DOI:10.32604/biocell.2021.017659
| BIOCELL DOI:10.32604/biocell.2021.017659 | |
| Review |
Implications of enolase in the RANKL-mediated osteoclast activity following spinal cord injury
1Department of Microbiology and Immunology, Medical University of South Carolina, Charleston, SC 29425, USA
2Department of Neurosurgery, Medical University of South Carolina, Charleston, SC 29425, USA
3Ralph H. Johnson Veterans Administration Medical Center, Charleston, SC 29401, USA
*Address correspondence to: Naren L. Banik, baniknl@musc.edu; Azizul Haque, haque@musc.edu
Received: 05 June 2021; Accepted: 19 July 2021
Abstract: Spinal Cord Injury (SCI) is a debilitating condition characterized by damage to the spinal cord, resulting in loss of function, mobility, and sensation. Although increasingly prevalent in the US, no FDA-approved therapy exists due to the unfortunate complexity of the condition, and the difficulties of SCI may be furthered by the development of SCI-related complications, such as osteoporosis. SCI demonstrates two crucial stages for consideration: the primary stage and the secondary stage. While the primary stage is suggested to be immediate and irreversible, the secondary stage is proposed as a promising window of opportunity for therapeutic intervention. Enolase, a metabolic enzyme upregulated after SCI, performs non-glycolytic functions, promoting inflammatory events via extracellular degradative actions and increased production of inflammatory cytokines and chemokines. Neuron-specific enolase (NSE) serves as a biomarker of functional damage to neurons following SCI, and the inhibition of NSE has been demonstrated to reduce signs of secondary injury of SCI and to ameliorate dysfunction. This Viewpoint article involves enolase activation in the regulation of RANK-RANKL pathway and summarizes succinctly the mechanisms influencing osteoclast-mediated resorption of bone in SCI. Our laboratory proposes that inhibition of enolase activation may reduce SCI-induced inflammatory response and decrease osteoclast activity, limiting the chances of skeletal tissue loss in SCI.
Keywords: Enolase; RANK; RANKL; Osteoporosis; Neuronal death; Spinal cord injury
Spinal cord injury (SCI) is a condition in which trauma occurs to the spinal cord, resulting in compromised sensation, function, and mobility (Guilcher et al., 2019). The pathophysiology of SCI has two phases, a primary phase and a secondary phase. Primary injury involves mechanical trauma, such as compression, laceration, and contusion, and vascular injury to the spinal cord (Alizadeh et al., 2019; Cox et al., 2015; Hachem et al., 2017; Shams et al., 2021). Cellular damage at the lesion site is considered immediate and irreversible. In contrast, secondary trauma describes a cascade of molecular events occurring days to weeks following injury, consisting of neuroinflammation, cell death, and tissue degeneration (Hachem et al., 2017). SCI-associated inflammation influences the development of chronic ischemic hypoxia, upregulation of Calpain proteases, activation of inflammatory signaling proteins, degeneration of the blood brain barrier, and production of reactive oxygen species (Cox et al., 2015; Shams et al., 2021). Therefore, the magnitude of the secondary phase of SCI drives the extent of chronic morbidity in individuals with SCI (Cox et al., 2015; Hachem et al., 2017; Shams et al., 2021).
Individuals with SCI are likely to experience paralysis-related complications, such as osteoporosis, which can increase their risk of forming fractures and further the reduction of their quality of life (Hachem et al., 2017; Haider et al., 2018; Shams et al., 2021). Several multidisciplinary mechanisms have been proposed in the literature for their role in bone loss following SCI (Invernizzi et al., 2020a). Perturbations in the Wnt pathway have been implicated in bone loss (Shams et al., 2021; Zhao et al., 2018). The Wnt pathway inhibitor, sclerostin, is upregulated following SCI, significantly compromising bone mineral density, bone strength, and bone architecture in rodent models (Zhao et al., 2018). An additional factor in bone loss is mechanical unloading, such as with SCI-induced paralysis, as it has been strongly implicated in the upregulation of sclerostin, as well as the downregulation of periostin, a protein promoting osteoblast activity (Invernizzi et al., 2020a; Zhao et al., 2018). Mechanical unloading may also upregulate osteoblast secretion of RANKL, contributing to increased bone resorption (Invernizzi et al., 2020a; Shams et al., 2021). In SCI, mechanical unloading may contribute to muscle atrophy, having crucial crosstalk with skeletal bone tissue; both smooth muscle cells and osteoblasts secrete inflammatory cytokines and chemokines, such as interleukin-6 (IL-6), during physical inactivity (Invernizzi et al., 2020a; Invernizzi et al., 2020b). Chronic inflammatory condition is suggested to be a major culprit in cellular apoptosis, tissue damage, and oxidative damage (Orr and Gensel, 2018; Samantaray et al., 2016).
In the US, 17,730 new SCI cases are reported annually, and unfortunately, this devastating condition has no FDA-approved therapy and hence, no cure (Alabama, 2019). However, this secondary phase is suggested to be reversible, posing it as a promising window of opportunity for therapeutic intervention (Polcyn et al., 2017; Shams et al., 2021). This short review aims to summarize our viewpoints on the mechanisms influencing osteoporosis or skeletal tissue loss in SCI via enolase-mediated inflammation and promotion of the receptor activator of nuclear factor-κB (RANK) pathway, a modulator for osteoclastogenesis and bone resorption. Our laboratory proposes that enolase inhibition may attenuate activation of RANK ligand (RANKL) and reduce inflammatory response and skeletal loss in SCI.
Mechanisms of healthy bone remodeling
Bone remodeling is a coupled process between bone construction by osteoblasts and bone resorption by osteoclasts. Transcription factors such as NF-kB, c-Fos, and NFATc1 secreted by osteoblasts stimulate the maturation of osteoclast progenitors. Mature osteoclasts secrete cathepsin K, with the function to dissolve the bone matrix, and hydrochloric acid, with the role to dissolve the bone mineral (Armas and Recker, 2012). When osteoblasts are activated by Wnt signaling proteins, extracellular transmitters (prostaglandin E2, prostacyclin, insulin-like growth factor 1, insulin-like growth factor 2, transforming growth factor beta), and intracellular signal transmitters (calcium, inositol trisphosphate (IP3), cyclic adenosine monophosphate (cAMP), and cyclic guanosine monophosphate (cGMP), osteoblasts are immobilized to layer across the dissolved surface of the bone for calcification (Qin et al., 2010). When these processes are coupled, there is healthy bone remodeling (Shams et al., 2021). Bone remodeling is influenced by mechanical loading onto the bone via weight-bearing, and through biochemical signaling, osteocytes can regulate bone remodeling. Upon injury, these processes may become uncoupled, and the osteocyte’s ability to modulate osteoblast and osteoclast activity is perturbed (Bonewald and Johnson, 2008; Shams et al., 2021).
The RANK/RANKL/OPG pathway and SCI-dysregulation of bone remodeling
The RANK/RANKL/OPG pathway is a modulator for bone resorption (Jiang et al., 2014; Liu and Zhang, 2015). This pathway consists of the RANK receptor, RANK ligand (RANKL), and osteoprotegerin (OPG) (Jiang et al., 2014; Liu and Zhang, 2015). It is suggested that osteocytes demonstrate RANKL upregulation, activating osteoclastogenesis (Wu et al., 2017). RANKL is expressed in osteoblast, osteoblast precursors, and stromal cells (Udagawa et al., 1999; Yasuda et al., 1998). The RANK receptor is particularly expressed on the surface of osteoclast and pre-osteoclast cells. Osteoblasts or osteocyte cells present RANKL to the RANK receptor embedded on the membrane of osteoclastic progenitor cells (Shams et al., 2021). Successful binding of RANKL to the RANK receptor promotes osteoclast differentiation and activation through the stimulation of tumor necrosis factor receptor-associated factor (TRAF6) recruitment (Jiang et al., 2014; Liu and Zhang, 2015). TRAF6 recruitment initiates the activation of nuclear factor-κB (NF-κB), p38-extracellular signal-regulated kinase (ERK), and c-Jun NH(2)-terminal kinase (JNK), subsequently interacting with nuclear factor of activated T cells C1 (NFATc1) in the cell nucleus and stimulating the transcription of genes crucial for osteoclastogenesis (Chen et al., 2020; Liu et al., 2020; Qian et al., 2020).
In individuals with SCI, bone remodeling is no longer balanced due to perturbations of the RANK/RANKL/OPG pathway, resulting in osteoporosis (Jiang et al., 2014; Kovacs et al., 2019). Following SCI, RANK ligand (RANKL) is upregulated, binding to the RANK receptors on osteoclasts (Invernizzi et al., 2020b). Osteoclast activation promotes TRAF-6 recruitment and activation of transcription factors JNK, p38, ERK, Akt, IκB, and NF-κB (Chen et al., 2020; Kim et al., 2018; Lee et al., 2005). Phosphorylation of these factors activates the NF-κB, MAPK, and AKT cellular signaling pathways, inducing the increased activation and translocation of NFATc1 and subsequent osteoclastogenesis (Chen et al., 2020). Perturbed off-pathway influences have been suggested to dysregulate osteoblast proliferation and maturation. With increased osteoclastogenesis and decreased osteoblastogenesis, bone integrity is compromised, and osteoporosis results (Shams et al., 2021). In SCI, bone remodeling may become uncoupled due to SCI-related impairments such as paralysis, malnutrition, or side effects of pharmacological treatments (Jiang et al., 2006). SCI may stimulate osteoclastogenesis by promoting the secretion of IL-6, whose role in osteoclast cell fate determination has been implicated (Demulder et al., 1998; Francois et al., 2006; Kurihara et al., 1990).
Enolase activation has been implicated in SCI (Haque et al., 2016). Enolase is a highly conserved, glycolytic enzyme with ubiquitous expression; it functions to convert 2-phosphoglycerate to phosphoenolpyruvate (Day et al., 1993). Several enolase isoforms exist, both as homodimers and heterodimers of the α-, β-, and γ-enolase subunits (Marangos et al., 1978; Shimizu et al., 1983). While enolase primarily functions in glycolysis, it also exhibits nonglycolytic functions (Avilan et al., 2011). When expressed in the cytosol, enolase interacts with the cytoskeleton to mediate material trafficking and influence cell morphology (Keller et al., 2007). Enolase expression in the nucleus is implicated with the regulation of genes of cell proliferation and morphological transformation (Lung et al., 2010). In conditions leading to cellular injury, enolase appears on the cell surface and induces a cascade of inflammatory immune responses via the activation of the CD14-dependent Toll-Like Receptor 4 (TLR4) pathway and downstream activation of NF-κB, eliciting powerful stimulation of pro-inflammatory cytokines and chemokines (Guillou et al., 2016; Haque et al., 2016). NF-κB may stimulate IL-1β (interleukin-1β), tumor necrosis factor alpha (TNF-α), IL-6 (interleukin-6), macrophage colony-stimulating factor (m-CSF), and additional inflammatory molecules (Barnes and Karin, 1997) . These may contribute to reactive oxygen species (ROS) production, exasperated cellular functioning, and tissue degeneration (Barnes and Karin, 1997; Haque et al., 2017).
Neuron specific enolase (NSE) is used as a marker for neurons and neuroendocrine neurons, and studies suggest NSE may perform inflammatory and neurotrophic activities, mediating neuronal differentiation, proliferation, survival, and death (Bae et al., 2012; Hafner et al., 2012; Haque et al., 2018). Elevated levels of enolase may induce cellular death and tissue degeneration through the production of reactive oxygen species (ROS), nitric oxide (NO), and pro-inflammatory molecules (Haque et al., 2017; Haque et al., 2016; Sedoris et al., 2010). NSE is implicated in conditions involving ischemia, hypoxia, and inflammation, such as autoimmune and neuroinflammatory diseases. Although NSE is required for neuronal survival, high levels of NSE could be detrimental (Anand and Stead, 2005; Woodcock and Morganti-Kossmann, 2013). In further, increased secretion of NSE in serum and cerebrospinal fluid (CSF) is implicated with a higher magnitude of neurodegeneration and subsequent disease progression (Haque et al., 2018). In studies using SCI models, it has been suggested that rats with SCI have elevated NSE levels than rats with no SCI, and the inhibition of NSE has dual functions in secondary injury of SCI (Haque et al., 2017; Li et al., 2014).
The Haque lab proposed inhibition of enolase activation as an effective strategy against the detrimental inflammatory events in SCI (Haque et al., 2016; Polcyn et al., 2020). ENOblock is a novel, small enolase inhibitor, developed to study predominantly the non-glycolytic functions of enolase. In an SCI rat model, it has been demonstrated that ENOblock can attenuate inflammatory metabolic and signaling factors (Li et al., 2014; Polcyn et al., 2017). ENOblock administration can result in the inhibition of Iba-1/GFAP expression (gliosis), reducing pro-inflammatory events in SCI rat models. ENOblock may also reduce NSE and subsequent pro-inflammatory cytokine/chemokine levels in serum and spinal cord tissue (Haque et al., 2017).
Potential enolase and RANK/RANKL pathway interactions
LPS-stimulated cells may experience pro-inflammatory conditions via production of interferon gamma (IFN-γ) and tumor necrosis factor alpha (TNF-α) secretion (Guillou et al., 2016). α-enolase or ENO1 is preferentially expressed by monocyte and macrophage cells, Raw264.7 cells for example. α-enolase-stimulated cells experience similar pro-inflammatory response to LPS-stimulated cells (Guillou et al., 2016). It is suggested that, in the inflammatory process, cell-surface expression of α-enolase is dramatically upregulated in monocytes following LPS-stimulation, due to the translocation of ENO1 to the surface of the cell. Studies in our laboratory suggest that, when enolase is inhibited, the inflammatory process is perturbed in LPS-stimulated Raw 264.7 cells as previously suggested (Guillou et al., 2016). In the inflammatory state, LPS-stimulated Raw264.7 cells, a cellular model of osteoclast activity, demonstrate up-regulated RANKL expression. With enolase inhibition via ENOblock, we demonstrate that RANKL expression is decreased compared to untreated and LPS-stimulated Raw264.7 cells (Fig. 1). We suggest that, with enolase inhibition, osteoclast activation via RANKL binding can be perturbed following SCI, thereby preventing the recruitment of TRAF-6 and activation of JNK, p38, ERK, Akt and NF-κB (Fig. 2). Therefore, inhibition of enolase activation could mitigate SCI-induced osteoporosis or skeletal loss.
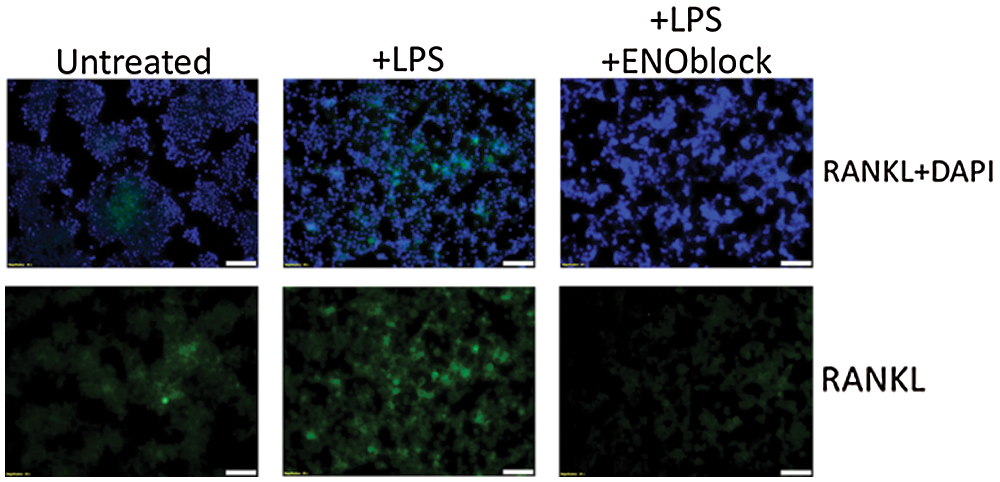
Figure 1: Enolase inhibition decreases RANKL expression in Raw264.7 cells. Raw264.7 macrophage cells were cultured in Dulbecco’s Modification of Eagle’s Medium (DMEM) with 4.5 g/L D-Gluvose, L-Glutamine, and 110 mg/L Sodium Pyruvate (11995-065, Gibco), supplemented with 10% Fetal Bovine Serum (FBS) (Thermo Scientific, Logan) and Penicillin-Streptomycin (P/S) (Mediatech, Inc., Herndon). Cells were treated with vehicle alone, LPS (100 ng/mL), and LPS + ENOblock (2.5 μM) for 72 hours. Immunofluorescence staining was performed to detect RANKL expression (sc-377079, Santa Cruz). Nuclear staining was performed using DAPI. Representative images of three trials are presented in 20x magnification.
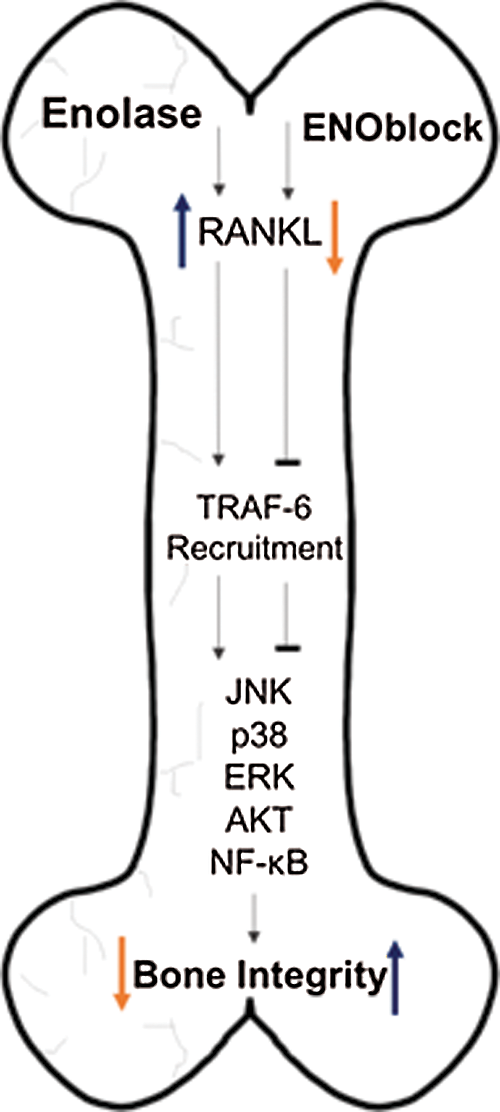
Figure 2: Enolase activation may trigger inflammatory responses leading to upregulation of RANKL after SCI. RANKL upregulation induces TRAF-6 recruitment, which in turn, promotes the activation of the transcription factors JNK, p38, ERK, Akt, and NF-κB. This causes an increase in osteoclast activity, leading to compromised bone integrity. Inversely, inhibition of enolase activation via ENOblock may block the upregulation of RANKL, and TRAF-6. Since the respective transcription factors would not be activated, osteoclast activity may be mitigated, preserving bone integrity.
The data is representative of preclinical studies using in vitro model. However, several multifactorial mechanisms influencing bone loss following SCI are described in the literature and may significantly skew the plausibility of these results. Additionally, with the novel nature of the study, the lack of systematic database search may hinder the reproducibility of the results and the strength of the conclusions.
This viewpoint short review attempts to summarize mechanisms mediating skeletal loss in SCI via osteoclast-mediated resorption of bone and the role of enolase activation on the RANK/RANKL pathway. This is the first viewpoint summarizing the current literature on this topic, aiming at paving the way to future works on this novel promising therapeutic target.
Author Contribution: R.S. performed the experiments, analyzed data, and wrote the manuscript. N.L.B. edited the manuscript. A.H. conceived the study, designed experiments, and edited the manuscript.
Funding Statement: This work was supported in part by funding from the Veterans Administration (1IOBX001262, 1I01 BX004269) and South Carolina State Spinal Cord Injury Research Fund (SCIRF #2018 I-01). Contents do not necessarily represent the policy of the SCIRF and do not imply endorsement by the funding agency. This work was also supported in part by funding from the National Institutes of Health (1R21NS118393-01).
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
Alabama UO (2019). Spinal cord injury facts and figures at a glance. https://www.nscisc.uab.edu/Public/Facts%20and%20Figures%202019%20-%20Final.pdf. [Google Scholar]
Alizadeh A, Dyck SM, Karimi-Abdolrezaee S (2019). Traumatic spinal cord injury: An overview of pathophysiology, models and acute injury mechanisms. Frontiers in Neurology 10: 282. [Google Scholar]
Anand N, Stead LG (2005). Neuron-specific enolase as a marker for acute ischemic stroke: A systematic review. Cerebrovascular Diseases 20: 213–219. [Google Scholar]
Armas LA, Recker RR (2012). Pathophysiology of osteoporosis: New mechanistic insights. Endocrinology and Metabolism Clinics of North America 41: 475–486. [Google Scholar]
Avilan L, Gualdron-Lopez M, Quinones W, Gonzalez-Gonzalez L, Hannaert V et al. (2011). Enolase: A key player in the metabolism and a probable virulence factor of trypanosomatid parasites-perspectives for its use as a therapeutic target. Enzyme Research 2011: 932549. [Google Scholar]
Bae S, Kim H, Lee N, Won C, Kim HR et al. (2012). Alpha-Enolase expressed on the surfaces of monocytes and macrophages induces robust synovial inflammation in rheumatoid arthritis. Journal of Immunology 189: 365–372. [Google Scholar]
Barnes PJ, Karin M (1997). Nuclear factor-kappaB: A pivotal transcription factor in chronic inflammatory diseases. New England Journal of Medicine 336: 1066–1071. [Google Scholar]
Bonewald LF, Johnson ML (2008). Osteocytes, mechanosensing and Wnt signaling. Bone 42: 606–615. [Google Scholar]
Chen H, Fang C, Zhi X, Song S, Gu Y et al. (2020). Neobavaisoflavone inhibits osteoclastogenesis through blocking RANKL signalling-mediated TRAF6 and c-Src recruitment and NF-kappaB. MAPK and Akt pathways. Journal of Cellular and Molecular Medicine 24: 9067–9084. [Google Scholar]
Cox A, Varma A, Banik N (2015). Recent advances in the pharmacologic treatment of spinal cord injury. Metabolic Brain Disease 30: 473–482. [Google Scholar]
Day IN, Peshavaria M, Quinn GB (1993). A differential molecular clock in enolase isoprotein evolution. Journal of Molecular Evolution 36: 599–601. [Google Scholar]
Demulder A, Guns M, Ismail A, Wilmet E, Fondu P, Bergmann P (1998). Increased osteoclast-like cells formation in long-term bone marrow cultures from patients with a spinal cord injury. Calcified Tissue International 63: 396–400. [Google Scholar]
Francois RJ, Neure L, Sieper J, Braun J (2006). Immunohistological examination of open sacroiliac biopsies of patients with ankylosing spondylitis: detection of tumour necrosis factor alpha in two patients with early disease and transforming growth factor beta in three more advanced cases. Annals of the Rheumatic Diseases 65: 713–720. [Google Scholar]
Guilcher SJT, Everall AC, Patel T, Packer TL, Hitzig SL, Lofters AK (2019). Medication adherence for persons with spinal cord injury and dysfunction from the perspectives of healthcare providers: A qualitative study. Journal of Spinal Cord Medicine 42: 215–225. [Google Scholar]
Guillou C, Freret M, Fondard E, Derambure C, Avenel G et al. (2016). Soluble alpha-enolase activates monocytes by CD14-dependent TLR4 signalling pathway and exhibits a dual function. Scientific Reports 6: 23796. [Google Scholar]
Hachem LD, Ahuja CS, Fehlings MG (2017). Assessment and management of acute spinal cord injury: From point of injury to rehabilitation. Journal of Spinal Cord Medicine 40: 665–675. [Google Scholar]
Hafner A, Obermajer N, Kos J (2012). gamma-Enolase C-terminal peptide promotes cell survival and neurite outgrowth by activation of the PI3K/Akt and MAPK/ERK signalling pathways. Biochemical Journal 443: 439–450. [Google Scholar]
Haider IT, Lobos SM, Simonian N, Schnitzer TJ, Edwards WB (2018). Bone fragility after spinal cord injury: reductions in stiffness and bone mineral at the distal femur and proximal tibia as a function of time. Osteoporosis International 29: 2703–2715. [Google Scholar]
Haque A, Capone M, Matzelle D, Cox A, Banik NL (2017). Targeting enolase in reducing secondary damage in acute spinal cord injury in rats. Neurochemical Research 42: 2777–2787. [Google Scholar]
Haque A, Polcyn R, Matzelle D, Banik NL (2018). New insights into the role of neuron-specific enolase in neuro-inflammation, neurodegeneration, and neuroprotection. Brain Sciences 8: 33. [Google Scholar]
Haque A, Ray SK, Cox A, Banik NL (2016). Neuron specific enolase: A promising therapeutic target in acute spinal cord injury. Metabolic Brain Disease 31: 487–495. [Google Scholar]
Invernizzi M, de Sire A, Carda S, Venetis K, Reno F, Cisari C, Fusco N (2020a). Bone muscle crosstalk in spinal cord injuries: Pathophysiology and implications for patients’ quality of life. Current Osteoporosis Reports 18: 422–431. [Google Scholar]
Invernizzi M, de Sire A, Reno F, Cisari C, Runza L, Baricich A, Carda S, Fusco N (2020b). Spinal cord injury as a model of bone-muscle interactions: Therapeutic implications from in vitro and in vivo studies. Frontiers in Endocrinology (Lausanne) 11: 204. [Google Scholar]
Jiang SD, Dai LY, Jiang LS (2006). Osteoporosis after spinal cord injury. Osteoporosis International 17: 180–192. [Google Scholar]
Jiang Y, Zhang Y, Chen W, Liu C, Li X et al. (2014). Achyranthes bidentata extract exerts osteoprotective effects on steroid-induced osteonecrosis of the femoral head in rats by regulating RANKL/RANK/OPG signaling. Journal of Translational Medicine 12: 334. [Google Scholar]
Keller A, Peltzer J, Carpentier G, Horvath I, Olah J, Duchesnay A, Orosz F, Ovadi J (2007). Interactions of enolase isoforms with tubulin and microtubules during myogenesis. Biochimical and Biophysical Acta 1770: 919–926. [Google Scholar]
Kim B, Lee KY, Park B (2018). Icariin abrogates osteoclast formation through the regulation of the RANKL-mediated TRAF6/NF-kappaB/ERK signaling pathway in Raw264.7 cells. Phytomedicine 51: 181–190. [Google Scholar]
Kovacs B, Vajda E, Nagy EE (2019). Regulatory effects and interactions of the Wnt and OPG-RANKL-RANK signaling at the bone-cartilage interface in osteoarthritis. International Journal of Molecular Sciences 20: 4653. [Google Scholar]
Kurihara N, Bertolini D, Suda T, Akiyama Y, Roodman GD (1990). IL-6 stimulates osteoclast-like multinucleated cell formation in long term human marrow cultures by inducing IL-1 release. Journal of Immunology 144: 4226–4230. [Google Scholar]
Lee NK, Choi YG, Baik JY, Han SY, Jeong DW et al. (2005). A crucial role for reactive oxygen species in RANKL-induced osteoclast differentiation. Blood 106: 852–859. [Google Scholar]
Li M, Wen H, Yan Z, Ding T, Long L, Qin H, Wang H, Zhang F (2014). Temporal-spatial expression of ENOLASE after acute spinal cord injury in adult rats. Neuroscience Research 79: 76–82. [Google Scholar]
Liu W, Zhang X (2015). Receptor activator of nuclear factor-kappaB ligand (RANKL)/RANK/osteoprotegerin system in bone and other tissues (review). Molecular Medicine Reports 11: 3212–3218. [Google Scholar]
Liu Y, Zeng W, Ma C, Wang Z, Wang C et al. (2020). Maackiain dampens osteoclastogenesis via attenuating RANKL-stimulated NF-kappaB signalling pathway and NFATc1 activity. Journal of Cellular and Molecular Medicine 24: 12308–12317. [Google Scholar]
Lung J, Liu KJ, Chang JY, Leu SJ, Shih NY (2010). MBP-1 is efficiently encoded by an alternative transcript of the ENO1 gene but post-translationally regulated by proteasome-dependent protein turnover. FEBS Journal 277: 4308–4321. [Google Scholar]
Marangos PJ, Zis AP, Clark RL, Goodwin FK (1978). Neuronal, non-neuronal and hybrid forms of enolase in brain: Structural, immunological and functional comparisons. Brain Research 150: 117–133. [Google Scholar]
Orr MB, Gensel JC (2018). Spinal cord injury scarring and inflammation: Therapies targeting glial and inflammatory responses. Neurotherapeutics 15: 541–553. [Google Scholar]
Polcyn R, Capone M, Hossain A, Matzelle D, Banik NL, Haque A (2017). Enolase and acute spinal cord injury. Journal of Clinical and Cellular Immunology 8: 536. [Google Scholar]
Polcyn R, Capone M, Matzelle D, Hossain A, Chandran R, Banik NL, Haque A (2020). Enolase inhibition alters metabolic hormones and inflammatory factors to promote neuroprotection in spinal cord injury. Neurochemistry International 139: 104788. [Google Scholar]
Qian Z, Zhong Z, Ni S, Li D, Zhang F et al. (2020). Cytisine attenuates bone loss of ovariectomy mouse by preventing RANKL-induced osteoclastogenesis. Journal of Cellular and Molecular Medicine 24: 10112–10127. [Google Scholar]
Qin W, Bauman WA, Cardozo C (2010). Bone and muscle loss after spinal cord injury: Organ interactions. Annals of the New York Academy of Sciences 1211: 66–84. [Google Scholar]
Samantaray S, Das A, Matzelle DC, Yu SP, Wei L et al. (2016). Administration of low dose estrogen attenuates persistent inflammation, promotes angiogenesis, and improves locomotor function following chronic spinal cord injury in rats. Journal of Neurochemistry 137: 604–617. [Google Scholar]
Sedoris KC, Thomas SD, Miller DM (2010). Hypoxia induces differential translation of enolase/MBP-1. BMC Cancer 10: 157. [Google Scholar]
Shams R, Drasites KP, Zaman V, Matzelle D, Shields DC et al. (2021). The pathophysiology of osteoporosis after spinal cord injury. International Journal of Molecular Sciences 22: 3057. [Google Scholar]
Shimizu A, Suzuki F, Kato K (1983). Characterization of alpha alpha, beta beta, gamma gamma and alpha gamma human enolase isozymes, and preparation of hybrid enolases (alpha gamma, beta gamma and alpha beta) from homodimeric forms. Biochimical and Biophysical Acta 748: 278–284. [Google Scholar]
Udagawa N, Takahashi N, Jimi E, Matsuzaki K, Tsurukai T et al. (1999). Osteoblasts/stromal cells stimulate osteoclast activation through expression of osteoclast differentiation factor/RANKL but not macrophage colony-stimulating factor. Bone 25: 517–523. [Google Scholar]
Woodcock T, Morganti-Kossmann MC (2013). The role of markers of inflammation in traumatic brain injury. Frontiers in Neurology 4: 18. [Google Scholar]
Wu Q, Zhou X, Huang D, Ji Y, Kang F (2017). IL-6 enhances osteocyte-mediated osteoclastogenesis by promoting JAK2 and RANKL activity in vitro. Cellular Physiology and Biochemistry 41: 1360–1369. [Google Scholar]
Yasuda H, Shima N, Nakagawa N, Yamaguchi K, Kinosaki M et al. (1998). Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proceedings of the National Academy of Sciences USA 95: 3597–3602. [Google Scholar]
Zhao W, Li X, Peng Y, Qin Y, Pan J et al. (2018). Sclerostin antibody reverses the severe sublesional bone loss in rats after chronic spinal cord injury. Calcified Tissue International 103: 443–454. [Google Scholar]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |