DOI:10.32604/biocell.2021.015817
| BIOCELL DOI:10.32604/biocell.2021.015817 | |
| Article |
Retinoic acid affects basic cellular processes and SOX2 and SOX18 expression in breast carcinoma cells
1Institute of Molecular Genetics and Genetic Engineering, University of Belgrade, Belgrade, Serbia
2Institute of Cell Biology, School of Biological Sciences, The University of Edinburgh, Edinburgh, UK
3Department of Radiation Oncology, Feinberg School of Medicine, Northwestern University, Chicago, IL, USA
4Faculty of Biology, University of Belgrade, Belgrade, Serbia
5Serbian Academy of Sciences and Arts, Belgrade, Serbia
*Address correspondence to: Milena Stevanovic, milenastevanovic@imgge.bg.ac.rs
Received: 16 January 2021; Accepted: 16 April 2021
Abstract: Genetic and molecular heterogeneity, together with intrinsic and acquired resistance to therapy, represent the major obstacles to the successful treatment of different types of breast carcinoma. Increasing evidence demonstrates that SOX transcription factors in breast carcinomas could act both as oncogenes and tumor suppressors and have been associated with tumor stage and grade, poor prognosis, and therapy resistance. Both SOX2 and SOX18 over-expression has been correlated with poor prognosis in breast carcinomas, and these genes are recognized as potential antitumor targets. Our aim was to evaluate the effect of retinoic acid (RA), a well-known cyto-differentiating agent, on breast carcinoma cells in vitro and to investigate the potential of RA treatment to modify the expression of SOX2 and SOX18 genes. By applying various experimental approaches, we evaluated the effect of RA on basic cellular processes in SK-BR-3 and MCF7 breast carcinoma cell lines. We have shown that RA inhibits cell growth, reduces the number of Ki-67 positive cells, and causes cell-cycle arrest. RA effect was more prominent in SK-BR-3 cell line that lacks SOX2 expression, including a higher decrease in cell viability, reduction in colony formation, and significant remodeling of cellular structure. We have shown that RA treatment led to the downregulation of SOX2 expression in MCF7 cells and to the reduction of SOX18 expression in both cell lines. By functional analysis, we showed that the anti-proliferative effect of RA in both cell lines was not based on the activity of stemness marker SOX2, pointing to a SOX2-independent mechanism of action. The ability of RA to reduce SOX2/SOX18 expression raises the possibility that these genes can be used as biomarkers to distinguish RA-responders from non-responders. Together, our study shows that the response of breast carcinoma cell lines to RA treatment may vary, highlighting that the development of RA-based therapy should consider differences in breast carcinoma subtypes.
Keywords: Breast carcinoma; Anti-proliferative activity; Transcription factors; MCF7 cell line; SK-BR-3 cell line
Breast cancer is a heterogeneous disease, with three major subtypes, estrogen and/or progesterone receptor-positive (ER+/PR+), HER-2 amplified (HER-2+), and triple-negative (ER−/PR−/HER2−) that respond differently to therapy and develop different mechanisms of chemotherapy resistance (Testa et al., 2020). In addition to surgical treatments, radiation and chemotherapy represent important parts of breast cancer therapies. However, many patients develop acquired resistance that prevents response to therapy that was initially effective (Housman et al., 2014). In some cases, intrinsic resistance to chemotherapy is a significant cause of treatment failure (Higgins and Baselga, 2011).
Since various mechanisms could contribute to the chemoresistance of breast cancer cells, the search for promising treatments that would overcome drug resistance is of high priority (Ji et al., 2019).
Breast cancer cells that survive various treatments often exhibit properties of cancer stem cells (Ji et al., 2019; Lee et al., 2011; Nedeljkovic and Damjanovic, 2019; Piva et al., 2014). It was shown that the level of SOX2 transcription factor, one of the well-known stem cells’ markers, is elevated in primary breast carcinomas that do not respond to endocrine therapy (Piva et al., 2014). Xiaoxian Li and co-workers showed that chemotherapy treatment increases the percentage of breast cancer cells that have unique properties and enhanced self-renewal ability and may display an increased propensity for tumor formation (Li et al., 2008). These cells with cancer stem cells’ properties are also more resistant to radiation therapy (Phillips et al., 2006). Therefore, targeting these chemo- or radio-resistant cells may provide a more specific approach to prevent cancer recurrence and improve long-term survival.
Overexpression of SOX2 has been documented in many types of solid tumors, including breast carcinomas (Li et al., 2004; Rodriguez-Pinilla et al., 2007; Sanada et al., 2006; Zhu et al., 2012). In a meta-analysis published by Wang and co-workers, the overexpression of SOX2 was correlated with poor prognosis in human solid tumors (Wang et al., 2020). The expression of SOX2, together with OCT4 and NANOG, was positively associated with poor pathological differentiation and advanced disease stage (Yang et al., 2018). Both gain-of-function and loss-of-function studies indicate that SOX2 contributes to breast cancer cell proliferation and tumorigenic properties both in vitro and in vivo (Chen et al., 2008). A significant positive correlation between SOX2 transcription factor expression and increased tumor size, higher histological grade, lymph node metastasis, and the highly aggressive triple-negative phenotype of breast cancer has been found in a meta-analysis performed by Yan Zheng and co-workers (Zheng et al., 2015). Another study revealed that SOX2 and ALDH1 could be considered as markers of poor prognosis, particularly in ER-negative breast cancer patients (Shima et al., 2016).
In addition to SOX2, SOX18 as another member of the SOX family of transcription factors has been shown to play an important role in tumor growth and metastasis, including breast carcinoma (Duong et al., 2012; Pula et al., 2013; Wang et al., 2015; Young et al., 2006; Zhang et al., 2016). Previously, we have revealed that SOX18 promotes migration and invasion of cancer cells in vitro, showing its potential in promoting metastatic phenotype (Petrovic et al., 2015). Jianxiang Zhang and co-workers have demonstrated that knockdown of SOX18 inhibits breast cancer cell growth (Zhang et al., 2016). In a mouse preclinical model of breast cancer, pharmacological inhibition of SOX18 significantly improved survival by reducing metastatic spread due to a reduction in tumor vascular density (Overman et al., 2017).
Both SOX2 and SOX18 transcription factors are recognized as potential targets in anticancer therapy (Pietrobono et al., 2016; Wang et al., 2016; Wuebben et al., 2016; Zhang et al., 2016). Therefore, it is important to explore the effects of modulation of their expression in cancer cells. Search for novel therapeutic strategies targeting transcription factors is challenging and one of the options is to employ already known agents that affect their expression/activity.
RA, an active metabolite of vitamin A, is the first clinically useful cyto-differentiating agent, being employed in the treatment of acute promyelocytic leukemia (APL) (Gianni et al., 2001). It has been shown that treatment of breast cancer cells with RA affects the expression of various miRNAs, inhibits invasive behavior, and deregulates the growth of cancer cells (Fisher et al., 2015; Khan et al., 2015; Terao et al., 2011). In the last two decades, the growing interest in the application of RA in the treatment of breast cancer led to many preclinical studies (Costantini et al., 2020). However, the results gathered in clinical trials are insufficient and seem to be rather disappointing (Bryan et al., 2011; Budd et al., 1998; Garattini et al., 2014; Sutton et al., 1997; Veronesi et al., 2006).
Our aim was to re-evaluate the effect of RA on breast carcinoma cells in vitro and to investigate the potential of RA treatment to modify the expression of SOX2 and SOX18 genes. We evaluated the effects of RA on the proliferation, apoptosis, cell cycle distribution, and migration of SK-BR-3 and MCF7 breast cancer cell lines. We showed that RA inhibits cell growth, reduces the number of Ki-67-positive cells, and causes cell-cycle arrest. Importantly, RA treatment led to the downregulation of SOX2 expression in MCF7 cells and SOX18 expression in both SK-BR-3 and MCF7 cells. Functional analysis showed that the anti-proliferative effect of RA in both cell lines is not related to the activity of stemness marker, SOX2, pointing to a SOX2-independent mechanism of action.
We have used two different breast carcinoma cell lines: MCF7 (ATCC HTB-22) and SK-BR-3 (ATCC HTB-30). These cells represent different breast cancer histotypes, MCF7 cells express estrogen receptor (ER+) and progesterone receptor (PR+) but lack HER-2 receptor (HER2−), while SK-BR-3 cells have HER2 amplification (HER2+). Both cell lines were maintained in high-glucose Dulbecco’s Modified Eagle Medium (Thermo Fisher Scientific, Waltham, Massachusetts, USA), supplemented with 10% fetal bovine serum (Thermo Fisher Scientific, Waltham, Massachusetts, USA) at 37°C in 5% CO2. Cells were treated with various concentrations of RA (Sigma-Aldrich/Merck, St. Louis, Missouri, USA), including 1 μM, 5 μM and 10 μM, or with DMSO (Serva, Heidelberg, Germany) as a vehicle.
Cells’ viability was tested using the MTT assay (Merck, St.Louis, Missouri, USA). A day before treatment with increasing concentrations of RA (1 μM, 5 μM and 10 μM), cells were seeded at a density of 1 × 104 cells per well for 3 days of treatment and 0.5 × 104 cells per well for 5 days of treatment in 96-well plates. The effect of RA on the cell viability was monitored using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) dye as described previously (Petrovic et al., 2015). Colorimetric quantification was performed by measuring absorbance at 550 nm using a microplate reader Infinite 200 PRO (Tecan, Mannedorf, Zurich, Switzerland).
For the LIVE/DEAD cytotoxicity test, SK-BR-3 and MCF7 cells were seeded on coverslips in 12-well plates at a density of 5 × 104 cells per well. The following day, DMSO or 1 μM RA were added to the cells and further incubated for 72 h. Cytotoxicity was measured using LIVE/DEAD™ Cell Imaging Kit (Thermo Fisher Scientific, Waltham, Massachusetts, USA) as previously described (Markovic et al., 2019). The kit enables a two-color discrimination system of live cells (green fluorescence) from dead cells (red fluorescence). The cells were visualized using Olympus BX51 fluorescence microscope (FITC filter for green fluorescence and Texas Red filter for red fluorescence) and analyzed using Cytovision 3.1 software (Applied Imaging Corporation, USA). All images were captured using a 20 × objective.
Clonogenic assay was performed as previously described (Buch et al., 2012). In particular, cells were seeded in triplicates in 12-well plates, 500 cells per well as single cells. Cells were maintained in the complete medium with DMSO as a negative control or with 1 μM RA for 8 days. Medium with DMSO or RA was replaced every second day. After colonies were formed (minimum 50 cells per colony), they were fixed and stained with crystal violet, performed as previously described (Petrovic et al., 2015). Upon staining, colonies were photographed and counted.
Cells were plated on coverslips and treated with 1 μM RA or 0.1% DMSO as a vehicle for 72 h. Following the treatment, cells were fixed in 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS) for 20 min at room temperature (RT), permeabilized in 0.2% Triton X-100 for 10 min, and blocked in 1% bovine serum albumin (BSA) and 10% normal goat serum in PBS for 1 h at RT. Primary antibodies were diluted in PBS containing 1% BSA, 0.1% Triton X-100 (PBT) and incubated overnight at 4°C as follows: mouse anti-tubulin, diluted 1:500 (ab56676, Abcam, Cambridge, UK), mouse anti-vimentin, diluted 1:300 (sc-6260, Santa Cruz Biotechnology, Inc., Heidelberg, Germany), rabbit anti-Ki67, diluted 1:250 (ab15580, Abcam, Cambridge, UK), rabbit anti-SOX2, diluted 1:1000 (39823, Active Motif, Carlsbad, California, USA), rabbit anti-SOX18, diluted 1:50 (orb163563, Biorbyt, Cambridge, UK). Appropriate secondary antibodies, biotinylated goat anti-rabbit IgG (BA-1000) or horse anti-mouse IgG (BA-2000) (Vector Laboratories, Burlingame, CA, USA) diluted 1:500 in 1% BSA and PBT were applied for 1 h at RT. For fluorescence detection, cells were incubated for 1 h on RT in DyLight 488 streptavidin (SA-5488) (Vector Laboratories, Burlingame, CA, USA) diluted 1:500 in PBS. Fluorescent staining of actin filaments (F-actin) was achieved by incubating cells for 1 h at RT in CytoPainter Phalloidin-iFluor 594 Reagent (ab176757, Abcam), diluted 1:3000 in PBS. Nuclei were stained with 0.1 mg/mL diamino phenylindole (DAPI) (Sigma-Aldrich/Merck, St. Louis, Missouri, USA). Images were taken by a Leica TCS SP8 confocal microscope and analyzed by Leica Microsystems LAS AF-TCS SP8 software (Leica Microsystems, Wetzlar, Germany). Images of tubulin and F-actin double staining were used to measure the cell area. The average cell area in each experiment was determined by measuring the surface area of individual cells. The relative levels of SOX2 and SOX18 nuclear staining intensity in each experiment were determined by measuring the mean fluorescence intensity (total fluorescence intensity per number of pixels) in the nuclei.
Apoptosis assay and cell cycle
For apoptosis assay, 6 ×105 cells were seeded in 35-mm cell culture dishes a day before treatment. The next day, they were treated with DMSO as a negative control, 1 μM doxorubicin as a positive control, or 1 μM RA for 24 h. Upon treatment, cells were collected from both media (detached cells) and dish surface (attached cells) and resuspended in 1 × Annexin binding buffer. 5 μL of Annexin V-AlexaFluor 488 conjugate (Thermo Fisher Scientific, Waltham, Massachusetts, USA) and 5 μL of propidium iodide (PI) (Thermo Fisher Scientific, Waltham, Massachusetts, USA) were added to resuspended cells. The cells were gently mixed, incubated for 15 min in the dark at RT, and examined by flow cytometer CyFlow space (Sysmex Partec, Gorlitz, Germany). The flow cytometer collected 100,000 events, which were analyzed using FloMax software for cytometry.
For flow cytometric analysis of the cell cycle, 1.25 × 105 cells (SK-BR-3 and MCF7) were seeded in 35-mm cell culture dishes and treated with 1 µM RA or vehicle (0.1% DMSO) for 72 h. Cells were harvested and collected by centrifugation, washed twice in PBS, and fixed with cold 70% ethanol at 4°C for 24 h. Cells were kept at −20°C until further analysis. After washing with PBS, the cells were incubated with RNase (250 µg/ml) and DNA-binding dye propidium iodide (PI, 20 µg/mL) in PBS for 30 min at 37°C in the dark. Flow cytometry analysis was performed on a FACSCalibur flow cytometer (BD Biosciences, Heidelberg, Germany), using CellQuest Pro software for acquisition and analysis. 10,000 cells were recorded, and the results presented as a percentage (± SEM) of cells in different phases of the cell cycle (G0/G1, S, G2/M) and in the sub G0 compartment (hypodiploid cells) from four independent experiments.
The potential of cells to migrate was investigated by wound-healing assay (wound-scratch assay) (Liang et al., 2007). Cells were plated in 35 mm-cell culture dishes, grown to confluence, and pre-treated with 1 μM RA or DMSO as a control. The confluent cell monolayer was scratched with a 200 μL tip, washed with serum-free medium to remove detached cells, and finally, fresh medium containing 1 µM RA or DMSO was added. Cell migration into the wounded area was monitored using DM IL LED Inverted Microscope (Leica Microsystems, Wetzlar, Germany), and closure of the gap distance was quantified using Leica Application Suite V4.3.0. The speed and the mode of cell migration were analyzed by capturing two different parts of the wounded area from three independent experiments.
Total RNA and cDNA synthesis were prepared as previously described (Popovic et al., 2014). For quantitative PCR analysis, cDNAs were subjected to real-time PCR using Power SYBR Green PCR Master Mix (Applied Biosystems, Carlsbad, Germany) in 7500 Real-Time PCR System (Applied Biosystems, Carlsbad, Germany). All samples were measured in triplicate, and the mean value was considered. The relative expression level of analyzed genes was determined using a comparative quantification algorithm where the resulting ΔΔCt value was incorporated to determine the fold difference in expression (2−ΔΔCt). The sequences of primers used in this study are SOX18 forward: 5’TTCCATGTCACAGCCCCCTAG3’; SOX18 reverse: 5’GACACGTGGGAACTCCAG3’; SOX2 forward: 5’CCCCTGGCATGGCTCTTGGC3’; SOX2 reverse: 5’TCGGCGCCGGGGAGATACAT3’; GAPDH forward: 5’GGACCTGACCTGCCGTCTAG3’; GAPDH reverse: 5’CCACCACCCTGTTGCTGTAG3’.
Whole-cell lysates (WCL) were prepared, proteins were separated, and Western blot was performed as previously described (Milivojevic et al., 2013). Antibodies used were: primary rabbit anti-SOX18, diluted 1:1000 (sc-20100, Santa Cruz Biotechnology, Texas, USA), rabbit anti-SOX2, diluted 1:1000 (39823, Active Motif, Carlsbad, California, USA), mouse anti-α-tubulin, diluted 1:10000 (CP06, Calbiochem, Massachusetts, USA).
Lentivirus production and infection of SK-BR-3 and MCF7 cell lines
Lentiviral particles were produced by transient co-transfection of HEK293T cells using PEI MAX™ transfection reagent (Polysciences Inc., Warrington, PA, USA) according to the manufacturer’s instructions. Co-transfection of HEK293T cells is performed in a 10 cm-cell culture dish with 8.6 µg of envelope plasmid (pCMV-VSV-G), 13 µg of packaging plasmid (delta 3.81), and 17 µg of either lentiviral pSin-SOX2 transduction construct or pSin-empty vector. The virus particles were harvested 48 h after transfection and filtered through a 0.2 μm-filtration unit. SK-BR-3 and MCF7 cell lines, cultured in 10 cm-cell culture dishes, were incubated overnight in a medium containing the lentiviral particles and 8 μg/mL of Polybrene (Sigma-Aldrich, St.Louis, Missouri USA). The following morning, the virus-containing medium was removed and replaced with growth medium. The cells were cultured for an additional two days before RA (1 μM) or DMSO treatments. The viability of the cells was measured upon 5 days of RA treatment.
Statistical analyses were performed with SPSS statistical software (version 20). The data represent means ± SEM from three to five independent experiments (indicated in figure legends). Statistical analyses were performed by Student’s t-test, and a P-value of 0.05 was considered significant.
Retinoic acid decreases the viability and proliferative capacity of breast cancer-derived cells and induces changes in their morphology
To examine the effect of RA on cell viability, we have treated SK-BR-3 cells (HER-2+) and MCF7 cells (ER+/PR+) with various concentrations of RA (1 μM, 5 μM and 10 μM) for 3 and 5 days. We have observed a significant decrease in the viability of the cells in both cell lines, with a more prominent effect on SK-BR-3 cells (Fig. 1A). Precisely, RA treatment led to a moderate reduction in MCF7 cell viability to approximately 70–80%, regardless of concentration used and treatment duration. On the other hand, SK-BR-3 showed a more pronounced time-dependent response. The maximal effect was achieved upon 5 days of treatment with 10 μM RA, which led to a decline of cell viability to approximately 20%. We did not observe any apparent cell death in both cell lines throughout the treatments.
In order to test whether a decline in cell viability detected by MTT assay is a result of RA cytotoxic activity, we have performed a LIVE/DEAD cytotoxicity test. Simultaneous staining with green and red-fluorescent dyes discriminates living (green) from dead (red) cells. The results of the analysis of three independent experiments revealed that RA has no significant cytotoxic effect on SK-BR-3 or MCF-7 cells as only sporadic dead cells were detected in RA-treated compared to DMSO-treated cultures (Fig. 1B). This indicates that treatment with RA does not induce considerable cell death.
The clonogenic assay revealed that treatment with 1 μM RA of both cell lines significantly impaired the capacity of cells to form colonies during the period of 8 days. In particular, both SK-BR-3 and MCF7 cells were capable of forming detectable colonies upon treatment with DMSO, while RA treatment led to the reduction in colony formation, both in number and size (Fig. 1C).
The remodeling of cellular structure (plasticity of cytoskeletal dynamics) is associated with cell turnover involved in metastasis. To evaluate the effects of RA on cell morphology of breast cancer cell lines, we analyzed the expression of cytoskeletal filaments tubulin, F-actin, and vimentin by immunocytochemistry and estimated cell surface area on images from immunostaining experiments. As presented in Fig. 2A, treatment with 1 μM RA for 72 h triggered rearrangements of tubulin, F-actin, and vimentin filaments in SK-BR-3 cells, causing significant changes in cell shape and size/volume, observed primarily as enlargement of cytoplasm (Fig. 2A). Quantitative analysis of treated (N = 178) and control (N = 198) cells revealed that RA induced an increase in the average cell area by approximately 3.6-fold (the calculated average cell area of RA-treated was 1771 µm2 compared to the DMSO-treated that was 495 µm2). Next, results of our immunocytochemistry analysis showed that treatment with 1 μM RA for 72 h also affected MCF7 cell morphology (Fig. 2B). Quantitative analysis of RA-treated cells (N = 195) and control cells (N = 206) showed that RA induced discrete reorganization of cytoskeletal proteins and enlargement of average cell area by approximately 1.5-fold (the calculated average cell area of RA-treated was 1227 µm2 compared to the DMSO-treated that was 810 µm2).
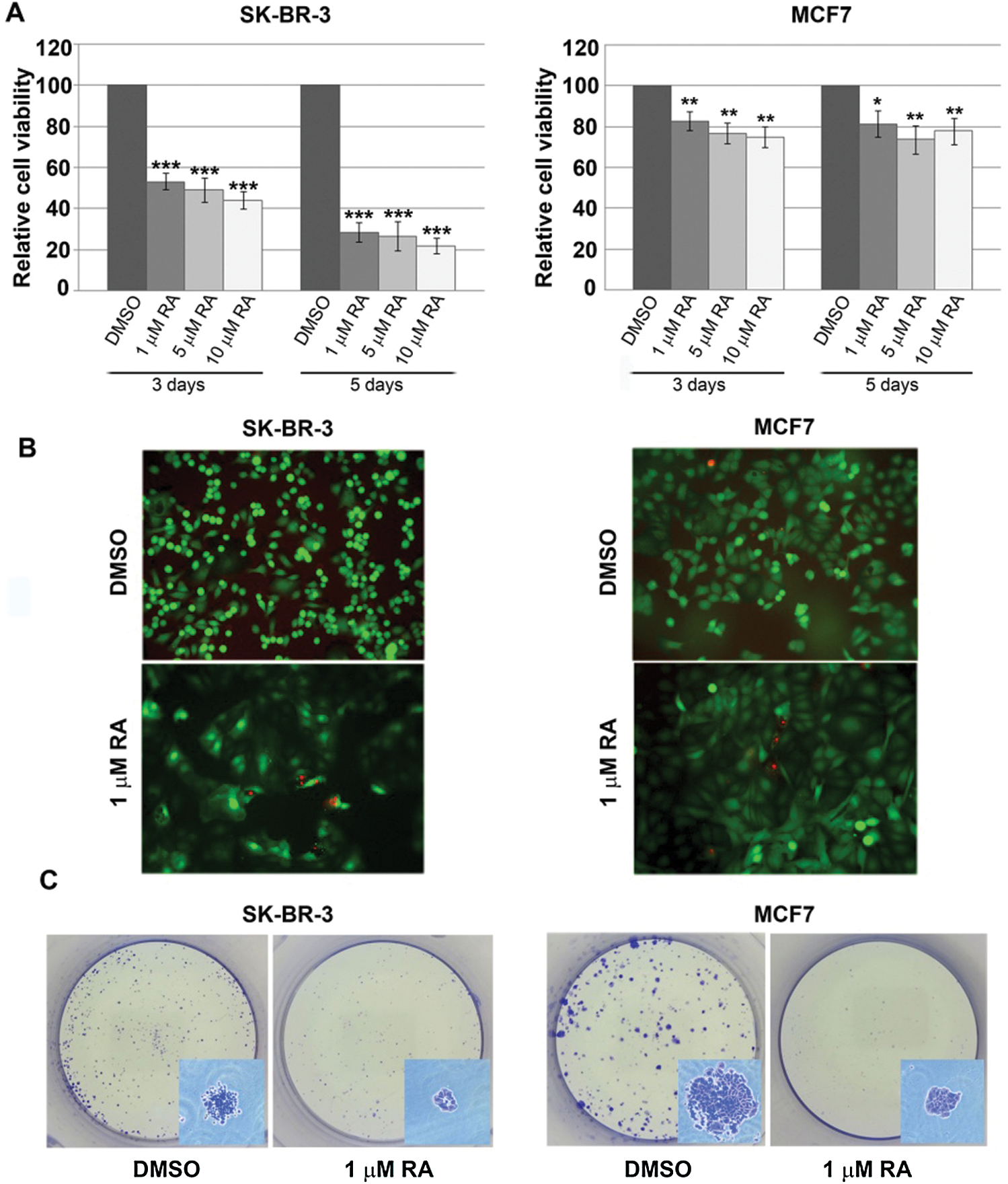
Figure 1: The effect of RA treatment on viability and proliferation capacity of SK-BR-3 and MCF7 cells.
Retinoic acid does not induce cell death but affects cell cycle distribution of breast cancer-derived cells
To further elucidate whether treatment of cells with RA induces apoptotic and/or necrotic cell death, we have treated cells either with 1 μM RA or 1 μM doxorubicin as a positive control for 24 h, and the proportion of apoptotic and necrotic cells was analyzed using Annexin V/PI staining. Obtained results revealed that treatment of SK-BR-3 and MCF7 cells with RA does not induce cell death by either apoptotic or necrotic processes (Fig. 3A).
Next, we tested whether RA treatment could affect cell cycle distribution of both SK-BR-3 and MCF7 cells. Flow cytometry analysis performed 72 h after treatment revealed a significant increase in the fraction of cells in the G0/G1 phase of the cell cycle, together with a corresponding decrease in S and G2/M phases (Fig. 3B). In particular, upon RA treatment, the percentage of SK-BR-3 and MCF7 cells in the G0/G1 phase is increased from 62% to 73% and from 55% to 78%, respectively. These results suggest that RA induced considerable accumulation of cells in the G0/G1 phase of the cell cycle, thus influencing the proliferative capacity of these cells.
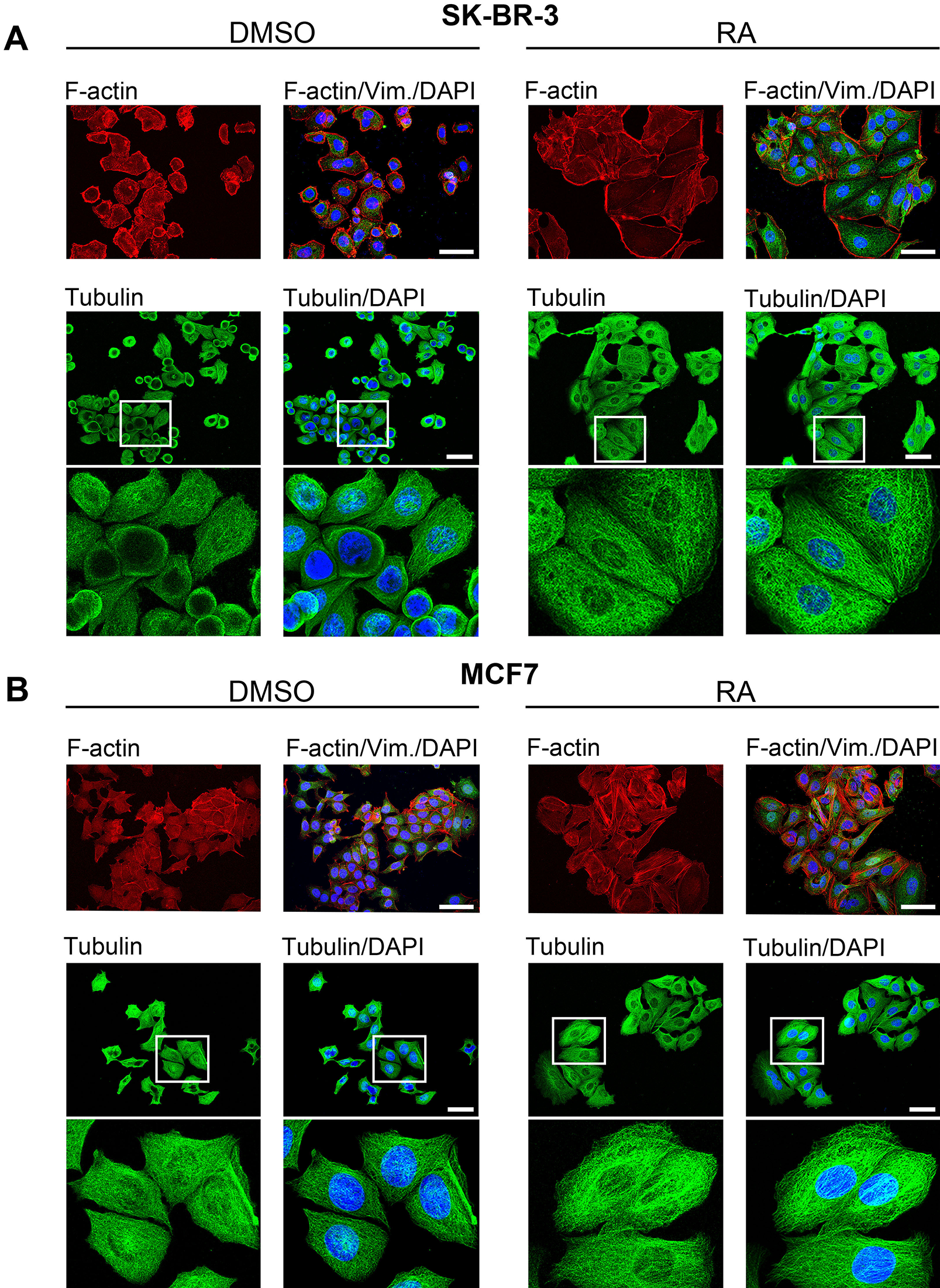
Figure 2: The effect of RA treatment on SK-BR-3 (A) and MCF7 (B) cell morphology.
Finally, we have investigated Ki-67 protein expression by immunofluorescence to determine the rate of cell growth of both cell lines upon treatment with 1 μM RA. It is well known that Ki-67 protein is present during all active phases of the cell cycle but is absent in resting cells, indicating a cell fraction in the cell population that is arrested in the G0 phase. As presented in Fig. 3C, the number of Ki-67-positive cells decreased by approximately 15% upon RA treatment in both cell lines, which correlates with the results obtained by cell cycle analyses where a similar increase in the percentage of cells arrested in the G0/G1 phase was detected.
Taken together, it was clearly demonstrated that RA does not induce cell death but influences cell cycle distribution and impairs the proliferation of both cell lines.
Retinoic acid does not affect the migration of breast carcinoma cell lines
The ability of cancer cells to migrate is closely related to their capacity to colonize distant organs (van Zijl et al., 2011). Therefore, we tested the migratory potential of SK-BR-3 and MCF7 cells upon treatment with 1 μM RA. Upon making a wound scratch, we have monitored the effect of RA treatment on the migratory potential of the cells at three time points (24 h, 48 h and 72 h). We have observed that RA treatment had no effect on the migration rate of both cell lines even upon three days. In Fig. 4, we have presented the statistical analysis of the results obtained 48 h upon treatment.
The effect of retinoic acid on SOX2 and SOX18 expression in breast carcinoma cell lines
Having in mind that both SOX2 and SOX18 overexpression have been correlated with poor prognosis in breast carcinoma, our next aim was to analyze the effects of RA treatment on their expression. SK-BR-3 and MCF7 display differences in the expression of these genes. MCF7 cells express both SOX2 and SOX18, while SK-BR-3 cells express only SOX18 to a lower extent (data not presented). Treatment with 1 μM RA reduced expression of SOX2 in MCF7 cells and SOX18 in both SK-BR-3 and MCF7 cells (Figs. 5A and 5B). This reduction was observed on both mRNA and protein levels.
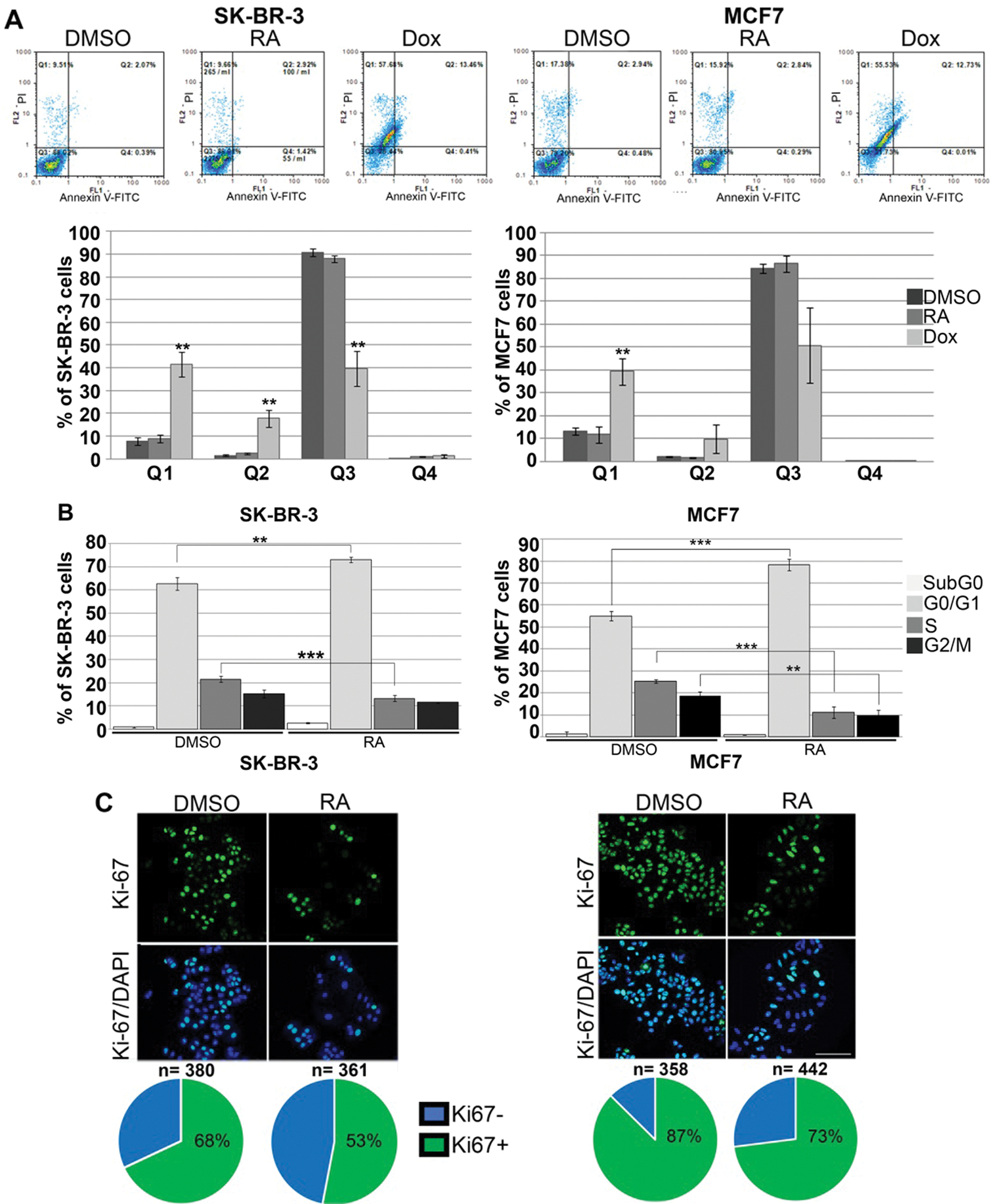
Figure 3: The effect of RA treatment on apoptosis and cell cycle distribution of SK-BR-3 and MCF7 cells.
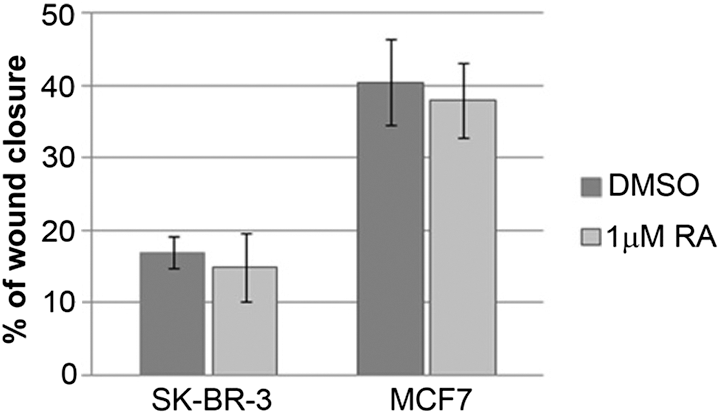
Figure 4: The effect of RA treatment on the migration of SK-BR-3 and MCF7 cells.
In order to gain further insight into the effect of RA, we performed immunofluorescence detection of SOX2 and SOX18 transcription factors in MCF7 cells (Fig. 5C) and quantified the relative level of fluorescence intensity of staining signal in the nuclei. As presented in Fig. 5, expression of SOX2 (Fig. 5C, left panel) and SOX18 (Fig. 5C, right panel) proteins were detected in all examined MCF7 cells. Quantitative analysis of treated (N = 196) and control (N = 234) cells showed that 72 h treatment with RA induced a decline in the average fluorescence intensity of SOX2 antibody staining in MCF7 cells’ nuclei by approximately 35% compared to fluorescence intensity in control cells that was set as 100%. Similarly, quantitative analysis of treated (N = 245) and control (N = 315) MCF7 cells’ nuclei revealed that RA caused a decrease in the mean fluorescence intensity of SOX18 antibody staining, by approximately 27%, compared to the fluorescence intensity in control cells that was set as 100%. In contrast to qPCR and Western blot analyses, by immunofluorescence, we were not able to detect any significant changes in SOX18 transcription factor level in SK-BR-3 cells following RA treatment, probably due to the low level of endogenous SOX18 in these cells (data not shown).
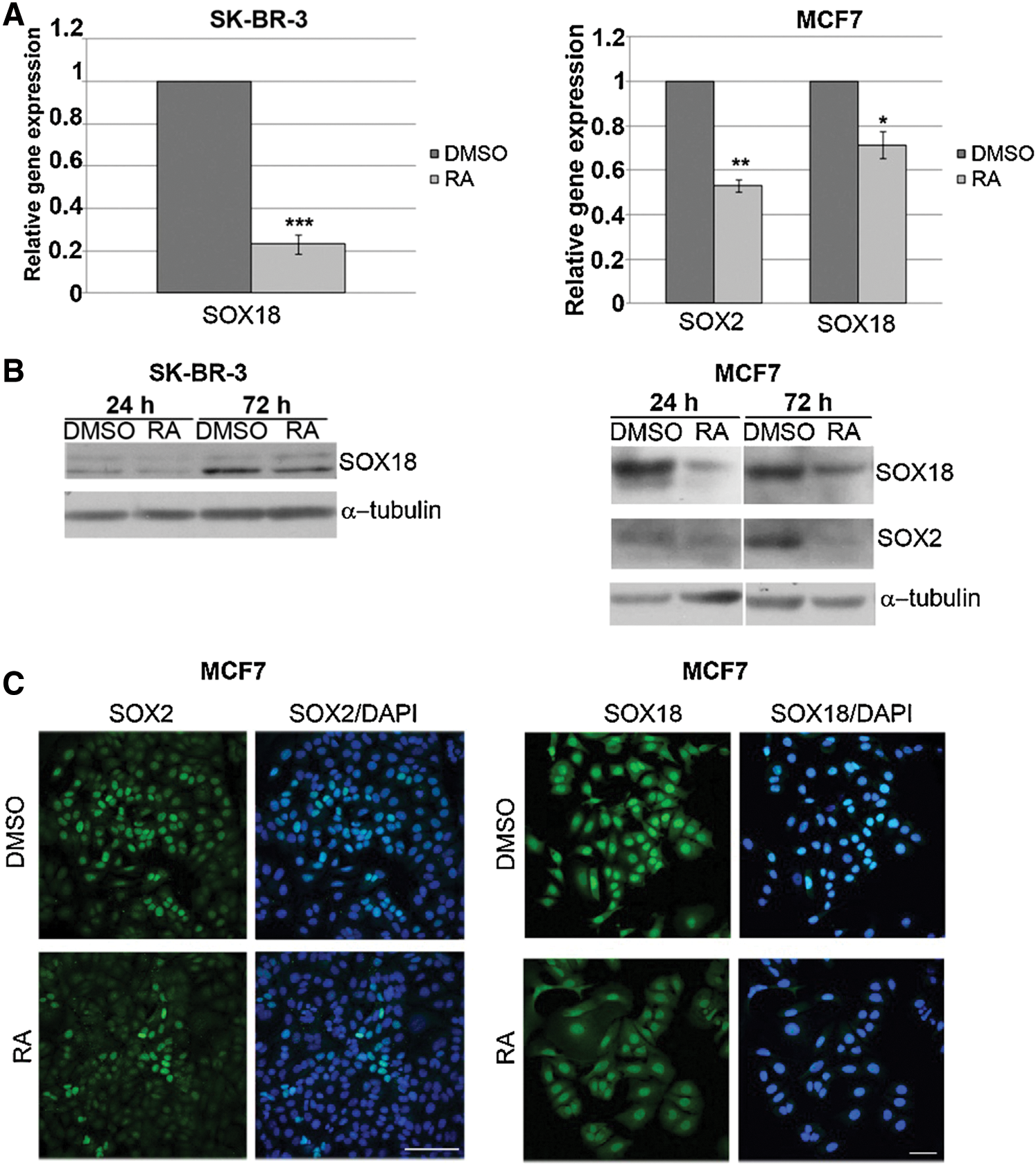
Figure 5: The effect of RA treatment on SOX2 and SOX18 expression in SK-BR-3 and MCF7 cells.
Anti-proliferative activity of RA is independent of SOX2 expression
The evident difference in response to RA treatment between two cell lines, with the more prominent effect on SK-BR-3 cells that lack SOX2 expression, raised the question of whether the reduction in cell viability and proliferation upon RA treatment is associated with the SOX2 expression level. To address that question, we have performed lentiviral transduction of both cell lines with SOX2 expression construct and treated the cells with 1 μM RA for 5 days. We have detected efficient ectopic overexpression of SOX2 in both cell lines (Fig. 6A). The analysis of cell viability showed that overexpression of SOX2 in both cell lines did not reverse the anti-proliferative effect of RA (Fig. 6B). Therefore, we concluded that RA affects cell viability in both cell lines by the mechanism that is SOX2-independent. However, reduction of the SOX2/SOX18 expression upon RA treatment in MCF7 cells deserves further investigation aimed to explore the combined effects of RA, chemotherapeutics, and endocrine therapeutics.
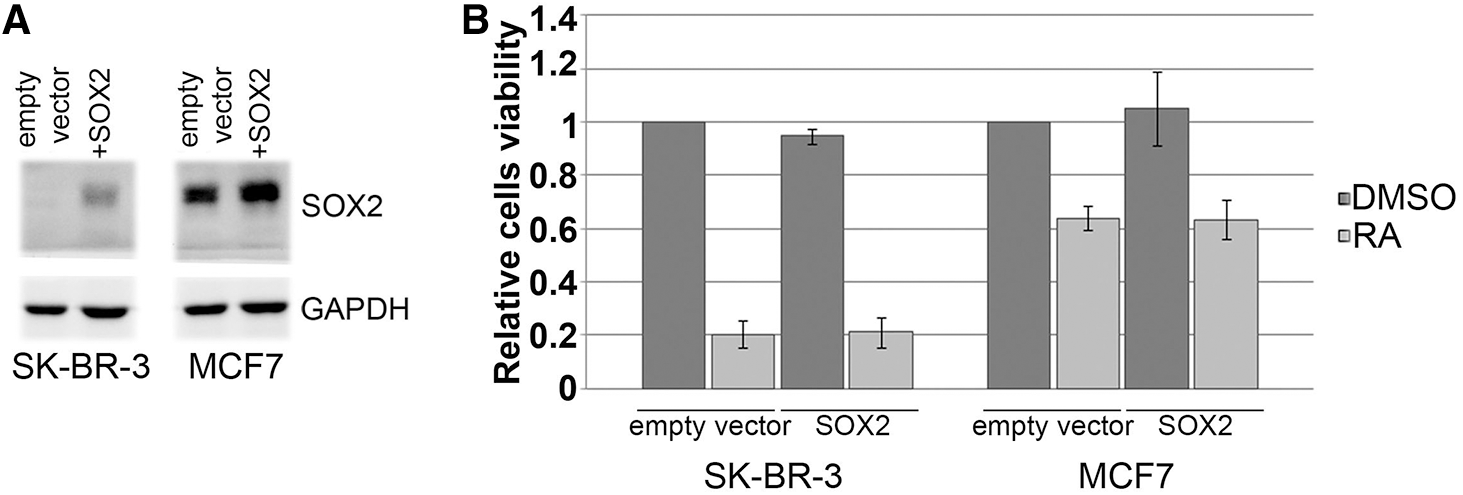
Figure 6: The effect of SOX2 overexpression on the outcome of RA treatment.
Cytotoxic and/or differentiating potential of RA on carcinoma cells was observed in many studies, including its therapeutic effects in certain hematologic malignancies and solid tumors such as breast cancer (de Thé and Chen, 2010; Garattini et al., 2012). Although it is well known that RA is endowed with antiproliferative, cyto-differentiating and apoptotic effects, the opposite function has also been described pointing that RA can antagonize cell differentiation or promote stemness (Mezquita and Mezquita, 2019). In the various breast carcinoma cells, both induction of stemness and cell differentiation by RA have been reported that are cell type-dependent. Thus, RA can produce pro-tumorigenic and anti-tumorigenic effects in different cancer cell types, leading to the poor clinical outcomes of RA treatments that might be associated with the types of breast cancer and heterogeneity of the cells within the tumor.
It has been demonstrated that approximately 30% of the human HER2-positive breast cancers show co-amplification of the gene encoding the retinoic acid nuclear receptor RARα, thus representing a subtype of breast cancer that has significant sensitivity to RA (Paroni et al., 2012). Also, in breast carcinoma cells, RARα expression is controlled by estrogens, and high levels of this nuclear receptor are observed in estrogen receptor-positive tumors (Sun et al., 1998). Herein, we have presented a moderate response of MCF7 cells (ER+) to RA treatment, represented by a reduction of both viability and proliferative capacity of these cells. On the other hand, SK-BR-3 cells (HER-2+) responded to RA treatment approximately twice as strong as MCF7 cells (Fig. 1A). This is in line with the fact that SK-BR-3 cells, in addition to HER-2 amplification, also have RXRα nuclear retinoid receptor co-amplification (Centritto et al., 2015; Li et al., 2004; Paroni et al., 2012). Retinoid receptor amplification affects the response to RA, and this, given the heterogeneity of the breast cancers, should be taken into consideration regarding therapy options.
Both SOX2 and SOX18 have been recognized as significant diagnostic and prognostic markers in various types of cancer, including breast carcinoma (Pietrobono et al., 2016; Wang et al., 2016; Wuebben et al., 2016; Zhang et al., 2016). Although chemotherapy is the primary treatment for breast carcinoma, as for many other malignant tumors, the resistance to chemotherapeutic drugs presents a major obstacle and limitation (Holohan et al., 2013). Many data are showing that SOX2-positive cells play a critical role in the progression of cancer due to, among other characteristics, high chemo-resistance to conventional drug treatments (Wuebben and Rizzino, 2017). Therefore, targeting SOX2-positive cells could be crucial for successful treatment. Many data points to RA being effective against tumor cells that have acquired cancer stem cell phenotype. Therefore, there is a possibility that RA treatment could be effective in the cases of patients that failed hormonal therapy since trials with hormonal therapy have shown enrichment of residual tumors with cancer stem cells (Creighton et al., 2009).
RA reduced the mammosphere-forming ability of cell lines that expressed higher levels of SOX2, suggesting that only the cancer cells that are dependent on SOX2 for self-renewal are responsive to RA (Bhat-Nakshatri et al., 2013). However, the results presented in this paper shed new light on the potential role of SOX2 in response to RA treatment. We showed that RA effect is more prominent in the SK-BR-3 cell line, pointing that this cell line represents a good RA responder. This effect includes a higher decrease in cell viability, accompanied by a reduction in colony formation and significant morphological changes (Figs. 1 and 2). This peculiar result led us to investigate whether SOX2 expression level has an impact on the outcome of RA treatment in these model systems. It is well known that SOX2, as a stem cell marker, achieves its effect by maintaining cell proliferation (Rizzino, 2008); therefore, we hypothesized that an increase in its expression level might, to some extent, compete with the antiproliferative effect of RA. We showed that the increased expression of SOX2 did not antagonize the antiproliferative effect of RA in both cell lines (Fig. 6). We have clearly presented that upon ectopic overexpression of SOX2 in both cell lines, RA retains its potential to significantly reduce the viability of breast carcinoma cells. However, overexpression of SOX2 in both cell lines did not lead to significant induction of cell viability as presented by MTT assay (Fig. 6). Although we detected a notable increase in the quantity of SOX2 protein upon lentiviral overexpression in both cell lines (Fig. 6A), we did not detect any changes in cell viability that could indicate changes in proliferative capacities of the cells. This might be explained with at least two previous findings. An important feature of SOX proteins is that they generally display their gene regulatory functions by forming complexes with transcription partners (Kamachi et al., 2000). Accordingly, SK-BR-3 cells that lack SOX2 are probably deficient in SOX2-partner factors; thus, SOX2 overexpression alone is not sufficient to activate transcription machinery necessary for increased proliferation. For instance, OCT4 as an important SOX2 binding partner has been reported to be undetectable in a large panel of breast tumors, including MCF7 cell line (Wu et al., 2012). The second explanation relies on the data of Wu and co-workers, who identified two novel phenotypically distinct cell subsets in two breast cancer cell lines, including MCF7, based on their differential activity of SOX2 transcription factor (Wu et al., 2012). Although both cell lines express SOX2 protein, its activity was confirmed in only a small proportion of cells. No significant difference in cell proliferation was found between cells showing SOX2 activity compared to those that lacked transcriptionally active SOX2. Moreover, the authors have shown no significant change in the number of viable cells despite considerable downregulation of SOX2 by siRNA in both phenotypically distinct MCF7 cell subsets. Authors have concluded that regardless of the total SOX2 protein level, only SOX2 protein that is transcriptionally active underlies the tumorigenicity and cancer stem cell-like phenotypes in breast cancers.
The reduction of viability of both SK-BR-3 and MCF7 cells was not governed by the induction of apoptosis but rather by interfering with cell cycle and proliferation processes. Obtained effect of RA on cell cycle distribution is in accordance with other reports showing RA-induced G0/G1 arrest in different cancer cell types, including breast carcinoma cells (Chen and Ross, 2004; Lin et al., 2014; Tighe and Talmage, 2004; Zhu et al., 1997). Various data indicate that RA interferes with breast carcinoma cell viability and proliferation, either by cytostatic or both cytostatic and cytotoxic mechanisms (Reinhardt et al., 2018). Clonal proliferation in the presence of RA, presented in this paper, revealed that RA led to a reduction of number, diameter, and cell density of respective colonies in both cell lines. Together with a lack of significant effect on apoptosis, this data indicates that RA exerts cytostatic and no cytotoxic effect in tested breast carcinoma cell lines. This is in correlation with previous data describing RA as a predominantly growth inhibitory and cyto-differentiating agent (Garattini et al., 2014; Garattini et al., 2007a, Garattini et al., 2007b). Also, the growth of SK-BR-3 and MCF7 cell lines was evaluated by Ki-67, an established biomarker of cell division that is routinely used in clinics to assess the proliferation rate of breast carcinoma cells. We have detected a reduction in the number of Ki-67-positive cells in both model systems upon treatment with RA.
The ability of RA to inhibit SOX2 expression in responsive cell lines raises the possibility that SOX2 gene can be used as a biomarker to distinguish RA responders from non-responders, as already discussed by Poornima Bhat-Nakshatri and co-workers (Bhat-Nakshatri et al., 2013). However, the results presented here demonstrated that SK-BR-3 cells, which are SOX2-negative, are RA responders, suggesting the existence of additional mechanisms that govern RA effect in these cells. Moreover, it has been postulated that ER+ breast cancer cells are more sensitive to RA treatment, and here we have shown that SK-BR-3 cells (ER−/HER2+) respond strongly to RA-induced growth inhibition. Taking together, we can make an assumption that even SOX2-negative breast carcinoma histotypes could benefit from RA treatment. Finally, for the first time, we have shown that expression of SOX18 could be altered by RA in breast carcinoma cell lines. The observed reduction of its expression, especially in MCF7 cells, deserves further investigation. Future research is needed to provide more insight into the mechanisms by which RA exerts its effects on cell viability and proliferative capacity of breast cancer-derived cells.
Although many SOX transcription factors have been shown to be essential for cell survival, tumor cell proliferation and growth, a number of limitations should be recognized in considering these genes as potential drug targets (Tighe and Talmage, 2004). There is a clear potential to deploy novels strategies aimed to regulate the activity of SOX transcription factors, SOX2 and SOX18 in particular, by developing specific inhibitors that will inhibit protein-DNA and protein-protein interactions (Chen and Ross, 2004; Overman et al., 2017). However, therapeutic targeting of transcription factors is still rather challenging. Today, the opinion prevails that retinoid monotherapy will probably never be recognized as a practicable option and that is of great importance to consider combined treatments with other agents. Therefore, further work is needed to investigate the potential options for modulation of SOX2/SOX18 expression by RA in breast carcinoma, enabling the further study of the combinatory effect of RA and chemotherapeutics and/or endocrine therapeutics.
Acknowledgement: We thank Prof Ivanka Markovic (Institute of Medical and Clinical Biochemistry, School of Medicine, University of Belgrade, Beograd) for the help in performing FACS analysis. Also, we would like to thank Prof Roberto Mantovani (Department of Biosciences, University of Milan, Italy) for a kind gift of plasmid vectors (pCMV-VSV-G, delta 3.81, pSin-SOX2, and pSin-empty vector).
Availability of Data and Materials: The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Author Contribution: IP designed the study; IP, MM, AA, AL, NKG, MaS and JP performed the experiments; IP, MiS, MM, AL, NKG, MaS and JP performed data analysis; IP, MiS, AL, NKG and MaS wrote the manuscript;. All authors reviewed the results and approved the final version of the manuscript.
Funding Statement: This work was supported by the Ministry of Education, Science and Technological Development of the Republic of Serbia (Agreement No. 451-03-9/2021-14/200042) and the Serbian Academy of Sciences and Arts (Grant No. F24).
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
Bhat-Nakshatri P, Goswami CP, Badve S, Sledge GW,Jr, Nakshatri H (2013). Identification of FDA-approved drugs targeting breast cancer stem cells along with biomarkers of sensitivity. Scientific Reports 3: 2530. [Google Scholar]
Bryan M, Pulte ED, Toomey KC, Pliner L, Pavlick AC, Saunders T, Wieder R (2011). A pilot phase II trial of all-trans retinoic acid (Vesanoid) and paclitaxel (Taxol) in patients with recurrent or metastatic breast cancer. Investigational New Drugs 29: 1482–1487. [Google Scholar]
Buch K, Peters T, Nawroth T, Sänger M, Schmidberger H, Langguth P (2012). Determination of cell survival after irradiation via clonogenic assay versus multiple MTT Assay-A comparative study. Radiation Oncology 7: 1. [Google Scholar]
Budd GT, Adamson PC, Gupta M, Homayoun P, Sandstrom SK, Murphy RF, McLain D, Tuason L, Peereboom D, Bukowski RM, Ganapathi R (1998). Phase I/II trial of all-trans retinoic acid and tamoxifen in patients with advanced breast cancer. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research 4: 635–642. [Google Scholar]
Centritto F, Paroni G, Bolis M, Garattini SK, Kurosaki M, Barzago MM, Zanetti A, Fisher JN, Scott MF, Pattini L, Lupi M, Ubezio P, Piccotti F, Zambelli A, Rizzo P, Gianni M, Fratelli M, Terao M, Garattini E (2015). Cellular and molecular determinants of all-trans retinoic acid sensitivity in breast cancer: Luminal phenotype and RARα expression. EMBO Molecular Medicine 7: 950–972. [Google Scholar]
Chen Q, Ross AC (2004). Retinoic acid regulates cell cycle progression and cell differentiation in human monocytic THP-1 cells. Experimental Cell Research 297: 68–81. [Google Scholar]
Chen Y, Shi L, Zhang L, Li R, Liang J, Yu W, Sun L, Yang X, Wang Y, Zhang Y, Shang Y (2008). The molecular mechanism governing the oncogenic potential of SOX2 in breast cancer. Journal of Biological Chemistry 283: 17969–17978. [Google Scholar]
Costantini L, Molinari R, Farinon B, Merendino N (2020). Retinoic acids in the treatment of most lethal solid cancers. Journal of Clinical Medicine 9: 360. [Google Scholar]
Creighton CJ, Li X, Landis M, Dixon JM, Neumeister VM, Sjolund A, Rimm DL, Wong H, Rodriguez A, Herschkowitz JI, Fan C, Zhang X, He X, Pavlick A, Gutierrez MC, Renshaw L, Larionov AA, Faratian D, Hilsenbeck SG, Perou CM, Lewis MT, Rosen JM, Chang JC (2009). Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proceedings of the National Academy of Sciences of the United States of America 106: 13820–13825. [Google Scholar]
de Thé H, Chen Z (2010). Acute promyelocytic leukaemia: novel insights into the mechanisms of cure. Nature Reviews Cancer 10: 775–783. [Google Scholar]
Duong T, Proulx ST, Luciani P, Leroux JC, Detmar M, Koopman P, Francois M (2012). Genetic ablation of SOX18 function suppresses tumor lymphangiogenesis and metastasis of melanoma in mice. Cancer Research 72: 3105–3114. [Google Scholar]
Fisher JN, Terao M, Fratelli M, Kurosaki M, Paroni G, Zanetti A, Gianni M, Bolis M, Lupi M, Tsykin A, Goodall GJ, Garattini E (2015). MicroRNA networks regulated by all-trans retinoic acid and Lapatinib control the growth, survival and motility of breast cancer cells. Oncotarget 6: 13176–13200. [Google Scholar]
Garattini E, Bolis M, Garattini SK, Fratelli M, Centritto F, Paroni G, Gianni M, Zanetti A, Pagani A, Fisher JN, Zambelli A, Terao M (2014). Retinoids and breast cancer: from basic studies to the clinic and back again. Cancer Treatment Reviews 40: 739–749. [Google Scholar]
Garattini E, Gianni M, Terao M (2007a). Cytodifferentiation by retinoids, a novel therapeutic option in oncology: rational combinations with other therapeutic agents. Vitamins and Hormones 75: 301–354. [Google Scholar]
Garattini E, Gianni M, Terao M (2007b). Retinoids as differentiating agents in oncology: a network of interactions with intracellular pathways as the basis for rational therapeutic combinations. Current Pharmaceutical Design 13: 1375–1400. [Google Scholar]
Garattini E, Paroni G, Terao M (2012). Retinoids and breast cancer: new clues to increase their activity and selectivity. Breast Cancer Research 14: 111. [Google Scholar]
Gianni M, Kalac Y, Ponzanelli I, Rambaldi A, Terao M, Garattini E (2001). Tyrosine kinase inhibitor STI571 potentiates the pharmacologic activity of retinoic acid in acute promyelocytic leukemia cells: effects on the degradation of RARα and PML-RARα. Blood 97: 3234–3243. [Google Scholar]
Higgins MJ, Baselga J (2011). Targeted therapies for breast cancer. Journal of Clinical Investigation 121: 3797–3803. [Google Scholar]
Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG (2013). Cancer drug resistance: an evolving paradigm. Nature Reviews Cancer 13: 714–726. [Google Scholar]
Housman G, Byler S, Heerboth S, Lapinska K, Longacre M, Snyder N, Sarkar S (2014). Drug resistance in cancer: an overview. Cancers 6: 1769–1792. [Google Scholar]
Ji X, Lu Y, Tian H, Meng X, Wei M, Cho WC (2019). Chemoresistance mechanisms of breast cancer and their countermeasures. Biomedicine & Pharmacotherapy 114: 108800. [Google Scholar]
Kamachi Y, Uchikawa M, Kondoh H (2000). Pairing SOX off: with partners in the regulation of embryonic development. Trends in Genetics 16: 182–187. [Google Scholar]
Khan S, Wall D, Curran C, Newell J, Kerin MJ, Dwyer RM (2015). MicroRNA-10a is reduced in breast cancer and regulated in part through retinoic acid. BMC Cancer 15: 345. [Google Scholar]
Lee HE, Kim JH, Kim YJ, Choi SY, Kim SW, Kang E, Chung IY, Kim IA, Kim EJ, Choi Y, Ryu HS, Park SY (2011). An increase in cancer stem cell population after primary systemic therapy is a poor prognostic factor in breast cancer. British Journal of Cancer 104: 1730–1738. [Google Scholar]
Li X, Lewis MT, Huang J, Gutierrez C, Osborne CK, Wu MF, Hilsenbeck SG, Pavlick A, Zhang X, Chamness GC, Wong H, Rosen J, Chang JC (2008). Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. Journal of the National Cancer Institute 100: 672–679. [Google Scholar]
Li XL, Eishi Y, Bai YQ, Sakai H, Akiyama Y, Tani M, Takizawa T, Koike M, Yuasa Y (2004). Expression of the SRY-related HMG box protein SOX2 in human gastric carcinoma. International Journal of Oncology 24: 257–263. [Google Scholar]
Liang CC, Park AY, Guan JL (2007). In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nature Protocols 2: 329. [Google Scholar]
Lin E, Chen MC, Huang CY, Hsu SL, Huang WJ, Lin MS, Wu JC, Lin H (2014). All-trans retinoic acid induces DU145 cell cycle arrest through Cdk5 activation. Cellular Physiology and Biochemistry: International Journal of Experimental Cellular Physiology, Biochemistry, and Pharmacology 33: 1620–1630. [Google Scholar]
Markovic ZM, Jovanovic SP, Maskovic PZ, Mojsin MM, Stevanovic MJ, Danko M, Micusík M, Jovanovic DJ, Kleinová A, Spitalský Z, Pavlovic VB, Todorović Marković BM (2019). Graphene oxide size and structure pro-oxidant and antioxidant activity and photoinduced cytotoxicity relation on three cancer cell lines. Journal of Photochemistry and Photobiology B: Biology 200: 111647. [Google Scholar]
Mezquita B, Mezquita C (2019). Two opposing faces of retinoic acid: induction of stemness or induction of differentiation depending on cell-type. Biomolecules 9: 567. [Google Scholar]
Milivojevic M, Petrovic I, Kovacevic-Grujicic N, Popovic J, Mojsin M, Stevanovic M (2013). Construction and functional analysis of novel dominant-negative mutant of human SOX18 protein. Biochemistry 78: 1287–1292. [Google Scholar]
Nedeljkovic M, Damjanovic A (2019). Mechanisms of chemotherapy resistance in triple-negative breast cancer-how we can rise to the challenge. Cells 8: 957. [Google Scholar]
Overman J, Fontaine F, Moustaqil M, Mittal D, Sierecki E, Sacilotto N, Zuegg J, Robertson AA, Holmes K, Salim AA, Mamidyala S, Butler MS, Robinson AS, Lesieur E, Johnston W, Alexandrov K, Black BL, Hogan BM, De Val S, Capon RJ, Carroll JS, Bailey TL, Koopman P, Jauch R, Smyth MJ, Cooper MA, Gambin Y, Francois M (2017). Pharmacological targeting of the transcription factor SOX18 delays breast cancer in mice. eLife 6: e21221. [Google Scholar]
Paroni G, Fratelli M, Gardini G, Bassano C, Flora M, Zanetti A, Guarnaccia V, Ubezio P, Centritto F, Terao M, Garattini E (2012). Synergistic antitumor activity of lapatinib and retinoids on a novel subtype of breast cancer with coamplification of ERBB2 and RARA. Oncogene 31: 3431–3443. [Google Scholar]
Petrovic I, Milivojevic M, Popovic J, Schwirtlich M, Rankovic B, Stevanovic M (2015). SOX18 is a novel target gene of Hedgehog signaling in cervical carcinoma cell lines. PLoS One 10: e0143591. [Google Scholar]
Phillips TM, McBride WH, Pajonk F (2006). The response of CD24(−/low)/CD44+ breast cancer-initiating cells to radiation. Journal of the National Cancer Institute 98: 1777–1785. [Google Scholar]
Pietrobono S, Morandi A, Gagliardi S, Gerlini G, Borgognoni L, Chiarugi P, Arbiser JL, Stecca B (2016). Down-regulation of SOX2 underlies the inhibitory effects of the triphenylmethane gentian violet on melanoma cell self-renewal and survival. Journal of Investigative Dermatology 136: 2059–2069. [Google Scholar]
Piva M, Domenici G, Iriondo O, Rabano M, Simoes BM, Comaills V, Barredo I, Lopez-Ruiz JA, Zabalza I, Kypta R, Vivanco M (2014). SOX2 promotes tamoxifen resistance in breast cancer cells. EMBO Molecular Medicine 6: 66–79. [Google Scholar]
Popovic J, Stanisavljevic D, Schwirtlich M, Klajn A, Marjanovic J, Stevanovic M (2014). Expression analysis of SOX14 during retinoic acid induced neural differentiation of embryonal carcinoma cells and assessment of the effect of its ectopic expression on SOXB members in HeLa cells. PLoS One 9: e91852. [Google Scholar]
Pula B, Olbromski M, Wojnar A, Gomulkiewicz A, Witkiewicz W, Ugorski M, Dziegiel P, Podhorska-Okolow M (2013). Impact of SOX18 expression in cancer cells and vessels on the outcome of invasive ductal breast carcinoma. Cellular Oncology 36: 469–483. [Google Scholar]
Reinhardt A, Liu H, Ma Y, Zhou Y, Zang C, Habbel JP, Possinger K, Eucker J (2018). Tumor cell-selective synergism of TRAIL- and ATRA-induced cytotoxicity in breast cancer cells. Anticancer Research 38: 2669–2682. [Google Scholar]
Rizzino A (2008). Transcription factors that behave as master regulators during mammalian embryogenesis function as molecular rheostats. Biochemical Journal 411: e5–7. [Google Scholar]
Rodriguez-Pinilla SM, Sarrio D, Moreno-Bueno G, Rodriguez-Gil Y, Martinez MA, Hernandez L, Hardisson D, Reis-Filho JS, Palacios J (2007). SOX2: a possible driver of the basal-like phenotype in sporadic breast cancer. Modern Pathology: An Official Journal of the United States and Canadian Academy of Pathology, Inc. 20: 474–481. [Google Scholar]
Sanada Y, Yoshida K, Ohara M, Oeda M, Konishi K, Tsutani Y (2006). Histopathologic evaluation of stepwise progression of pancreatic carcinoma with immunohistochemical analysis of gastric epithelial transcription factor SOX2: comparison of expression patterns between invasive components and cancerous or nonneoplastic intraductal components. Pancreas 32: 164–170. [Google Scholar]
Shima H, Kutomi G, Satomi F, Maeda H, Hirohashi Y, Hasegawa T, Mori M, Torigoe T, Takemasa I (2016). SOX2 and ALDH1 as predictors of operable breast cancer. Anticancer Research 36: 2945–2953. [Google Scholar]
Sun G, Porter W, Safe S (1998). Estrogen-induced retinoic acid receptor α1 gene expression: role of estrogen receptor-Sp1 complex. Molecular Endocrinology 12: 882–890. [Google Scholar]
Sutton LM, Warmuth MA, Petros WP, Winer EP (1997). Pharmacokinetics and clinical impact of all-trans retinoic acid in metastatic breast cancer: a phase II trial. Cancer Chemotherapy and Pharmacology 40: 335–341. [Google Scholar]
Terao M, Fratelli M, Kurosaki M, Zanetti A, Guarnaccia V, Paroni G, Tsykin A, Lupi M, Gianni M, Goodall GJ, Garattini E (2011). Induction of miR-21 by retinoic acid in estrogen receptor-positive breast carcinoma cells: biological correlates and molecular targets. Journal of Biological Chemistry 286: 4027–4042. [Google Scholar]
Testa U, Castelli G, Pelosi E (2020). Breast cancer: a molecularly heterogenous disease needing subtype-specific treatments. Medical Sciences 8: 18. [Google Scholar]
Tighe AP, Talmage DA (2004). Retinoids arrest breast cancer cell proliferation: retinoic acid selectively reduces the duration of receptor tyrosine kinase signaling. Experimental Cell Research 301: 147–157. [Google Scholar]
van Zijl F, Krupitza G, Mikulits W (2011). Initial steps of metastasis: cell invasion and endothelial transmigration. Mutation Research 728: 23–34. [Google Scholar]
Veronesi U, Mariani L, Decensi A, Formelli F, Camerini T, Miceli R, Di Mauro MG, Costa A, Marubini E, Sporn MB, De Palo G (2006). Fifteen-year results of a randomized phase III trial of fenretinide to prevent second breast cancer. Annals of Oncology: Official Journal of the European Society for Medical Oncology 17: 1065–1071. [Google Scholar]
Wang G, Wei Z, Jia H, Zhao W, Yang G, Zhao H (2015). Knockdown of SOX18 inhibits the proliferation, migration and invasion of hepatocellular carcinoma cells. Oncology Reports 34: 1121–1128. [Google Scholar]
Wang S, Liu X, Chen Y, Zhan X, Wu T, Chen B, Sun G, Yan S, Xu L (2020). The role of SOX2 overexpression in prognosis of patients with solid tumors: a meta-analysis and system review. Medicine 99: e19604. [Google Scholar]
Wang Y, Guo H, Zhang D, Yu X, Leng X, Li S, Zhu W (2016). Overexpression of SOX18 correlates with accelerated cell growth and poor prognosis in human pancreatic ductal adenocarcinoma. Biochemical and Biophysical Research Communications 479: 510–516. [Google Scholar]
Wu F, Zhang J, Wang P, Ye X, Jung K, Bone KM, Pearson JD, Ingham RJ, McMullen TP, Ma Y, Lai R (2012). Identification of two novel phenotypically distinct breast cancer cell subsets based on SOX2 transcription activity. Cellular Signalling 24: 1989–1998. [Google Scholar]
Wuebben EL, Rizzino A (2017). The dark side of SOX2: cancer-A comprehensive overview. Oncotarget 8: 44917–44943. [Google Scholar]
Wuebben EL, Wilder PJ, Cox JL, Grunkemeyer JA, Caffrey T, Hollingsworth MA, Rizzino A (2016). SOX2 functions as a molecular rheostat to control the growth, tumorigenicity and drug responses of pancreatic ductal adenocarcinoma cells. Oncotarget 7: 34890–34906. [Google Scholar]
Yang F, Zhang J, Yang H (2018). OCT4, SOX2, and NANOG positive expression correlates with poor differentiation, advanced disease stages, and worse overall survival in HER2+ breast cancer patients. Oncotargets and Therapy 11: 7873–7881. [Google Scholar]
Young N, Hahn CN, Poh A, Dong C, Wilhelm D, Olsson J, Muscat GE, Parsons P, Gamble JR, Koopman P (2006). Effect of disrupted SOX18 transcription factor function on tumor growth, vascularization, and endothelial development. Journal of the National Cancer Institute 98: 1060–1067. [Google Scholar]
Zhang J, Ma Y, Wang S, Chen F, Gu Y (2016). Suppression of SOX18 by siRNA inhibits cell growth and invasion of breast cancer cells. Oncology Reports 35: 3721–3727. [Google Scholar]
Zheng Y, Qin B, Li F, Xu S, Wang S, Li L (2015). Clinicopathological significance of SOX2 expression in patients with breast cancer: a meta-analysis. International Journal of Clinical and Experimental Medicine 8: 22382–22392. [Google Scholar]
Zhu WY, Jones CS, Kiss A, Matsukuma K, Amin S, De Luca LM (1997). Retinoic acid inhibition of cell cycle progression in MCF-7 human breast cancer cells. Experimental Cell Research 234: 293–299. [Google Scholar]
Zhu Y, Li Y, Jun Wei JW, Liu X (2012). The role of Sox genes in lung morphogenesis and cancer. International Journal of Molecular Sciences 13: 15767–15783. [Google Scholar]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |