DOI:10.32604/biocell.2021.013770
| Biocell DOI:10.32604/biocell.2021.013770 | |
| Article |
iPhosD-PseAAC: Identification of phosphoaspartate sites in proteins using statistical moments and PseAAC
1Faculty of Computing and Information Technology, Department of Information Systems, King Abdulaziz University, Jeddah, 80200, Saudi Arabia
2Department of Computer Science, School of Systems and Technology, University of Management and Technology, Lahore, 54770, Pakistan
3Department of Computer Sciences, Abdul Wali Khan University Mardan, Mardan, 23200, Pakistan
*Address correspondence to: Sher Afzal Khan, sher.afzal@awkum.edu.pk
Received: 20 August 2020; Accepted: 18 February 2021
Abstract: Phosphoaspartate is one of the major components of eukaryotes and prokaryotic two-component signaling pathways, and it communicates the signal from the sensor of histidine kinase, through the response regulator, to the DNA alongside transcription features and initiates the transcription of correct response genes. Thus, the prediction of phosphoaspartate sites is critical, and its experimental identification can be expensive, time-consuming, and tedious. For this purpose, we propose iPhosD-PseAAC, a new computational model for predicting phosphoaspartate sites in a particular protein sequence using Chou’s 5-steps rues: (1) Benchmark dataset. (2) The feature extraction techniques such as pseudo amino acid composition (PseAAC), statistical moments, and position relative features. (3) For the classification, artificial neural network AAN will be used. (4) In this step, 10-fold cross-validation and self-consistency testing will be used for validation. For self-consistency testing, 100% Acc is achieved, whereas, for 10-fold cross-validation 95.14% Acc, 95.58% Sn, 94.70% Sp and 0.95 MCC are observed. (5). The final step is the development of a user-friendly web server for the ease of users. Thus, the iPhosD-PseAAC is the first and novel predictor for accurate and efficient identification of phosphoaspartate sites.
Keywords: Phosphorylation; Phosphoaspartate; Prediction; 5-step rule; Statistical moments; PseAAC
Proteins are the basic and key part of the human body and perform many kinds of major functions in and outside of a cell. The proteins are translated or synthesized from messenger RNA, which is first codified into ribosomes and makes a chain of amino acids called a polypeptide chain. Later, the polypeptide passes through the process of folding and makes it an active protein. In the translation process, some of the amino acids can experience chemical changes at the C- or N- terminal of amino acid side chains, this process of alteration is called post-translation modifications (PTLM or PTM). The post-translation modification can modify or may introduce a new functional group to the protein, such as phosphate, so it plays a key role in the making of protein products (Xu et al., 2017).
Among all PTMs, phosphorylation is of great importance. It deactivates or activates the protein target and affects the speed at which a protein can be degraded. Also, it enables translocation of the protein from one subcellular compartment to another and helps protein binding (Mok and Snyder, 2009). Phosphorylation has exhibited pathological implications in diseases like Parkinson’s and Alzheimer’s alongside other neurodegenerative disorders.
Various eukaryotic proteins experience phosphorylation which causes modifications in localization, conformation, stability, function, and so forth (Hubbard and Cohen, 1993). It occurs on threonine (T), serine (S), histidine (H), tyrosine (Y), and aspartate (D) residuals in eukaryotes usually. However, as mentioned above, histidine (H) and aspartate (D) are unusual and least studied (Khan et al., 2018; Mann et al., 2002; Thomason and Kay, 2000).
The phosphorylated form of aspartate is known as phosphoaspartate (PhosD) and has a key role in multiple biological processes (Fig. 1). Phosphorylation of active-site aspartate residues also supports many enzyme-catalyzed reactions. It is observed that sometimes both dephosphorylation and phosphorylation of aspartate residues occur in proteins (Attwood et al., 2011; Knowles, 1980).
Phosphoaspartate is one of the major components of eukaryotic and prokaryotic two-component signaling pathways. The two-component signaling pathways, which have two parts, the histidine kinase and response regulator protein (RR), communicate from the sensor of histidine kinase to the DNA alongside transcription features and begin transcription of correct response genes (Capra and Laub, 2012; Falke et al., 1997).
These two signaling pathways are common in microorganisms and also in plants in order to regulate ripening and the circadian rhythm. Cytokinin and ethylene are the key components of plant-specific hormones, which normally regulate the entire life cycle of a plant from the germination of seeds to the development of flowers to set the new seeds. During evolution, plants follow the bacterial type of basic signal transduction strategy to regulate such important and mature biological mechanisms (Lohrmann and Harter, 2002; Mizuno, 2005).
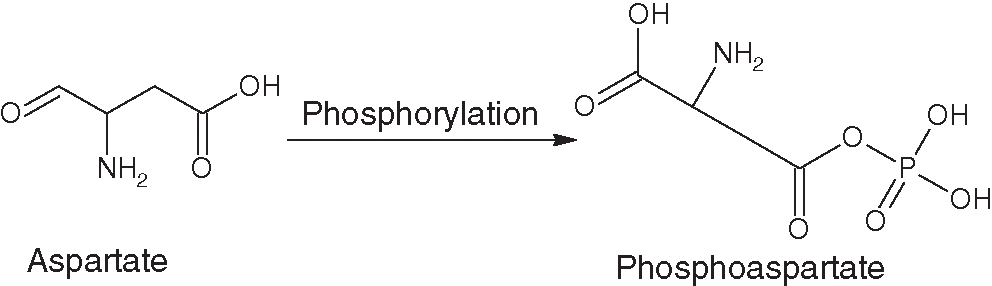
Figure 1: Structural transformation of aspartate to phosphoaspartate.
Phosphoaspartate is involved in many important biological processes and plays an important role in both prokaryotic and eukaryotic organisms, especially in plants. Finding these positions is, therefore, a fundamental task. There are several experimental approaches to determine these positions, and of these approaches, high throughput mass spectrometry (MS) is one of the most common techniques (Akmal et al., 2017). However, the test results are time-consuming, tedious, and expensive.
To overcome these problems, various machine learning methods, algorithms, and techniques for predicting phosphorylation sites such as neural NN, SVM, and ANN have been proposed (Jiang et al., 2016; Li et al., 2016). However, no procedure has been proposed for PhosD sites.
Several studies have been proposed for other types of phosphorylation. Ingrell et al. (2007) developed NetPhosYeast with ANN. Huang et al. (2005) developed the KinasePhos web server using Hidden Markov Models (HMM) to predict specific phosphorylation sites of kinases. Lin et al. (2015) developed a server called Rice_Phospho to predict the phosphorylation sites of rice using SVM. The cluster-based Phosphorylation Scoring and Prediction (GPS) webserver was developed for the prediction of kinase-specific phosphorylation sites. He can find almost 70 types of phosphorylation, kinase-specific (Senawongse et al., 2005; Xue et al., 2008).
Senawongse et al. (2005) used an HMM-based model that focused on extracting features based on the proteomic primary structure for forming function-based clusters of proteins. The feature vector was formulated from both positive and negative samples (Cheng et al., 2018b). In 2018, Khan et al. (2018) proposed a strategy named iPhosT-PseAAC for an expectation of phosphothreonine destinations utilizing PSeAAC, measurable minutes, and different position relative highlights. ANN was utilized for classification while testing was performed by 10-overlay Cross-Validation and Jackknife testing.
Until this point in time, no predictor so far has been proposed for the identification of phosphoaspartate sites in proteins. This study proposes a novel prediction methodology iPhosD-PseAAC for the identification of phosphoaspartate sites in a given protein arrangement. The essential objectives for the proposed model are to outline a predictor that features the significance of phosphoaspartate and formulation of a methodology for in-silico examinations to their pertinent sequences. To address these objectives, we pursue Chou’s 5-step rule (Chou, 2011) as established in a progression of various studies (Cai et al., 2018; Chen et al., 2018; Cheng et al., 2018a; Cheng et al., 2018c, Cheng et al., 2018d; Cheng et al., 2018e; Chou et al., 2018; Liu et al., 2015; Liu et al., 2016a; Liu et al., 2016b; Xiao et al., 2017; Xuao et al., 2018). Formulation of the proposed predictor based on the 5-step rule brings a large dividend. It renders the model clarity of rationale, sets a benchmark for improvements, and makes it easily accessible to the wide-spread scientific community. The rules of Chou’s 5-step model are given as: (1) benchmark dataset development; (2) Transformation to equivalent mathematical form; (3) prediction algorithm; (4) Model Validation; and (5) development of a webserver. From here onwards, let us address these strategies one by one.
In this study, we used Chou’s peptide formulation (Chou, 2001c) to facilitate the description of samples in the dataset. In computational biology, Chou’s peptide formulation has been widely used for the prediction of phosphothreonine sites (Khan et al., 2018), methylation sites (Qiu et al., 2014b), lysine ubiquitination sites (Qiu et al., 2015), signal peptide cleavage sites (Shen and Chou, 2007), hydroxyproline and hydroxylysine sites (Qiu et al., 2016a; Xu et al., 2014a), lysine succinylation sites (Jia et al., 2016b), phosphorylation sites (Qiu et al., 2016c), lysine PTM sites (Qiu et al., 2016b) and protein-protein binding sites (Jia et al., 2016a).
Uniprot is a huge database of proteins that contain annotated descriptions based on several experimental studies. The advanced query option of Uniprot was used to query PTM annotated protein sequences to form a benchmark dataset. The term 4-aspartyl phosphate was specified as modified residue to filter the protein having phosphoaspartate sites. Subsequently, to increase the reliability of data, only the proteins which have been reviewed and their post-translation modification identification is based on experimental assertion, were selected. As a result of this query, 1043 proteins were listed having 1052 phosphoaspartate sites. After removing duplicate sequences and reducing homology using CD-HIT, only 985 proteins were left. A negative dataset was generated using exactly the converse query, which yielded a multitude of samples. Negative samples were only collected from 1043 proteins which were identified to have positive sites as well. Even limiting the number of proteins yielded 7193 unique sequences having low homology. Out of these 7193 sites, only 1000 were randomly filtered out. For each occurrence of aspartic acid residue in a sequence, that particular residue and its associated upstream and downstream amino acids were extracted. Hence, for every selected instance, the length was 41 generated, comprising D residue, 20 residues upstream, and 20 residues downstream. For better results, both negative and positive datasets were pre-processed to remove duplicate occurrences. The dataset is constructed using only sequences that have been experimentally proven. Computationally annotated data and uncategorized data is left out as it may yield even more redundancies. In pre-processing all special characters, spaces, characters that are not amino acids (Z, X, U, O, J, and B) were removed. After pre-processing, the benchmark dataset contained 1994 samples (994 positive and 1000 negative samples).
By following Chou’s method (Chou, 2001b), the peptide having a potential PhosD can be expressed generally by
where
In Eq. (2),
In prediction models, the selected benchmark dataset normally contains both training data and testing data: the purpose of training and testing datasets are training the prediction model and testing of the prediction model, respectively. As extensively reviewed (Chou and Shen, 2008), it is noted that there is no need for these two separate datasets if one is validating the model using extensive validation methods, i.e., k-fold cross-validation or jackknife as its outcome is obtained by using a number of the different exclusive dataset for multiple tests. Moreover, unbiased results are proved by curating datasets that have the least homology. A dataset encompassing significant homologous samples will only yield biased results, and the predictor hence derived may not be as assiduous. Furthermore, during the initial exploration, there was found that the best value for
where
Today, biological data and sequences are growing enormously. The most difficult task for us is to express the biological data and sequences into a vector or discrete form without dropping sequence pattern information and its characteristics for final analysis. As because the machine learning algorithms such as Covariance Discrimination or CD algorithm (Chou and Elrod, 2002; Lin et al., 2012), KNN (Cai and Chou, 2004; Chou and Cai, 2006), SVM (Feng et al., 2013a; Feng et al., 2013b) and RF- Algorithms (Jia et al., 2016b; Lin et al., 2011) can only process vectors (Chou, 2015). However, a vector characterized by a discrete model can lose all design data completely. To avoid completely losing protein grouping design data, the composition of pseudo amino acids (Chou, 2001a) or PseAAC (Chou, 2005) has been proposed. At this point, Chou’s PseAAC was already used in almost all regions of computer-aided proteomics (see, e.g., Akbar and Hayat, 2018; Arif et al., 2018; Contreras-Torres, 2018; Chou, 2017; Javed and Hayat, 2019; Ju and Wang, 2018; Krishnan, 2018; Liang and Zhang, 2018; Mei et al., 2018; Mei and Zhao, 2018a, 2018b; Qiu et al., 2018; Rahman et al., 2018; Sabooh et al., 2018; Sankari and Manimegalai, 2018; Srivastava et al., 2018; Zhang and Kong, 2018; Zhang and Duan, 2018; Zhang and Liang, 2018; Zhao et al., 2018). According to the general PseAAC (Chou, 2011), a sample protein sequence can be formulated as follows:
where
In Eq. (5)
Statistical moments’ calculation
Here we use the statistical moments’ approach for the sequence to define the dimensions and its components of Eq. (4). Using different orders of moments, multiple kinds of data features are described; from those moments, few can be used to indicate the eccentricity and orientation of data, and others can be used for data size evaluation. Numerous moments were defined and described by statisticians and mathematicians primarily based on distribution functions and polynomials, actually well know (Khan et al., 2012; Khan et al., 2014a).
For the iPhosD-PseAAC prediction model, Hahn, raw, and central moments are computed. The Hahn moments are calculated, which are location and scale variant, using the Hahn polynomial (Khan et al., 2014c). Raw moments calculated, which can be a place and scale variant, are used for the mean, asymmetry, and variance calculation, using the probability distribution of the benchmark dataset. Besides, the central moment calculates the asymmetry, mean, and variance, but those are vicinity invariant because the calculation is done concerning the centroid (Butt et al., 2016; Butt et al., 2017), and these calculations are scale variant.
There is a particular reason for working with statistical moments; it maintains the sequence order sensitive information, which is an important point as described above. Furthermore, moments based on scale variants were not used. In the form of quantified value, data is defined on its own by every method (Khan et al., 2014b). In this current method, moments were used in 2-dimensional (2D) matrix P′ having k*k dimensions to accommodate all the residues from a protein P.
The function ω used for matrix transformation into P′ as described by Akmal et al. (2017). Using the elements of P′ all statistical moments were calculated up to 3rd degree. First of all, the (a+b)th order raw moments are calculated as
where the degree of moments is x+y and M00, M01, M02, M10, M11, M12, M20, M21, M30, and M03 are the calculated raw moments and
The P matrix transformed into 2-dimensional square matrix P′ which helps to calculate Hahn moments because Hahn moments can be easily calculated from square dimensional data. The distinct Hahn moments are the orthogonal moments of 2-dimension, which requires an even matrix in the input. The orthogonal Hahn moments have the feature that they can be reversed by using the inverse method of Hahn moments; it is feasible to reconstruct the material, and consequently, the data regarding relative positions, and the sequence composition, can also be conserved in those moments.
N order Hahn polynomial is calculated by using this equation
The Eq. (9), Gamma operator and pochhammer symbol are explained in Akmal et al. (2017). The following equation used to calculate the orthogonal normalized Hahn moments
The model based on residue information of protein related to relative positions and the primary protein sequence is the central paradigm. It is necessary to quantize the relative positions of amino acids, thus the 20 × 20 matrix of Position Relative Incidence Matrix (PRIM) is constructed to extract the information, from all the instances of the benchmark dataset, about the relative position of amino acids residue of proteins as
In the DPRIM matrix, each Ai→j holds the sum of the relative position of the ith element in accordance with the first appearance of the jth element is represented. Similarly, the Reverse Position Relative Incidence Matrix (DRPRIM) is calculated with the reverse protein sequence sample. The DRPRIM is calculated as
Both DPRIM and DRPRIM yield the 400 coefficients. To reduce the number of coefficients, the statistical moments are calculated for both, which yield the 30 coefficients.
Determination of Frequency Matrix (FM)
To find out the frequency of each amino acid residue and mine compositional information from the sequence in benchmark dataset instances, the frequency matrix is calculated. The frequency matrix is calculated as
Herein, f1 is the frequency of each amino acid residue occurring in the sequence arranged by their alphabetical order.
To accumulate the positional information, the Accumulative Absolute Position Incidence Vector is calculated for the length of 20 native amino acids as
Here,
Similarly, the Reverse Accumulative Absolute Position Incidence Vector (RAAPIV) is computed by using the reverse sequence of the protein as
Billions of neurons in the human brain process and transmit information about a certain aspect when they are activated. Whenever you learn things through patterns without having a certain inclination, you take action based on the situation. The artificial neural network (ANN) inspired by the human processing system is made up of several highly interconnected neurons that work together to find a specific solution to a problem. It takes the information from the neuron and processes it using different patterns in the examples given. The ANN has two modes of working: training the ANN on the given input labeled data. In the second mode, when the input pattern matches the learned pattern, it becomes the current output, when the input pattern does not match any of the learned patterns it finds the closest and outputs according to that pattern (Khan et al., 2018), as shown in Fig. 2.
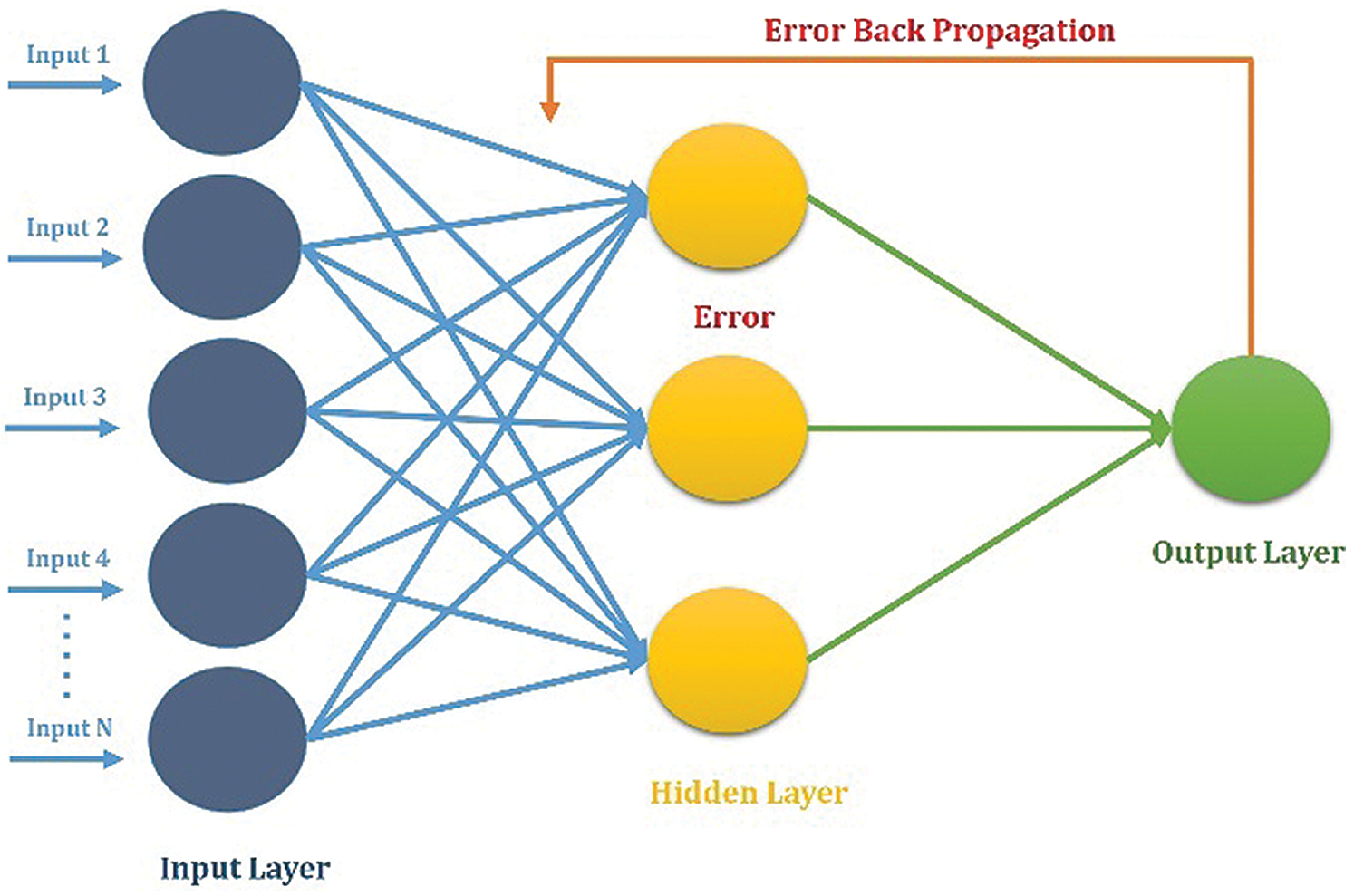
Figure 2: ANN for the proposed prediction model.
In this study, we also used AAN with backpropagation to reduce the output error. Using the benchmarks dataset, which contains 994 positive data samples and 1000 negative samples, the features vector was calculated for all data samples. Every feature vector contains Hahn, central, and raw moments of two-dimensional protein sample representation, PRIM, and RPRIM. For positional and compositional information, the calculated FM, AAPIV, RAAPIV, and Site Vicinity Vector also in the feature vector. The final feature vector contains 194 features based on positional and compositional information for each instance of the benchmark dataset. Thus, both an input matrix comprising all feature vectors and a label matrix were used to train an ANN (Jiang et al., 2016; Khan et al., 2018). Furthermore, extensive probing and experimentation showed that the ANN exhibited optimal results with 50 neurons in the hidden layer while adaptive gradient descent was used for learning.
The important process for the new predicting model is, how to justifiably measure the success rate (Chou, 2011). We have to consider two issues to address the justifiable evaluation of the model: (1) to reflect the model quality, what kind of metrics should be used? (2) to score the metrics, what kind of test methods should be used?
Generally, the following metrics are used from four different viewpoints to evaluate the prediction model accuracy: (1) MCC for model stability (2) Sp for model specificity (3) Sn for model sensitivity (4) Acc to measure the total accuracy of a prediction model. Thus, a set of four intuitive metrics were derived (Feng et al., 2013a; Xu et al., 2013) as given in Eq. (17).
For PhosD sites prediction,
The set of intuitive metrics have been concurred and applauded by a series of recent publications see, e.g., Chen et al. (2016a); Chen et al. (2017b); Ehsan et al. (2018); Feng et al. (2013a); Feng et al. (2017); Feng et al. (2018); Jia et al. (2016c); Lin et al. (2014); Liu et al. (2017a); Liu et al. (2017b); Liu et al. (2018); Xu et al. (2014b); Zhang et al. (2016).
In Eq. (17), defined equations set are only effective for single-label data. For multi-label, which becomes more popular in biological (Chou et al., 2012; Lin et al., 2013; Xiao et al., 2011) and biomedicine (Xiao et al., 2013), is a completely different problem and need different metrics (Chou, 2013).
A self-consistency test on the same dataset was performed to validate the iPhosD-PseAAC predictive model. The validation method was carried on the already known actual positive and negative sample dataset, and the result is shown in Tab. 1.
Commonly, the experimentally proven datasets are used for model prediction; sometimes for testing, we do not have an experimentally proven dataset to test the model against the actual available data. By chance, if the data is available, it might be possible that data are not sufficient to test the accuracy of the predicting model. Only those samples could be incorporated into the model which exists in nature as for such a biological problem, it is not possible to build hypothetical datasets. To score the four metrics of Eq. (17), what kind of testing should be done to meet sufficient accuracy reliability? Usually, the dataset is split into 3 partitions. One partition is used for training while another is used for testing, and the leftover partition is used for validation. However, if the dataset is limited, then it is recommended that a predictor should be tested against k-folds (Subsampling), jackknife, and independent test (Chou and Zhang, 1995). Prediction model testing against jackknife is very exhausted and can always give a different outcome for a given benchmark dataset. It has been widely used to validate the prediction model by investigators (Chen et al., 2017a; Chen et al., 2017b; Dehzangi et al., 2015; Dou et al., 2014; Feng et al., 2005; Kumar et al., 2015; Mondal and Pai, 2014; Nanni et al., 2014; Qiu et al., 2014a; Shen et al., 2007; Wu et al., 2011; Zhou and Doctor, 2003). If an obvious dataset is not available to validate the model prediction, cross-validation is the best option to choose and to give the validation that the developed model is working fine.
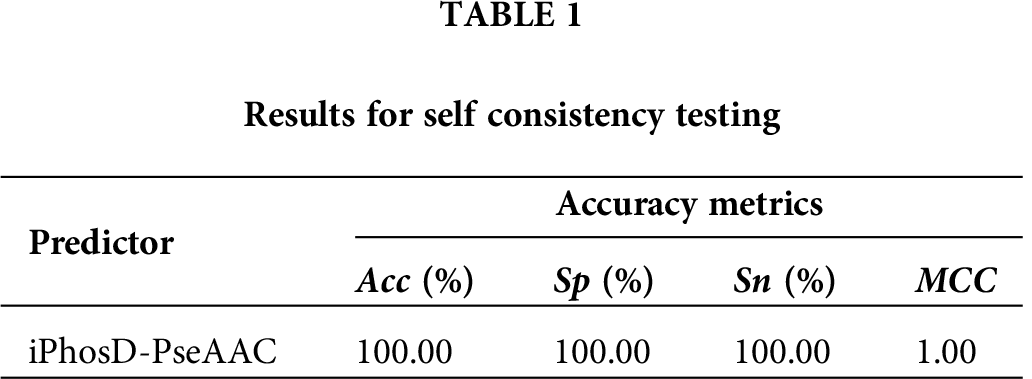
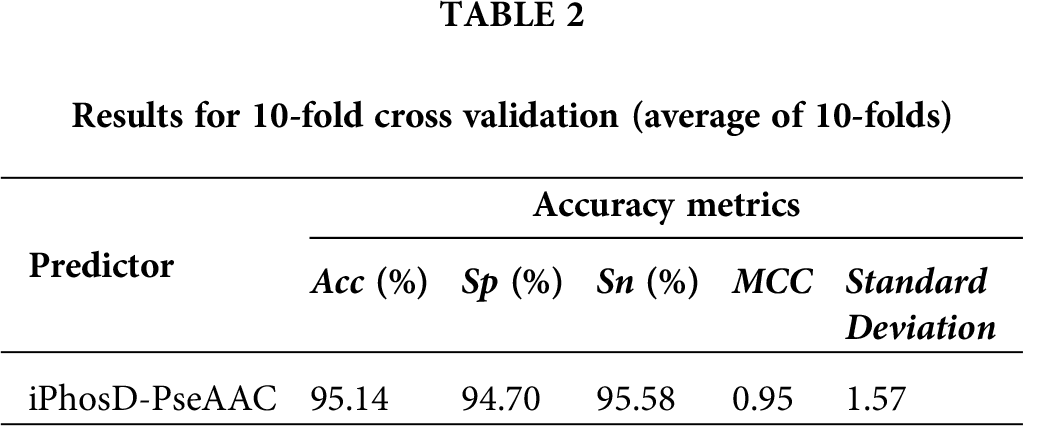
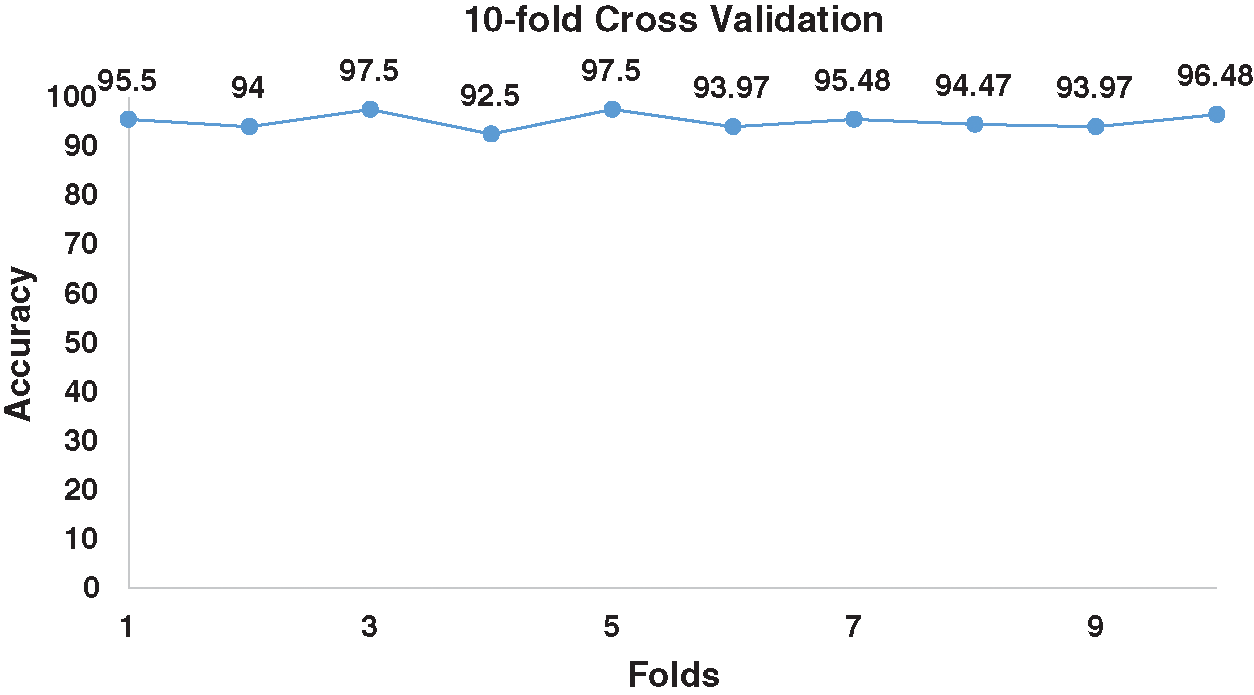
Figure 3: 10-fold cross validation for benchmark dataset.
Herein, we performed 10-fold cross-validation and calculated accumulated accuracy by adding the accuracy of each fold. The average accuracy was 95.14%, as shown in Tab. 2 and Fig. 3. We also validate the prediction model using jackknife to verify the quality of iPhosD-PseAAC. For jackknife validation training, every instance of both the datasets is used for training and testing for unique output and received 94.46% of the prediction validation accuracy.
In a comparative analysis, the results of iPhosD-PseAAC for the metrics are compared with already existing PTM site prediction models, i.e., iPhosT-PseAAC (Khan et al., 2018) and PhosphoSVM (Dou et al., 2014). Both the models iPhosT-PseAAC and PhosphoSVM are merely used benchmarks for comparison of accuracy metrics. Since no earlier model for identification phosphorylation sites of aspartic acid has been found in texts. Considering these benchmark values, the metrics yielded by iPhosD-PseAAC has higher values than iPhosT-PseAAC and PhosphoSVM for all Acc, Sp, Sn, and MCC. This indicates better prediction as compared to others.
From Tab. 3, there can be seen that iPhosD-PseAAC outperforms other benchmark models. iPhosD-PseAAC uses different kinds of compositional and positional features to perform the prediction of PhosD sites. Firstly, it uses PseAAC and trims the modified residue by 20 downstream and upstream, then construct AAPIV, RAAPIV, PRIM, RPIRM, and moments are calculated, using the compositional and positional features. The feature extraction technique derived to furnish the ANN bears great significance in the vibrant performance of the predictor. Results showing high accuracy are a testament to its potential. High accuracy rate yielded in all validation tests compared with other state-of-the-art methods connote that the feature extraction technique is proficient in extracting obscure and pivotal traits of data peculiar to each class. Subsequently, the multilayer ANN is also well equipped to partition classes based on these intricate features.
The 5th step is the user-friendly public web server, as documented in many recent publications (Cheng et al., 2017a, 2017b; Cheng et al., 2017c; Cheng et al., 2016; Feng et al., 2017; Liu et al., 2017b; Qiu et al., 2017a; Qiu et al., 2017b). The webservers are of great importance; thus, the webserver for iPhosD-PseAAC is available at https://www.biopred.org/iphosd, which is developed in Django framework with Python 3.6 and scikit-Learn (Fig. 4).
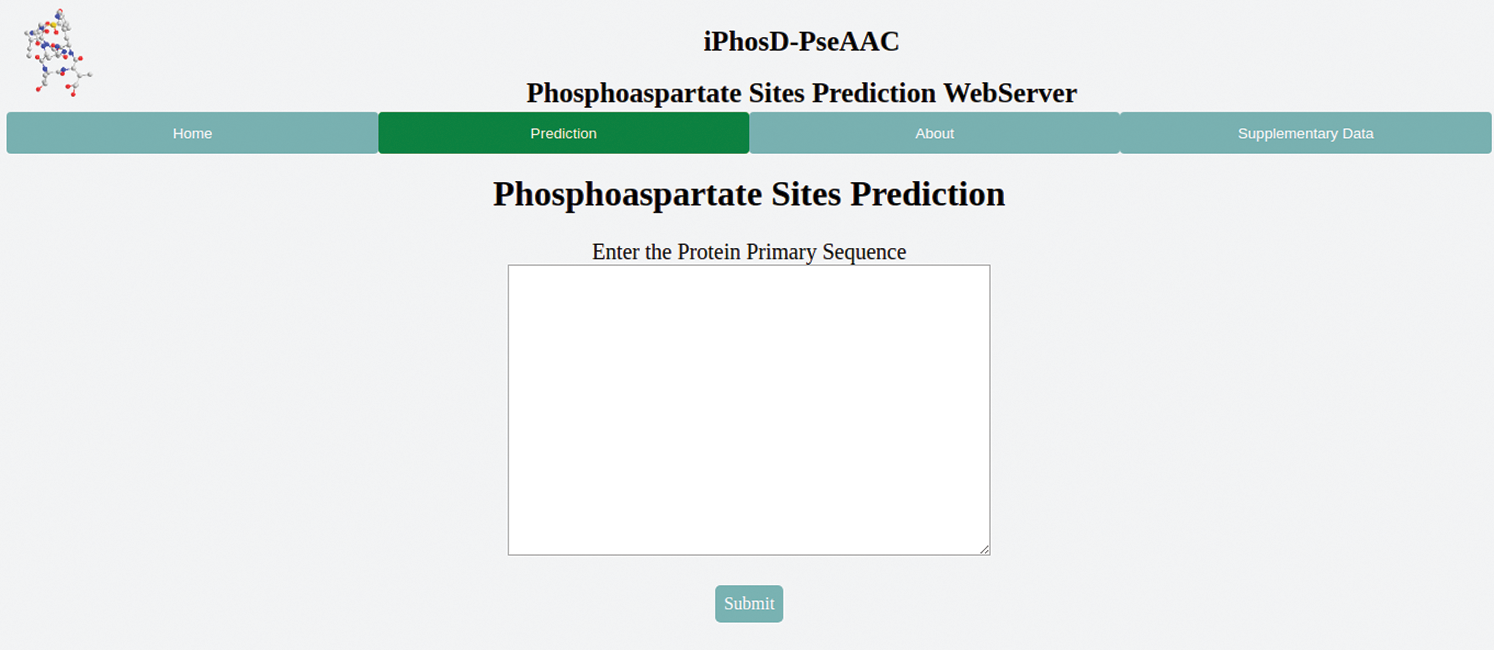
Figure 4: The graphical user interface of the iPhosD-PseAAC available at biopred.org/iphosd.
In this study, we have proposed a prediction model named iPhosD-PseAAC for phosphoaspartate site prediction using Chou’s 5-steps rule. Phosphoaspartate plays many fundamental roles in a number of biological processes, including signal transduction pathways, energy metabolism, various cellular processes, and ripening and circadian rhythms in plants. The aim of the current study was to propose a new and more accurate phosphoaspartate sites predictor and make it easy-to-use, user-friendly, and publicly available to experimental biologists to get their desired results. In PseAAC, it uses the various compositional and positional features of the protein sequence. The proposed model was validated against different exhaustive validation techniques, i.e., self-consistency, jackknife, and cross-validation. Using self-consistency, the accuracy is 100%, for cross-validation 95.14%, and jackknife gives 94.46% accuracy. The overall accuracy of the proposed model is 95.14%, sensitivity value 95.58%, and specificity 94.70%. It is concluded that the proposed model for the prediction of the PhosD sites has the ability of accurate and efficient predictions for phosphoaspartate sites in proteins, but it still can be improved in computational ways as the protein sequences may rapidly increase, day by day.
Acknowledgement: This project was funded by the Deanship of Scientific Research (DSR) at King Abdulaziz University, Jeddah, under Grant No. G:136-611-1441. The authors, therefore, acknowledge with thanks DSR for technical and financial support.
Availability of Data and Materials: The data that support the findings of this study are available in https://www.biopred.org/iphosd/supl.
Author Contribution: AO ALMAGRABI and YD Khan designed the model and the computational framework and analyzed, preprocessed, and extracted the data by SA Khan and YD Khan. The classification was carried out by AO ALMAGRABI and YD Khan. The evaluation of the model was carried out by AO ALMAGRABI and YD Khan under the overall supervision of SA Khan.
Ethics Approval: The work/experiments do not involve human subjects, animals, or plants.
Funding Statement: This project was funded by the Deanship of Scientific Research (DSR) at King Abdulaziz University, Jeddah, https://dsr.kau.edu.sa/Default-305-EN under Grant No. G:136-611-1441.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
Akbar S, Hayat M (2018). iMethyl-STTNC: Identification of N6-methyladenosine sites by extending the idea of SAAC into Chou’s PseAAC to formulate RNA sequences. Journal of Theoretical Biology 455: 205–211. DOI 10.1016/j.jtbi.2018.07.018. [Google Scholar] [CrossRef]
Akmal MA, Rasool N, Khan YD (2017). Prediction of N-linked glycosylation sites using position relative features and statistical moments. PLoS One 12: e0181966. DOI 10.1371/journal.pone.0181966. [Google Scholar] [CrossRef]
Arif M, Hayat M, Jan Z (2018). iMem-2LSAAC: a two-level model for discrimination of membrane proteins and their types by extending the notion of SAAC into Chou’s pseudo amino acid composition. Journal of Theoretical Biology 442: 11–21. DOI 10.1016/j.jtbi.2018.01.008. [Google Scholar] [CrossRef]
Attwood P, Besant P, Piggott MJ (2011). Focus on phosphoaspartate and phosphoglutamate. Amino Acids 40: 1035–1051. DOI 10.1007/s00726-010-0738-5. [Google Scholar] [CrossRef]
Butt AH, Khan SA, Jamil H, Rasool N, Khan YD (2016). A prediction model for membrane proteins using moments based features. BioMed Research International 2016: 1–7. DOI 10.1155/2016/8370132. [Google Scholar] [CrossRef]
Butt AH, Rasool N, Khan YD (2017). A treatise to computational approaches towards prediction of membrane protein and its subtypes. Journal of Membrane Biology 250: 55–76. DOI 10.1007/s00232-016-9937-7. [Google Scholar] [CrossRef]
Cai L, Huang T, Su J, Zhang X, Chen W, Zhang F, He L, Chou KC (2018). Implications of newly identified brain eQTL genes and their interactors in Schizophrenia. Molecular Therapy-Nucleic Acids 12: 433–442. DOI 10.1016/j.omtn.2018.05.026. [Google Scholar] [CrossRef]
Cai YD, Chou KC (2004). Predicting subcellular localization of proteins in a hybridization space. Bioinformatics 20: 1151–1156. DOI 10.1093/bioinformatics/bth054. [Google Scholar] [CrossRef]
Capra EJ, Laub MT (2012). Evolution of two-component signal transduction systems. Annual Review of Microbiology 66: 325–347. DOI 10.1146/annurev-micro-092611-150039. [Google Scholar] [CrossRef]
Contreras-Torres E (2018). Predicting structural classes of proteins by incorporating their global and local physicochemical and conformational properties into general Chou’s PseAAC. Journal of Theoretical Biology 454: 139–145. DOI 10.1016/j.jtbi.2018.05.033. [Google Scholar] [CrossRef]
Chen W, Ding H, Feng P, Lin H, Chou KC (2016a). iACP: a sequence-based tool for identifying anticancer peptides. Oncotarget 7: 16895–16909. DOI 10.18632/oncotarget.7815. [Google Scholar] [CrossRef]
Chen W, Ding H, Zhou X, Lin H, Chou KC (2018). iRNA(m6A)-PseDNC: identifying N6-methyladenosine sites using pseudo dinucleotide composition. Analytical Biochemistry 561–562: 59–65. DOI 10.1016/j.ab.2018.09.002. [Google Scholar] [CrossRef]
Chen W, Feng P, Ding H, Lin H, Chou KC (2016b). Using deformation energy to analyze nucleosome positioning in genomes. Genomics 107: 69–75. DOI 10.1016/j.ygeno.2015.12.005. [Google Scholar] [CrossRef]
Chen W, Feng P, Yang H, Ding H, Lin H, Chou KC (2017). iRNA-AI: identifying the adenosine to inosine editing sites in RNA sequences. Oncotarget 8: 4208–4217. DOI 10.18632/oncotarget.13758. [Google Scholar] [CrossRef]
Cheng X, Xiao X, Chou KC (2017a). pLoc-mPlant: predict subcellular localization of multi-location plant proteins by incorporating the optimal GO information into general PseAAC. Molecular BioSystems 13: 1722–1727. DOI 10.1039/C7MB00267J. [Google Scholar] [CrossRef]
Cheng X, Xiao X, Chou KC (2017b). pLoc-mVirus: predict subcellular localization of multi-location virus proteins via incorporating the optimal GO information into general PseAAC. Gene 628: 315–321. DOI 10.1016/j.gene.2017.07.036. [Google Scholar] [CrossRef]
Cheng X, Lin WZ, Xiao X, Chou KC (2018a). pLoc_bal-mAnimal: predict subcellular localization of animal proteins by balancing training dataset and PseAAC. Bioinformatics 35: 398–406. DOI 10.1093/bioinformatics/bty628. [Google Scholar] [CrossRef]
Cheng X, Xiao X, Chou KC (2018b). pLoc-mHum: predict subcellular localization of multi-location human proteins via general PseAAC to winnow out the crucial GO information. Bioinformatics 34: 1448–1456. DOI 10.1093/bioinformatics/btx711. [Google Scholar] [CrossRef]
Cheng X, Xiao X, Chou KC (2018c). pLoc-mGneg: predict subcellular localization of Gram-negative bacterial proteins by deep gene ontology learning via general PseAAC. Genomics 110: 231–239. DOI 10.1016/j.ygeno.2017.10.002. [Google Scholar] [CrossRef]
Cheng X, Xiao X, Chou KC, Hancock J (2018d). pLoc-mHum: predict subcellular localization of multi-location human proteins via general PseAAC to winnow out the crucial GO information. Bioinformatics 34: 1448–1456. DOI 10.1093/bioinformatics/btx711. [Google Scholar] [CrossRef]
Cheng X, Xiao X, Chou KC (2018e). pLoc_bal-mGneg: predict subcellular localization of Gram-negative bacterial proteins by quasi-balancing training dataset and general PseAAC. Journal of Theoretical Biology 458: 92–102. DOI 10.1016/j.jtbi.2018.09.005. [Google Scholar] [CrossRef]
Cheng X, Zhao SG, Lin WZ, Xiao X, Chou KC, Hancock J (2017c). pLoc-mAnimal: predict subcellular localization of animal proteins with both single and multiple sites. Bioinformatics 33: 3524–3531. DOI 10.1093/bioinformatics/btx476. [Google Scholar] [CrossRef]
Cheng X, Zhao SG, Xiao X, Chou KC (2016). iATC-mISF: a multi-label classifier for predicting the classes of anatomical therapeutic chemicals. Bioinformatics 33: 341–346. [Google Scholar]
Chou KC (2001a). Prediction of protein cellular attributes using pseudo-amino acid composition. Proteins: Structure, Function, and Genetics 43: 246–255. DOI 10.1002/prot.1035. [Google Scholar] [CrossRef]
Chou KC (2001b). Prediction of signal peptides using scaled window. Peptides 22: 1973–1979. DOI 10.1016/S0196-9781(01)00540-X. [Google Scholar] [CrossRef]
Chou KC (2001c). Using subsite coupling to predict signal peptides. Protein Engineering, Design and Selection 14: 75–79. DOI 10.1093/protein/14.2.75. [Google Scholar] [CrossRef]
Chou KC (2005). Using amphiphilic pseudo amino acid composition to predict enzyme subfamily classes. Bioinformatics 21: 10–19. DOI 10.1093/bioinformatics/bth466. [Google Scholar] [CrossRef]
Chou KC (2011). Some remarks on protein attribute prediction and pseudo amino acid composition. Journal of Theoretical Biology 273: 236–247. DOI 10.1016/j.jtbi.2010.12.024. [Google Scholar] [CrossRef]
Chou KC (2013). Some remarks on predicting multi-label attributes in molecular biosystems. Molecular BioSystems 9: 1092–1100. DOI 10.1039/c3mb25555g. [Google Scholar] [CrossRef]
Chou KC (2015). Impacts of bioinformatics to medicinal chemistry. Medicinal Chemistry 11: 218–234. DOI 10.2174/1573406411666141229162834. [Google Scholar] [CrossRef]
Chou KC (2017). An unprecedented revolution in medicinal chemistry driven by the progress of biological science. Current Topics in Medicinal Chemistry 17: 2337–2358. DOI 10.2174/1568026617666170414145508. [Google Scholar] [CrossRef]
Chou KC, Cai YD (2006). Prediction of protease types in a hybridization space. Biochemical and Biophysical Research Communications 339: 1015–1020. DOI 10.1016/j.bbrc.2005.10.196. [Google Scholar] [CrossRef]
Chou KC, Elrod DW (2002). Bioinformatical analysis of G-protein-coupled receptors. Journal of Proteome Research 1: 429–433. DOI 10.1021/pr025527k. [Google Scholar] [CrossRef]
Chou KC, Shen HB (2008). Cell-PLoc: a package of Web servers for predicting subcellular localization of proteins in various organisms. Nature Protocols 3: 153–162. DOI 10.1038/nprot.2007.494. [Google Scholar] [CrossRef]
Chou KC, Wu ZC, Xiao X (2012). iLoc-Hum: using the accumulation-label scale to predict subcellular locations of human proteins with both single and multiple sites. Molecular Biosystems 8: 629–641. DOI 10.1039/C1MB05420A. [Google Scholar] [CrossRef]
Chou KC, Zhang CT (2008). Prediction of protein structural classes. Critical Reviews in Biochemistry and Molecular Biology 30: 275–349. DOI 10.3109/10409239509083488. [Google Scholar] [CrossRef]
Chou KC, Cheng X, Xiao X (2019). pLoc_bal-mHum: predict subcellular localization of human proteins by PseAAC and quasi-balancing training dataset. Genomics 111: 1274–1282. DOI 10.1016/j.ygeno.2018.08.007. [Google Scholar] [CrossRef]
Dehzangi A, Heffernan R, Sharma A, Lyons J, Paliwal K, Sattar A (2015). Gram-positive and Gram-negative protein subcellular localization by incorporating evolutionary-based descriptors into Chou’s general PseAAC. Journal of Theoretical Biology 364: 284–294. DOI 10.1016/j.jtbi.2014.09.029. [Google Scholar] [CrossRef]
Dou Y, Yao B, Zhang C (2014). PhosphoSVM: prediction of phosphorylation sites by integrating various protein sequence attributes with a support vector machine. Amino Acids 46: 1459–1469. DOI 10.1007/s00726-014-1711-5. [Google Scholar] [CrossRef]
Ehsan A, Mahmood K, Khan YD, Khan SA, Chou KC (2018). A novel modeling in mathematical biology for classification of signal peptides. Scientific Reports 8: 502. DOI 10.1038/s41598-018-19491-y. [Google Scholar] [CrossRef]
Falke JJ, Bass RB, Butler SL, Chervitz SA, Danielson MA (1997). The two-component signaling pathway of bacterial chemotaxis: a molecular view of signal transduction by receptors, kinases, and adaptation enzymes. Annual Review of Cell and Developmental Biology 13: 457–512. DOI 10.1146/annurev.cellbio.13.1.457. [Google Scholar] [CrossRef]
Feng KY, Cai YD, Chou KC (2005). Boosting classifier for predicting protein domain structural class. Biochemical and Biophysical Research Communications 334: 213–217. DOI 10.1016/j.bbrc.2005.06.075. [Google Scholar] [CrossRef]
Feng PM, Ding H, Chen W, Lin H (2013a). Naive Bayes classifier with feature selection to identify phage virion proteins. Computational and Mathematical Methods in Medicine 2013: 1–6. DOI 10.1155/2013/530696. [Google Scholar] [CrossRef]
Feng PM, Lin H, Chen W (2013b). Identification of antioxidants from sequence information using naive Bayes. Computational and Mathematical Methods in Medicine 2013: 567529–5. DOI 10.1155/2013/567529. [Google Scholar] [CrossRef]
Feng P, Ding H, Yang H, Chen W, Lin H, Chou KC (2017). iRNA-PseColl: identifying the occurrence sites of different RNA modifications by incorporating collective effects of nucleotides into PseKNC. Molecular Therapy-Nucleic Acids 7: 155–163. DOI 10.1016/j.omtn.2017.03.006. [Google Scholar] [CrossRef]
Feng P, Yang H, Ding H, Lin H, Chen W, Chou KC (2018). iDNA6mA-PseKNC: identifying DNA N6-methyladenosine sites by incorporating nucleotide physicochemical properties into PseKNC. Genomics 110: 239–246. DOI 10.1016/j.ygeno.2017.10.008. [Google Scholar] [CrossRef]
Huang HD, Lee TY, Tzeng SW, Horng JT (2005). KinasePhos: a web tool for identifying protein kinase-specific phosphorylation sites. Nucleic Acids Research 33: W226–W229. DOI 10.1093/nar/gki471. [Google Scholar] [CrossRef]
Hubbard MJ, Cohen P (1993). On target with a new mechanism for the regulation of protein phosphorylation. Trends in Biochemical Sciences 18: 172–177. DOI 10.1016/0968-0004(93)90109-Z. [Google Scholar] [CrossRef]
Ingrell CR, Miller ML, Jensen ON, Blom N (2007). NetPhosYeast: prediction of protein phosphorylation sites in yeast. Bioinformatics 23: 895–897. DOI 10.1093/bioinformatics/btm020. [Google Scholar] [CrossRef]
Javed F, Hayat M (2019). Predicting subcellular localization of multi-label proteins by incorporating the sequence features into Chou’s PseAAC. Genomics 111: 1325–1332. DOI 10.1016/j.ygeno.2018.09.004. [Google Scholar] [CrossRef]
Jia J, Liu Z, Xiao X, Liu B, Chou KC (2016a). Identification of protein-protein binding sites by incorporating the physicochemical properties and stationary wavelet transforms into pseudo amino acid composition. Journal of Biomolecular Structure and Dynamics 34: 1946–1961. DOI 10.1080/07391102.2015.1095116. [Google Scholar] [CrossRef]
Jia J, Liu Z, Xiao X, Liu B, Chou KC (2016b). pSuc-Lys: Predict lysine succinylation sites in proteins with PseAAC and ensemble random forest approach. Journal of Theoretical Biology 394: 223–230. DOI 10.1016/j.jtbi.2016.01.020. [Google Scholar] [CrossRef]
Jia J, Liu Z, Xiao X, Liu B, Chou KC (2016c). pSuc-Lys: Predict lysine succinylation sites in proteins with PseAAC and ensemble random forest approach. Journal of Theoretical Biology 394: 223–230. DOI 10.1016/j.jtbi.2016.01.020. [Google Scholar] [CrossRef]
Jiang L, Zhang J, Xuan P, Zou Q (2016). BP neural network could help improve pre-miRNA identification in various species. BioMed Research International 2016: 1–11. DOI 10.1155/2016/9565689. [Google Scholar] [CrossRef]
Ju Z, Wang SY (2018). Prediction of citrullination sites by incorporating k-spaced amino acid pairs into Chou’s general pseudo amino acid composition. Gene 664: 78–83. DOI 10.1016/j.gene.2018.04.055. [Google Scholar] [CrossRef]
Khan YD, Ahmad F, Anwar MW (2012). A neuro-cognitive approach for iris recognition using back propagation. World Applied Sciences Journal 16: 678–685. [Google Scholar]
Khan YD, Ahmed F, Khan SA (2014a). Situation recognition using image moments and recurrent neural networks. Neural Computing and Applications 24: 1519–1529. DOI 10.1007/s00521-013-1372-4. [Google Scholar] [CrossRef]
Khan YD, Khan NS, Farooq S, Abid A, Khan SA, Ahmad F, Mahmood MK (2014b). An efficient algorithm for recognition of human actions. Scientific World Journal 2014: 1–11. DOI 10.1155/2014/875879. [Google Scholar] [CrossRef]
Khan YD, Khan SA, Ahmad F, Islam S (2014c). Iris recognition using image moments and k-means algorithm. The Scientific World Journal 2014: 1–9. DOI 10.1155/2014/723595. [Google Scholar] [CrossRef]
Khan YD, Rasool N, Hussain W, Khan SA, Chou KC (2018). iPhosT-PseAAC: identify phosphothreonine sites by incorporating sequence statistical moments into PseAAC. Analytical Biochemistry 550: 109–116. DOI 10.1016/j.ab.2018.04.021. [Google Scholar] [CrossRef]
Knowles JR (1980). Enzyme-catalyzed phosphoryl transfer reactions. Annual Review of Biochemistry 49: 877–919. DOI 10.1146/annurev.bi.49.070180.004305. [Google Scholar] [CrossRef]
Krishnan SM (2018). Using Chou’s general PseAAC to analyze the evolutionary relationship of receptor associated proteins (RAP) with various folding patterns of protein domains. Journal of Theoretical Biology 445: 62–74. DOI 10.1016/j.jtbi.2018.02.008. [Google Scholar] [CrossRef]
Kumar R, Srivastava A, Kumari B, Kumar M (2015). Prediction of β-lactamase and its class by Chou’s pseudo-amino acid composition and support vector machine. Journal of Theoretical Biology 365: 96–103. DOI 10.1016/j.jtbi.2014.10.008. [Google Scholar] [CrossRef]
Li D, Ju Y, Zou Q (2016). Protein folds prediction with hierarchical structured SVM. Current Proteomics 13: 79–85. DOI 10.2174/157016461302160514000940. [Google Scholar] [CrossRef]
Liang Y, Zhang S (2018). Identify Gram-negative bacterial secreted protein types by incorporating different modes of PSSM into Chou’s general PseAAC via Kullback–Leibler divergence. Journal of Theoretical Biology 454: 22–29. DOI 10.1016/j.jtbi.2018.05.035. [Google Scholar] [CrossRef]
Lin H, Deng EZ, Ding H, Chen W, Chou KC (2014). iPro54-PseKNC: a sequence-based predictor for identifying sigma-54 promoters in prokaryote with pseudo k-tuple nucleotide composition. Nucleic Acids Research 42: 12961–12972. DOI 10.1093/nar/gku1019. [Google Scholar] [CrossRef]
Lin H, Ding C, Song Q, Yang P, Ding H, Deng KJ, Chen W (2012). The prediction of protein structural class using averaged chemical shifts. Journal of Biomolecular Structure and Dynamics 29: 1147–1153. DOI 10.1080/07391102.2011.672628. [Google Scholar] [CrossRef]
Lin S, Song Q, Tao H, Wang W, Wan W, Huang J, Xu C, Chebii V, Kitony J, Que S, Harrison A, He H (2015). Rice_Phospho 1.0: a new rice-specific SVM predictor for protein phosphorylation sites. Scientific Reports 1: 5. DOI 10.1038/srep11940. [Google Scholar] [CrossRef]
Lin WZ, Fang JA, Xiao X, Chou KC (2011). iDNA-Prot: identification of DNA binding proteins using random forest with grey model. PLoS One 6: e24756. DOI 10.1371/journal.pone.0024756. [Google Scholar] [CrossRef]
Lin WZ, Fang JA, Xiao X, Chou KC (2013). iLoc-Animal: a multi-label learning classifier for predicting subcellular localization of animal proteins. Molecular BioSystems 9: 634–644. DOI 10.1039/c3mb25466f. [Google Scholar] [CrossRef]
Liu B, Fang L, Long R, Lan X, Chou KC (2015). iEnhancer-2L: a two-layer predictor for identifying enhancers and their strength by pseudo k-tuple nucleotide composition. Bioinformatics 32: 362–369. DOI 10.1093/bioinformatics/btv604. [Google Scholar] [CrossRef]
Liu B, Long R, Chou KC (2016a). iDHS-EL: identifying DNase I hypersensitive sites by fusing three different modes of pseudo nucleotide composition into an ensemble learning framework. Bioinformatics 32: 2411–2418. DOI 10.1093/bioinformatics/btw186. [Google Scholar] [CrossRef]
Liu B, Wang S, Long R, Chou KC (2016b). iRSpot-EL: identify recombination spots with an ensemble learning approach. Bioinformatics 33: 35–41. DOI 10.1093/bioinformatics/btw539. [Google Scholar] [CrossRef]
Liu B, Wang S, Long R, Chou KC (2017a). iRSpot-EL: identify recombination spots with an ensemble learning approach. Bioinformatics 33: 35–41. DOI 10.1093/bioinformatics/btw539. [Google Scholar] [CrossRef]
Liu B, Yang F, Chou KC (2017b). 2L-piRNA: a two-layer ensemble classifier for identifying piwi-interacting RNAs and their function. Molecular Therapy-Nucleic Acids 7: 267–277. DOI 10.1016/j.omtn.2017.04.008. [Google Scholar] [CrossRef]
Liu B, Yang F, Huang DS, Chou KC (2018). iPromoter-2L: a two-layer predictor for identifying promoters and their types by multi-window-based PseKNC. Bioinformatics 34: 33–40. DOI 10.1093/bioinformatics/btx579. [Google Scholar] [CrossRef]
Lohrmann J, Harter K (2002). Plant two-component signaling systems and the role of response regulators. Plant Physiology 128: 363–369. DOI 10.1104/pp.010907. [Google Scholar] [CrossRef]
Mann M, Ong SE, Grønborg M, Steen H, Jensen ON, Pandey A (2002). Analysis of protein phosphorylation using mass spectrometry: deciphering the phosphoproteome. Trends in Biotechnology 20: 261–268. DOI 10.1016/S0167-7799(02)01944-3. [Google Scholar] [CrossRef]
Mei J, Fu Y, Zhao J (2018). Analysis and prediction of ion channel inhibitors by using feature selection and Chou’s general pseudo amino acid composition. Journal of Theoretical Biology 456: 41–48. DOI 10.1016/j.jtbi.2018.07.040. [Google Scholar] [CrossRef]
Mei J, Zhao J (2018a). Analysis and prediction of presynaptic and postsynaptic neurotoxins by Chou’s general pseudo amino acid composition and motif features. Journal of Theoretical Biology 447: 147–153. DOI 10.1016/j.jtbi.2018.03.034. [Google Scholar] [CrossRef]
Mei J, Zhao J (2018b). Prediction of HIV-1 and HIV-2 proteins by using Chou’s pseudo amino acid compositions and different classifiers. Scientific Reports 8: 661. DOI 10.1038/s41598-018-20819-x. [Google Scholar] [CrossRef]
Mizuno T (2014). Two-component phosphorelay signal transduction systems in plants: from hormone responses to circadian rhythms. Bioscience, Biotechnology, and Biochemistry 69: 2263–2276. DOI 10.1271/bbb.69.2263. [Google Scholar] [CrossRef]
Mok J, Snyder M (2010). Global analysis of phosphoregulatory networks. Handbook of cell signaling, pp. 645–655. [Google Scholar]
Mondal S, Pai PP (2014). Chou’s pseudo amino acid composition improves sequence-based antifreeze protein prediction. Journal of Theoretical Biology 356: 30–35. DOI 10.1016/j.jtbi.2014.04.006. [Google Scholar] [CrossRef]
Nanni L, Brahnam S, Lumini A (2014). Prediction of protein structure classes by incorporating different protein descriptors into general Chou’s pseudo amino acid composition. Journal of Theoretical Biology 360: 109–116. DOI 10.1016/j.jtbi.2014.07.003. [Google Scholar] [CrossRef]
Qiu WR, Jiang SY, Xu ZC, Xiao X, Chou KC (2017a). Identifying RNA 5-methylcytosine sites by incorporating physical-chemical properties into pseudo dinucleotide composition. Oncotarget 8: 41178–41188. DOI 10.18632/oncotarget.17104. [Google Scholar] [CrossRef]
Qiu WR, Sun BQ, Xiao X, Xu ZC, Chou KC (2016a). iHyd-PseCp: Identify hydroxyproline and hydroxylysine in proteins by incorporating sequence-coupled effects into general PseAAC. Oncotarget 7: 44310–44321. DOI 10.18632/oncotarget.10027. [Google Scholar] [CrossRef]
Qiu WR, Sun BQ, Xiao X, Xu ZC, Chou KC (2016b). iPTM-mLys: identifying multiple lysine PTM sites and their different types. Bioinformatics 32: 3116–3123. DOI 10.1093/bioinformatics/btw380. [Google Scholar] [CrossRef]
Qiu WR, Xiao X, Chou KC (2014a). iRSpot-TNCPseAAC: identify recombination spots with trinucleotide composition and pseudo amino acid components. International Journal of Molecular Sciences 15: 1746–1766. DOI 10.3390/ijms15021746. [Google Scholar] [CrossRef]
Qiu WR, Xiao X, Lin WZ, Chou KC (2014b). iMethyl-PseAAC: identification of protein methylation sites via a pseudo amino acid composition approach. BioMed Research International 2014: 1–12. DOI 10.1155/2014/947416. [Google Scholar] [CrossRef]
Qiu WR, Xiao X, Lin WZ, Chou KC (2014). iUbiq-Lys: prediction of lysine ubiquitination sites in proteins by extracting sequence evolution information via a gray system model. Journal of Biomolecular Structure and Dynamics 33: 1731–1742. DOI 10.1080/07391102.2014.968875. [Google Scholar] [CrossRef]
Qiu WR, Xiao X, Xu ZC, Chou KC (2016c). iPhos-PseEn: identifying phosphorylation sites in proteins by fusing different pseudo components into an ensemble classifier. Oncotarget 7: 51270–51283. DOI 10.18632/oncotarget.9987. [Google Scholar] [CrossRef]
Qiu W, Li S, Cui X, Yu Z, Wang M, Du J, Peng Y, Yu B (2018). Predicting protein submitochondrial locations by incorporating the pseudo-position specific scoring matrix into the general Chou’s pseudo-amino acid composition. Journal of Theoretical Biology 450: 86–103. DOI 10.1016/j.jtbi.2018.04.026. [Google Scholar] [CrossRef]
Qiu WR, Sun BQ, Xiao X, Xu D, Chou KC (2017b). iPhos-PseEvo: identifying human phosphorylated proteins by incorporating evolutionary information into general PseAAC via grey system theory. Molecular Informatics 36: 1600010. DOI 10.1002/minf.201600010. [Google Scholar] [CrossRef]
Rahman MS, Shatabda S, Saha S, Kaykobad M, Rahman MS (2018). Dpp-pseaac: a DNA-binding protein prediction model using Chou’s general pseaac. Journal of Theoretical Biology 452: 22–34. DOI 10.1016/j.jtbi.2018.05.006. [Google Scholar] [CrossRef]
Sabooh MF, Iqbal N, Khan M, Khan M, Maqbool HF (2018). Identifying 5-methylcytosine sites in RNA sequence using composite encoding feature into Chou’s PseKNC. Journal of Theoretical Biology 452: 1–9. DOI 10.1016/j.jtbi.2018.04.037. [Google Scholar] [CrossRef]
Sankari ES, Manimegalai D (2018). Predicting membrane protein types by incorporating a novel feature set into Chou’s general PseAAC. Journal of Theoretical Biology 455: 319–328. DOI 10.1016/j.jtbi.2018.07.032. [Google Scholar] [CrossRef]
Senawongse P, Dalby AR, Yang ZR (2005). Predicting the phosphorylation sites using hidden Markov models and machine learning methods. Journal of Chemical Information and Modeling 45: 1147–1152. DOI 10.1021/ci050047+. [Google Scholar] [CrossRef]
Shen HB, Chou KC (2007). Signal-3L: A 3-layer approach for predicting signal peptides. Biochemical and Biophysical Research Communications 363: 297–303. DOI 10.1016/j.bbrc.2007.08.140. [Google Scholar] [CrossRef]
Shen HB, Yang J, Chou KC (2007). Euk-PLoc: an ensemble classifier for large-scale eukaryotic protein subcellular location prediction. Amino Acids 33: 57–67. DOI 10.1007/s00726-006-0478-8. [Google Scholar] [CrossRef]
Srivastava A, Kumar R, Kumar M (2018). BlaPred: Predicting and classifying β-lactamase using a 3-tier prediction system via Chou’s general PseAAC. Journal of Theoretical Biology 457: 29–36. DOI 10.1016/j.jtbi.2018.08.030. [Google Scholar] [CrossRef]
Thomason P, Kay R (2000). Eukaryotic signal transduction via histidine-aspartate phosphorelay. Journal of Cell Science 113: 3141–3150. [Google Scholar]
Wu ZC, Xiao X, Chou KC (2011). iLoc-Plant: a multi-label classifier for predicting the subcellular localization of plant proteins with both single and multiple sites. Molecular BioSystems 7: 3287–3297. DOI 10.1039/c1mb05232b. [Google Scholar] [CrossRef]
Xiao X, Cheng X, Su S, Nao Q, Chou KC (2017). pLoc-mGpos: Incorporate key gene ontology information into general PseAAC for predicting subcellular localization of Gram-positive bacterial proteins. Natural Science 09: 330–349. DOI 10.4236/ns.2017.99032. [Google Scholar] [CrossRef]
Xiao X, Wang P, Lin WZ, Jia JH, Chou KC (2013). iAMP-2L: a two-level multi-label classifier for identifying antimicrobial peptides and their functional types. Analytical Biochemistry 436: 168–177. DOI 10.1016/j.ab.2013.01.019. [Google Scholar] [CrossRef]
Xiao X, Wu ZC, Chou KC (2011). iLoc-Virus: a multi-label learning classifier for identifying the subcellular localization of virus proteins with both single and multiple sites. Journal of Theoretical Biology 284: 42–51. DOI 10.1016/j.jtbi.2011.06.005. [Google Scholar] [CrossRef]
Xiao X, Ye HX, Liu Z, Jia JH, Chou KC (2016). iROS-gPseKNC: predicting replication origin sites in DNA by incorporating dinucleotide position-specific propensity into general pseudo nucleotide composition. Oncotarget 7: 34180–34189. DOI 10.18632/oncotarget.9057. [Google Scholar] [CrossRef]
Xu Y, Shao XJ, Wu LY, Deng NY, Chou KC (2013). iSNO-AAPair: incorporating amino acid pairwise coupling into PseAAC for predicting cysteine S-nitrosylation sites in proteins. PeerJ 1: e171. DOI 10.7717/peerj.171. [Google Scholar] [CrossRef]
Xu Y, Wang Z, Li C, Chou KC (2017). iPreny-PseAAC: identify C-terminal cysteine prenylation sites in proteins by incorporating two tiers of sequence couplings into PseAAC. Medicinal Chemistry 13: 544–551. DOI 10.2174/1573406413666170419150052. [Google Scholar] [CrossRef]
Xu Y, Wen X, Shao XJ, Deng NY, Chou KC (2014a). iHyd-PseAAC: predicting hydroxyproline and hydroxylysine in proteins by incorporating dipeptide position-specific propensity into pseudo amino acid composition. International Journal of Molecular Sciences 15: 7594–7610. DOI 10.3390/ijms15057594. [Google Scholar] [CrossRef]
Xu Y, Wen X, Wen LS, Wu LY, Deng NY, Chou KC (2014b). iNitro-Tyr: prediction of nitrotyrosine sites in proteins with general pseudo amino acid composition. PLoS One 9: e105018. DOI 10.1371/journal.pone.0105018. [Google Scholar] [CrossRef]
Xuao X, Cheng X, Chen G, Mao Q, Chou KC (2019). pLoc_bal-mGpos: predict subcellular localization of Gram-positive bacterial proteins by quasi-balancing training dataset and PseAAC. Genomics 111: 886–892. DOI 10.1016/j.ygeno.2018.05.017. [Google Scholar] [CrossRef]
Xue Y, Ren J, Gao X, Jin C, Wen L, Yao X (2008). GPS 2. 0, a tool to predict kinase-specific phosphorylation sites in hierarchy. Molecular & Cellular Proteomics 7: 1598–1608. DOI 10.1074/mcp.M700574-MCP200. [Google Scholar] [CrossRef]
Zhang CJ, Tang H, Li WC, Lin H, Chen W, Chou KC (2016). iOri-Human: identify human origin of replication by incorporating dinucleotide physicochemical properties into pseudo nucleotide composition. Oncotarget 7: 69783–69793. DOI 10.18632/oncotarget.11975. [Google Scholar] [CrossRef]
Zhang L, Kong L (2018). iRSpot-ADPM: identify recombination spots by incorporating the associated dinucleotide product model into Chou’s pseudo components. Journal of Theoretical Biology 441: 1–8. DOI 10.1016/j.jtbi.2017.12.025. [Google Scholar] [CrossRef]
Zhang S, Duan X (2018). Prediction of protein subcellular localization with oversampling approach and Chou’s general PseAAC. Journal of Theoretical Biology 437: 239–250. DOI 10.1016/j.jtbi.2017.10.030. [Google Scholar] [CrossRef]
Zhang S, Liang Y (2018). Predicting apoptosis protein subcellular localization by integrating auto-cross correlation and PSSM into Chou’s PseAAC. Journal of Theoretical Biology 457: 163–169. DOI 10.1016/j.jtbi.2018.08.042. [Google Scholar] [CrossRef]
Zhao W, Wang L, Zhang TX, Zhao ZN, Du PF (2018). A brief review on software tools in generating Chou’s pseudo-factor representations for all types of biological sequences. Protein & Peptide Letters 25: 822–829. DOI 10.2174/0929866525666180905111124. [Google Scholar] [CrossRef]
Zhou GP, Doctor K (2003). Subcellular location prediction of apoptosis proteins. Proteins: structure, Function, and Bioinformatics 50: 44–48. DOI 10.1002/prot.10251. [Google Scholar] [CrossRef]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |