leads to mitochondrial dysfunction and caspase activation (which carries out apoptosis). PUMA inhibition causes apoptotic deficiency and increases the risk of cancer development. According to a few reports, PUMA can directly activate BAX/BAK to induce mitochondrial function (Bean, 2014). Cancer cells with compromised functions of PUMA were observed in many studies. For instance, the reduction in PUMA expression in malignant cutaneous melanoma was reported by Karst et al. (2005). Later, Adams and Cory (2007) stated that “frequent overexpression of anti-apoptotic BCL-2 family proteins and other anti-apoptotic oncoproteins in tumor antagonizes PUMA-induced apoptosis”. Additionally, mutant p53 in human tumors repudiates induction of PUMA by irradiation and by chemotherapeutic drugs (Yu and Zhang, 2008). Garrison et al. (2008) also found lower expression of PUMA in approximately 40% of human Burkitt’s lymphomas. Loss of PUMA in the hypoxia-induced tumor model causes chromosomal instability and promotes tumorigenesis (Nelson et al., 2004). Further research is needed to prove the role of PUMA in tumorigenesis because few studies suggest that PUMA is not directly inactivated in cancers, rather its loss is the outcome of alterations in other genes (Yoo et al., 2007).
The TP53 gene codes for the transcriptional factor p53, which plays important roles in apoptosis. It is observed that TP53 is the most frequently mutated or inactivated tumor suppressor gene in cancer. More than half of human tumors contain TP53 mutations (Vogelstein and Kinzler, 2004; Oren and Rotter, 2010). In normal cells, in response to stress, stabilized p53 combines with other p53 subunits and functions as a critical transcription factor that modulates the expression of an array of genes known to trigger apoptosis. p53 promotes activation of GADD45 (Growth Arrest and DNA Damage 45) and P53R2 (p53 controlled Ribonucleotide reductase 2) genes, which repairs damaged DNA, due to which mutation (this DNA damage) is not accumulated and is not passed to the daughter cells. However, when DNA changes are irreparable, p53 activates the genes involved in apoptosis, such as PUMA, BAX, FAS, PIG3 (p53 Inducible Gene 3), and Killer/DR5 (Death Receptor 5), which leads to apoptotic death of the cells with damaged DNA.
The p53 protein regulates glycolysis, PPP (pentose phosphate pathway), mitochondrial oxidative phosphorylation, lipids and nucleotides metabolism, and the cell response to oxidative stress (Perri et al., 2016). Tumor cells depend mainly on glycolysis for energy (referred to as the Warburg effect). p53 down-regulates glycolysis by inducing genes TIGAR (TP53 Induced Glycolysis and Apoptosis Regulator), which inhibits glycolysis, and by inducing genes SCO2 (synthesis of cytochrome oxidase 2) and GLS2 (mitochondrial glutaminase 2), which promotes oxidative phosphorylation (Jiang et al., 2011). Moreover, glycolysis is down-regulated by inhibiting the expression of GLUT1 (Glucose Transporter 1) and GLUT4 (Glucose Transporter 4) due to which glucose uptake is blocked (Jiang et al., 2011). Other than glycolysis, PPP (Pentose Phosphate Pathway) is an alternative pathway for tumor cells; p53 inhibits PPP as well, by reducing the activity of enzyme G6PD (glucose-6-phosphate dehydrogenase) (Perri et al., 2016). p53 also arrests fatty acids biosynthesis by acting on FASN (fatty acids synthase) and ACLY (ATP citrate lyase) and inhibits the formation of the membrane in tumor cells (Freed-Pastor et al., 2012).
In cancer cells, a mutation in TP53 leads to the formation of mutant p53 protein. Liu et al. (2014) stated that “most p53 mutations in human cancer are missense mutations, which result in the production of full length mutant p53 proteins”. 10–15% of TP53 mutations (termed as ‘disruptive mutations’) produce inactive proteins, and the remaining 85–90% of mutations form functioning proteins. Thus, mutations affect the ordinary function of p53 as a transcriptional factor, which imparts survival advantage to cancer cells and eventual down-regulation of apoptosis. Furthermore, several studies have demonstrated that mutant p53 is not only unable to perform tumor suppressor function, but also gains new oncogenic functions (termed as “gain-of-function”) (Oren and Rotter, 2010; Liu et al., 2014). A recent study on myelodysplastic syndromes reported the role of TP53 dysfunction in cancer outcomes and suggested its use in diagnosis (Bernard et al., 2020).
Caspase genes encode protease enzymes called caspases. They are named caspase because of their specific cysteine protease activity, and they have an essential role to play in apoptosis. Based on their pro-apoptotic functions, caspases are divided into two groups: initiators and effectors. The activated first group of initiator (or apical) caspases (-2, -8, -9, -10, and -11) in turn activate the second group of caspases (-3, -6, and -7) (Li and Yuan, 2008). At times, initiator caspases equally act as effector caspases to turn up weak suicide signals (Ghavami et al., 2009). Playing a crucial role in apoptosis, caspases are obliged to be targeted in uncontrolled cell division. P53 transcriptionally regulates the expression of some caspase genes (-2, -7, -8, and -9) (Ghavami et al., 2009); in cancer cells where p53 is mutated, expression of these caspases could be compromised, resulting in apoptotic protection.
According to Soung et al. (2005), various cancers showed the presence of mutated caspase-8 (mutations such as missense, in-frame deletion, frame-shift mutations in the coding sequences, mutations in the initiation codon, mutations in the introns, and mutation in the untranslated region), and there was a remarkable decrease in the apoptosis-inducing activity of all mutated caspase-8. For instance, missense mutations resulted in the substitution of amino acids, and frame-shift mutations caused premature terminations of caspase-8 protein synthesis (Ghavami et al., 2009; Li et al., 2014). Caspase-8 mutations were mostly detected in gastric cancers (Soung et al., 2005), though rarely observed in other types of cancer. Sun et al. (2007) identified a 6 bp deletion polymorphism (2652 6N del) in the promoter of the CASP8 (Caspase 8) gene, which lowers caspase-8 protein level, which is associated with 25% increased risk of lung-, esophageal-, stomach-, colorectal-, breast– and cervical– cancers. The absence of caspase-8 protein precursor in cancer cells was reported by Hopkins-Donaldson et al. (2003). CASP8 variants were observed in breast cancer (Cox et al., 2007). Yang et al. (2008) examined the association of CASP8 deletion polymorphism with pancreatic cancer. Silent mutations of CASP9 (Caspase-9) are found in gastric- and colorectal- carcinoma (Soung et al., 2006). A case-control study found an association of CASP9 polymorphism with multiple myeloma (Hosgood III et al., 2008).
Caspase-3 is an effector caspase playing an essential role in the execution phase of cell apoptosis. Soung et al. (2004) studied CASP3 (Caspase-3) mutation in carcinomas, myelomas, and lymphomas, suggesting the presence of CASP3 mutation in human cancer tissues. They reported CASP3 mutations in several cancers (stomach carcinoma, colon carcinoma, breast carcinoma, non-small cell lung cancer, laryngeal carcinomas, hepatocellular carcinoma, esophagus carcinomas, renal cell carcinomas, urinary bladder carcinomas, medulloblastomas, Wilms’ tumors, non-Hodgkin lymphomas, acute leukemias, and multiple myeloma) and compared seven healthy malignant tissues from the same patients; they found no CASP3 mutations in normal samples and concluded that CASP3 mutations arise somatically. They also found that CASP3 mutations consisted of missense mutations, silent mutations, mutations in the introns, and mutation in the untranslated regions in few cancers (stomach adenocarcinoma, lung cancer, colon cancer, hepatocellular carcinoma, and multiple myeloma). SCCHN (Squamous Cell Carcinoma of the Head and Neck) is one of the most common cancers in the world (Li et al., 2014), and association of CASP3 polymorphism with an increased risk of SCCHN has been reported (Chen et al., 2008; John et al., 2015). Caspase-7 is another effector caspase playing a central role in executing apoptosis. Some common cancers (such as SCCHN, Urinary bladder carcinoma, esophageal carcinoma, lung cancer, colon adenocarcinoma, and gastric carcinoma) displayed CASP7 (Caspase-7) somatic polymorphism (Soung et al., 2003; Yoo et al., 2004). CASP7 mutations found in this study were non-sense mutations causing premature termination of protein synthesis forming loss-of-function protein. Arg-43 is a highly conserved amino acid in caspase-7; alteration of Arg-43 decreases the protease function of caspase-7 that has a defect in the induction of apoptosis (Soung et al., 2003).
DR5 (Death Receptor 5)- The KILLER/DR5, also known as TRAILR2 (TNF-related apoptosis-inducing ligand receptor 2), has been identified as a potent inducer of apoptosis. KILLER/DR5 gene codes for cell surface receptors having a death domain, and over-expressed KILLER/DR5 gene is an effective apoptotic inducer (Wu et al., 2000). Binding of DR5 to its specific ligand TRAIL (TNF-related apoptosis-inducing ligand) mediates the apoptotic effects of TRAIL by activating caspase cascade (Griffith and Lynch, 1998). According to Takimoto and El-Deiry (2000), the expression of the KILLER/DR5 gene prevents cancer development and helps to carry out p53-induced apoptosis in cells with wild type p53, but this gene is affected in cells with inhibited p53 function. They also suggested that KILLER/DR5 alterations have a role in tumorigenesis.
Park et al. (2001) showed frequent allelic loss of KILLER/DR5 in cancers, mostly in gastric cancer. There is accumulating evidence suggesting that the mutation of the primary structure of DR5 might be one of the possible mechanisms that disrupt apoptosis in tumor cells. Cancer cells have escaping mechanisms from DR5-mediated apoptosis such as by imitating the expression of DR5 receptor, by loss of DR5 expression, overexpression of inhibitory proteins like FLICE (FADD-like Interleukin-1beta- Converting Enzyme), and mutation in structural genes of DR5 (Park et al., 2001; Karbasi et al., 2015). By analyzing the genetic alterations of KILLER/DR5 in a number of gastric cancers, Park et al. (2001) found the loss of function of KILLER/DR5 mutants. Further, they found all the mutants inhibited apoptotic cell death in transfection studies, suggesting that inactivation of KILLER/DR5 due to mutations might be one of the possible escaping mechanisms against KILLER/DR5 (extrinsic) mediated apoptosis, and this might contribute to the development of gastric cancers. Pai et al. (1998) performed a sequence analysis of KILLER/ DR5 and found mutations localized to the functional cytoplasmic death domain; alterations in this domain resulted in a truncated protein. This KILLER/DR5 mutation was also present in the germline of the affected patient. Gene coding for DR5 was found on chromosome 8p21-22 (MacFarlane et al., 1997), and several cancers such as gastric cancer (Yustein et al., 1999), lung cancer (Sug Hyung Lee et al., 1999), head and neck cancer (El-Naggar et al., 1998) showed frequent allelic losses in this chromosome.
FAS gene-FAS codes for FAS receptor (containing death domain), which is also known as CD95 (Cluster of Differentiation 95) or APO-1(Apoptosis Antigen-1) (Wajant, 2002). In the extrinsic pathway, FAS ligand binds to FAS receptor or TNF ligand binds with TNF receptor, resulting in binding of TRADD (TNF Receptor type 1-Associated Death Domain) with FADD, and RIP (Receptor Interacting Protein) (Kelliher et al., 1998; Wajant, 2002). Subsequently, the FADD–procaspase-8 association leads to the formation of DISC (Wang et al., 2010). Apart from its crucial role in apoptosis, FAS was observed to have non-apoptotic functions such as differentiation or proliferation of the cell and facilitating the pathogenesis of various malignancies (Krammer, 2000). Edathara et al. (2016) found that FAS gene polymorphism discouraged the apoptotic activity of its receptor, and this altered FAS receptor was associated with tumorigenesis. Further study by Cavalcante et al. (2019) found an association of FAS polymorphisms with ATL (Adult T-cell Leukemia). According to O’Reilly et al. (2009), a mutation in FASl (FAS ligand) prevents its binding to the receptor and retracts the function of membrane-bound FASl and soluble FASl. Studies reported that membrane-bound FASl-FAS and soluble FASl-FAS can enhance the development of cancer (Kaufmann et al., 2012; Waring and Mullbacher, 1999).
APAF-1 is trans-activated by p53 (Steele et al., 1998; Soengas et al., 2001). This gene codes for a cytoplasmic protein APAF-1. APAF-1 acts downstream of p53 to induce apoptosis. It forms one of the important units, “apoptosome”, in the apoptosis process, which contains a CARD (Caspase Recruitment Domain). The apoptosome binds to Procaspase-9, cleaves it, and releases caspase-9 (Shakeri et al., 2017). Salvesen and Duckett (2002) pointed out that despite the current understanding of the mitochondrial pathway of apoptosis, many details are yet to be described clearly, such as the molecular mechanism of APAF-1 oligomerization and activation of caspase-9 by APAF-1.
A study on mice reported APAF-1 deficient cells displayed resistance to apoptotic stimuli and showed reduced apoptosis (Yoshida, 2003). Hence, APAF-1 is considered an essential tumor suppressor downstream of p53. A study on drug-resistant tumor cells showed that anti-cancer drugs released cytochrome-c but failed to activate caspase-9; APAF-1 expression was down-regulated in these drug-resistant malignant melanomas, suggesting that impairment in APAF-1 provides proliferative benefit to malignant cells (Soengas et al., 2001). Few other studies also proved the role of APAF-1 in tumorigenicity (Soengas et al., 1999; Paik et al., 2007), while the loss of APAF-1 expression was reported in several types of cancers, such as colorectal cancer (Zlobec et al., 2007), gastric cancer (Wang et al., 2007), bladder cancer (Hinz et al., 2007), and melanoma (Dai et al., 2004).
A study on human neuroblastoma cell lines found that overexpression of miRNA-3613-3p decreased transcription of APAF-1 mRNA, resulting in down-regulation of APAF-1, which indicates that the key molecule inhibiting apoptosis in BE(2)-C human neuroblastoma cells is APAF-1 (Nowak et al., 2018).
Proto-oncogenes are highly regulated protein-coding genes regulating cell growth and cell survival signals (Wang et al., 2018). Proto-oncogenes can be altered by chromosomal translocation, mutation, gene amplification, or retroviral insertion. If it gets mutated, it becomes oncogene and contributes to cancerogenesis, i.e., gain-of-function mutation (McDuff and Turner, 2011). Oncogenes abrogate cell cycle checkpoints, thus promote irregular cell proliferation (Lam and Schmidt, 2012).
Proto-oncogene MYC codes for multifunctional phosphoprotein that acts as a transcription factor and plays important role in cell cycle progression, apoptosis, and cellular transformation. According to a study, MYC trans-activates its target genes to deploy its oncogenic effect (Grandori et al., 2005; Hoffman and Liebermann, 2008). Miller et al. (2012) found that mediators of metabolism, biosynthesis, and cell cycle progression are genes targeted by MYC, and irregularity in MYC expression leads to uncontrolled cell growth, uncontrolled division, and metastasis. MYC is frequently aberrated in human cancers, and overexpression of MYC confers the progression of 40% of tumors (Mohamed, 2017). Various genetic alterations, such as chromosomal translocations, amplifications, point mutations, and epigenetic reprogramming, are involved in the deregulation of MYC expression.
Studies found the association of translocations and/or amplification of MYC with Burkitt lymphoma, follicular lymphoma, mantle cell lymphoma, hematopoietic and non-hematopoietic tumors (like lung, breast, colon, and prostate cancers), and multiple myeloma; insertional mutagenesis in avian leucosis virus (ALV)-induced hematopoietic tumors were also observed (Mohamed, 2017; Nguyen et al., 2017).
Non-protein coding microRNAs (miRNA) are also MYC target genes; MYC represses miRNA-15A/miRNA16-1 (tumor suppressor) and miR-34 (Apoptotic regulator) (O’Donnell et al., 2005). This MYC-miRNA interaction promotes malignant phenotype (Jackstadt and Hermeking, 2015) (Details are given under “Proto-oncogene”). MYC plays an important role in mitochondrial biogenesis by inducing nuclear-encoded mitochondrial genes and by binding to the promoters of genes encoding proteins involved in mitochondrial function. Additionally, the genes involved with mitochondrial biogenesis are among the MYC target genes (Gao et al., 2009). The MYC oncogene contributes to the genesis of many human malignancies (Wang et al., 2014).
miRNAs are non-coding RNAs made up of approximately 22 nucleotides (Hwang and Mendell, 2006), which were first discovered in C. elegans (Bagga et al., 2005). Later on, human genome studies discovered more than 300 miRNAs and estimated up to 1000 miRNA genes (Hwang and Mendell, 2006). MicroRNAs are involved in post-transcriptional gene regulation (Nowak et al., 2018) and important mediators in critical cellular pathways. miRNAs function by base-pairing with their complementary sites within 3’UTR of their target mRNAs (Hwang and Mendell, 2006; Cui and Placzek, 2018). Apart from playing an important role in cancer progression (Slattery et al., 2018), miRNAs act as regulators in crucial cellular events like cell proliferation, cell differentiation, and cell death during normal development. Genomic studies of C. elegans and Drosophila suggested that miRNAs have a crucial role in regulating cancer-relevant cellular phenotypes (Hwang and Mendell, 2006). miRNAs influence apoptosis either as an anti-apoptotic molecule (targeting pro-apoptotic mRNAs) or as tumor-suppressors (by targeting anti-apoptotic mRNAs) (Lima et al., 2011). Likewise, some miRNAs function as oncogenes, while others behave as tumor suppressor genes in a cell-typed manner (Mohamed, 2017). miRNAs with pro-proliferative and anti-apoptotic activity likely promote oncogenesis and thus may be overexpressed in cancer cells.
Abnormal miRNA expressions were observed to be very common in malignancies. In the study on Chronic Lymphocytic Leukemia (B-CLL), Cimmino et al. (2005) had found that the most common chromosomal abnormality in this disorder was the deletion of chromosome 13q14. They also found that the tumor suppressor activity was imparted by miRNAs (specifically miR-15a & miR-16-1) as they were localized in the chromosomal deleted regions in B-CLL patients. They concluded that, in the human genome, miRNAs were mostly located at sites that are frequently amplified, deleted, or rearranged in cancer. Dysregulation of miRNA expression was observed in other cancer types as well, such as Burkitt’s lymphoma, colorectal cancer, lung cancer, breast cancer, and glioblastoma (Hwang and Mendell, 2006).
miRNAs are targets of the MYC gene; it has been established that MYC regulates the expression of many miRNAs leading to their repression. Concurrently, MYC is also regulated by miRNAs, and the combined effect of sustained MYC expression and repressed miRNAs regulates oncogenesis by MYC (Chang et al., 2008). For instance, MYC up-regulates expression of a set of miRNAs (miR-17-92 clusters) (O’Donnell et al., 2005; Mohamed, 2017); from this cluster, miR-19 was identified as the key oncogenic factor (Wang et al., 2014), which is commonly amplified in several human cancers such as carcinomas, medulloblastomas, neuroblastomas, B-cell lymphoma, and several subtypes of aggressive lymphomas, thus suggesting that miR-17-92 might contribute to the oncogenic properties of MYC.
miRNAs also display their oncogenic effect by inhibiting apoptosis, which is attained by down-regulation of TP53, E2F1 (E2 Factor transcriptional factor 1), and PTEN (Phosphate and Tensin homolog), leading to the activation of AKT (protein kinase B) pathway and eventual inhibition of apoptosis (Lima et al., 2011). The onset of cancer in eµ-MYC B-cell lymphoma can be dramatically accelerated by the insertion of a truncated miR-17 cluster (eµ-MYC transgenic mice develop B-cell lymphoma late in life). It is also found that miRNAs expressing lymphomas show a high mitotic index with lesser apoptosis (Wang et al., 2014). A decreased level of apoptosis was observed in the eµ-MYC miRNA-expressing tumors. E2F1 is a pro-apoptotic factor, which is transcriptionally activated by c-MYC (Fernandez et al., 2003); at the same time, its translational efficiency is compromised by miR-17. This simultaneous effect of c-MYC and miR-17 modulates the activity of E2F1 to disapprove apoptosis and favors proliferation (Coller et al., 2007). miR-21 is another member of the family displaying anti-apoptotic activity; overexpression of this miRNA was observed in glioblastoma tumor cell-lines (Chan et al., 2005) and breast cancer tissue (Kumar et al., 2013).
The term engulfment is used to describe the removal of apoptotic cells (Hsu and Wu, 2010). An instant removal of apoptotic cells is very crucial to ensure that cells induced to undergo apoptosis must die rather than recovering themselves after the initial stages of apoptosis. Engulfment of an apoptotic cell is different from phagocytosis as it does not evoke inflammatory response because cells being removed carry self-antigens; therefore, apoptosis is commonly termed as “immunologically silent” cell death.
Apoptotic clearance is a complex process accomplished in multiple steps with the help of abundant surface receptors and signaling molecules. The “find-me” signals sent by the apoptotic cells are sensed by the engulfing cells which recognize the “eat-me” signals on the apoptotic cell surface; this leads to intracellular signaling, causing engulfing cup formation around the apoptotic cell which is then ingested before its processing (Ravichandran and Lorenz, 2007; Elliott and Ravichandran, 2010). Administration of antitumor chemotherapeutics mostly induces apoptosis of tumor cells, efficient engulfment of apoptotic cells, and the characteristic release of anti-inflammatory mediators such as TGFβ (the antitumor immune response is suppressed by the encounter of TGFβ with eat-me signals) (Fond and Ravichandran, 2016).
Mutations in engulfment genes enhance the frequency of cell survival. It appears feasible that apoptotic cell clearance could have a profound impact on carcinogenesis, but there are very few genetic studies to prove the specific role of engulfment signaling pathways in tumorigenesis. Although engulfment of apoptotic cells has been extensively studied, much less is known about how mutations in engulfment genes provide a proliferative advantage to dying apoptotic cells and how do they assist cancer progression. With the recent discoveries that apoptotic cells release many “find-me” factors that attract phagocytes towards apoptotic cells, new insights have been gained on the connection of cell clearance with tumorigenesis (Elliott and Ravichandran, 2010).
Studies on Burkitt lymphoma determined how macrophages sense apoptotic tumor cells and eventually engulf them, and how this mechanism impacts tumor progression (Truman et al., 2017; Ogden et al., 2019). By studying fractalkine receptor-deficient mice, Truman et al. (2017) found that fractalkine is a potent macrophage attractant as they observed overexpression of fractalkine in neoplastic B cells. However, Ogden et al. (2019) found that macrophages produce IL-10 that seems to suppress tumor immunity, and simultaneously release B-cell survival factor that appears to promote tumor growth. Few studies reported that chronic inflammation is a key factor in tumorigenesis, but the removal of apoptotic cells does not elicit an immune response; hence, its role in cancer progression remains mysterious (Condeelis and Pollard, 2006).
Post translational modification
Cancer cells also employ (or distort the regular) post-translational modifications of apoptotic regulatory proteins to evade apoptosis. Either functions or stability (or both) of anti-apoptotic and/ or pro-apoptotic proteins are altered by post-translational modification processes such as phosphorylation and ubiquitination (Dai and Gu, 2010; Vucic et al., 2011; Kim et al., 2012). Kinases and acetyltransferases promote the post-translational modification of apoptotic modulators (Declercq et al., 2009).
Post-translational modification ubiquitination/ubiquitylation modifies protein by adding ubiquitin; it can be mono- or poly-ubiquitination (depending on the number of ubiquitin molecules linked) (Vucic et al., 2011). Ubiquitination plays important role in cellular processes like cell cycle regulation, and is a step-wise sequential action of ubiquitin-activating enzymes (E1), ubiquitin-conjugating enzymes (E2), and ubiquitin ligases (E3); each catalyzing transfer of covalent bond with ubiquitin, from one enzyme to other until it reaches the target protein. Improper ubiquitination affects protein degradation, sequentially causing protein accumulation, deregulated apoptosis, and cancer (Vucic et al., 2011).
Following apoptotic stimuli, the MCL-1 (anti-apoptotic) level drops, which accelerates MOMP. So far, studies have found five E3 ligases that ubiquitinate MCL-1 for degradation. BAX is an important pro-apoptotic BCL-2 family member, which has a direct impact on MOMP. In response to apoptotic stimuli, BAX moves towards the mitochondrial outer membrane to initiate MOMP. The recently identified p53RFP ligase (one of the E3 ligases) was found to ubiquitinate BAX on the mitochondrial outer membrane, leading to its degradation (Benard et al., 2010). Degradation of BAX is linked with high tumor grades in prostate cancer (Agrawal et al., 2008).
Phosphorylation is the most prominent post-translational modification, which can promote protein folding to improve stability as well as serve regulatory functions. Phosphorylation is found to be most frequently damaged in cancer. By phosphorylation of anti-apoptotic and/or pro-apoptotic proteins, cancer cells modify their functions and prevent apoptosis; or stabilize anti-apoptotic proteins and/or destabilize pro-apoptotic proteins to inhibit apoptosis. Phosphorylation can also influence ubiquitination by enabling a docking site for ubiquitin-E3-ligase (Kaleigh and Kurokawa, 2013).
In cancer cells, apoptosis is also down-regulated by inhibiting activation of APAF-1 by phosphorylation; for example, in prostate cancer, RSK (Ribosomal S6 Kinase) is overexpressed and it phosphorylates APAF-1, resulting in inhibition of its activation (Kim et al., 2012; Clark et al., 2005). Phosphorylation can alter the function of apoptotic regulators; if phosphorylation is carried out by a pro-apoptotic kinase JNK (c-Jun N-terminal Kinase), pro-apoptotic proteins are activated and anti-apoptotic proteins are inhibited; conversely, if phosphorylation is done by a pro-survival kinase AKT or ERK, pro-apoptotic proteins are inhibited and anti-apoptotic proteins are activated. Phosphorylation also plays a key role in the stability of MCL-1 protein. Phosphorylation of MCL-1 by JNK occurs at the T163 site, followed by other sites S155/ S159, eventually leading to proteasomal degradation of MCL-1. On the contrary, phosphorylation by AKT or ERK (Extracellular signal Regulated Kinase) occurs only at the T163 site imparting substantial stabilization to MCL-1. As the AKT/ERK level is high in cancer cells, they phosphorylate MCL-1 at only T163 (leading to MCL-1 stabilization), whereas in normal cells, the AKT level is lowered and JNK is activated, which phosphorylates MCL-1 at S155/S159/T163 and causes degradation of MCL-1 (Lei and Davis, 2003; Maurer et al., 2006; Kopper and Peták, 2008; Nifoussi et al., 2012). Similarly, phosphorylation of BIM by JNK promotes the pro-apoptotic activity of BIM, while ERK phosphorylation of BIM prevents the interaction between BIM and BAX, thereby inhibiting apoptosis (Kaleigh and Kurokawa, 2013).
The serine-threonine kinase AKT is also found to phosphorylate pro-apoptotic protein, thereby inhibiting apoptosis and exerting survival advantage to the cell (Datta et al., 1997). The BH-3 only protein PMAIP1 (Phorbol-12-myristate-13-Acetate-Induced Protein 1 - also known as Noxa), is a pro-apoptotic protein that interacts with MCL-1 and supports its degradation. In cancer cells where higher glucose levels are observed, CDK5 (cyclin-dependent kinase) is activated, which phosphorylates Noxa at Ser13, thereby stabilizing Noxa and suppressing its pro-apoptotic activity (Alves et al., 2006). Phosphorylation of BAD inhibits its pro-apoptotic function. When BAD is phosphorylated at Ser112, Ser136, and Ser155 sites, it provides a docking site for adaptor protein 14-3-3. The binding of adaptor protein at this site prevents the interactivity of BAD and BCL-XL, which results in a lack of MOMP, leading to apoptosis inhibition (Zha et al., 1996). Even caspases involved in apoptosis can be suppressed by their direct phosphorylation (Seifert and Clarke, 2009), or substrates of these caspases are phosphorylated, which protects these substrates from proteolytic cleavage by caspases, e.g., phosphorylation of BID by CK1/CK2 (Casein Kinases) protects BID from proteolytic degradation by caspase-8 (Kurokawa et al., 2008).
Some tumor cells use altered metabolism compared to normal cells, as they need to fulfill their nutrient requirement (to build new cells) from nutrient-deprived surroundings. However, recent studies suggested that altered metabolism is one of the ways cancer cells avoid apoptosis, as metabolic alterations modulate apoptotic machinery to protect the cell from death (Heiden et al., 2009; Matsuura et al., 2016; Pavlova and Thompson, 2016).
Tumor cells adapt their metabolism to their microenvironment and take up more biomolecules (such as glucose, lipids, nucleotides, amino acids) to meet the requirements of rapidly dividing cells. Thus, glucose uptake is intensified in cancer cells, and unlike normal cells (which depend on oxidative phosphorylation for energy generation), cancer cells ferment the glucose to lactate by aerobic glycolysis, even in presence of an ample amount of oxygen. This is called as “Warburg effect” (Heiden et al., 2009), and also enhanced anaerobic glycolysis is observed in cancer cells (Matsuura et al., 2016). Cellular metabolites such as glucose, lipids, amino acids, and nucleic acids; are found to regulate functions of pro-apoptotic and anti-apoptotic proteins (Kaleigh and Kurokawa, 2013). Among the metabolic pathways, glucose and lipid metabolism are specifically altered in cancer cells, which can impact the apoptotic pathways (Matsuura et al., 2016).
Glucose taken up by cells is converted to Glucose-6-phosphate with the help of hexokinases, which further undergoes either glycolysis or pentose phosphate pathway (PPP). If it undergoes glycolysis, energy is generated (32 ATP molecules) at the end via Kreb’s cycle and oxidative phosphorylation, whereas PPP generates fatty acids, ribose-5-phosphate (precursor for nucleotide), and NADPH (Nicotinamide Adenine Dinucleotide Phosphate). This is why PPP is considered to be essential for biomass production; as cancer cells (being highly proliferative cells) require much biomass, PPP is enhanced in cancer cells to meet their requirement. Solid tumor development increases the cell-density, which creates hypoxic conditions (low oxygen) due to which oxidative phosphorylation is suppressed and PPP is enhanced. In hypoxic conditions, gene expression is regulated by HIF-1 (Hypoxia-Inducible Factor-1), which is overexpressed in several types of cancers (Semenza, 2002). HIF-1 up-regulates its target genes, and many of these gene products encourage glycolysis. Studies showed the correlation between HIF-1 activity and tumor formation (Unruh et al., 2003; Fulda and Debatin, 2007). Besides, HIF-1 suppresses pro-apoptotic protein BID (Seenath et al., 2008).
AKT activated in cancer cells increases glycolysis and lactate production (Elstrom et al., 2004). AKT promotes the union of outer mitochondrial membrane and hexokinase in presence of glucose, which inhibits the release of cytochrome-c (Majewski et al., 2004). Moreover, AKT down-regulates MCL-1 degradation (discussed under phosphorylation section). In presence of glucose AKT suppresses the function of p53 (Yang et al., 2006), which indicates that AKT conceals PUMA induction in presence of glucose. Pro-apoptotic protein BAD is also a substrate for AKT (Datta et al., 1997).
p53 also plays role in cellular metabolism (Bensaad et al., 2006); the p53 protein represses the transcription of the GLUT1 (Dang, 1999), and loss of p53 activity leads to the Warburg effect (Levine and Puzio-Ku, 2010). TIGAR (TP53-Induced Glycolysis & Apoptosis Regulator) is a p53 target gene, which regulates glucose metabolism; it lowers the intracellular concentrations of fructose 2,6 bisphosphate and decreases glycolysis by switching to the PPP (Bensaad et al., 2006). TIGAR also has functional similarities to the bisphosphate domain of PFK-2/FBPase-2 (Phosphofructo Kinase-2/ Fructose Biphosphatase-2) in regulating glycolysis, and it protects the cell from ROS (Reactive Oxygen Species)-associated apoptosis (Levine and Puzio-Ku, 2010; Dai and Gu, 2010). As p53 is genetically mutated or functionally inactivated in the majority of cancers, these cells are expected to become highly glycolytic following loss of normal p53 activity (Muller and Vousden, 2013). Therefore, p53-mediated glucose metabolism is likely dysregulated in most cancers.
Glucose metabolism regulates Noxa phosphorylation (Lowman et al., 2010). In cancer cells, higher glucose levels aid in the phosphorylation of Noxa, leading to suppression of its pro-apoptotic activity (discussed in phosphorylation). NADPH produced in PPP contributes to metabolic inhibition of apoptosis by controlling the redox state of cytochrome-c, as reduced cytochrome cannot form apoptosome. However, oxidized cytochrome-c (oxidized by ROS) forms apoptosome. This shows that glucose metabolism negatively regulates apoptosis by producing NADPH (Chandra et al., 2006). Cancer cells alter metabolism to make maximum use of available elements to synthesize macromolecules needed in large amounts in rapidly proliferating cells; for example, catabolism and disposal of nitrogen and carbon are altered in cancer cells (Keshet et al., 2018). As in the urea cycle, enzymes ASL (Argininosuccinate Lyase) and ASS1 (Argininosuccinate Synthase-1) are altered, which leads to the accumulation of ornithine that is used to produce polyamines. Polyamines are nitrogenous compounds that can inhibit apoptosis and assist tumor progression. Even arginine acts as a precursor for polyamines in the tumor microenvironment (Pavlova and Thompson, 2016).
Ca++ also plays role in apoptosis and cancer progression. When there is a high concentration of Ca++ in ER, Ca++ are released from ER (Endoplasmic Reticulum) due to apoptotic stimuli; consequently, the level of Ca++ in mitochondria increases, which increases the permeability of the mitochondrial membrane and facilitates the flow of pro-apoptotic molecules into the cytoplasm (Chami et al., 2004; Jeong and Seol, 2008). Recent studies suggest that the connecting points between ER and mitochondria are the target sites for oncogenes and tumor suppressor genes. This Ca++ flow between ER and mitochondria determines the metabolism of cancer cells (Pedriali et al., 2017).
Mitochondrial metabolic alterations have been observed in cancer cells; alteration of the physiological mitochondrial metabolism might act as an oncogenic trigger (details under “Mitochondrial alterations”).
Recent studies have found that in most of the cancer cells, mitochondria drive the malignant phenotype (Riganti and Donadelli, 2019). As mentioned earlier, mitochondria play an important role in apoptosis.
In metabolic aspects of apoptosis, mitochondria provide energy for the apoptosis. As cancer cells are dividing rapidly, they need energy, and mitochondria are energy suppliers of the cell, hence in cancer cells, mitochondrial functions are accelerated. For example, mitochondria-related metabolic pathways are exploited to produce ATP and building blocks, mitochondrial ROS functions as signaling molecules for anti-apoptotic pathways. Alterations of specific mitochondrial enzymes exert oncogenic properties e.g. SDH (succinate dehydrogenase) loses its function in cancer cells because of inactivating mutations due to which its substrate succinate (oncometabolite) gets accumulated that promotes cell proliferation (Dalla Pozza et al., 2019). Hence loss in function of SDH aids cell proliferation, whereas overexpression of SDH suppresses cell proliferation and promotes apoptosis (Dong et al., 2008). Similarly, FH (fumarate hydratase) function is lost in cancer cells leading to the concentration of its substrate fumarate. How FH reduction leads to cancer is still elusive (Riganti and Donadelli, 2019).
Mitochondria have an essential role in mediating intrinsic apoptosis induced by diverse stimuli. In cancer cells, mitochondrial apoptosis is inhibited either by obstructing MOMP (by affecting BCL-2 family proteins and/or by loss of p53 function) or by inactivating caspases (by suppressing Apoptosome with the help of inhibitory phosphorylation of APAF-1, or by cytochrome c ubiquitination) (Lopez and Tait, 2015). As mitochondria play a substantial role in apoptosis, defects of mtDNA might have the capacity to alter cellular response towards apoptotic stimuli, which might provide a survival advantage to the cell. Studies have observed mtDNA mutations in several cancers, such as breast cancer, ovarian cancer, colorectal cancer, gastric cancer, hepatic cancer, esophageal cancer, pancreatic cancer, prostate cancer, thyroid cancer, renal cell carcinoma, hematologic malignancies, brain tumor, and other solid tumors (Carew and Huang, 2002). Also, the nuclei-encoded long non-coding RNA MEG3 (Maternally Expressed 3) was found in mitochondria enhancing apoptosis by reducing the expression of Bcl-2 and promoting the release of Cyt-c to the cytoplasm (Wang et al., 2015). ncRNAs (Non-coding RNAs) like ASncmtRNA-1 and ASncmtRNA-2 were found inducing apoptosis in several human and mouse tumor cell lines and involved in aging and replicative senescence in normal human cells (Landerer et al., 2011).
According to Chinnery and Turnbull (2000), different levels of heteroplasmy in mtDNA (within a cell, mitochondria with wild-mtDNA and mitochondria with mutant mtDNA co-exist) might have a significant importance in the expression of mtDNA genes and nuclear genes, including genes involved directly and indirectly in apoptosis and tumorigenesis. According to the studies on several diseases from a mito-epigenetic point of view, abnormal mtDNA methylation is associated with pathological conditions, and minute alterations in these ncRNAs might affect their apoptosis-inducing ability. It has been established that both intrinsic and extrinsic pathways require mitochondria in certain cell types, where death receptor proteins interact with mitochondria (Khosravi-Far, 2004; Zeng et al., 2012).
Escaping immune system scrutiny
Interactions between the immune system and malignant cells play an important role in tumorigenesis. Many mechanisms of tumor resistance to immune rejection have been studied, but very few of them involve immune escape by using alteration in apoptosis. As already mentioned, tumor cells somehow alter the expression of molecules involved in apoptosis signaling, which can result in resistance of tumor cells to immune-rejection (e.g., mutations in surface receptors like DR5 or FAS); paradoxically, molecules with altered expression might also serve as antigens of tumor cells (e.g., overexpressed non-mutated p53 in tumor cells may act as tumor antigen for T-cell) (Lee Peter et al., 1999).
Another strategy used by tumor cells to evade anti-tumor immunological response is by inducing apoptosis of host lymphocytes, termed as “tumor counterattack” (Igney and Krammer, 2002; Horton and Gajewski, 2018); for instance, in colon cancer, soluble mediator sFASl (released by membrane-bound FASl on colon cancer cell) induces apoptosis of host lymphocytes and allows tumor progression (Song et al., 2001). However, the induction of apoptosis by sFASl is not clearly understood and hence needs further elucidation.
Macrophages constitute the immune system, which forage for apoptotic bodies. They can induce apoptosis (Diez-Roux and Lang, 1997) and in most cases, macrophage cytotoxicity results in the death of target cells through apoptosis (Aliprantis et al., 1996). In an attempt to treat treatment-resistant tumor cells, Whitworth et al. (1990) suggested that systematic activation of tumoricidal properties of the host’s macrophages could help eliminate such resistant tumor cells. A study on laboratory animals clearly indicated that inhibition/elimination of macrophages disrupts apoptosis (Diez-Roux and Lang, 1997). Thus, it demonstrates that macrophages play important role in completing the apoptosis process, and inhibition or elimination of macrophages may hinder apoptosis.
Epigenetic modifications are DNA modifications that do not change the DNA sequence but affect gene activity. Epigenetics is greatly involved in gene dysregulation in cancer cells. Even mutation-free cells can be induced with pro-cancer characteristics, due to epigenetic changes (Sarkar et al., 2013). Progressive epigenetic alterations in advancing tumors result in an aberrant apoptotic pathway, thereby promoting tumor development (Pal et al., 2010). Epigenetics affects the pro- and anti-apoptotic protein expressions and provides apoptosis resistance to cancer cells (Hervouet et al., 2013). Recent progress in cancer epigenetics found modifications such as DNA methylation and hydroxymethylation, histone acetylation and methylation, nucleosome positioning, and changes in small ncRNAs and miRNAs (Sarkar et al., 2013; Qu et al., 2013). The most widely focused epigenetic changes in cancer are DNA methylation. Evidence suggests that the downregulation of apoptotic genes due to DNA methylation (e.g., aberrant DNA methylation patterns, hypermethylation, and hypomethylation) is one of the reasons for cancer cells to evade apoptosis (Hervouet et al., 2013; Gopisetty et al., 2006).
DNA methylation confers stable gene silencing thus plays important role in gene expression regulation and chromatin architecture (Qu et al., 2013). Methylation plays a crucial role in tumorigenesis. The DNA methylation results from a transfer of a CH3 (methyl group) from S-adenosyl methionine to the fifth carbon of cytosine in CpG (Cytosine triphosphate deoxynucleotide-phosphodiester bond-Guanine triphosphate deoxynucleotide) motifs. It occurs in about 50% CpG in the genome; the CpG-rich areas, referred to as CpG islands (CGI), are more frequently methylated than isolated CpG (Hervouet et al., 2013). DNA methylation between cell division is maintained by DNMT1 (DNA-methyltransferases 1) that is found to be highly expressed in cancer cells (Robertson, 2000). As reported, DNA methylation occurs either after replication on neosynthesized strands of DNA by DNMT1, or de novo on unmethylated DNA strands by DNMT3a and DNMT3b (Chen et al., 2003; Bird, 2002). Several apoptosis-linked genes are silenced by methylation, such as APAF1, p14, BCL2, EDNRB (Endothelin Receptor B), in bladder cancer DAPK (Death Associated Protein Kinase), PUMA (in lymphoma), FADD, TNFRSF25 (TNF Receptor Superfamily member 25), TNFRSF21 (TNF Receptor Superfamily member 21), TRAILR3 (TNF Receptor Superfamily member 10c), LITAF (Lipopolysaccharide Induced TNF Factor), BAX, CASP8, CASP3, CASP9, DR4 (Death Receptors 4), DR5; cell adherence CDH1 (Cadherin-1), CDH3 (Cadherin-3); tumor suppressor p53, RASSF1A (Ras Associated Domain Family-1 isoform A), BLU; cell cycle regulator CHFR (Checkpoint protein with Fork-head and Ring-finger domain), p16, p21, p27; DNA repair MGMT (Methyl Guanine Methyl Transferase), ARH1 (ADP-Ribosylarginine Hydrolase-1) (breast and ovarian cancer); cellular growth suppressor RIL (Reversion-Induced LIM domain); and TMS1 (Target of Methylation-induced Silencing)/ASC (Apoptosis associated Speck-like protein containing a CARD); each gene harbors a CpG island in its 5’ region (Hervouet et al., 2013; Friedrich et al., 2004; Martinez et al., 2007; Sarkar et al., 2013; Das et al., 2006; Boumber et al., 2007). The Knudson’s “two-hit hypothesis” proposes promoter methylation as a pathway of carcinogenesis (Jones and Laird, 1999). The promotors of TSG (tumor suppressor gene) in cancer cells are silenced epigenetically, which leads to tumor progression through apoptosis inhibition (Sarkar et al., 2013). It is important to note that methylation of CpG sites after transcription of initiation site does not block expression; thus, it may not lead to gene silencing, but it can be used as a tumor marker.
Aberrant methylation: Transcriptional silencing of TSG promotors by aberrant methylation is another way of cancer development (Das et al., 2006), and this is found to be a hallmark of gastric cancer (Qu et al., 2013). Expression of silenced TSGs can be regained by demethylation, which results in cell cycle inhibition and apoptosis (Mataga et al., 2012). Usually, aberrant methylation leads to loss of gene function, thus providing survival advantage to neoplastic cells (Jones and Baylin, 2002). DNA coding for miRNAs also undergoes aberrant methylation, causing abnormal levels of miRNAs in various cancers from solid tumors to blood cancers (Lujambio et al., 2007).
Hyper-methylation: DNA hypermethylation carries out pro-apoptotic gene silencing (Hatziapostolou and Iliopoulos, 2011). Many apoptotic genes were found to be hypermethylated in cancer cells; few are more specific to tumor type, while others are found to be commonly hypermethylated in cancer (Garrison et al., 2008). Hyper-methylation of the TSG promoter is the prominent epigenetic change in human neoplasia (Friedrich et al., 2004). In ovarian cancer, decreased expression of TRAIL due to hypermethylation was observed (Horak et al., 2005). Hypermethylation of apoptosis-related genes covers large promotor sequences, and it is found to be irregular (Mohammad et al., 2015). Further study is needed to design demethylations therapies to treat hypermethylation-dependent silencing of TSGs.
Hypomethylation: Hypomethylation abnormally activates oncogenes. DNA hypomethylation plays important role in tumor formation and progression (Ehrlich, 2009). However, less is known about apoptotic genes hypomethylation in cancer.
Tumor dormancy came into light when disease relapse was observed in several cancer patients, years after their surgical treatment. Tumor dormancy is crucial in understanding the metastatic disease. It is contrived that few cancer cells survive even after the supposedly successful treatment. These residual cancer cells may dwell in distant organs and contribute to disease recurrence; these are termed as disseminated tumor cells (DTCs), and a subgroup of DTCs circulating in the blood is termed as circulating tumor cells (CTCs) (Mohme et al., 2017; Gao et al., 2017). DTCs and CTCs get exposed to diverse microenvironments during dissemination and circulation, which constrains tumor growth; moreover, when they arrive on a secondary site, they must undergo apoptosis, but they maintain long-term survival. The key mechanism behind this resistance is found to be tumor dormancy (Townson and Chambers, 2006).
Dormancy can be of two types, dormant solitary cells and dormant micrometastases (Naumov et al., 2001; Naumov et al., 2002). In dormant micrometastases, there is a balance between proliferation and apoptosis, resulting in no net increase in tumor size; this may be due to angiogenic/immunologic dormancy (Kareva, 2016). On the other hand, dormant solitary cells are quiescent cells with no proliferation and apoptosis; this might be due to G0-G1 arrest, or they might release certain factors and modulate growth-related signaling pathways to accommodate themselves in the new microenvironment (Gao et al., 2017; Aguirre-Ghiso, 2007). However, they can exit dormancy and proliferate again, yet the clear mechanism as to how is it decided is unknown, which cell would continue to be dormant, which would undergo apoptosis, and which would start proliferating (Kareva, 2016). Thus, these dormant cells are potentially resistant to therapies that rely on cell cycle progression and apoptosis induction (Naumov et al., 2003). Studies were reported on detecting DTCs (in the bone marrow and lymph node) and CTCs (in the blood) in asymptomatic patients at the early stage so that it could be used as selection markers and monitoring tools (Mohme et al., 2017; Lianidou et al., 2015).
It is highly conceived that solitary dormant cells are responsible for tumor recurrence, but the clear mechanism is yet to be elucidated, regarding how this dormant cell localizes in the tissue for a long time, how it remains unnoticed by the immune system, and how does it give rise to large metastasis.
Cancer clonal theory and stem cells
Regarding the origin of cancer, the two basic theories being considered are cancer clonal theory and cancer stem cells (CSCs) hypothesis. According to the cancer clonal theory, random mutations lead to malignant transformation and subsequent clonal selection of cancer cells with the potential to regenerate tumor growth (Tysnes, 2010). The clonal theory is being widely accepted and is gaining attention for the development of therapies that attack individual clones with genetic survival traits. One of the areas of these beneficial mutations was found to be in apoptotic genes (Messerschmidt et al., 2017).
The CSCs hypothesis suggests that tumor arises from a small population of self-renewing stem-like cells with unlimited proliferation potential, which are dissimilar to differentiated progeny (Reya et al., 2001; Garcia et al., 2012). Both theories are acceptable, and it is worth noting that these two paradigms are not disparate, as CSCs go through clonal evolution (Barabé et al., 2007). The CSC model is currently well-established in the context of developing therapies. Studies suggest that CSCs are disseminated tumor cells (DTCs) holding stem cell-like phenotype, and they are dormant cells causing metastasis and relapse (Chaffer and Weinberg, 2011; Mascré et al., 2012; Naik et al., 2016; Kleffel and Schatton, 2013). The cancer stem cells’ apoptosis resistance may involve innate cellular mechanisms and extrinsic factors such as self-renewal, differentiation pathways (e.g., Wnt, Notch, and Hedgehog), tumor suppressor genes (PTEN, and TP53), asymmetric cell division, indefinite self-replication, genetic instability, epigenetics, chromatin changes, mobilization, and modified microenvironment (e.g., secreted survival factors, adhesion-mediated apoptosis resistance, and hypoxic conditions) (Barabé et al., 2007; Medina et al., 2009; Kruyt and Schuringa, 2010; Garcia et al., 2012). Overexpression of anti-apoptotic BCL-2 molecules is associated with CSCs (Garcia et al., 2012). Upregulation of ABC (ATP-Binding Cassette) transporters confers resistance to toxic agents and multi-drugs (Tysnes, 2010; Fulda and Pervaiz, 2010). In certain types of cancers, CSCs showed elevated expression of death receptors such as DR4 (in colon cancer) and DR5(in bladder cancer) (Signore et al., 2013; Sussman et al., 2007; Szliszka et al., 2009). CSCs are found to be TRAIL-resistant due to overexpressed c-FLIP (cellular FLICE Inhibitory Protein) (Piggott et al., 2013). Certain miRNAs are found to have an impact on CSCs fate; for example, miR-34 suppresses CSCs tumor formation by apoptosis regulation (Ji et al., 2009).
These studies are yet under investigation and need rigorous research, as the possibility of dedifferentiation of differentiated cells into CSCs cannot be denied. Evidence is needed to prove tumor formation from early progenitors (normal stem cells) (Kruyt and Schuringa, 2010).
It has already been mentioned that MOMP might be a point of no return, but cells could survive MOMP in few situations (Lopez and Tait, 2015). The early stages of apoptosis are reversible upon removal of the apoptotic stimulus. Many studies found that when apoptosis is induced in cancer cells via chemotherapy, apoptosis is reversed after initial response and the cancer cells are returned back; this is due to inefficient apoptosis (Geske et al., 2001; Kim and Tannock, 2005; Letai, 2008; Tang et al., 2009). Tang et al. (2009) showed that when various cancer cell lines (cervical carcinoma, skin cancer, liver cancer, breast cancer, prostate cancer) were exposed to apoptotic inducers (e.g., jasplakinolide, ethanol, staurosporine), they started to undergo apoptosis and displayed apoptotic features (such as mitochondrial dysfunction and fragmentation, nuclear condensation, cell shrinkage, and caspase activation); however, upon removal of these inducers, they reverted back apoptosis and survived. This study also found that these features of apoptosis were not point-of-no-return, as apoptosis could reverse even after these stages; however, it could not be reversed after nuclear fragmentation. According to Geske et al. (2001), DNA repair is activated early in the p53-induced apoptotic process, which might be involved in reversing the cell death pathway in some circumstances.
It is obvious from the present review that cancer cells apply multiple mechanisms to evade apoptosis, which influence cancer progression. Apoptosis is halted by cancer cells using various mechanisms simultaneously, but we cannot exclusively separate any mechanism. Apoptosis can be disturbed by mutation of the genes coding for proteins involved, metabolic alterations, mitochondrial changes, or axing of immunity. These provide a survival advantage to the cell and help cancer development. Few points deduced from the review of extensive research in this field are summarized below:
1. Collectively, the transcriptional network is found to be dysregulated in cancer cells, remarkably affecting the expression of apoptotic regulators, thus resulting in resistance to drugs and favors tumor development (Fig. 3). For instance:
• Cancer cells modulate BCL-2 and TP53 genes to escape apoptosis, and targeting these genes would be a valuable treatment strategy for a wide variety of tumors.
• Mutation in caspase genes is one of the ways to escape apoptosis by cancer cells.
• Cancer cells affect the DR5 receptor to escape apoptosis, which results in tumor cell survival.
• miRNAs play essential roles in many basic physiologic processes, including apoptosis; thus, abnormalities in miRNA function influence tumorigenesis.
• The expression of several key engulfment players is upregulated in neoplastic cells, but the importance of this observation is unclear.
• The cell clearance process can influence antitumor immune responses, and apoptotic cell clearance promotes tumorigenesis, or it suppresses tumorigenesis.
• APAF-1 is the key regulator molecule of apoptosis, and its suppression can mediate tumorigenesis.
2. Post-translational modifications play a critical role in the regulation of apoptosis by modulating apoptotic regulatory protein stabilities and functions.
3. Metabolic alterations in cancer cells affect apoptosis.
4. Mitochondria play important role in almost all aspects of apoptosis, such as gene expression regulation, metabolism, and release of pro-apoptotic proteins.
5. Macrophage cytotoxicity results in apoptosis in the target cell population and could treat cancers. Cancers can be treated by immune therapies that activate macrophages.
6. Dormant tumor cells are found to resist apoptosis and cause metastasis. Mechanisms through which each cell achieves dormancy might be different.
7. Reversibility of apoptosis plays role in cancer progression.
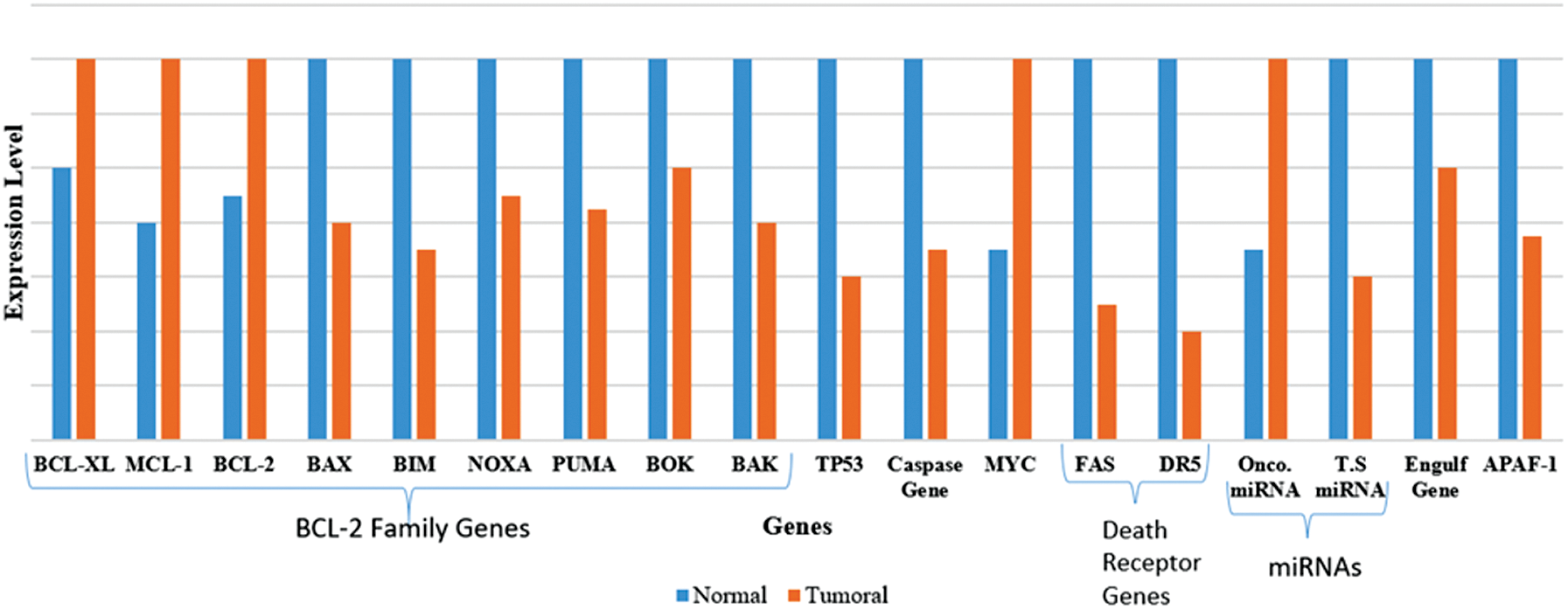
Figure 3: Gene expression level in normal and cancer cells (Karbasi et al., 2015).
As apoptosis is found to be affected in cancers and playing role in tumorigenesis, targeting apoptosis to treat cancer is a valid strategy. However, there are many challenges that need to be addressed, as summarized below:
• Apoptosis-based therapies: Although many therapies targeting apoptosis are already at the stage of human clinical trials, most of the therapeutic agents remain at the pre-clinical stage, due to the lack of their specificity and resistance developed towards them before the completion of treatment. Thus, future studies should focus on identifying the predominant apoptosis evasion mechanism specific to each type of cancer and designing therapeutic agents that can specifically target that mechanism to treat the particular type of cancer.
• Improving the efficacy of existing drugs: Apart from inducing apoptosis in normal cells, the existing drugs are resisted by cancer cells and have side-effects that limit the required dose for treatment. A further detailed study is needed to improve these drugs. As mutations in apoptotic genes also cause resistance, these mutations must be studied further as they become new therapeutic targets.
• Development of new drugs: Deciphering apoptosis knowledge into clinical practices will be remarkable as it will treat not only cancer but also a broad range of disorders such as Alzheimer’s, AIDS, and ischemia-associated injuries. Drugs that modulate either by altering actions of anti-apoptotic and pro-apoptotic proteins or by stumbling RNA transcription (for example by using mimetic of pro-apoptotic transcription factors), holds the potential for use in cancer therapy.
• Apoptotic stimuli in cancer treatment: Stimuli that causes DNA damage in the cell and how the cell responds to the DNA damage play important role in cell death and cell survival. If detailed mechanistic is studied, we can manipulate it to use it in treatments. For example, in p53-induced apoptosis, p53 repairs the DNA damage due to which the cell survives death. What are the factors that decide damage is irreparable and the cell must die? What death pathway to choose; apoptosis or inflammatory response? If how cells take these decisions is known, manipulation of these decisions would be helpful in treatment.
• A detailed study of macrophages: It has been suggested to treat cancer by activating the tumoricidal properties of macrophages. We need to find out a detailed model of macrophage subsets that are actually involved in tumor killing as these subsets possess therapeutic importance.
• Tumor counterattack: Rigorous work is needed to find out the specific and unique antigens displayed by cancer cells so that these antigens can be targeted. Tumor cells display a counterattack which is killing tumor-infiltrating lymphocytes by expressing apoptosis inducing ligand CD95L. Extended research on tumor counterattack is needed to use this phenomenon in treatment.
• Mitochondrial alteration: As mitochondria play important role in apoptosis and aberration in mitochondrial functions facilitates tumorigenesis, many things in mitochondria are yet to be explored; for example, small molecules, used as a sensor in response to different stimuli, must be found out.
• Use of genetic engineering in therapies: As genetic engineering is rapidly advancing, we need to explore the potential means of employing it in cancer treatment, such as introducing anti-oncogene, replacing altered or mutated genes, cutting-off mutated genes, repairing mutation by insertion or deletion of base-pairs.
• Further studies are needed to find out, if genes coding for surface receptors and signaling molecules involved in cell clearance, get mutated because of which cell clearance will fail, then what role this failure will play in tumor progression.
• Metabolic alteration: Further studies are needed to completely elucidate the link between metabolic alteration and apoptosis and its connection to tumorigenesis.
• Immunity escape: Detailed study is required to find how tumor cells alter the immune system to escape apoptosis.
• Cancer dormancy: Rigorous research is needed to explore mechanisms of cancer dormancy and to find specific markers to detect dormant cells, as it has prime clinical importance in metastasis treatment. A clear understanding of chemo-resistant mechanisms by dormant cells might completely change our reflection on tumorigenesis, tumor diagnosis, and their treatment.
A comprehensive review of the various mechanisms used by cancer cells to evade apoptosis is presented. Apoptosis is found to be frequently deregulated in almost all types of cancers studied so far. There has been dramatic progress to explore apoptosis evasion mechanisms by cancer cells, which attracted researchers and health professionals. As the world is facing threats of cancer, there is a growing demand for successful cancer treatment. Killing a cancer cell is the best cancer treatment, for which induction of apoptosis can be a remarkably effective therapy. As most of the compounds that successfully induce apoptosis are usually non-toxic to normal cells, they can be used in cancer therapies. Though many existing cancer therapies treat cancers by inducing apoptosis either by activating apoptotic pathway or by removing apoptosis inhibitors, they are not fully successful with several cancer types and different stages of cancer; moreover, cancer cells gradually develop resistance towards them and block apoptosis induced by these therapeutic agents. The cancer cells’ response to these therapies depends on the differences in apoptosis evasion methods and molecular strategies used to escape apoptosis.
Thus, to provide new molecular targets for future therapeutic approaches, a detailed study of apoptosis evasion mechanisms is needed, which involves identifying the escape mechanism predominant in a particular type of cancer and its stage of occurrence. We can develop specifically targeting gene therapies; for example, if it is found that a particular anti-oncogene has been altered to a high extent or lost in a certain type of cancer, introducing oncogene to normal chromosome can be achieved. Moreover, as cancer cells use multiple mechanisms to evade apoptosis, we need agents that can alter these multiple pathways and treat cancer without leaving a single reason for a cancer cell to survive.
Author Contribution: The author confirms sole responsibility for the following: study conception and design, data collection, analysis and interpretation of results, and manuscript preparation.
Ethics Approval: This article does not contain any experiment that involve human subjects, animals, or plants.
Availability of Data and Materials: Studies reviewed during this study are included in the manuscript.
Funding Statement: The author received no specific funding for this study.
Conflicts of Interest: The author declares that there are no conflicts of interest to report regarding the present study.
Adams JM, Cory S (2007). Bcl-2-regulated apoptosis: mechanism and therapeutic potential. Current Opinion in Immunology 19: 488–496. DOI 10.1016/j.coi.2007.05.004. [Google Scholar] [CrossRef]
Agrawal SG, Liu FT, Wiseman C, Shirali S, Liu H, Lillington D, Du MQ, Syndercombe-Court D, Newland AC, Gribben JG, Jia L (2008). Increased proteasomal degradation of Bax is a common feature of poor prognosis chronic lymphocytic leukemia. Blood 111: 2790–2796. DOI 10.1182/blood-2007-10-110460. [Google Scholar] [CrossRef]
Aguirre-Ghiso JA (2007). Models, mechanisms and clinical evidence for cancer dormancy. Nature Reviews Cancer 7: 834–846. DOI 10.1038/nrc2256. [Google Scholar] [CrossRef]
Aleksakhina SN, Kashyap A, Imyanitov EN (2019). Mechanisms of acquired tumor drug resistance. Biochimica et Biophysica Acta-Reviews on Cancer 1872: 188310. DOI 10.1016/j.bbcan.2019.188310. [Google Scholar] [CrossRef]
Aliprantis AO, Diez-Roux G, Mulder LCF, Zychlinsky A, Lang RA (1996). Do macrophages kill through apoptosis? Immunology Today 17: 573–576. DOI 10.1016/S0167-5699(96)10071-2. [Google Scholar] [CrossRef]
Alves NL, Derks IAM, Berk E, Spijker R, van Lier RAW, Eldering E (2006). The Noxa/Mcl-1 axis regulates susceptibility to apoptosis under glucose limitation in dividing T cells. Immunity 24: 703–716. DOI 10.1016/j.immuni.2006.03.018. [Google Scholar] [CrossRef]
Bagga S, Bracht J, Hunter S, Massirer K, Holtz J, Eachus R, Pasquinelli AE (2005). Regulation by let-7 and lin-4 miRNAs results in target mRNA degradation. Cell 122: 553–563. DOI 10.1016/j.cell.2005.07.031. [Google Scholar] [CrossRef]
Barabé F, Kennedy JA, Hope KJ, Dick JE (2007). Modeling the initiation and progression of human acute leukemia in mice. Science 316: 600–604. DOI 10.1126/science.1139851. [Google Scholar] [CrossRef]
Bean GR (2014). PUMA and BIM are required for oncogene inactivation-induced apoptosis and anoikis (Doctoral Thesis). Graduate School of Arts and Sciences, Washington University in St. Louis. [Google Scholar]
Benard G, Neutzner A, Peng G, Wang C, Livak F, Youle RJ, Karbowski M (2010). IBRDC2, an IBR-type E3 ubiquitin ligase, is a regulatory factor for Bax and apoptosis activation. EMBO Journal 29: 1458–1471. DOI 10.1038/emboj.2010.39. [Google Scholar] [CrossRef]
Bensaad K, Tsuruta A, Selak MA, Vidal MNC, Nakano K, Bartrons R, Gottlieb E, Vousden KH (2006). TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 126: 107–120. DOI 10.1016/j.cell.2006.05.036. [Google Scholar] [CrossRef]
Bernard E, Nannya Y, Hasserjian RP, Devlin SM, Tuechler H, Medina-martinez JS, Yoshizato T, Shiozawa Y, Saiki R, Malcovati L, Levine MF, Arango JE, Zhou Y, Sole F, Cargo CA, Haase D, Creignou M, Germing U, Zhang Y, Gundem G, Sarian A, van de Loosdrecht AA, Jadersten M, Tobiasson M, Kosmider O, Follo MY, Thol F, Pinheiro RF, Santini V, Kotsianidis I, Boultwood J, Santos FPS, Schanz J, Kasahara S, Ishikawa T, Tsurumi H, Takaori-Kondo A, Kiguchi T, Polprasert C, Bennett JM, Klimek VM, Savona MR, Belickova M, Ganster C, Palomo L, Sanz G, Ades L, Porta MGD, Smith AG, Werner Y, Patel M, Viale A, Vanness K, Neuberg DS, Stevenson KE, Menghrajani K, Bolton KL, Fenaux P, Pellagatti A, Platzbecker U, Henser M, Valent P, Chiba S, Miyazaki Y, Finelli C, Voso MT, Shih LY, Fontenay M, Jansen JH, Cervera J, Atsuta Y, Gattermann N, Ebert BL, Bejar R, Greenberg PL, Cazzola M, Hellstrom-Lindberg E, Ogawa S, Papaemmanuil E (2020). Implications of TP53 allelic state for genome stability, clinical presentation and outcomes in myelodysplastic syndromes. Nature Medicine 26: 1549–1556. DOI 10.1038/s41591-020-1008-z. [Google Scholar] [CrossRef]
Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, Mc Henry KT, Pinchback RM, Ligon AH, Cho YJ, Haery L, Greulich H, Reich M, Winckler W, Lawrence MS, Weir BA, Tanaka KE, Chiang DY, Bass AJ, Loo A, Hoffman C, Prensner J, Liefeld T, Gao Q, Yecies D, Signoretti S, Maher E, Kaye FJ, Sasaki H, Tepper JE, Fletcher JA, Tabernero J, Baselga J, Tsao MS, Demichelis F, Rubin MA, Janne PA, Daly MJ, Nucera C, Levine RL, Ebert BL, Gabriel S, Rustgi AK, Antonescu CR, Ladanyi M, Letai A, Garraway LA, Loda M, Beer DG, True LD, Okamoto A, Pomeroy SL, Singer S, Golub TR, Lander ES, Getz G, Seller WR, Meyerson M (2010). The landscape of somatic copy-number alteration across human cancers. Nature 463: 899–905. DOI 10.1038/nature08822. [Google Scholar] [CrossRef]
Bird A (2002). DNA methylation patterns and epigenetic memory. Genes & Development 16: 6–21. DOI 10.1101/gad.947102. [Google Scholar] [CrossRef]
Boumber YA, Kondo Y, Chen X, Shen L, Gharibyan V, Konishi K, Estey E, Kantarjian H, Garcia-Manero G, Issa JPJ (2007). RIL, a LIM gene on 5q31, is silenced by methylation in cancer and sensitizes cancer cells to apoptosis. Cancer Research 67: 1997–2005. DOI 10.1158/0008-5472.CAN-06-3093. [Google Scholar] [CrossRef]
Brimmell M, Mendiola R, Mangion J, Packham G (1998). BAX frameshift mutations in cell lines derived from human haemopoietic malignancies are associated with resistance to apoptosis and microsatellite instability. Oncogene 16: 1803–1812. DOI 10.1038/sj.onc.1201704. [Google Scholar] [CrossRef]
Carew JS, Huang P (2002). Mitochondrial defects in cancer. Molecular Cancer 1: 9. DOI 10.1186/1476-4598-1-9. [Google Scholar] [CrossRef]
Cavalcante GC, Schaan AP, Cabral GF, Santana-da-Silva MN, Pinto P, Vidal AF, Ribeiro-dos-Santos A (2019). A cell’s fate: An overview of the molecular biology and genetics of apoptosis. International Journal of Molecular Sciences 20: 4133. DOI 10.3390/ijms20174133. [Google Scholar] [CrossRef]
Cesari IM, Carvalho E, Figueiredo Rodrigues M, Mendonça BDS, Amôedo ND, Rumjanek FD (2014). Methyl jasmonate: Putative mechanisms of action on cancer cells cycle, metabolism, and apoptosis. International Journal of Cell Biology 2014: 1–25. DOI 10.1155/2014/572097. [Google Scholar] [CrossRef]
Chaffer CL, Weinberg RA (2011). A perspective on cancer cell metastasis. Science 331: 1559–1564. DOI 10.1126/science.1203543. [Google Scholar] [CrossRef]
Chami M, Prandini A, Campanella M, Pinton P, Szabadkai G, Reed JC, Rizzuto R (2004). Bcl-2 and Bax exert opposing effects on Ca++ signaling, which do not depend on their putative pore-forming region. Journal of Biological Chemistry 279: 54581–54589. DOI 10.1074/jbc.M409663200. [Google Scholar] [CrossRef]
Chan JA, Krichevsky AM, Kosik KS (2005). MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Research 65: 6029–6033. DOI 10.1158/0008-5472.CAN-05-0137. [Google Scholar] [CrossRef]
Chandra D, Bratton SB, Person MD, Tian Y, Martin AG, Ayres M, Fearnhead HO, Gandhi V, Tang DG (2006). Intracellular nucleotides act as critical prosurvival factors by binding to cytochrome C and inhibiting apoptosome. Cell 125: 1333–1346. DOI 10.1016/j.cell.2006.05.026. [Google Scholar] [CrossRef]
Chang TC, Yu D, Lee YS, Wentzel EA, Arking DE, West KM, Dang CV, Thomas-Tikhonenko A, Mendell JT (2008). Widespread microRNA repression by Myc contributes to tumorigenesis. Nature Genetics 40: 43–50. DOI 10.1038/ng.2007.30. [Google Scholar] [CrossRef]
Chen K, Zhao H, Hu Z, Wang L, Zhang W, Sturgis EM, Wei Q (2008). CASP3 polymorphisms and risk of squamous cell carcinoma of the head and neck. Cancer Prevention and Susceptibility 14: 6343–6350. [Google Scholar]
Chen T, Ueda Y, Dodge JE, Wang Z, Li E (2003). Establishment and maintenance of genomic methylation patterns in mouse embryonic stem cells by Dnmt3a and Dnmt3b. Molecular and Cellular Biology 23: 5594–5605. DOI 10.1128/MCB.23.16.5594-5605.2003. [Google Scholar] [CrossRef]
Chinnery PF, Turnbull DM (2000). Mitochondrial DNA mutations in the pathogenesis of human disease. Molecular Medicine Today 6: 425–432. DOI 10.1016/S1357-4310(00)01805-0. [Google Scholar] [CrossRef]
Chipuk JE, Bouchier-Hayes L, Green DR (2006). Mitochondrial outer membrane permeabilization during apoptosis: The innocent bystander scenario. Cell Death & Differentiation 13: 1396–1402. DOI 10.1038/sj.cdd.4401963. [Google Scholar] [CrossRef]
Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, Wojcik SE, Aqeilan RI, Zupo S, Dono M, Rassenti L, Alder H, Volinia S, Liu C, Kipps TJ, Negrini M, Croce CM (2005). miR-15 and miR-16 induce apoptosis by targeting BCL2. Proceedings of the National Academy of Sciences of the United States of America 102: 13944–13949. [Google Scholar]
Clark DE, Errington TM, Smith JA, Frierson HF, Weber MJ, Lannigan DA (2005). The serine/threonine protein kinase, p90 ribosomal S6 kinase, is an important regulator of prostate cancer cell proliferation. Cancer Research 65: 3108–3116. DOI 10.1158/0008-5472.CAN-04-3151. [Google Scholar] [CrossRef]
Coller HA, Forman JJ, Legesse-miller A (2007). ‘Myc’ed Messages’’: Myc induces transcription of E2F1 while inhibiting its translation via a microRNA polycistron. PLoS Genetics 3: e146. DOI 10.1371/journal.pgen.0030146. [Google Scholar] [CrossRef]
Condeelis J, Pollard JW (2006). Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell 124: 263–266. DOI 10.1016/j.cell.2006.01.007. [Google Scholar] [CrossRef]
Costa DB, Halmos B, Kumar A, Schumer ST, Huberman MS, Boggon TJ, Tenen DG, Kobayashi S (2007). BIM mediates EGFR tyrosine kinase inhibitor-induced apoptosis in lung cancers with oncogenic EGFR mutations. PLoS Medicine 4: e315. DOI 10.1371/journal.pmed.0040315. [Google Scholar] [CrossRef]
Cox A, Dunning AM, Balasubramanian S, Reed MWR, Pooley KA, Scollen S, Baynes C, Ponder BAJ, Chanock S, Lissowska J, brinton L, Peplonska B, Southey MC, Hopper JL, McCredie MRE, Giles GG, Fletcher O, Johnson N, Silva IS, Gibson L, Bojesen SE, Nordestgaard BG, Axelsson CK, Torres D, Hamann U, Justenhoven C, Brauch H, Chang-Claude J, Kropp S, Risch A, Wang-Gohrke S, Schurmann P, Bogdanova N, Dork T, Fagerholm R, Aaltonen K, Blomqvist C, Nevanlinna H, Seal S, Renwick A, Stratton MR, Rahman N, Sangrajrang S, Hughes D, Odefrey F, Brennan P, Spurdle A, Chenevix-Trench G, Beesley J, Mannermaa A, Hartikainen J, Kataja V, Kosma VM, Couch FJ, Olson JE, Goode EL, Broeks A, Schmidt MK, Hogervorst FBL, Veer LJV, Kang D, Yoo KY, Noh DY, Ahn SH, Wedren S, Hall P, Low YL, Liu J, Milne RL, Ribas G, Gonzalez-Neira A, Benitez J, Sigurdson AJ, Stredrick DL, Alexander BH, Struewing JP, Pharoah PDP, Easton DF (2007). A common coding variant in CASP8 is associated with breast cancer risk. Nature Genetics 39: 352–358. DOI 10.1038/ng1981. [Google Scholar] [CrossRef]
Cui J, Placzek WJ (2018). Post-transcriptional regulation of anti-apoptotic BCL2 family members. International Journal of Molecular Sciences 19: 308. DOI 10.3390/ijms19010308. [Google Scholar] [CrossRef]
Dai C, Gu W (2010). p53 post-translational modification: Eregulated in tumorigenesis. Trends in Molecular Medicine 16: 528–536. DOI 10.1016/j.molmed.2010.09.002. [Google Scholar] [CrossRef]
Dai DL, Martinka M, Bush JA, Li G (2004). Reduced Apaf-1 expression in human cutaneous melanomas. British Journal of Cancer 91: 1089–1095. DOI 10.1038/sj.bjc.6602092. [Google Scholar] [CrossRef]
Dalla Pozza E, Dando I, Pacchiana R, Liboi E, Scupoli MT, Donadelli M, Palmieri M (2020). Regulation of succinate dehydrogenase and role of succinate in cancer. Seminars in Cell and Developmental Biology 98: 4–14. DOI 10.1016/j.semcdb.2019.04.013. [Google Scholar] [CrossRef]
Dang CV (1999). c-Myc target genes involved in cell growth, apoptosis, and metabolism. Molecular and Cellular Biology 19: 1–11. DOI 10.1128/MCB.19.1.1. [Google Scholar] [CrossRef]
Das PM, Ramachandran K, VanWert J, Ferdinand L, Gopisetty G, Reis IM, Singal R (2006). Methylation mediated silencing of TMS1/ASC gene in prostate cancer. Molecular Cancer 5: 28. DOI 10.1186/1476-4598-5-28. [Google Scholar] [CrossRef]
Dasgupta A, Nomura M, Shuck R, Yustein J (2017). Cancer’s achilles’ heel: apoptosis and necroptosis to the rescue. International Journal of Molecular Sciences 18: 23. DOI 10.3390/ijms18010023. [Google Scholar] [CrossRef]
Datta SR, Dudek H, Xu T, Masters S, Haian F, Gotoh Y, Greenberg ME (1997). Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 91: 231–241. DOI 10.1016/S0092-8674(00)80405-5. [Google Scholar] [CrossRef]
Declercq W, Vanden Berghe T, Vandenabeele P (2009). RIP Kinases at the crossroads of cell death and survival. Cell 138: 229–232. DOI 10.1016/j.cell.2009.07.006. [Google Scholar] [CrossRef]
Diez-Roux G, Lang RA (1997). Macrophages induce apoptosis in normal cells in vivo. Development 124: 3633–3638. [Google Scholar]
Dong LF, Low P, Dyason JC, Wang XF, Prochazka L, Witting PK, Freeman R, Swettenham E, Valis K, Liu J, Zobalova R, Turanek J, Spitz DR, Domann FE, Scheffler IE, Ralph SJ, Neuzil J (2008). α-Tocopheryl succinate induces apoptosis by targeting ubiquinone-binding sites in mitochondrial respiratory complex II. Oncogene 27: 4324–4335. DOI 10.1038/onc.2008.69. [Google Scholar] [CrossRef]
Edathara PM, Gorre M, Kagita S, Vuree S, Cingeetham A, Nanchari SR, Meka PB, Annamaneni S, Digumarthi RR, Satti V (2016). Association of promoter polymorphisms of Fas –FasL genes with development of Chronic Myeloid Leukemia. Tumor Biology 37: 5475–5484. DOI 10.1007/s13277-015-4295-0. [Google Scholar] [CrossRef]
Ehrlich M (2009). DNA hypomethylation in cancer cells. Epigenomics 1: 239–259. DOI 10.2217/epi.09.33. [Google Scholar] [CrossRef]
El-Naggar AK, Coombes MM, Batsakis JG, Hong WK, Goepfert H, Kagan J (1998). Localization of chromosome 8p regions involved in early tumorigenesis of oral and laryngeal squamous carcinoma. Oncogene 16: 2983–2987. DOI 10.1038/sj.onc.1201808. [Google Scholar] [CrossRef]
Slee E A, Adrain C, Martin S J (2001). Executioner caspase-3, -6, and -7 perform distinct, non-redundant roles during the demolition phase of apoptosis. Journal of Biological Chemistry 276: 7320–7326. DOI 10.1074/jbc.M008363200. [Google Scholar] [CrossRef]
Elliott MR, Ravichandran KS (2010). Clearance of apoptotic cells: Implications in health and disease. Journal of Cell Biology 189: 1059–1070. DOI 10.1083/jcb.201004096. [Google Scholar] [CrossRef]
Elmore S (2016). Apoptosis: A review of programmed cell death. Toxicologic Pathology 35: 495–516. DOI 10.1080/01926230701320337. [Google Scholar] [CrossRef]
Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR, Zhuang H, Cinalli RM, Alavi A, Rudin CM, Thompson CB (2004). Akt stimulates aerobic glycolysis in cancer cells. Cancer Research 64: 3892–3899. DOI 10.1158/0008-5472.CAN-03-2904. [Google Scholar] [CrossRef]
Faqar-Uz-Zaman SF, Heinicke U, Meister MT, Vogler M, Fulda S (2018). BCL-xL-selective BH3 mimetic sensitizes rhabdomyosarcoma cells to chemotherapeutics by activation of the mitochondrial pathway of apoptosis. Cancer Letters 412: 131–142. DOI 10.1016/j.canlet.2017.09.025. [Google Scholar] [CrossRef]
Fernandez PC, Frank SR, Wang L, Schroeder M, Liu S, Greene J, Cocito A, Amati B (2003). Genomic targets of the human c-Myc protein. Genes & Development 17: 1115–1129. DOI 10.1101/gad.1067003. [Google Scholar] [CrossRef]
Fond AM, Ravichandran KS (2016). Clearance of dying cells by phagocytes: mechanisms and implications for disease pathogenesis. In: GregoryCD (ed.Apoptosis in Cancer Pathogenesis and Anti-Cancer Therapy -New Perspectives and Opportunities, pp. 25–50. London, UK: Springer Nature. [Google Scholar]
Freed-Pastor WA, Mizuno H, Zhao X, Langerød A, Moon SH, Rodriguez-Barrueco R, Barsotti A, Chicas A, Li W, Polotskaia A, Bissell MJ, Osborne TF, Tian B, Lowe SW, Silva JM, Borresen-Dale AL, Levine AJ, Bargonetti J, Prives C (2012). Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 148: 244–258. DOI 10.1016/j.cell.2011.12.017. [Google Scholar] [CrossRef]
Friedrich MG, Weisenberger DJ, Cheng JC, Chandrasoma S, Siegmund KD, Gonzalgo ML, Toma MI, Huland H, Yoo C, Tsai YC, Nichols PW, Bochner BH, Jones PA, Liang G (2004). Detection of methylated apoptosis-associated genes in urine sediments of bladder cancer patients. Clinical Cancer Research 10: 7457–7465. DOI 10.1158/1078-0432.CCR-04-0930. [Google Scholar] [CrossRef]
Fulda S (2010). Evasion of apoptosis as a cellular stress response in cancer. International Journal of Cell Biology 2010: 1–6. DOI 10.1155/2010/370835. [Google Scholar] [CrossRef]
Fulda S, Debatin K (2014). HIF-1-regulated glucose metabolism: a key to apoptosis resistance? Cell Cycle 6: 790–792. DOI 10.4161/cc.6.7.4084. [Google Scholar] [CrossRef]
Fulda S, Pervaiz S (2010). Apoptosis signaling in cancer stem cells. International Journal of Biochemistry and Cell Biology 42: 31–38. DOI 10.1016/j.biocel.2009.06.010. [Google Scholar] [CrossRef]
Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, Zeller KI, De Marzo AM, Eyk JEV, Mendell JT, Dang CV (2009). c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 458: 762–765. DOI 10.1038/nature07823. [Google Scholar] [CrossRef]
Gao XL, Zhang M, Tang YL, Liang XH (2017). Cancer cell dormancy: mechanisms and implications of cancer recurrence and metastasis. OncoTargets and Therapy 10: 5219–5228. DOI 10.2147/OTT.S140854. [Google Scholar] [CrossRef]
Garcia MA, Carrasco E, Ramirez A, Jimenez G, Lopez-Ruiz E, Peran M, Picon M, Compos J, Boulaiz H, Marchal JA (2012). Apoptosis as a therapeutic target in cancer and cancer stem cells: novel strategies and futures perspectives, Apoptosis and Medicine. London, UK: IntechOpen, 111–154. [Google Scholar]
Garrison SP, Jeffers JR, Yang C, Nilsson JA, Hall MA, Rehg JE, Yue W, Yu J, Zhang L, Onciu M, Sample JT, Cleveland JL, Zambetti GP (2008). Selection against PUMA gene expression in Myc-driven B-cell lymphomagenesis. Molecular and Cellular Biology 28: 5391–5402. DOI 10.1128/MCB.00907-07. [Google Scholar] [CrossRef]
Geske FJ, Lieberman R, Strange R, Gerschenson L (2001). Early stages of p53-induced apoptosis are reversible. Cell Death & Differentiation 8: 182–191. DOI 10.1038/sj.cdd.4400786. [Google Scholar] [CrossRef]
Ghavami S, Hashemi M, Ande SR, Yeganeh B, Xiao W, Eshraghi M, Bus CJ, Kadkhoda K, Wiechec E, Halayko AJ, Los M (2009). Apoptosis and cancer: mutations within caspase genes. Journal of Medical Genetics 46: 497–510. DOI 10.1136/jmg.2009.066944. [Google Scholar] [CrossRef]
Gopisetty G, Ramachandran K, Singal R (2006). DNA methylation and apoptosis. Molecular Immunology 43: 1729–1740. DOI 10.1016/j.molimm.2005.11.010. [Google Scholar] [CrossRef]
Grandori C, Gomez-Roman N, Felton-Edkins ZA, Ngouenet C, Galloway DA, Eisenman RN, White RJ (2005). c-Myc binds to human ribosomal DNA and stimulates transcription of rRNA genes by RNA polymerase I. Nature Cell Biology 7: 311–318. DOI 10.1038/ncb1224. [Google Scholar] [CrossRef]
Griffith TS, Lynch DH (1998). TRAIL: A molecule with multiple receptors and control mechanisms. Current Opinion in Immunology 10: 559–563. DOI 10.1016/S0952-7915(98)80224-0. [Google Scholar] [CrossRef]
Hatziapostolou M, Iliopoulos D (2011). Epigenetic aberrations during oncogenesis. Cellular and Molecular Life Sciences 68: 1681–1702. DOI 10.1007/s00018-010-0624-z. [Google Scholar] [CrossRef]
Heiden MGV, Cantley LC, Thompson CB (2009). Understanding the warburg effect: the metabolic requirements of cell proliferation. Science 324: 1029–1033. DOI 10.1126/science.1160809. [Google Scholar] [CrossRef]
Hervouet E, Cheray M, Vallette F, Cartron PF (2013). DNA methylation and apoptosis resistance in cancer cells. Cells 2: 545–573. DOI 10.3390/cells2030545. [Google Scholar] [CrossRef]
Hinz S, Kempkensteffen C, Weikert S, Schostak M, Schrader M, Miller K, Christoph F (2007). EZH2 polycomb transcriptional repressor expression correlates with methylation of the APAF-1 gene in superficial transitional cell carcinoma of the bladder. Tumor Biology 28: 151–157. DOI 10.1159/000103380. [Google Scholar] [CrossRef]
Hoeppner DJ, Hengartner MO, Schnabel R (2001). Engulfment genes cooperate with ced-3 to promote cell death in Caenorhabditis elegans. Nature 412: 202–206. DOI 10.1038/35084103. [Google Scholar] [CrossRef]
Hoffman B, Liebermann DA (2008). Apoptotic signaling by c-MYC. Oncogene 27: 6462–6472. DOI 10.1038/onc.2008.312. [Google Scholar] [CrossRef]
Hopkins-Donaldson S, Ziegler A, Kurtz S, Bigosch C, Kandioler D, Ludwig C, Zangemeister-Wittke U, Stahel R (2003). Silencing of death receptor and caspase-8 expression in small cell lung carcinoma cell lines and tumors by DNA methylation. Cell Death & Differentiation 10: 356–364. DOI 10.1038/sj.cdd.4401157. [Google Scholar] [CrossRef]
Horak P, Pils D, Haller G, Pribill I, Roessler M, Tomek S, Horvat R, Zeillinger R, Zielinski C, Krainer M (2005). Contribution of epigenetic silencing of tumor necrosis factor-related apoptosis inducing ligand receptor 1 (DR4) to TRAIL resistance and ovarian cancer. Molecular Cancer Research 3: 335–343. DOI 10.1158/1541-7786.MCR-04-0136. [Google Scholar] [CrossRef]
Horton BL, Gajewski TF (2018). Back from the dead: TIL apoptosis in cancer immune evasion. British Journal of Cancer 118: 309–311. DOI 10.1038/bjc.2017.483. [Google Scholar] [CrossRef]
Hosgood III HD, Baris D, Zhang Y, Zhu Y, Zheng T, Yeager M, Welch R, Zahm S, Chanock S, Rothman N, Lan Q (2008). Caspase polymorphisms and genetic susceptibility to multiple myeloma. Hematological Oncology 26: 148–151. DOI 10.1002/hon.852. [Google Scholar] [CrossRef]
Hsu TY, Wu YC (2010). Engulfment of apoptotic cells in C. elegans is mediated by integrin α/SRC signaling. Current Biology 20: 477–486. DOI 10.1016/j.cub.2010.01.062. [Google Scholar] [CrossRef]
Hübner A, Barrett T, Flavell RA, Davis RJ (2008). Multisite phosphorylation regulates Bim stability and apoptotic activity. Molecular Cell 30: 415–425. DOI 10.1016/j.molcel.2008.03.025. [Google Scholar] [CrossRef]
Hwang HW, Mendell JT (2006). MicroRNAs in cell proliferation, cell death, and tumorigenesis. British Journal of Cancer 94: 776–780. DOI 10.1038/sj.bjc.6603023. [Google Scholar] [CrossRef]
Igney FH, Krammer PH (2002). Immune escape of tumors: apoptosis resistance and tumor counterattack. Journal of Leukocyte Biology 71: 907–920. [Google Scholar]
Jackstadt R, Hermeking H (2015). MicroRNAs as regulators and mediators of c-MYC function. Biochimica et Biophysica Acta 1849: 544–553. DOI 10.1016/j.bbagrm.2014.04.003. [Google Scholar] [CrossRef]
Jain MV, Paczulla AM, Klonisch T, Dimgba FN, Rao SB, Roberg K, Schweizer F, Lengerke C, Davoodpour P, Palicharla VR, Maddika S, Los M (2013). Interconnections between apoptotic, autophagic and necrotic pathways: Implications for cancer therapy development. Journal of Cellular and Molecular Medicine 17: 12–29. DOI 10.1111/jcmm.12001. [Google Scholar] [CrossRef]
Jan R, Chaudhry GS (2019). Understanding apoptosis and apoptotic pathways targeted cancer therapeutics. Advanced Pharmaceutical Bulletin 9: 205–218. DOI 10.15171/apb.2019.024. [Google Scholar] [CrossRef]
Jeong SY, Seol DW (2008). The role of mitochondria in apoptosis. Journal of Biochemistry and Molecular Biology 41: 11–22. [Google Scholar]
Ji Q, Hao X, Zhang M, Tang W, Meng Y, Li L, Xiang D, DeSano JT, Bommer GT, Fan D, Fearon ER, Lawrence TS, Xu L (2009). MicroRNA miR-34 inhibits human pancreatic cancer tumor-initiating cells. PLoS One 4: e6816. DOI 10.1371/journal.pone.0006816. [Google Scholar] [CrossRef]
Jiang P, Du W, Wang X, Mancuso A, Gao X, Wu M, Yang X (2011). p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nature Cell Biology 13: 310–316. DOI 10.1038/ncb2172. [Google Scholar] [CrossRef]
John K, Rosner I, Keilholz U, Gauler T, Bantel H, Grunwald V (2015). Baseline caspase activity predicts progression free survival of temisirolimus-treated head neck cancer patients. European Journal of Cancer 51: 1596–1602. DOI 10.1016/j.ejca.2015.05.021. [Google Scholar] [CrossRef]
Jones PA, Baylin SB (2002). The fundamental role of epigenetic events in cancer. Nature Reviews Genetics 3: 415–428. DOI 10.1038/nrg816. [Google Scholar] [CrossRef]
Jones PA, Laird PW (1999). Cancer-epigenetics comes of age. Nature Genetics 21: 163–167. DOI 10.1038/5947. [Google Scholar] [CrossRef]
Kaleigh F, Kurokawa M (2013). Evading apoptosis in cancer. Trends in Cell Biology 23: 620–633. DOI 10.1016/j.tcb.2013.07.006. [Google Scholar] [CrossRef]
Karbasi A, Borhani N, Daliri K, Kazemi B, Manoochehri M (2015). Downregulation of external death receptor genes FAS and DR5 in colorectal cancer samples positive for human papillomavirus infection. Pathology–Research and Practice 211: 444–448. DOI 10.1016/j.prp.2015.02.001. [Google Scholar] [CrossRef]
Kareva I (2016). Escape from tumor dormancy and time to angiogenic switch as mitigated by tumor-induced stimulation of stroma. Journal of Theoretical Biology 395: 11–22. DOI 10.1016/j.jtbi.2016.01.024. [Google Scholar] [CrossRef]
Karst AM, Dai DL, Martinka M, Li G (2005). PUMA expression is significantly reduced in human cutaneous melanomas. Oncogene 24: 1111–1116. DOI 10.1038/sj.onc.1208374. [Google Scholar] [CrossRef]
Kaufmann T, Strasser A, Jost PJ (2012). Fas death receptor signalling: Roles of Bid and XIAP. Cell Death & Differentiation 19: 42–50. DOI 10.1038/cdd.2011.121. [Google Scholar] [CrossRef]
Kelliher M A, Grimm S, Ishida Y, Kuo F, Stanger B Z, Leder P (1998). The death domain kinase RIP mediates the TNF-induced NF-κB signal. Immunity 8: 297–303. DOI 10.1016/S1074-7613(00)80535-X. [Google Scholar] [CrossRef]
Keshet R, Szlosarek P, Carracedo A, Erez A (2018). Rewiring urea cycle metabolism in cancer to support anabolism. Nature Reviews Cancer 18: 634–645. DOI 10.1038/s41568-018-0054-z. [Google Scholar] [CrossRef]
Kessel D (2015). Apoptosis and associated phenomena as a determinants of the efficacy of photodynamic therapy. Photochemical & Photobiological Sciences 14: 1397–1402. DOI 10.1039/C4PP00413B. [Google Scholar] [CrossRef]
Khosravi-Far R (2014). Death receptor signals to the mitochondria. Cancer Biology & Therapy 3: 1051–1057. DOI 10.4161/cbt.3.11.1173. [Google Scholar] [CrossRef]
Kim J, Parrish AB, Kurokawa M, Matsuura K, Freel CD, Andersen JL, Johnson CE, Kornbluth S (2012). Rsk-mediated phosphorylation and 14-3-3ε binding of Apaf-1 suppresses cytochrome c-induced apoptosis. EMBO Journal 31: 1279–1292. DOI 10.1038/emboj.2011.491. [Google Scholar] [CrossRef]
Kim JJ, Tannock IF (2005). Repopulation of cancer cells during therapy: An important cause of treatment failure. Nature Reviews Cancer 5: 516–525. DOI 10.1038/nrc1650. [Google Scholar] [CrossRef]
Kleffel S, Schatton T (2013). Tumor dormancy and cancer stem cells: two sides of the same coin? In: Enderling H, Almog N, Hlatky L (eds.Systems Biology of Tumor Dormancy, pp. 145–180. Berlin, Germany: Springer. [Google Scholar]
Konstantinidou AE, Korkolopoulou P, Patsouris E (2002). Apoptotic markers for tumor recurrence: a minireview. Apoptosis 7: 461–470. DOI 10.1023/A:1020091226673. [Google Scholar] [CrossRef]
Kopper L, Peták I 2008. Apoptosis and cancer. In: Kaiser HE, Nasir A (eds.Selected Aspects of Cancer Progression: Metastasis, Apoptosis and Immune Response. vol. 11, pp. 103–114. Berlin, Germany: Springer. [Google Scholar]
Krammer PH (2000). CD95’s deadly mission in the immune system. Nature 407: 789–795. DOI 10.1038/35037728. [Google Scholar] [CrossRef]
Kruyt FAE, Schuringa JJ (2010). Apoptosis and cancer stem cells: Implications for apoptosis targeted therapy. Biochemical Pharmacology 80: 423–430. DOI 10.1016/j.bcp.2010.04.010. [Google Scholar] [CrossRef]
Kumar S, Cakouros D (2004). Transcriptional control of the core cell-death machinery. Trends in Biochemical Sciences 29: 193–199. DOI 10.1016/j.tibs.2004.02.001. [Google Scholar] [CrossRef]
Kumar S, Keerthana R, Pazhanimuthu A, Perumal P (2013). Overexpression of circulating miRNA-21 and miRNA-146a in plasma samples of breast cancer patients. Indian Journal of Biochemistry and Biophysics 50: 210–214. [Google Scholar]
Kurokawa M, Zhao C, Reya T, Kornbluth S (2008). Inhibition of apoptosome formation by suppression of Hsp90 phosphorylation in tyrosine kinase-induced leukemias. Molecular and Cellular Biology 28: 5494–5506. DOI 10.1128/MCB.00265-08. [Google Scholar] [CrossRef]
Lam DK, Schmidt BL 2012. Molecular biology of head and neck cancer: therapeutic implications. In: John Dolan CS (ed.Current Therapy in Oral and Maxillofacial Surgery. Georgia: Saunders, 92–101. [Google Scholar]
Landerer E, Villegas J, Burzio VA, Oliveira L, Villota C, Lopez C, Restovic F, Martinez R, Castillo O, Burzio LO (2011). Nuclear localization of the mitochondrial ncRNAs in normal and cancer cells. Cellular Oncology 34: 297–305. DOI 10.1007/s13402-011-0018-8. [Google Scholar] [CrossRef]
Lee JH, Soung YH, Lee JW, Park WS, Kim SY, Cho YG, Kim CJ, Seo SH, Nam SW, Yoo NJ, Lee SH, Lee JY (2004). Inactivating mutation of the pro-apoptic gene BID in gastric cancer. Journal of Pathology 202: 439–445. DOI 10.1002/path.1532. [Google Scholar] [CrossRef]
Lee Peter P, Yee C, Savage PA, Fong L, Brockstedt D, Weber JS, Johnson D, Swetter S, Thompson J, Greenberg PD, Roederer M, Davis MM (1999). Characterization of circulating T cells specific for tumor-associated antigens in melanoma patients. Nature Medicine 5: 677–685. DOI 10.1038/9525. [Google Scholar] [CrossRef]
Lee SH, Shin MS, Park WS, Kim SY, Kim HS, Han JY, Park GS, Dong SM, Pi JH, Kim CS, Kim SH, Lee JY, Yoo NJ (1999). Alterations of Fas (Apo-1/CD95) gene in non-small cell lung cancer. Oncogene 18: 3754–3760. DOI 10.1038/sj.onc.1202769. [Google Scholar] [CrossRef]
Lei K, Davis RJ (2003). JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proceedings of the National Academy of Sciences of the United States of America 100: 2432–2437. DOI 10.1073/pnas.0438011100. [Google Scholar] [CrossRef]
Leone K, Poggiana C, Zamarchi R (2018). The interplay between circulating tumor cells and the immune system: From immune escape to cancer immunotherapy. Diagnostics 8: 59. DOI 10.3390/diagnostics8030059. [Google Scholar] [CrossRef]
Lessene G, Czabotar PE, Colman PM (2008). BCL-2 family antagonists for cancer therapy. Nature Reviews Drug Discovery 7: 989–1000. DOI 10.1038/nrd2658. [Google Scholar] [CrossRef]
Letai AG (2008). Diagnosing and exploiting cancer’s addiction to blocks in apoptosis. Nature Reviews Cancer 8: 121–132. DOI 10.1038/nrc2297. [Google Scholar] [CrossRef]
Levine AJ, Puzio-Ku AM (2010). The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science 330: 1340–1344. DOI 10.1126/science.1193494. [Google Scholar] [CrossRef]
Li C, Egloff AM, Sen M, Grandis JR, Johnson DE (2014). Caspase-8 mutations in head and neck cancer confer resistance to death receptor-mediated apoptosis and enhance migration, invasion, and tumor growth. Molecular Oncology 8: 1220–1230. DOI 10.1016/j.molonc.2014.03.018. [Google Scholar] [CrossRef]
Li J, Yuan J (2008). Caspases in apoptosis and beyond. Oncogene 27: 6194–6206. DOI 10.1038/onc.2008.297. [Google Scholar] [CrossRef]
Lianidou ES, Markou A, Strati A (2015). The role of CTCs as tumor biomarkers, Advances in Experimental Medicine and Biology 867: 341–367. DOI 10.1007/978-94-017-7215-0_21. [Google Scholar] [CrossRef]
Lima RT, Busacca S, Almeida GM, Gaudino G, Fennell DA, Vasconcelos MH (2011). MicroRNA regulation of core apoptosis pathways in cancer. European Journal of Cancer 47: 163–174. DOI 10.1016/j.ejca.2010.11.005. [Google Scholar] [CrossRef]
Liu J, Zhang C, Feng Z (2014). Tumor suppressor p53 and its gain-of-function mutants in cancer. Acta Biochimica et Biophysica Sinica 46: 170–179. DOI 10.1093/abbs/gmt144. [Google Scholar] [CrossRef]
Liu S, Zhang P, Chen Z, Liu M, Li X, Tang H (2013). MicroRNA-7 downregulates XIAP expression to suppress cell growth and promote apoptosis in cervical cancer cells. FEBS Letters 587: 2247–2253. DOI 10.1016/j.febslet.2013.05.054. [Google Scholar] [CrossRef]
Lopez J, Tait SWG (2015). Mitochondrial apoptosis: Killing cancer using the enemy within. British Journal of Cancer 112: 957–962. DOI 10.1038/bjc.2015.85. [Google Scholar] [CrossRef]
Lowe SW, Lin AW (2000). Apoptosis in cancer. Carcinogenesis 21: 485–495. DOI 10.1093/carcin/21.3.485. [Google Scholar] [CrossRef]
Lowman XH, McDonnell MA, Kosloske A, Odumade OA, Jenness C, Karim CB, Jemmerson R, Kelekar A (2010). The proapoptotic function of noxa in human leukemia cells is regulated by the kinase Cdk5 and by glucose. Molecular Cell 40: 823–833. DOI 10.1016/j.molcel.2010.11.035. [Google Scholar] [CrossRef]
Lujambio A, Ropero S, Ballestar E, Fraga MF, Cerrato C, Setién F, Casado S, Suarez-Gauthier A, Sanchez-Cespedes M, Gitt A, Spiteri I, Das PP, Caldas C, Miska E, Esteller M (2007). Genetic unmasking of an epigenetically silenced microRNA in human cancer cells. Cancer Research 67: 1424–1429. DOI 10.1158/0008-5472.CAN-06-4218. [Google Scholar] [CrossRef]
MacFarlane M, Ahmad M, Srinivasula SM, Fernandes-Alnemri T, Cohen GM, Alnemri ES (1997). Identification and molecular cloning of two novel receptors for the cytotoxic ligand TRAIL. Journal of Biological Chemistry 272: 25417–25420. DOI 10.1074/jbc.272.41.25417. [Google Scholar] [CrossRef]
Majewski N, Nogueira V, Robey RB, Hay N (2004). Akt inhibits apoptosis downstream of BID cleavage via a glucose-dependent mechanism involving mitochondrial hexokinases. Molecular and Cellular Biology 24: 730–740. DOI 10.1128/MCB.24.2.730-740.2004. [Google Scholar] [CrossRef]
Martinez R, Setien F, Voelter C, Casado S, Quesada MP, Schackert G, Esteller M (2007). CpG island promoter hypermethylation of the pro-apoptotic gene caspase-8 is a common hallmark of relapsed glioblastoma multiforme. Carcinogenesis 28: 1264–1268. DOI 10.1093/carcin/bgm014. [Google Scholar] [CrossRef]
Mascré G, Dekoninck S, Drogat B, Youssef KK, Brohée S, Sotiropoulou PA, Simons BD, Blanpain C (2012). Distinct contribution of stem and progenitor cells to epidermal maintenance. Nature 489: 257–262. DOI 10.1038/nature11393. [Google Scholar] [CrossRef]
Mashima T, Tsuruo T (2005). Defects of the apoptotic pathway as therapeutic target against cancer. Drug Resistance Updates 8: 339–343. DOI 10.1016/j.drup.2005.11.001. [Google Scholar] [CrossRef]
Mataga MA, Rosenthal S, Heerboth S, Devalapalli A, Kokolus S, Evans LR, Longacre M, Housman G, Sarkar S (2012). Anti-breast cancer effects of histone deacetylase inhibitors and calpain inhibitor. Anticancer Research 32: 2523–2530. [Google Scholar]
Matsuura K, Canfield K, Feng W, Kurokawa M (2016). Metabolic regulation of apoptosis in cancer. International Review of Cell and Molecular Biology 327: 43–87. [Google Scholar]
Maurer U, Charvet C, Wagman AS, Dejardin E, Green DR (2006). Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Molecular Cell 21: 749–760. DOI 10.1016/j.molcel.2006.02.009. [Google Scholar] [CrossRef]
McDuff FKE, Turner SD (2011). Jailbreak: Oncogene-induced senescence and its evasion. Cellular Signalling 23: 6–13. DOI 10.1016/j.cellsig.2010.07.004. [Google Scholar] [CrossRef]
Medina V, Calvo MB, Díaz-Prado S, Espada J (2009). Hedgehog signalling as a target in cancer stem cells. Clinical and Translational Oncology 11: 199–207. DOI 10.1007/s12094-009-0341-y. [Google Scholar] [CrossRef]
Messerschmidt JL, Bhattacharya P, Messerschmidt GL (2017). Cancer clonal theory, immune escape, and their evolving roles in cancer multi-agent therapeutics. Current Oncology Reports 19: 23. DOI 10.1007/s11912-017-0625-2. [Google Scholar] [CrossRef]
Miller DM, Thomas SD, Islam A, Muench D, Sedoris K (2012). c-Myc and cancer metabolism. Clinical Cancer Research 18: 5546–5553. DOI 10.1158/1078-0432.CCR-12-0977. [Google Scholar] [CrossRef]
Mohamed AN (2017). MYC (MYC proto-oncogene, bHLH transcription factor). Atlas of Genetics and Cytogenetics in Oncology and Haematology 22: 227–232. [Google Scholar]
Mohammad RM, Muqbil I, Lowe L, Yedjou C, Hsu HY, Lin LT, Siegelin MD, Fimognari C, Kumar NB, Don QP, Yang H, Samadi AK, Russo GL, Spagnuolo C, Ray SK, Chakrabarti M, Morre JD, Coley HM, Honoki K, Fujii H, Georgakilas AG, Amedei A, Niccolai E, Amin A, Ashraf SS, Helferich WG, Yang X, Boosani CS, Guha G, Bhakta D, Ciriolo MR, Aquilano K, Chen S, Mohammed SI, Keith WN, Bilsland A, Halicka D, Nowsheen S, Azmi AS (2015). Broad targeting of resistance to apoptosis in cancer. Seminars in Cancer Biology 35: S78–S103. DOI 10.1016/j.semcancer.2015.03.001. [Google Scholar] [CrossRef]
Mohme M, Riethdorf S, Pantel K (2017). Circulating and disseminated tumour cells—mechanisms of immune surveillance and escape. Nature Reviews Clinical Oncology 14: 155–167. DOI 10.1038/nrclinonc.2016.144. [Google Scholar] [CrossRef]
Muller PAJ, Vousden KH (2013). p53 mutations in cancer. Nature Cell Biology 15: 2–8. DOI 10.1038/ncb2641. [Google Scholar] [CrossRef]
Naik PP, Das DN, Panda PK, Mukhopadhyay S, Sinha N, Praharaj PP, Agarwal R, Bhutia SK (2016). Implications of cancer stem cells in developing therapeutic resistance in oral cancer. Oral Oncology 62: 122–135. DOI 10.1016/j.oraloncology.2016.10.008. [Google Scholar] [CrossRef]
Naumov GN, MacDonald IC, Chambers AF, Groom AC (2001). Solitary cancer cells as a possible source of tumour dormancy? Seminars in Cancer Biology 11: 271–276. DOI 10.1006/scbi.2001.0382. [Google Scholar] [CrossRef]
Naumov GN, MacDonald IC, Weinmeister PM, Kerkvliet N, Nadkarni KV, Wilson SM, Morris VL, Groom AC, Chambers AF (2002). Persistence of solitary mammary carcinoma cells in a secondary site: A possible contributor to dormancy. Cancer Research 62: 2162–2168. [Google Scholar]
Naumov GN, Townson JL, MacDonald IC, Wilson SM, Bramwell VHC, Groom AC, Chambers AF (2003). Ineffectiveness of doxorubicin treatment on solitary dormant mammary carcinoma cells or late-developing metastases. Breast Cancer Research and Treatment 82: 199–206. DOI 10.1023/B:BREA.0000004377.12288.3c. [Google Scholar] [CrossRef]
Nelson DA, Tan TT, Rabson AB, Anderson D, Degenhardt K, White E (2004). Hypoxia and defective apoptosis drive genomic instability and tumorigenesis. Genes & Development 18: 2095–2107. DOI 10.1101/gad.1204904. [Google Scholar] [CrossRef]
Nguyen L, Papenhausen P, Shao H (2017). The role of c-MYC in B-cell lymphomas: Diagnostic and molecular aspects. Genes 8: 116. DOI 10.3390/genes8040116. [Google Scholar] [CrossRef]
Nifoussi SK, Vrana JA, Domina AM, de Biasio A, Gui J, Gregory MA, Hann SR, Craig RW (2012). Thr 163 Phosphorylation causes Mcl-1 stabilization when degradation is independent of the adjacent GSK3-targeted phosphodegron, promoting drug resistance in cancer. PLoS One 7: e47060. DOI 10.1371/journal.pone.0047060. [Google Scholar] [CrossRef]
Nowak I, Boratyn E, Durbas M, Horwacik I, Rokita H (2018). Exogenous expression of miRNA-3613-3p causes APAF1 downregulation and affects several proteins involved in apoptosis in BE(2)-C human neuroblastoma cells. International Journal of Oncology 53: 1787–1799. [Google Scholar]
O’Reilly LA, Tai L, Lee L, Kruse EA, Grabow S, Fairlie WD, Haynes NM, Tarlinton DM, Zhang JG, Belz GT, Smyth MJ, Bouillet P, Robb L, Strasser A (2009). Membrane-bound Fas ligand only is essential for Fas-induced apoptosis. Nature 461: 659–663. DOI 10.1038/nature08402. [Google Scholar] [CrossRef]
O’Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT (2005). c-Myc-regulated microRNAs modulate E2F1 expression. Nature 435: 839–843. DOI 10.1038/nature03677. [Google Scholar] [CrossRef]
Ogden CA, Pound JD, Batth BK, Owens S, Johannessen I, Wood K, Gregory CD (2005). Enhanced apoptotic cell clearance capacity and B cell survival factor production by IL-10-activated macrophages: Implications for Burkitt’s Lymphoma. The Journal of Immunology 174: 3015–3023. DOI 10.4049/jimmunol.174.5.3015. [Google Scholar] [CrossRef]
Oren M, Rotter V (2010). Mutant p53 gain-of-function in cancer. Cold Spring Harbor Perspectives in Biology 2: a001107–a001107. DOI 10.1101/cshperspect.a001107. [Google Scholar] [CrossRef]
Pai SI, Wu GS, Özören N, Wu L, Jen J, Sidransky D, El-Deiry WS (1998). Rare loss-of-function mutation of a death receptor gene in head and neck cancer. Cancer Research 58: 3513–3518. [Google Scholar]
Paik SS, Jang KS, Song YS, Jang SH, Min KW, Han HX, Na W, Lee KH, Choi D, Jang SJ (2007). Reduced expression of Apaf-1 in colorectal adenocarcinoma correlates with tumor progression and aggressive phenotype. Annals of Surgical Oncology 14: 3453–3459. DOI 10.1245/s10434-007-9541-2. [Google Scholar] [CrossRef]
Pal R, Srivastava N, Chopra R, Gochhait S, Gupta P, Prakash N, Agarwal G, Bamezai RNK (2010). Investigation of DNA damage response and apoptotic gene methylation pattern in sporadic breast tumors using high throughput quantitative DNA methylation analysis technology. Molecular Cancer 9: 303. DOI 10.1186/1476-4598-9-303. [Google Scholar] [CrossRef]
Park WS, Lee JH, Shin MS, Park JY, Kim HS, Kim YS, Park CH, Lee SK, Lee SH, Lee SN, Kim H, Yoo NJ, Lee JY (2001). Inactivating mutations of KILLER/DR5 gene in gastric cancers. Gastroenterology 121: 1219–1225. DOI 10.1053/gast.2001.28663. [Google Scholar] [CrossRef]
Pavlova NN, Thompson CB (2016). The emerging hallmarks of cancer metabolism. Cell Metabolism 23: 27–47. DOI 10.1016/j.cmet.2015.12.006. [Google Scholar] [CrossRef]
Pedriali G, Rimessi A, Sbano L, Giorgi C, Wieckowski MR, Previati M, Pinton P (2017). Regulation of endoplasmic reticulum–mitochondria Ca2+ transfer and its importance for anti-cancer therapies. Frontiers in Oncology 7: 245. DOI 10.3389/fonc.2017.00180. [Google Scholar] [CrossRef]
Perri F, Pisconti S, Scarpati V, Della G (2016). P53 mutations and cancer: a tight linkage. Annals of Translational Medicine 4: 522. DOI 10.21037/atm.2016.12.40. [Google Scholar] [CrossRef]
Piggott L, Omidvar N, Pérez S, Eberl M, Clarkson RWE (2011). Suppression of apoptosis inhibitor c-FLIP selectively eliminates breast cancer stem cell activity in response to the anti-cancer agent. TRAIL Breast Cancer Research 13: 6. DOI 10.1186/bcr2945. [Google Scholar] [CrossRef]
Porter A, Jänicke R (1999). Emerging roles of caspase-3 in apoptosis. Cell Death Differ 6: 99–104. DOI 10.1038/sj.cdd.4400476. [Google Scholar] [CrossRef]
Qu Y, Dang S, Hou P (2013). Gene methylation in gastric cancer. Clinica Chimica Acta 424: 53–65. DOI 10.1016/j.cca.2013.05.002. [Google Scholar] [CrossRef]
Ravichandran KS, Lorenz U (2007). Engulfment of apoptotic cells: Signals for a good meal. Nature Reviews Immunology 7: 964–974. DOI 10.1038/nri2214. [Google Scholar] [CrossRef]
Real PJ, Sierra A, De Juan A, Segovia JC, Lopez-Vega JM, Fernandez-Luna JL (2002). Resistance to chemotherapy via Stat3-dependent overexpression of Bcl-2 in metastatic breast cancer cells. Oncogene 21: 7611–7618. DOI 10.1038/sj.onc.1206004. [Google Scholar] [CrossRef]
Reya T, Morrison SJ, Clarke MF, Weissman IL (2001). Stem cells, cancer, and cancer stem cells. Nature 414: 105–111. DOI 10.1038/35102167. [Google Scholar] [CrossRef]
Riganti C, Donadelli M (2020). Mitochondrial metabolic alterations in cancer cells and related therapeutic targets. Seminars in Cell & Developmental Biology 98: 1–3. DOI 10.1016/j.semcdb.2019.10.005. [Google Scholar] [CrossRef]
Robertson KD (2000). Differential mRNA expression of the human DNA methyltransferases (DNMTs) 1, 3a and 3b during the G0/G1 to S phase transition in normal and tumor cells. Nucleic Acids Research 28: 2108–2113. DOI 10.1093/nar/28.10.2108. [Google Scholar] [CrossRef]
Rodriguez C, Martín V, Herrera F, García-Santos G, Rodriguez-Blanco J, Casado-Zapico S, Sanchez-Sanchez AM, Suarez S, Puente-Moncada N, Anitua MJ, Antolin I (2013). Mechanisms involved in the pro-apoptotic effect of melatonin in cancer cells. International Journal of Molecular Sciences 14: 6597–6613. DOI 10.3390/ijms14046597. [Google Scholar] [CrossRef]
Rodriguez JA (2017). HLA‐mediated tumor escape mechanisms that may impair immunotherapy clinical outcomes via T-cell activation. Oncology Letters 14: 4415–4427. DOI 10.3892/ol.2017.6784. [Google Scholar] [CrossRef]
Salvesen GS, Duckett CS (2002). IAP proteins: Blocking the road to death’s door. Nature Reviews Molecular Cell Biology 3: 401–410. DOI 10.1038/nrm830. [Google Scholar] [CrossRef]
Salvesen GS, Walsh CM (2014). Functions of caspase 8: The identified and the mysterious. Seminars in immunology 26: 246–252. DOI 10.1016/j.smim.2014.03.005. [Google Scholar] [CrossRef]
Sarkar S, Horn G, Moulton K, Oza A, Byler S, Kokolus S, Longacre M (2013). Cancer development, progression, and therapy: An epigenetic overview. International Journal of Molecular Sciences 14: 21087–21113. DOI 10.3390/ijms141021087. [Google Scholar] [CrossRef]
Schafer ZT, Kornbluth S (2006). The apoptosome: physiological, developmental, and pathological modes of regulation. Developmental Cell 10: 549–561. DOI 10.1016/j.devcel.2006.04.008. [Google Scholar] [CrossRef]
Schmitt CA (2003). Senescence, apoptosis and therapy—cutting the lifelines of cancer. Nature Reviews Cancer 3: 286–295. DOI 10.1038/nrc1044. [Google Scholar] [CrossRef]
Seenath M, Roberts D, Cawthrone C, Saunders M, Armstrong G, O’Dwyer S, Stratford IJ, Dive C, Renehan AG (2008). Reciprocal relationship between expression of hypoxia inducible factor 1α (HIF-1α) and the pro-apoptotic protein Bid in ex vivo colorectal cancer. British Journal of Cancer 99: 459–463. DOI 10.1038/sj.bjc.6604474. [Google Scholar] [CrossRef]
Seifert A, Clarke PR (2009). p38α- and DYRK1A-dependent phosphorylation of caspase-9 at an inhibitory site in response to hyperosmotic stress. Cellular Signalling 21: 1626–1633. DOI 10.1016/j.cellsig.2009.06.009. [Google Scholar] [CrossRef]
Semenza GL (2002). HIF-1 and tumor progression: pathophysiology and therapeutics. Trends in Molecular Medicine 8: 62–67. DOI 10.1016/S1471-4914(02)02317-1. [Google Scholar] [CrossRef]
Shakeri R, Kheirollahi A, Davoodi J (2017). Apaf-1: Regulation and function in cell death. Biochimie 135: 111–125. DOI 10.1016/j.biochi.2017.02.001. [Google Scholar] [CrossRef]
Signore M, Ricci-Vitiani L, De Maria R (2013). Targeting apoptosis pathways in cancer stem cells. Cancer Letters 332: 374–382. DOI 10.1016/j.canlet.2011.01.013. [Google Scholar] [CrossRef]
Sjöström J, Bergh J (2001). How apoptosis is regulated, and what goes wrong in cancer. British Medical Journal 322: 1538–1539. DOI 10.1136/bmj.322.7301.1538. [Google Scholar] [CrossRef]
Slattery ML, Mullany LE, Sakoda LC, Wolff RK, Samowitz WS, Herrick JS (2018). Dysregulated genes and miRNAs in the apoptosis pathway in colorectal cancer patients. Apoptosis 23: 237–250. DOI 10.1007/s10495-018-1451-1. [Google Scholar] [CrossRef]
Soderquist RS, Crawford L, Liu E, Lu M, Agarwal A, Anderson GR, Lin KH, Winter PS, Cakir M, Wood KC (2018). Systematic mapping of BCL-2 gene dependencies in cancer reveals molecular determinants of BH3 mimetic sensitivity. Nature Communications 9: 121. DOI 10.1038/s41467-018-05815-z. [Google Scholar] [CrossRef]
Soengas MS, Alarco RM, Yoshida H, Giaccia AJ, Hakem R, Mak TW, Lowe SW (1999). Apaf-1 and Caspase-9 in p53-dependent apoptosis and tumor inhibition. Science 284: 156–159. DOI 10.1126/science.284.5411.156. [Google Scholar] [CrossRef]
Soengas MS, Capodieci P, Polsky D, Mora J, Esteller M, Opitz-Araya X, McCombie R, Herman JG, Gerald WL, Lazebnik YA, Cordon-Cardo C, Lowe SW (2001). Inactivation of the apoptosis effector Apaf-1 in malignant melanoma. Nature 409: 207–211. DOI 10.1038/35051606. [Google Scholar] [CrossRef]
Song E, Chen J, Ouyang N, Su F, Wang M, Heemann U (2001). Soluble Fas ligand released by colon adenocarcinoma cells induces host lymphocyte apoptosis: An active mode of immune evasion in colon cancer. British Journal of Cancer 85: 1047–1054. DOI 10.1054/bjoc.2001.2042. [Google Scholar] [CrossRef]
Soung YH, Lee JW, Kim HS, Park WS, Kim SY, Lee JH, Park JY, Cho YG, Kim CJ, Park YG, Nam SW, Jeong SW, Kim SH, Lee JY, Yoo NJ, Lee SH (2003). Inactivating mutations of CASPASE-7 gene in human cancers. Oncogene 22: 8048–8052. DOI 10.1038/sj.onc.1206727. [Google Scholar] [CrossRef]
Soung YH, Lee JW, Kim SY, Jang J, Park YG, Park WS, Nam SW, Lee JY, Yoo NJ, Lee SH (2005). Caspase-8 gene is inactivated by somatic mutations in gastric carcinomas. Cancer Research 65: 815–822. [Google Scholar]
Soung YH, Lee JW, Kim SY, Park WS, Nam SW, Lee JY, Yoo NJ, Lee SH (2004). Somatic mutations of CASP3 gene in human cancers. Human Genetics 115: 112–115. DOI 10.1007/s00439-004-1129-3. [Google Scholar] [CrossRef]
Soung YH, Lee JW, Kim SY, Park WS, Nam SW, Lee JY, Yoo NJ, Lee SH (2006). Mutational analysis of proapoptotic caspase-9 gene in common human carcinomas. APMIS 114: 292–297. DOI 10.1111/j.1600-0463.2006.apm_364.x. [Google Scholar] [CrossRef]
Steele RJC, Thompson AM, Hall PA, Lane DP (1998). The p53 tumour suppressor gene. British Journal of Surgery 85: 1460–1467. DOI 10.1046/j.1365-2168.1998.00910.x. [Google Scholar] [CrossRef]
Steven A, Seliger B (2018). The role of immune escape and immune cell infiltration in breast cancer. Breast Care 13: 16–21. DOI 10.1159/000486585. [Google Scholar] [CrossRef]
Sun HR, Wang S, Yan SC, Zhang Y, Nelson PJ, Jia HL, Qin LX, Dong QZ (2019). Therapeutic strategies targeting cancer stem cells and their microenvironment. Frontiers in Oncology 9: 1104. DOI 10.3389/fonc.2019.01104. [Google Scholar] [CrossRef]
Sun T, Gao Y, Tan W, Ma S, Shi Y, Yao J, Guo Y, Yang M, Zhang X, Zhang Q, Zeng C, Lin D (2007). A six-nucleotide insertion-deletion polymorphism in the CASP8 promoter is associated with susceptibility to multiple cancers. Nature Genetics 39: 605–613. DOI 10.1038/ng2030. [Google Scholar] [CrossRef]
Sussman RT, Ricci MS, Hart LS, Shi YS, El-Deiry WS (2014). Chemotherapy-resistant side-population of colon cancer cells has a higher sensitivity to TRAIL than the non-SP, a higher expression of c-Myc and TRAIL-receptor DR4. Cancer Biology & Therapy 6: 1486–1491. DOI 10.4161/cbt.6.9.4905. [Google Scholar] [CrossRef]
Szliszka E, Mazur B, Zdowicz G, Czuba ZP, Król W (2009). TRAIL-induced apoptosis and expression of death receptor TRAIL-R1 and TRAIL-R2 in bladder cancer cells. Folia Histochemica et Cytobiologica 47: 579–585. [Google Scholar]
Takimoto R, El-Deiry WS (2000). Wild-type p53 transactivates the KILLER/DR5 gene through an intronic sequence-specific DNA-binding site. Oncogene 19: 1735–1743. DOI 10.1038/sj.onc.1203489. [Google Scholar] [CrossRef]
Tang HL, Yuen KL, Tang HM, Fung MC (2009). Reversibility of apoptosis in cancer cells. British Journal of Cancer 100: 118–122. DOI 10.1038/sj.bjc.6604802. [Google Scholar] [CrossRef]
Timofeev O, Klimovich B, Schneikert J, Wanzel M, Pavlakis E, Noll J, Mutlu S, Elmshäuser S, Nist A, Mernberger M, Lamp B, Wenig U, Brobeil A, Gattenlöhner S, Köhler K, Stiewe T (2019). Residual apoptotic activity of a tumorigenic p53 mutant improves cancer therapy responses. EMBO Journal 38: 8664. DOI 10.15252/embj.2019102096. [Google Scholar] [CrossRef]
Townson JL, Chambers AF (2006). Dormancy of solitary metastatic cells. Cell Cycle 5: 1744–1750. DOI 10.4161/cc.5.16.2864. [Google Scholar] [CrossRef]
Trapani JA, Smyth MJ (2002). Functional significance of the perforin/granzyme cell death pathway. Nature Reviews Immunology 2: 735–747. DOI 10.1038/nri911. [Google Scholar] [CrossRef]
Truman LA, Ford CA, Pasikowska M, Pound JD, Wilkinson SJ, Dumitriu IE, Melrose ML, Ogden LA, Nibbs CA, Graham R, Combadiere G, Gregory CD (2017). CX3CL1/fractalkine is released from apoptotic lymphocytes to stimulate macrophage chemotaxis. Immunobiology 112: 5026–5036. [Google Scholar]
Tysnes BBO (2010). Tumor-initiating and -propagating cells: Cells that we would like to identify and control. Neoplasia 12: 506–515. DOI 10.1593/neo.10290. [Google Scholar] [CrossRef]
Unruh A, Ressel A, Mohamed HG, Johnson RS, Nadrowitz R, Richter E, Katschinski DM, Wenger RH (2003). The hypoxia-inducible factor-1α is a negative factor for tumor therapy. Oncogene 22: 3213–3220. DOI 10.1038/sj.onc.1206385. [Google Scholar] [CrossRef]
Vogelstein B, Kinzler KW (2004). Cancer genes and the pathways they control. Nature Medicine 10: 789–799. DOI 10.1038/nm1087. [Google Scholar] [CrossRef]
Vucic D, Dixit VM, Wertz IE (2011). Ubiquitylation in apoptosis: A post-translational modification at the edge of life and death. Nature Reviews Molecular Cell Biology 12: 439–452. DOI 10.1038/nrm3143. [Google Scholar] [CrossRef]
Wajant H (2002). The Fas signaling pathway: more than a paradigm. Science 296: 1635–1636. DOI 10.1126/science.1071553. [Google Scholar] [CrossRef]
Wang HL, Bai H, Li Y, Sun J, Wang XQ (2007). Rationales for expression and altered expression of apoptotic protease activating factor-1 gene in gastric cancer. World Journal of Gastroenterology 13: 5060–5064. DOI 10.3748/wjg.v13.i38.5060. [Google Scholar] [CrossRef]
Wang JM, Chao JR, Chen W, Kuo ML, Yen JJY, Yang-Yen HF (1999). The antiapoptotic gene mcl-1 is up-regulated by the phosphatidylinositol 3-kinase/Akt signaling pathway through a transcription factor complex containing CREB. Molecular and Cellular Biology 19: 6195–6206. DOI 10.1128/MCB.19.9.6195. [Google Scholar] [CrossRef]
Wang L, Zhang X, Jia LT, Hu SJ, Jing Zhao, Yang JD, Wen WH, Wang Z, Wang T, Zhao J, Wang RA, Meng YL, Nie YZ, Dou KF, Chen SY, Yao LB, Fan DM, Zhang R, Yang AG (2014). C-Myc-mediated epigenetic silencing of MicroRNA-101 contributes to dysregulation of multiple pathways in hepatocellular carcinoma. Hepatology 59: 1850–1863. DOI 10.1002/hep.26720. [Google Scholar] [CrossRef]
Wang LH, Wu CF, Rajasekaran N, Shin YK (2019). Loss of tumor suppressor gene function in human cancer: An overview. Cellular Physiology and Biochemistry 51: 2647–2693. DOI 10.1159/000495956. [Google Scholar] [CrossRef]
Wang L, Yang JK, Kabaleeswaran V, Rice AJ, Cruz AC, Park AY, Yin Q, Damko E, Jang SB, Raunser S, Robinson CV, Siegel RM, Walz T, Wu H (2010). The Fas–FADD death domain complex structure reveals the basis of DISC assembly and disease mutations. Nature Structural & Molecular Biology 17: 1324–1329. DOI 10.1038/nsmb.1920. [Google Scholar] [CrossRef]
Wang M, Huang T, Luo G, Huang C, Xiao X, Wang L, Jiang G, Zeng F (2015). Long non-coding RNA MEG3 induces renal cell carcinoma cells apoptosis by activating the mitochondrial pathway. Journal of Huazhong University of Science and Technology [Medical Sciences] 35: 541–545. DOI 10.1007/s11596-015-1467-5. [Google Scholar] [CrossRef]
Wang S, Yang D, Lippman ME (2003). Targeting Bcl-2 and Bcl-XL with nonpeptidic small-molecule antagonists. Seminars in Oncology 30: 133–142. DOI 10.1053/j.seminoncol.2003.08.015. [Google Scholar] [CrossRef]
Waring P, Mullbacher A (1999). Cell death induced by the Fas/Fas ligand pathway and its role in pathology. Immunology and Cell Biology 77: 312–317. DOI 10.1046/j.1440-1711.1999.00837.x. [Google Scholar] [CrossRef]
Whitworth PW, Pak CC, Esgro J, Kleinerman ES, Fidler IJ (1990). Macrophages and cancer. Cancer and Metastasis Reviews 8: 319–351. DOI 10.1007/BF00052607. [Google Scholar] [CrossRef]
Wong RSY (2011). Apoptosis in cancer: From pathogenesis to treatment. Journal of Experimental and Clinical Cancer Research 30: 14. DOI 10.1186/1756-9966-30-14. [Google Scholar] [CrossRef]
Wu GS, Kim K, El-Deiry WS 2000. Killer/DR5, a novel DNA-damage inducible death receptor gene, links the p53-tumor suppressor to caspase activation and apoptotic death. In: Habib NA (ed.Cancer Gene Therapy Past Achievements and Future Challenges. London UK: Kluwer Academic Publishers, 143–152. [Google Scholar]
Yan X, Qi M, Li P, Zhan Y, Shao H (2017). Apigenin in cancer therapy: anti-cancer effects and mechanisms of action. Cell & Bioscience 7: 190. DOI 10.1186/s13578-017-0179-x. [Google Scholar] [CrossRef]
Yang M, Sun T, Wang L, Yu D, Zhang X, Miao X, Liu J, Zhao D, Li H, Tan W, Lin D (2008). Functional variants in cell death pathway genes and risk of pancreatic cancer. Clinical Cancer Research 14: 3230–3237. DOI 10.1158/1078-0432.CCR-08-0177. [Google Scholar] [CrossRef]
Yang X, Fraser M, Moll UM, Basak A, Tsang BK (2006). Akt-mediated cisplatin resistance in ovarian cancer: Modulation of p53 action on caspase-dependent mitochondrial death pathway. Cancer Research 66: 3126–3136. DOI 10.1158/0008-5472.CAN-05-0425. [Google Scholar] [CrossRef]
Yip KW, Reed JC (2008). Bcl-2 family proteins and cancer. Oncogene 27: 6398–6406. DOI 10.1038/onc.2008.307. [Google Scholar] [CrossRef]
Yoo NJ, Lee JW, Jeong EG, Lee SH (2007). Immunohistochemical analysis of pro-apoptotic PUMA protein and mutational analysis of PUMA gene in gastric carcinomas. Digestive and Liver Disease 39: 222–227. DOI 10.1016/j.dld.2006.11.006. [Google Scholar] [CrossRef]
Yoo N, Lee JW, Kim Y, Soung YH, Kim SY, Nam SW, Park WS, Lee JY, Lee SH (2004). Loss of caspase-2, -6 and -7 expression in gastric cancers. APMIS 112: 330–335. DOI 10.1111/j.1600-0463.2004.t01-1-apm1120602.x. [Google Scholar] [CrossRef]
Yoshida H (2003). The role of Apaf-1 in programmed cell death: From worm to tumor. Cell Structure and Function 28: 3–9. DOI 10.1247/csf.28.3. [Google Scholar] [CrossRef]
Yu J, Zhang L (2008). PUMA, a potent killer with or without p53. Oncogene 27: S71–S83. DOI 10.1038/onc.2009.45. [Google Scholar] [CrossRef]
Yustein AS, Harper JC, Petroni GR, Cummings OW, Moskaluk CA, Powell SM (1999). Allelotype of gastric adenocarcinoma. Cancer Research 59: 1437–1441. [Google Scholar]
Zeng L, Li T, Xu DC, Liu J, Mao G, Cui MZ, Fu X, Xu X (2012). Death receptor 6 induces apoptosis not through type I or type II pathways, but via a unique mitochondria-dependent pathway by interacting with bax protein. Journal of Biological Chemistry 287: 29125–29133. DOI 10.1074/jbc.M112.362038. [Google Scholar] [CrossRef]
Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ (1996). Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-XL. Cell 87: 619–628. DOI 10.1016/S0092-8674(00)81382-3. [Google Scholar] [CrossRef]
Zlobec I, Minoo P, Baker K, Haegert D, Khetani K, Tornillo L, Terracciano L, Jass JR, Lugli A (2007). Loss of APAF-1 expression is associated with tumour progression and adverse prognosis in colorectal cancer. European Journal of Cancer 43: 1101–1107. DOI 10.1016/j.ejca.2007.01.029. [Google Scholar] [CrossRef]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |