DOI:10.32604/biocell.2021.014873
www.techscience.com/journal/biocell
| Biocell DOI:10.32604/biocell.2021.014873 | www.techscience.com/journal/biocell |
| Article |
Three-month effects of corneal cross-linking on corneal fibroblasts
1School of Biomedical Engineering, Capital Medical University, Beijing, 100069, China
2Beijing Key Laboratory of Fundamental Research on Biomechanics in Clinical Application, Capital Medical University, Beijing, 100069, China
*Address correspondence: Lin Li, lil@ccmu.edu.cn
Received: 20 November 2020; Accepted: 10 January 2021
Abstract: Corneal collagen crosslinking (CXL) has revolutionized the treatment of keratoconus in the past decade. In order to evaluate the 3-month effects of CXL on corneal fibroblasts, a longitudinal study at the tissue and cellular level was carried out with a total of 16 rabbits that underwent CXL, deepithelialization (DEP), or non-treatment (control) and kept for 1 to 3 months. The duration of corneal stromal remodeling after CXL was determined by examining the differentiation, apoptosis, and number changes of keratocytes in tissue sections from animals 1, 2, or 3 months post-treatment. Upon the finish of tissue remodeling, separate rabbits were used to extract keratocytes and set up cell culture for vimentin immunofluorescence staining. The same cell culture was used for (1) migration measurement through the wound-healing assay; (2) elastic modulus measurement by atomic force microscope (AFM); (3) the proliferation, apoptosis, cytoskeleton and α-SMA expression tests through EdU (5-ethynyl-2’-deoxyuridine) assay, TUNEL (TdT-mediated dUTP Nick-End Labeling) assay, phalloidin and α-SMA (alpha-smooth muscle actin) immunofluorescence analysis, respectively. Results showed that the migratory activity, elastic modulus, and α-SMA expression of the corneal fibroblasts increased after CXL treatment, while apoptosis, proliferation, and morphology of F-actin cytoskeleton of the fibroblasts had no significant change after 3 months. In contrast, measured cellular parameters (migratory, elastic moduli, α-SMA expression, apoptosis, proliferation, and morphology of F-actin cytoskeleton of fibroblasts) did not change significantly after DEP. In conclusion, the dynamic changes of keratocytes were nearly stable 3 months after CXL treatment. CXL has an impact on corneal fibroblasts, including migration, elastic modulus and α-SMA expression, while epithelialization may not alter the biological behavior of cells significantly.
Keywords: CXL; Elastic modulus; Stromal remodeling; Mechanobiological behavior
Keratoconus often occurs in adolescents, is an ectasia disease with documented changes in corneal thickness and geometric shape (Hayes et al., 2007). The etiology of keratoconus remains unclear, and effective treatment has become the focus of ophthalmology. Corneal collagen crosslinking (CXL) has revolutionized the treatment of keratoconus in the past decade (Subasinghe et al., 2018). The increased covalent bonds between adjacent collagen fibers induced by CXL can increase corneal mechanical strength and regulate corneal shape (Khan et al., 2015; Subasinghe et al., 2018). The standard epithelium-off CXL protocols include epithelium removal, riboflavin soak, and ultraviolet irradiation, which are the classic protocols of crosslinking. Improving protocols, such as iontophoresis-assisted (Bikbova and Bikbov, 2014) and trans-epithelial CXL (Kocak et al., 2014), have been proposed in the hope of improving safety and reducing complications. However, the improvement of corneal mechanical properties of these new protocols has been shown not as good as that of the standard epithelium-off CXL protocols (Baiocchi et al., 2009; Kymionis et al., 2017). The mechanism of CXL on the cornea needs to be further understood.
For now, the vast majority of the published studies about CXL are focusing on its biomechanical effects. While the micro-level alterations, such as cellular changes, may be critical information of CXL (Sharif et al., 2018a). Keratocytes reside between the corneal collagen lamellae and maintain the extracellular matrix (Jester et al., 2012; Zhang et al., 2019). Activated keratocytes can transform into fibroblasts or even myofibroblasts. Researchers extracted and cultured fibroblasts in vitro, then treated them with riboflavin-UVA to show some short-term cellular changes (Covre et al., 2016; Stachon et al., 2015). While the corneal tissue underwent an enormously complex process as a result of multiple biochemical cues acting at the cellular level (Raghunathan et al., 2017), which cannot be recapitulated by only cross-linked treatment in vitro.
In addition, the increase in corneal stiffness after CXL potentially lasts longer than the time of actual collagen turnover (Raiskup et al., 2015), which suggests that CXL has long-term and permanent effects on tissues (Kling et al., 2017). The in vitro tests are difficult to reveal the changes of cells after prolonged corneal remodeling. Understanding changes of corneal fibroblasts after CXL may provide a new basis for explaining how CXL intervenes in keratoconus progression, why postoperative complications occur, and the guidance for the selection of specific CXL procedures. Further longitudinal studies at the cellular level are needed.
At present, there is no unified direction for improving the protocols of CXL. Whether to carry out the deepithelialization (DEP) in CXL still remains controversial. To understand the effects of CXL on keratocytes, as well as the effects of DEP, this study constructed CXL and DEP rabbit animal models. We determined the time span of corneal stromal remodeling by monitoring the differentiation, apoptosis, and number changes of keratocytes in corneal sections from animals at different time points after CXL treatment. Then we extracted keratocytes from the corneas of the CXL-, DEP- and control-group animals after corneal remodeling was completed. These cells were tested for their migration activity, elastic modulus, proliferation, apoptosis, and cytoskeleton morphology.
Normal healthy New Zealand white rabbits (N = 16, aged 7 months) of specific pathogen-free (SPF) grade were obtained from the Experimental Animal Department of the Capital Medical University. The animals were kept in temperature-controlled rooms with a 12-h light/dark cycle and provided with standard food and water. All animals were given at least 48 h of adaptation time before treatment. The study was approved and monitored by the Institutional Animal Care and Use Committee of the Capital Medical University of Beijing. All surgeries were performed under systemic anesthesia, and all efforts were made to minimize animal suffering.
All rabbits were randomized into 4 groups (At, t = 1, 2, 3 and B). The detailed assignment of treatments and sampling of corneas is summarized in Tab. 1. Three animals from each of the first three groups (A1, A2 and A3) were sacrificed at the 1st, 2nd, and 3rd months after treatment, respectively. Three CXL corneas and the fellow healthy untreated corneas from A1, A2 and A3 at each time point were obtained. The other 2 DEP-rabbits (both eyes) in A3 were kept up to 3 months to obtain 4 DEP corneas. Five animals in Group B, where 3 animals were treated with CXL on one eye and DEP on the other, and two animals as blank control, were sacrificed after corneal remodeling has completed (3 months post-treatment) to extract keratocytes.
Table 1: Number of experimental animals in each group

The standard epithelium-off crosslinking protocols were used to conduct animal models. The central region of each cornea in the CXL group was deepithelialized, then dropped into 0.1% riboflavin solution (Avedro, Waltham, MA, USA) every 2 min to keep the cornea fully immersed and permeated in riboflavin solution for 20 min. Then the cornea was irradiated by UVA (IROC, Zurich, Switzerland) for 30 min with riboflavin solution infiltration. The beam diameter, the wavelength, and the energy density of UVA were 5 mm, 365 nm, and 3 mW/cm2, respectively. The corneas of the DEP group were deepithelialized only. Erythromycin Eye Ointment was used twice a day as the anti-infection treatment during the first postoperative week.
The corneal tissues were extracted and fixed in 4% paraformaldehyde (Solabio, Beijing, China). After dehydrated in 30% sucrose solution (Solabio, Beijing, China), tissues were embedded and frozen with liquid nitrogen, and cut into 10 μm sections at −20°C.
Cell differentiation of tissue sections
Sections were blocked in PBS (Hyclone, Logan, Utah, USA) containing 5% BSA (Sigma, Shanghai, China), 0.3% Triton X-100 (Solabio, Beijing, China) for 1 h at room temperature. After incubated with primary antibody anti-alpha-smooth muscle actin (α-SMA) (1:500 dilution; Boster, Wuhan, China) at 4°C overnight, sections were then incubated with goat anti-mouse IgG secondary antibody (1:500 dilution; Abcam, Cambridge, MA, USA) for 2 h at 37°C. Nuclei were counterstained with DAPI (Solabio, Beijing, China). Negative controls were included with secondary antibody alone.
Cell apoptosis of tissue sections
Sections were first treated with PBS containing 0.3% Triton X-100 for 5 min, then incubated with TUNEL working solution (1 TdT Enzyme: 9 TRITC-Dutp Labeling Mix; Meilune, Dalian, China) for 1 h at 37°C. Nuclei were counterstained with DAPI. No less than 3 sections were selected at each time point, 3 different areas in each section were photographed and counted. The apoptosis ratios were calculated as the number positive for both nuclear and TUNEL stain divided by the total positive of nuclear stain at each photo, normalized to per mm2.
Sections with DAPI staining at the same magnification were used to count the number of keratocytes in the corneal matrix by Image-Pro Plus software (Media Cybernetics, Rockville, MD, USA). Average keratocytes number was calculated for each time point from 3 randomly selected regions in each section, no less than 10 sections for each group. After counting, the area and thickness of the stroma were used to normalize the keratocytes density to per mm3.
The corneal tissue was extracted along the limbus, and the epithelium and endothelium were scraped in a sterile environment. After being repeatedly rinsed by PBS, corneal matrix tissue was cut into small pieces and first digested by 0.5 g/L Type Ι collagenase solution (Invitrogen, Carlsbad, California, U.S.) for nearly 2 h to remove necrotic cells and residual epithelial/endothelial cells. Then residual corneal matrix tissue was digested by 1 g/L Type Ι collagenase solution for 4 h to obtain primary fibroblasts.
Primary fibroblasts were cultured at a humidified incubator at 37°C in 5% CO2 in DMEM/F12 medium (Hyclone, Logan, Utah, USA) with 10% FBS (Fetal Bovine Serum, Gibco, Carlsbad, California, USA) and 1% Penicillin-Streptomycin-Gentamicin solution (Hyclone, Logan, Utah, USA). Culture media were first changed 24 h after cell seeding, then every 2 to 3 days. At cell confluence, 0.05% trypsin-EDTA (Hyclone, Logan, Utah, USA) was used for passage, and the 2nd to 5th generations of cells were used for experiments in this study.
Fibroblasts were fixed in 4% paraformaldehyde and blocked in PBS containing 5% BSA, 0.3% Triton X-100. After that, cells were incubated overnight with vimentin primary antibodies (1:500; Boster, Wuhan, China), and then incubated with goat anti-mouse IgG secondary antibody (1:500; Abcam, Cambridge, MA, USA) for 2 h at 37°C. Nuclei were counterstained with DAPI. Negative controls were included with secondary antibody alone.
Fibroblasts were seeded in 6-well plates, 3 wells for each group, allowed to grow until reaching confluence. Then the culture medium was removed, cells were rinsed and changed to the culture medium without FBS for 24 h. After that, fibroblasts monolayers were scratched with 200 mL yellow pipette tips, 6 images (2 for each well) of each group of cell’s scratch wound were taken every 12 h over a period of 72 h. The opening of the scratched wound was measured by the Image-Pro software, an average wound width was calculated for each scratch wound from the 6 images taken at each time point for each group.
Fibroblasts in each group were inoculated into 3 dishes for atomic force microscopy (AFM, Karlsruhe, Germany) test. Fibroblasts were gently rinsed with pre-warmed PBS solution, immersed in the PBS solution, and placed under the AFM cantilever tip. The spring constant of the conical tip used in this study is 0.07 N/m. After calibrated at the blank base, the cells were tested through the force-volume mode of AFM to obtain the height and the force-indentation curve in selected regions. The elastic modulus of the central region of the cell was selected by the height sensor. The Snedden model was used to fit the curve to obtain the elastic modulus:
where F denotes Young’s elastic modulus (fitting parameter), and ν denotes the Poisson’s ratio, α is the half-angle of the AFM cantilever tip, and δ is the indentation depth in the test.
Fibroblasts in each group were inoculated into 3 dishes for EdU assay. Fibroblasts were incubated with EdU medium (Ribobio, Guangzhou, China) for 2 h, then rinsed with PBS and fixed in 4% paraformaldehyde. After treated with PBS containing 0.3% Triton X-100 for 10 min, cells were incubated with Apollo staining solution for 30 min. Nuclei were counterstained with DAPI. No less than 12 random regions per group were photographed at the same magnification to calculate the ratios. The proliferative ratios were calculated as the number positive for both nuclear and EdU stain divided by the total positive of nuclear stain at each photo. Then the results were normalized to per mm2 after calculated.
Fibroblasts in each group were inoculated into 3 dishes for TUNEL assay. After fixation, fibroblasts were carried out similar experimental steps as described in “Cell apoptosis of tissue sections”. No less than 12 random regions per group were photographed at the same magnification to calculate the ratios. The apoptosis ratios were calculated as the number positive for both nuclear and TUNEL stain divided by the total positive of nuclear stain at each photo. Then the results were normalized to per mm2 after calculated.
Fibroblasts in each group were inoculated into 3 dishes for cytoskeleton assay. After fixation, fibroblasts were carried out similar experimental steps as described in “Cell differentiation of tissue sections”. To visualize F-actin, fibroblasts were then stained with phalloidin (Abbkine, Wuhan, China) for 30 min at room temperature. Nuclei were counterstained with DAPI. No less than 15 random regions per group were photographed at the same magnification to calculate the proportion. The proportion of α-SMA expressing-cells is calculated as the number positive for α-SMA stain divided by the total positive of nuclear stain at each photo, then normalized to per mm2 after calculation.
The normal distribution of data was detected first. Then the Student’s t-test was used for the data that met the normal distribution, and Mann–Whitney U-test for the data that did not. The numbers of cells, E, EdU positive rates, TUNEL positive rates, and α-SMA expression between the experimental group and the control group were compared. All statistical analyses were performed using SPSS (IBM, Armonk, New York, USA) with a significance cutoff of P-value at 0.05.
Corneal remodeling completed three months after CXL
To establish the time course of corneal remodeling, we measured keratocytes differentiation, apoptosis, and cell number. Differentiation (α-SMA, green fluorescence) was visible under the corneal epithelium 1 and 2 month(s) after CXL treatment (group A1 and A2, as shown in Figs. 1B and 1C), but not after 3 months (group A3, as shown in Fig. 1D) or in the control corneas (as shown in Fig. 1A). This indicates that a part of keratocytes near the epithelium differentiated into myofibroblasts one and two month(s) after CXL treatments, but not after three months or in the control corneas. Apoptosis (TUNEL staining, red fluorescence) was only observed one and two months after CXL treatment (as shown in Figs. 1G and 1H), and the TUNEL ratio was 10.02% and 8.03%, respectively (as shown in Fig. 1L). As shown in Fig. 1, cell loss occurred mainly in about 1/3 of the anterior stroma at 1 and 2 month(s) post-treatment, and non-cell areas appeared near the epithelium. Three months after the treatment, cells reappeared in the anterior stroma. The numbers of cells in corneal sections were 25094.68 ± 5693.95, 18150.63 ± 3239.55, 21738.24 ± 3378.1, 23569.09 ± 3339.05, and 24064.92 ± 3253.05 per mm3 in the control corneas, at the 1st month, 2nd month, 3rd month after CXL treatment, and 3rd month after DEP treatment, respectively (as shown in Fig. 1M). Compared with control corneas, cell counts first decreased, then increased gradually with time after CXL treatment. Significant differences were found between control and the experimental corneas at the 1st month (A1, P < 0.0001, N = 30) and 2nd month (A2, P = 0.0049, N = 30), respectively. However, no significant difference was found between the control corneas and the CXL-corneas at the 3rd month (A3, P = 0.1939, N = 30), so as the DEP-corneas (P = 0.3769, N = 30). Compared to the control corneas, changes in keratocytes differentiation, apoptosis or cell number disappeared 3 months after CXL or DEP treatment. Therefore, we inferred that the corneal remodeling is completed 3 months after CXL or DEP treatment. Consequently, keratocytes from corneas of CXL, DEP rabbits were extracted at the 3rd month after the treatments for further tests.
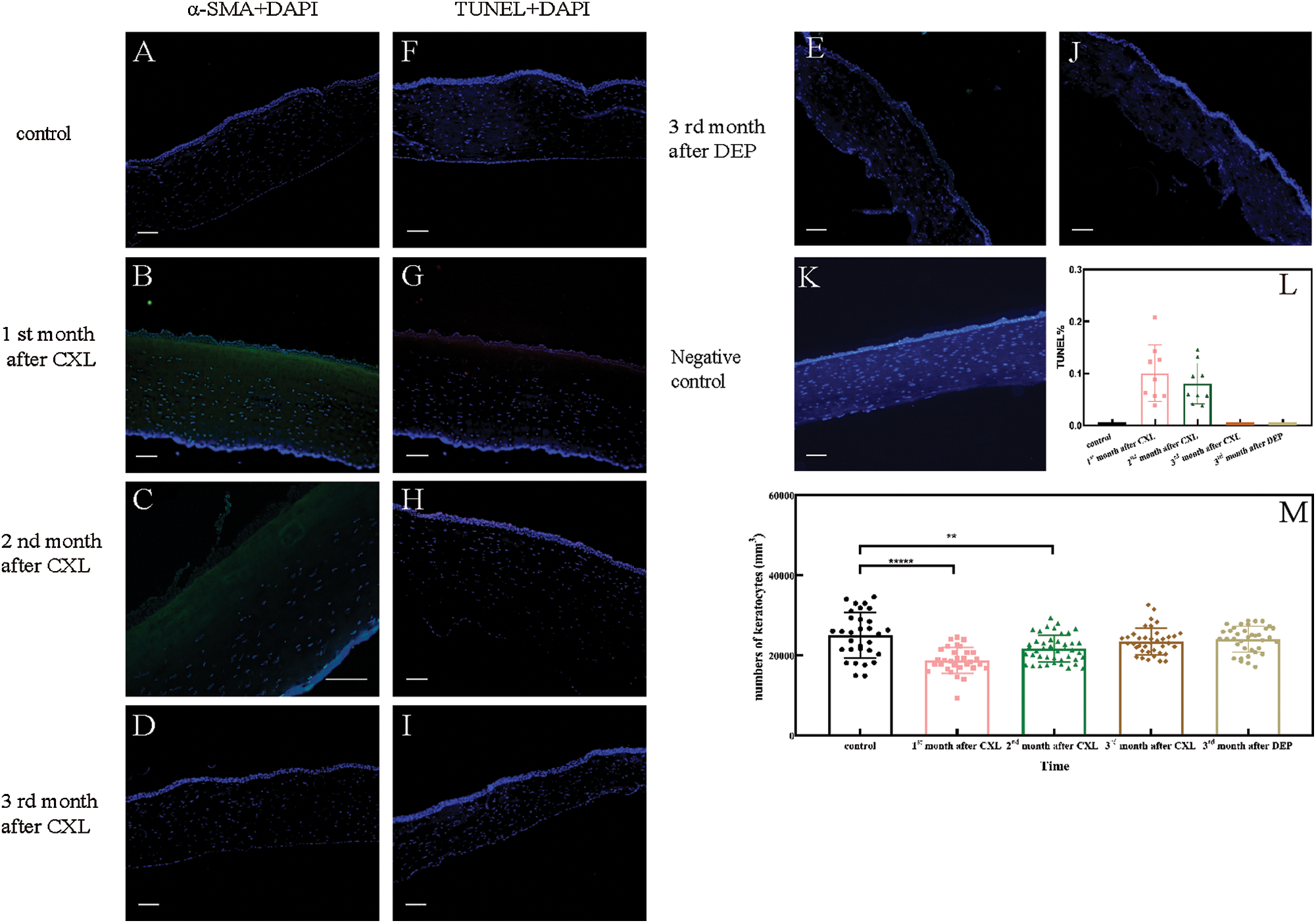
Figure 1: (A–E) Keratocytes differentiation after treatment. (F–J) Keratocytes apoptosis after treatment. (K) Negative control. (L) The apoptotic ratios of corneal stroma sections. (M) Numbers of keratocytes.
Fibroblasts migration increased after CXL
We confirmed the identity of the extracted corneal fibroblasts in group B by vimentin staining (green fluorescence, as shown in Fig. 2). Then we performed a wound-healing assay on the extracted fibroblasts from corneas after CXL, DEP, or non-treatment.

Figure 2: Fibroblasts identification.
Fibroblasts stained with vimentin (green) and DAPI (blue) in three groups, all cells showed positive reactions, confirmed that the extracted and cultured cells were fibroblasts. No positive reaction was shown in the photo of the negative control. Scale bars are 50 µm.
After the scratch, the widths of scratched areas in three groups all decreased with time (as shown in Fig. 3). At 12 h post-scratch, the CXL group showed faster migration than the control group (P = 0.0029, N = 6), then the difference shrank. No statistically significant difference was shown between the DEP group and the control group at all time points.

Figure 3: Fibroblasts migration of three groups.
Fibroblasts elastic moduli increased after CXL
The average of elastic moduli (E) of fibroblasts in the control, DEP, and CXL groups were 6.37 ± 2.63 kPa, 7.05 ± 1.97 kPa, and 12.37 ± 4.08 kPa, respectively (data was shown in Fig. 4). The E of fibroblasts in the CXL group were significantly higher than those of the control group (P < 0.0001, N = 100), whereas no significant difference was found between the DEP and control group (P = 0.0561, N = 100).

Figure 4: (A) Indentation-F curves of fibroblasts. (B) Elastic moduli of fibroblasts.
Fibroblasts proliferation and apoptosis did not change after CXL and DEP
The results of proliferative and apoptotic tests were shown in Fig. 5A. The EdU-based proliferative ratios of fibroblasts in the control, DEP and CXL group were 1.74 ± 1.4%, 1.36 ± 0.8%, 1.39 ± 1.2%, respectively (data was shown in Fig. 5B). The TUNEL-based apoptotic ratios were 0.25 ± 0.1%, 0.19 ± 0.1% and 0.43 ± 0.3%, respectively (data was shown in Fig. 5C). There was no significant difference between the three groups in either proliferation or apoptosis (P > 0.05, N = 12).

Figure 5: (A) Proliferation and apoptosis of fibroblasts in each group. (B and C) The proliferative and apoptotic ratios of fibroblasts in each group.
The expression of α-SMA increased after CXL
Fibroblasts were stained with phalloidin and α-SMA antibodies to visualize the F-actin and α-SMA expression. As shown in Fig. 6A, the F-actin fibers of the three groups were uniformly arranged along the long axis of the cell with clear stress fibers. The synthesis of α-SMA was observed in some fibroblasts in the CXL group. The proportion of α-SMA expressing-cells was shown in Fig. 6B. There was a significant difference in the proportion of α-SMA expressing-cells between the CXL and control group (P < 0.0001, N = 15), but not between the DEP and control group (P = 0.1803, N = 15).

Figure 6: (A) Fibroblasts cytoskeleton in each group. (B) Positive ratios of α-SMA expression in each group.
Keratocytes maintain the extracellular matrix, which is essential for transparency. After CXL, a series of dynamic changes occurred in the corneal stroma. Keratocytes-matrix mechanical interactions may trigger long-term changes in cell mechanics and biology. Meanwhile, the current understanding is relatively limited. This research aimed to explain the 3-month effects of CXL on keratocytes.
In order to determine the duration of corneal stromal remodeling due to CXL- or DEP-treatment, we collected data on keratocytes number, differentiation and apoptosis via corneal sections from animals 1, 2, or 3 months after treatment. In the first two months after CXL, keratocytes underwent differentiation, apoptosis and a corresponding drop in cell number when compared with the control corneas. The keratocyte density of the normal rabbit corneas was 25094.68 ± 5693.95 cells/mm3 in this study, within the range of 20000–50000 cells/mm3 in the literature (Patel et al., 1999; Petroll et al., 1995; Twa and Giese, 2011). The keratocyte density of CXL-corneas rose gradually over time after treatment, which is consistent with the results reported (Hovakimyan et al., 2011; Tang et al., 2019; Wollensak et al., 2007). For now, the time of remodeling completion is still controversial. Bradford et al. (2018) observed the migration of keratocytes appeared in rabbit corneas from 1 to 3 months after CXL, which means the recovery of corneal stroma was not yet completed in the 3rd month. In other studies, keratocyte differentiation (α-SMA) and apoptosis (TUNEL) were observed in rabbits’ tissue sections 4 weeks after CXL (Salomao et al., 2011), the density of cells did not recover until 6 weeks after CXL (Wollensak et al., 2007). Changes in keratocytes were basically disappeared at the 3rd month in this study; therefore, we believed that the corneal remodeling nearly completed at that time. Then we extracted fibroblasts at this time point, observed the migration activity, elastic moduli, proliferation, apoptosis, and cytoskeleton morphology of fibroblasts in vitro.
We first observed that fibroblasts of the CXL group showed faster migration at 12 h post-scratch compared with the control group. Fibroblasts cross-linked in vitro also showed a slight increase in cell migration (Sharif et al., 2017), which is concordant with our result. A number of factors can accelerate the migration. CXL protocols may cause some related growth factors and cytokines, like TGF-β and IL-1 (Kaur et al., 2009), enter into the corneal matrix and promote the migratory activity of keratocytes (Andresen et al., 2000; Jester et al., 2003; Jester et al., 2002; Kim et al., 2010). The treatment of UVA-riboflavin causes a certain degree of stiffness and topographic change in the corneal stroma (Khan et al., 2015; Sharif et al., 2018a; Subasinghe et al., 2018), which also can change the migration (Chou et al., 2016). In addition, we noticed some slight differences between group DEP and control, which may also be due to the release of related cytokines induced by deepithelialization treatment (Andresen et al., 2000; Jester et al., 2003; Jester et al., 2002; Kim et al., 2010).
Then, we tested the elastic moduli of fibroblasts through AFM and detected that E of the CXL group significantly higher than those of the DEP and control groups. To the authors’ knowledge, there are few reports about changes in cellular mechanical properties after CXL treatment. While we noticed that fibroblasts are mechanical-sensitive cells. The mechanical stimulation via the Flexcell system can change fibroblast morphology, adhesion, and cytokines expression in vitro (Du et al., 2017). Studies also confirmed that the stiffer the substrate, the stiffer the cells (Li et al., 2018; Sharif et al., 2017). Consequently, the stiffer corneal matrix after CXL treatment may be the cause of increased E. In addition, our results also showed that E in the control group (6.37 ± 2.63) was slightly larger than the reported result (Miyagi et al., 2018; Sharif et al., 2017), which may due to the conical tip (Park et al., 2010) and the increased water flow resistance caused by rapid scanning test (Berthold et al., 2017) used in this study.
In the next, no significant change was observed in proliferation and apoptosis of fibroblasts compared with the control, either in CXL or DEP group. For now, the observations of the cellular cytotoxicity after corneal stromal remodeling are rare, while the short-term effects of CXL treatment have been confirmed in the literature. In vivo, proliferation and apoptosis increased immediately after CXL treatment (Raiskup et al., 2015; Wollensak et al., 2007). In vitro, 0.1% riboflavin with UVA irradiation (3–10 min) did not cause apoptosis, while decreased the proliferation of fibroblasts in the short term (Sharif et al., 2017). Other researches related to changed stroma after CXL also supported the short-term cellular cytotoxicity (Danciu et al., 2004; Jester and Ho-Chang, 2003; Li et al., 2018; Yang et al., 2004). Our results may indicate that CXL does not change the cellular cytotoxicity after corneal stromal remodeling, although the short-term effects exist.
Besides, CXL and DEP treatment had no significant effect on F-actin, while the expression of α-SMA increased after CXL treatment in this study. CXL treatment on fibroblasts in vitro also slightly increase α-SMA expression (Sharif et al., 2017), which closely with our results. An increase in α-SMA expression represents an increase in myofibroblasts number (Jester et al., 1996). Namely, the increased α-SMA expression in the CXL group means the increased transformation of keratocytes, which may trigger by the change of matrix after CXL treatment (Dreier et al., 2013; Jester and Ho-Chang, 2003; Karamichos et al., 2007; Myrna et al., 2012). Myofibroblasts have faster migratory activity and larger elastic moduli than keratocytes (Jester et al., 1996; Jester and Ho-Chang, 2003; Miyagi et al., 2018). Therefore, the increased migration and E in the CXL group possibility be a result of increased myofibroblasts. Subsequent experiments on α-SMA, such as Western Blot or PCR, may further explain the changes in protein expression. In addition, in this study, no α-SMA expression was observed in the sections at the 3rd month after CXL treatment, while α-SMA was shown in some cells in vitro. This may be the results of the stimulation of serum in culture medium and experimental operation, which increase α-SMA-expression of cells (Vishwanath et al., 2003). More α-SMA-expressed cells were observed in the CXL group than those of the other two groups under the similar stimulation, which may indicate that cells after CXL treatment are more sensitive to stimulation and more prone to transformation.
Based on the results observed in vitro, we speculate that such 3-month effects may indicate changes in cellular gene expression. CXL treatment has been confirmed to affect fibroblast metabolism (Sharif et al., 2018b). Sabine Kling et al. (2017) researched the differential genes of corneal stroma in the short term after CXL treatment and found that several target genes might be related to the biomechanical stability and shape of the cornea. Differential expressions of many genes have also been found between normal corneas and keratoconus (Sharif et al., 2018b). These genes are associated with the cellular changes (Kabza et al., 2017), such as the increased migration, elastic moduli, and α-SMA expression of fibroblasts in this study. The reported research, together with our findings, suggest a new perspective on corneal remodeling after surgery. That is to say, CXL may not only increase the link between collagen fibers (Hayes et al., 2013) but also regulate the biological behavior of keratocytes and then act on keratoconus through gene transcription.
Furthermore, DEP treatment had no adverse effects on fibroblasts in this study. This indicates that deepithelialization itself probably does not cause keratocytes-related complications after CXL. UVA may damage endothelial cells through the thinner corneal stroma after deepithelialization (Kymionis et al., 2012) or even induce corneal virus infection by the direct irradiation to corneal stroma (Price et al., 2012). That is to say, deemed DEP-related complications (Mazzotta et al., 2007) could possibly be due to the losses of epithelial cells, which have a protective effect against UVA-riboflavin. Moreover, compared with the DEP group, the increase of E and α-SMA expression of fibroblasts in the CXL group suggest that besides deepithelialization, both riboflavin-UVA and cell-matrix mechanical interactions may have a more pronounced effect on cells.
A number of modifications of CXL protocols have been proposed to shorten the duration or reduce complications (Kymionis et al., 2017). Meanwhile, there is still no consensus on the direction of modifications, and there is still a debate about whether DEP should be carried out during the surgery. A systemic review revealed that haze and scar after trans-epithelial CXL decreased, nevertheless, the treatment efficacy (Zhang et al., 2018) and mechanical property improvement (Scarcelli et al., 2013) also reduced compared with the standard epithelium-off CXL. In addition, trans-epithelial CXL even has a toxic effect, which can change the regeneration of epithelial cells (Mazzotta et al., 2015). Hence, taking both the treatment efficacy and the related complications into account, we suggest that it might be a better way to improve the cross-linking technique through the concentration of riboflavin and the energy of UVA.
The one limitation of this study is that despite the rabbit and human have similar corneal characteristics, there still some differences like Bowman’s layer, which may potentially affect the cell-matrix interactions after CXL (Naranjo et al., 2019). The other is that the keratocytes were extracted and tested in vitro, which might not completely correspond to the environment in vivo. In addition, this study only observed α-SMA by immunofluorescent staining, further Western blots experiments were needed to confirm the specific differences in its expression.
Our results suggest that CXL has 3-month effects on fibroblasts, including migration, elastic moduli, and α-SMA expression. It may provide a new basis for explaining how CXL hinders keratoconus progression and a new reference for the selection of CXL protocols.
Author Contribution: The authors confirm contribution to the paper as follows: Study conception and design: Xinyan Chen, Lin Li; data collection: Xinyan Chen; analysis and interpretation of results: Xinyan Chen, Haixia Zhang; draft manuscript preparation: Xinyan Chen, Lin Li. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
Ethics Approval: The study was approved and monitored by the Institutional Animal Care and Use Committee of the Capital Medical University of Beijing. The approval code is AEEI-2014-066, and the approval date is July 2014.
Funding Statement: This work was supported by the National natural science foundation of China (Nos. 31370952, 31470914).
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
Andresen JL, Ledet T, Hager H, Josephsen K, Ehlers N (2000). The influence of corneal stromal matrix proteins on the migration of human corneal fibroblasts. Experimental Eye Research 71: 33–43. DOI 10.1006/exer.2000.0850. [Google Scholar] [CrossRef]
Baiocchi S, Mazzotta C, Cerretani D, Caporossi T, Caporossi A (2009). Corneal crosslinking: Riboflavin concentration in corneal stroma exposed with and without epithelium. Journal of Cataract & Refractive Surgery 35: 893–899. DOI 10.1016/j.jcrs.2009.01.009. [Google Scholar] [CrossRef]
Berthold T, Benstetter G, Frammelsberger W, Rodriguez R, Nafria M (2017). Numerical study of hydrodynamic forces for AFM operations in liquid. Scanning 2017: 1–12. DOI 10.1155/2017/6286595. [Google Scholar] [CrossRef]
Bikbova G, Bikbov M (2014). Transepithelial corneal collagen cross-linking by iontophoresis of riboflavin. Acta Ophthalmologica 92: e30–34. [Google Scholar]
Bradford SM, Mikula ER, Juhasz T, Brown DJ, Jester JV (2018). Collagen fiber crimping following in vivo UVA-induced corneal crosslinking. Experimental Eye Research 177: 173–180. DOI 10.1016/j.exer.2018.08.009. [Google Scholar] [CrossRef]
Covre JL, Cristovam PC, Loureiro RR, Hazarbassanov RM, Campos M, Sato ÉH, Gomes JÁ (2016). The effects of riboflavin and ultraviolet light on keratocytes cultured in vitro. Arquivos Brasileiros de Oftalmologia 79: 180–185. DOI 10.5935/0004-2749.20160052. [Google Scholar] [CrossRef]
Chou SF, Lai JY, Cho CH, Lee CH (2016). Relationships between surface roughness/stiffness of chitosan coatings and fabrication of corneal keratocyte spheroids: Effect of degree of deacetylation. Colloids and Surfaces B: Biointerfaces 142: 105–113. DOI 10.1016/j.colsurfb.2016.02.051. [Google Scholar] [CrossRef]
Danciu TE, Gagari E, Adam RM, Damoulis PD, Freeman MR (2004). Mechanical strain delivers anti-apoptotic and proliferative signals to gingival fibroblasts. Journal of Dental Research 83: 596–601. DOI 10.1177/154405910408300803. [Google Scholar] [CrossRef]
Dreier B, Thomasy SM, Mendonsa R, Raghunathan VK, Russell P, Murphy CJ (2013). Substratum compliance modulates corneal fibroblast to myofibroblast transformation. Investigative Ophthalmology & Visual Science 54: 5901–5907. DOI 10.1167/iovs.12-11575. [Google Scholar] [CrossRef]
Du GL, Chen WY, Li XN, He R, Feng PF (2017). Induction of MMP1 and 3 by cyclical mechanical stretch is mediated by IL6 in cultured fibroblasts of keratoconus. Molecular Medicine Reports 15: 3885–3892. DOI 10.3892/mmr.2017.6433. [Google Scholar] [CrossRef]
Hayes S, Boote C, Tuft SJ, Quantock AJ, Meek KM (2007). A study of corneal thickness, shape and collagen organisation in keratoconus using videokeratography and X-ray scattering techniques. Experimental Eye Research 84: 423–434. DOI 10.1016/j.exer.2006.10.014. [Google Scholar] [CrossRef]
Hayes S, Kamma-Lorger CS, Boote C, Young RD, Quantock AJ, Rost A, Khatib Y, Harris J, Yagi N, Terrill N, Meek KM, Chakravarti S (2013). The effect of riboflavin/UVA collagen cross-linking therapy on the structure and hydrodynamic behaviour of the ungulate and rabbit corneal stroma. PLoS One 8: e52860. DOI 10.1371/journal.pone.0052860. [Google Scholar] [CrossRef]
Hovakimyan M, Guthoff R, Reichard M, Wree A, Nolte I, Stachs O (2011). In vivo confocal laser-scanning microscopy to characterize wound repair in rabbit corneas after collagen cross-linking. Clinical & Experimental Ophthalmology 39: 899–909. DOI 10.1111/j.1442-9071.2011.02634.x. [Google Scholar] [CrossRef]
Jester JV, Barry-Lane PA, Cavanagh HD, Petroll WM (1996). Induction of alpha-smooth muscle actin expression and myofibroblast transformation in cultured corneal keratocytes. Cornea 15: 505–516. DOI 10.1097/00003226-199609000-00011. [Google Scholar] [CrossRef]
Jester JV, Brown D, Pappa A, Vasiliou V (2012). Myofibroblast differentiation modulates keratocyte crystallin protein expression, concentration, and cellular light scattering. Investigative Ophthalmology & Visual Science 53: 770–778. DOI 10.1167/iovs.11-9092. [Google Scholar] [CrossRef]
Jester JV, Ho-Chang J (2003). Modulation of cultured corneal keratocyte phenotype by growth factors/cytokines control in vitro contractility and extracellular matrix contraction. Experimental Eye Research 77: 581–592. DOI 10.1016/S0014-4835(03)00188-X. [Google Scholar] [CrossRef]
Jester JV, Huang J, Fisher S, Spiekerman J, Chang JH, Wright WE, Shay WE, Shay JW (2003). Myofibroblast differentiation of normal human keratocytes and hTERT, extended-life human corneal fibroblasts. Investigative Ophthalmology & Visual Science 44: 1850–1858. DOI 10.1167/iovs.02-0973. [Google Scholar] [CrossRef]
Jester JV, Huang J, Petroll WM, Cavanagh HD (2002). TGFbeta induced myofibroblast differentiation of rabbit keratocytes requires synergistic TGFbeta, PDGF and integrin signaling. Experimental Eye Research 75: 645–657. DOI 10.1006/exer.2002.2066. [Google Scholar] [CrossRef]
Kabza M, Karolak JA, Rydzanicz M, Szcześniak MłW, Nowak DM, Ginter-Matuszewska B, Polakowski P, Ploski R, Szaflik JP, Gajecka M (2017). Collagen synthesis disruption and downregulation of core elements of TGF-beta, Hippo, and Wnt pathways in keratoconus corneas. European Journal of Human Genetics 25: 582–590. DOI 10.1038/ejhg.2017.4. [Google Scholar] [CrossRef]
Karamichos D, Lakshman N, Petroll WM (2007). Regulation of corneal fibroblast morphology and collagen reorganization by extracellular matrix mechanical properties. Investigative Ophthalmology & Visual Science 48: 5030–5037. DOI 10.1167/iovs.07-0443. [Google Scholar] [CrossRef]
Kaur H, Chaurasia SS, Agrawal V, Suto C, Wilson SE (2009). Corneal myofibroblast viability: Opposing effects of IL-1 and TGF beta1. Experimental Eye Research 89: 152–158. DOI 10.1016/j.exer.2009.03.001. [Google Scholar] [CrossRef]
Khan WA, Zaheer N, Khan S (2015). Corneal collagen cross-linking for keratoconus: Results of 3-year follow-up in Pakistani population. Canadian Journal of Ophthalmology 50: 143–150. DOI 10.1016/j.jcjo.2014.11.003. [Google Scholar] [CrossRef]
Kim A, Lakshman N, Karamichos D, Petroll WM (2010). Growth factor regulation of corneal keratocyte differentiation and migration in compressed collagen matrices. Investigative Ophthalmology & Visual Science 51: 864–875. DOI 10.1167/iovs.09-4200. [Google Scholar] [CrossRef]
Kling S, Hammer A, Neettoa EAT, Hafezi F (2017). Differential gene transcription of extracellular matrix components in response to in vivo corneal crosslinking (CXL) in rabbit corneas. Translational Vision Science & Technology 6: 8. DOI 10.1167/tvst.6.6.8. [Google Scholar] [CrossRef]
Kocak I, Aydin A, Kaya F, Koc H (2014). Comparison of transepithelial corneal collagen crosslinking with epithelium-off crosslinking in progressive keratoconus. Journal Français d’Ophtalmologie 37: 371–376. DOI 10.1016/j.jfo.2013.11.012. [Google Scholar] [CrossRef]
Kymionis GD, Kontadakis GA, Hashemi KK (2017). Accelerated versus conventional corneal crosslinking for refractive instability: An update. Current Opinion in Ophthalmology 28: 343–347. DOI 10.1097/ICU.0000000000000375. [Google Scholar] [CrossRef]
Kymionis GD, Portaliou DM, Diakonis VF, Kounis GA, Panagopoulou SI, Grentzelos MA (2012). Corneal collagen cross-linking with riboflavin and ultraviolet-A irradiation in patients with thin corneas. American Journal of Ophthalmology 153: 24–28. DOI 10.1016/j.ajo.2011.05.036. [Google Scholar] [CrossRef]
Li X, Wei J, Lan W, Rong S, Wang X, He R, Chen W, Qin YX (2018). Non-enzymatic glycation crosslinking affects human keratoconus fibroblasts behavior in vitro. Journal of Biomaterials and Tissue Engineering 8: 1334–1341. DOI 10.1166/jbt.2018.1883. [Google Scholar] [CrossRef]
Mazzotta C, Balestrazzi A, Baiocchi S, Traversi C, Caporossi A (2007). Stromal haze after combined riboflavin-UVA corneal collagen cross-linking in keratoconus: In vivo confocal microscopic evaluation. Clinical & Experimental Ophthalmology 35: 580–582. DOI 10.1111/j.1442-9071.2007.01536.x. [Google Scholar] [CrossRef]
Mazzotta C, Hafezi F, Kymionis G, Caragiuli S, Jacob S, Traversi C, Barabino S, Randleman JB (2015). In vivo confocal microscopy after corneal collagen crosslinking. Ocular Surface 13: 298–314. DOI 10.1016/j.jtos.2015.04.007. [Google Scholar] [CrossRef]
Miyagi H, Jalilian I, Murphy CJ, Thomasy SM (2018). Modulation of human corneal stromal cell differentiation by hepatocyte growth factor and substratum compliance. Experimental Eye Research 176: 235–242. DOI 10.1016/j.exer.2018.09.001. [Google Scholar] [CrossRef]
Myrna KE, Mendonsa R, Russell P, Pot SA, Liliensiek SJ, Jester JV, Nealey PF, Brown D, Murphy CJ (2012). Substratum topography modulates corneal fibroblast to myofibroblast transformation. Investigative Ophthalmology & Visual Science 53: 811–816. DOI 10.1167/iovs.11-7982. [Google Scholar] [CrossRef]
Naranjo A, Pelaez D, Arrieta E, Salero-Coca E, Martinez JD, Sabater AL, Amescua G, Parel JM (2019). Cellular and molecular assessment of rose bengal photodynamic antimicrobial therapy on keratocytes, corneal endothelium and limbal stem cell niche. Experimental Eye Research 188: 107808. DOI 10.1016/j.exer.2019.107808. [Google Scholar] [CrossRef]
Park S, Duong CT, Lee JH, Lee SS, Son K (2010). Effect of tip geometry of atomic force microscope on mechanical responses of bovine articular cartilage and agarose gel. International Journal of Precision Engineering and Manufacturing 11: 129–136. DOI 10.1007/s12541-010-0016-1. [Google Scholar] [CrossRef]
Patel SV, Mclaren JW, Camp JJ, Nelson LR, Bourne WM (1999). Automated quantification of keratocyte density by using confocal microscopy in vivo. Investigative Ophthalmology & Visual Science 40: 320–326. [Google Scholar]
Petroll WM, Boettcher K, Barry P, Cavanagh HD, Jester JV (1995). Quantitative assessment of anteroposterior keratocyte density in the normal rabbit cornea. Cornea 14: 3–9. DOI 10.1097/00003226-199501000-00002. [Google Scholar] [CrossRef]
Price MO, Tenkman LR, Schrier A, Fairchild KM, Trokel SL, Price FWJr (2012). Photoactivated riboflavin treatment of infectious keratitis using collagen cross-linking technology. Journal of Refractive Surgery 28: 706–713. DOI 10.3928/1081597X-20120921-06. [Google Scholar] [CrossRef]
Raghunathan VK, Thomasy SM, Strom P, Yanez-Soto B, Garland SP, Sermeno J, Reilly CM, Murphy CJ (2017). Tissue and cellular biomechanics during corneal wound injury and repair. Acta Biomaterialia 58: 291–301. DOI 10.1016/j.actbio.2017.05.051. [Google Scholar] [CrossRef]
Raiskup F, Theuring A, Pillunat LE, Spoerl E (2015). Corneal collagen crosslinking with riboflavin and ultraviolet-A light in progressive keratoconus: Ten-year results. Journal of Cataract & Refractive Surgery 41: 41–46. DOI 10.1016/j.jcrs.2014.09.033. [Google Scholar] [CrossRef]
Salomao MQ, Chaurasia SS, Sinha-Roy A, Ambrosio R,Jr., Esposito A, Sepulveda R, Agrawal V, Wilson SE (2011). Corneal wound healing after ultraviolet-A/riboflavin collagen cross-linking: A rabbit study. Journal of Refractive Surgery 27: 401–407. DOI 10.3928/1081597X-20101201-02. [Google Scholar] [CrossRef]
Scarcelli G, Kling S, Quijano E, Pineda R, Marcos S, Yun SH (2013). Brillouin microscopy of collagen crosslinking: Noncontact depth-dependent analysis of corneal elastic modulus. Investigative Ophthalmology & Visual Science 54: 1418–1425. DOI 10.1167/iovs.12-11387. [Google Scholar] [CrossRef]
Sharif R, Fowler B, Karamichos D (2018a). Collagen cross-linking impact on keratoconus extracellular matrix. PLoS One 13: e0200704. DOI 10.1371/journal.pone.0200704. [Google Scholar] [CrossRef]
Sharif R, Hjortdal J, Sejersen H, Frank G, Karamichos D (2017). Human in vitro model reveals the effects of collagen cross-linking on keratoconus pathogenesis. Scientific Reports 7: 12517. DOI 10.1038/s41598-017-12598-8. [Google Scholar] [CrossRef]
Sharif R, Sejersen H, Frank G, Hjortdal J, Karamichos D (2018b). Effects of collagen cross-linking on the keratoconus metabolic network. Eye 32: 1271–1281. DOI 10.1038/s41433-018-0075-6. [Google Scholar] [CrossRef]
Stachon T, Wang J, Song X, Langenbucher A, Seitz B, Szentmary N (2015). Impact of crosslinking/riboflavin-UVA-photodynamic inactivation on viability, apoptosis and activation of human keratocytes in vitro. Journal of Biomedical Research 29: 321–325. [Google Scholar]
Subasinghe SK, Ogbuehi KC, Dias GJ (2018). Current perspectives on corneal collagen crosslinking (CXL). Graefe’s Archive for Clinical and Experimental Ophthalmology 256: 1363–1384. DOI 10.1007/s00417-018-3966-0. [Google Scholar] [CrossRef]
Tang Y, Song W, Qiao J, Rong B, Wu Y, Yan X (2019). A study of corneal structure and biomechanical properties after collagen crosslinking with genipin in rabbit corneas. Molecular Vision 25: 574–582. [Google Scholar]
Twa MD, Giese MJ (2011). Assessment of corneal thickness and keratocyte density in a rabbit model of laser in situ keratomileusis using scanning laser confocal microscopy. American Journal of Ophthalmology 152: 941–953 e941. DOI 10.1016/j.ajo.2011.05.023. [Google Scholar] [CrossRef]
Vishwanath M, Ma L, Otey CA, Jester JV, Petroll WM (2003). Modulation of corneal fibroblast contractility within fibrillar collagen matrices. Investigative Ophthalmology & Visual Science 44: 4724–4735. DOI 10.1167/iovs.03-0513. [Google Scholar] [CrossRef]
Wollensak G, Iomdina E, Dittert DD, Herbst H (2007). Wound healing in the rabbit cornea after corneal collagen cross-linking with riboflavin and UVA. Cornea 26: 600–605. DOI 10.1097/ICO.0b013e318041f073. [Google Scholar] [CrossRef]
Yang G, Crawford RC, Wang JH (2004). Proliferation and collagen production of human patellar tendon fibroblasts in response to cyclic uniaxial stretching in serum-free conditions. Journal of Biomechanics 37: 1543–1550. DOI 10.1016/j.jbiomech.2004.01.005. [Google Scholar] [CrossRef]
Zhang K, Ren XX, LI P, Pang KP, Wang H (2019). Construction of a full-thickness human corneal substitute from anterior acellular porcine corneal matrix and human corneal cells. International Journal of Ophthalmology 12: 351–362. [Google Scholar]
Zhang X, Zhao J, Li M, Tian M, Shen Y, Zhou X (2018). Conventional and transepithelial corneal cross-linking for patients with keratoconus. PLoS One 13: e0195105. DOI 10.1371/journal.pone.0195105. [Google Scholar] [CrossRef]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |