DOI:10.32604/biocell.2021.09528
| BIOCELL DOI:10.32604/biocell.2021.09528 | |
| Article |
Targeted editing of intronic-splicing silencer enhancement of SMN2 Exon 7 inclusion by CRISPR/Case 9
1Laboratory Animal Center, Nantong University, Nantong, 226001, China
2Medical College, Nantong University, Nantong, 226001, China
3Institute of Neuroscience, Soochow University, Suzhou, 215123, China
*Address correspondence to: Shunxing Zhu, zsx@ntu.edu.cn
#These authors contributed equally to this work
Received: 27 December 2019; Accepted: 22 April 2020
Abstract: Spinal muscular atrophy (SMA) is an autosomal recessive hereditary neuromuscular disease. Exon 7 and 8 of survival of motor neuron 1 (SMN1) gene or only exon 7 homology deletion leads to the failure to produce a full-length SMN gene. The copy number of SMN2 gene with high homology of SMN1 affects the degree of disease and was the target gene for targeting therapy, in which splicing silencer in intron 7 was the key to suppress the inclusion of exon 7. In this study, we projected to use CRISPR/Case 9 for the targeted editing of intronic-splicing silencer (ISS) sequence to promote the inclusion of SMN2 exon 7 and increase the production of SMN2 full-length (FL) gene expression. It happens that there was a protospacer adjacent motif (PAM) at one end of the ISS sequence according to the design of sgRNA. The recombinant vector of sgRNA HSMN2 CRISPR/Case 9 was constructed and transfected into HEK293 cells. Sequencing results showed that the ISS sequence could be edited accurately and targeting in the predicted direction, in which deleting small fragments, inserting small amounts and mutation. Quantitative analysis of RT-PCR products by restriction enzyme of DdeI digestion showed that the FL of SMN2 increased by 8% (P < 0.05). In the primary cultured chondrocytes of SMA mice, in which sgRNA HSMN2 CRISPR/Case9 recombinant vector transfection could increase the SMN2 FL gene by 23% (P < 0.05) and significantly improve SMN protein levels (P < 0.05). CRISPR/Case 9 is an effective tool for gene editing and therapy of hereditary diseases, but it is rarely reported in the treatment of SMA diseases. This study shows that CRISPR/Case 9 was first used for the precision target of ISS sequence editing, which can effectively promote the production of SMN2 FL gene expressions, in which there was an important clinical reference value.
Keywords: Spinal muscular atrophy; ISS sequence; sgRNA HSMN2; CRISPR/Case 9; Inclusion of Exon 7; SMN2 full length
Spinal muscular atrophy (SMA) is a rare autochromatic recessive neuromuscular disorder characterized by degeneration of alpha motor neurons in the anterior horn of the spinal cord (Burghes and Beattie, 2009; Tisdale and Pellizzoni, 2015). Its clinical manifestation is muscle weakness and muscle atrophy (Crawford and Pardo, 1996). The mortality rate is very high due to respiratory complications. It is the most common genetic cause of infant mortality. One of about 6000 newborns is affected, and about one in 40 newborns is a genetic carrier (Alías et al., 2018; Kolb and Kissel, 2011; Pearn, 1978). The pathogenic gene of SMA is survival of motor neuron 1 (SMN1), located at 5q13, in which exon 7 and 8 or only exon 7 homologous deletion, and then it was failure to expression FL gene and protein (Brzustowicz et al., 1990; García-Cabezas et al., 2004; Greensmith and Vrbová, 1997; Miskovic et al., 2011).
The SMN2 gene, the only one high homology with human SMN1 in this repeat fragment region, might be a multi-copy gene produced by gene replication in the genetic process (Baker et al., 2013). Nucleotide 6 of exon 7 was transformed from C in SMN1 to T (C6T) in SMN2. Although it did not change the amino acid in translation, it seriously affected the inclusion of exon 7 in SMN2 (Benchaouir et al., 2015; Lefebvre et al., 1995; Wu et al., 2017). The limited amount of FL SMN protein expressed by SMN2 is not enough to compensate for the defect of the SMN1 gene, but it is essential for the survival of patients (Prior et al., 2009). It is critical for the therapeutic of SMA that increase the extent of exon 7 inclusion during splicing of SMN2 (Arnold and Fischbeck, 2018).
An important ISS in intron 7 in the human SMN1/2 genes (Singh et al., 2006, 2017; Wan and Dreyfuss, 2017). Using antisense oligonucleotide (ASO) blocks the ISS 10-27 in the intron 7 in SMA mice, which effectively corrects SMN2 splicing and restores SMN expression in motor neurons after intracerebroventricular injection (Hua et al., 2010; Passini et al., 2011). Above all, the ISS 10-27 is a crucial inhibitor for exon 7 inclusion of SMN2. Antisense oligonucleotides (ASOs) can efficiently silence proteins with gain-of-function mutations. However, naked ASOs have a short circulation half-life and are unable to cross the blood–brain barrier (BBB), warranting the use of a drug carrier for effective delivery. Recently years, CRISPR/Case 9, the gene-editing tools, were gradually used to correct gene mutation and treat disease (Burnight et al., 2017; Demirci et al., 2019; Long et al., 2016; Miao et al., 2019).
Here, this study aimed to edit the SMN2 ISS sequence by CRISPR/Case 9, and investigate gene editing location about the ISS sequence, which enhance the Exon 7 inclusion of SMN2 and improve the SMN protein levels or not.
Target sgRNA was designed by online software (https://zlab.bio/guide-design-resources) to construct the CRISPR/Cas9 targeting vector. Input SMN2 gene sequence in the corresponding area on the website, select species, submit, search for sgRNA in the target area of ISS, find sgRNA sequence and input the specificity of gene bank blast detection sequence to ensure the uniqueness of sgRNA, so as to reduce the Miss effect of sgRNA as much as possible. HSMN2 sgRNA oligo F: 5’–CACCG AAGAT TCACT TTCAT AATGC–3’, R: 5’–AAACG CATTA TGAAA GTGAA TCTTC–3’. The vector used in this experiment was lentivirul CRISPR/Cas9 (pXPR_001). Referring to the Zhang lab recombinant vector construction method operation (Shalem et al., 2014). Finally, the sgRNA HSMN2 CRISPR/Cas9 recombinant vector was successfully constructed by sequencing and analysis in positive clones (Fig. 1C).
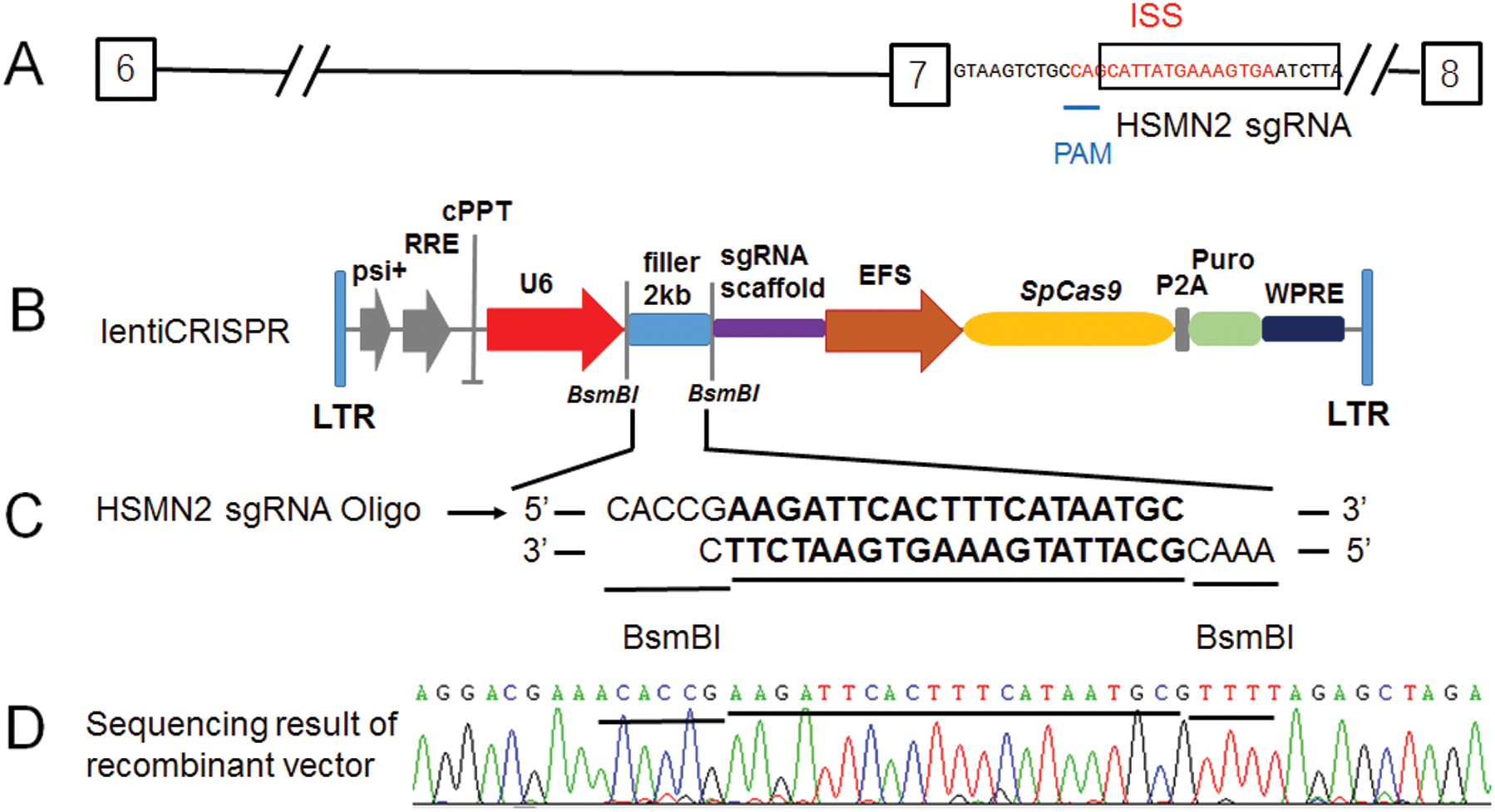
Figure 1: The lenti-CRISPR/Case 9 sgRNA recombinant vector constructed target for the ISS sequence in intron 7 of SMN2.
(A) Diagram of a partial segment of SMN2 allele (from exon 6 to 8) and in which the ISS sequence in red, the sgRNA HSMN2 sequence in the black box, PAM corresponding sequence was underlined. (B) Diagram of the lentiCRISPR, in which the 2 kb filler was enzyme digested by BsmBI and recombinant with sgRNA HSMN2 Oligo. (C) After transfection, the positive clones of recombinant lentiCRISPR sgRNA HSMN2 were screened with ampicillin LB medium. (D) Identification by cloning sequencing.
Cell culture, transfection and sequencing
The human embryonic kidney 293 (HEK293) cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% (v/v) fetal bovine serum (FBS), 100 U/mL penicillin and 100 μg/mL streptomycin (HyClone), which were cultured at 37°C in a humidified 5% CO2 atmosphere.
The HSMN2 sgRNA recombinant vector was transfected in HEK293 cell line with transfection reagent of petroleum equipment institute (PEI) (Sigma) in a 6-cm dish. After 48 h of transfection, puromycin was added into the medium, and the cells were cultured for 7 days formation of stable cell lines and continuing to expand the culture reserve. The cells were harvested by Dzup Genomic DNA Isolation Reagent (Sangon). After adding anhydrous ethanol, the white flocculent was picked up by tips, which was washed with 75% ethanol, and dissolved in sterilized deionized water as template.
The primers design was based on the SMN2 genome as a template, and the left and right wings of exon 7 contained some introns of the target sequence. Primer SMN2 G7 F: 5’–CAAGG GCACA TTCAC AGCT–3’, R: 5’–TTAGA ACCAG AGGCT TGACG–3’, and the length of the PCR product was 1277 bp. In vitro amplification system by PCR, 2 × Taq Plus Master Mix (Vazyma), 12.5 μL; primers F/R (10 μM), each 1 μL; template DNA, 1.25 ng; sterilized deionized water, to a total volume of 25 μL. PCR reaction procedure was 35 cycles 94°C for 30 s, 55°C for 30 s, and 72°C for 40 s, the PCR product was detected by 1% agarose gel electrophoresis, purified, and sent to the company for sequencing.
Cloning and Sequencing of PCR Products. Connection of purified products of PCR in the above steps with T vector (Sangon), transforming into TOP10 Active Cells (Sangon), which were cultured in LB Luria-Bertani medium with IPTG, X-Gal and AMP, and after culture 16 h in 37°C using “white-blue plaque selection” for sscreening 10 positive clones sequencing.
In vitro assessment of gene expression
The stable cell lines HSMN2 sgRNA vector transfection were cultured 48 h in 6 cells plate with HEK293 as control. The cells were harvested, and total RNA was extracted by TRIzol reagent following the protocol of the manufacturer (BBI). Fist-strand cDNA was synthesized with an M-MuLV reverse transcriptase kit (Vazyma) and 1 μg RNA template per 20 μL reaction.
Splicing products were amplified semi-quantitatively with forward primer E6-F: 5’–ATAAT TCCCC CACCA CCTCC C–3’ and E8–467R: 5’–Cy5–TTGCC ACATA CGCCT CACAT AC–3’ by 28 cycles (94°C for 30 s, 55°C for 30 s, and 72°C for 25 s). PCR products restriction enzyme digestion by DdeI (Promega) and which were separated on 2% agarose gels, followed signals were quantitated by image software, and exon 7 inclusion was expressed as a percentage of the total amount of spliced transcripts.
Primary cell culture of SMA mice and the effect of CRISPR editing SMN2 gene on the FL expressions
The mice were cared for in a barrier system at 23 ± 2°C with a humidity of 55 ± 5% following a 12 h light/12 h dark cycle with free access to radiation-sterilized food and sterile water and Use of Animals Committee of Nantong University (China). The Taiwanese mouse model (Smn-/-, SMN22TG/0) (Hsieh-Li et al., 2000) in FVB background was generated by crosses as described (Sheng et al., 2018), which was a gift from Krainer lab at Cold Spring Harbor Laboratory.
Neonatal mice were tail-clipped and genotypes were identified by genotype rapid identification kit (Vazyma). The smn-/-, SMN22TG/0 mice were anesthetized on ice and disinfected with 75% ethanol for 10 min. The cartilage tissue of the limbs was cut and digested with collagenase II at 37°C for 3 h and then mixed every half hour. The digestion was terminated by adding three times the volume of collagenase II (Sigma) into the complete medium. Using a 40-μm diameter sterile filter, 1000 rpm, 5 min centrifugation, discarded supernatant added PBS suspension, centrifugation again discarded supernatant. After weight drop with 1 mL complete medium, the cells were transplanted in a 6-cm culture dish. After homogeneous and stable morphology, chondrocytes were implanted into 6-well plates with density 3 × 105 cells/L. Three well of which transfected empty CRISPR/Case9 vector as control, and other three well transfected HSMN2 sgRNA CRISPR/Case9 vector. Cultured for 48 h, harvested cells one of the 6-plate, the total RNA isolation, and first-strand cDNA methods were similar to the above described. The SMN2 FL expressions detected by real-time PCR with primers SMN1/2-E5-F: 5’–ACCAC CCCAC TTACT ATCAT GC–3’ and SMN1/2-E7-R: 5’–GAATG TGAGC ACCTT CCTTC TT–3’ using SYBGreen Mix (Roche). Harvested cells another 6-plate extracted total protein by lysis buffer (Sangon) for the western blotting samples.
Protein samples separated by 10% SDS-PAGE gel and electroblotted onto PVDF membranes (Roche). The blots were probed with primary mAbs or polyclonal antibodies (pAbs), after that by secondary rabbit anti-mouse or goat anti-rabbit antibody (Sangon). Anti-β actin mAb (1:1000) was generated at ZSGB-BIO, anti-SMN pAbs (1:200) was purchased from Abcam. The membrane incubated with Tanon™ High-sig ECL Western Blotting Substrate detection reagents (Tanon Science & Technology) and then exposed to X-ray film in Tanon-5200Multi Gel Imaging System (Tanon Science & Technology). The scanned images were imported into ImageJ software. The ratio of each band divided by actin was calculated for the final results.
Data were graphed as mean ± SD. Statistical significance was analyzed by student’s t-test and one-way ANOVA with the software SPSS 13.0. P < 0.05 was considered as significant.
The CRISPR/Case 9 sgRNA recombinant vector constructed target for the ISS sequence in intron 7 of SMN2
For targeting the ISS sequence (intronic-splicing silencers) of intron 7 of the SMN2 gene, the sgRNA sequence was designed (Fig. 1A). It happened that there was a PAM sequence at one end of the ISS sequence (Fig. 1A). Moreover, this sgRNA sequence is specific in the genome and appropriate for the requirements of sgRNA design. More importantly, PAM is only 9 bases close to exon 7 at the 5’ end of the ISS sequence. It happens the reverse sequence CCA of TGG (Fig. 1A). According to the editing characteristics of Case 9, which guides Case 9 to delete and edit the 3’ end of CCA. It exactly targets the ISS sequence (Fig. 1A). We constructed the SMN2 sgRNA using the lentiCRISPR V2 vector (Figs. 1B and 1C). The recombinant vector lentiCRISPR V2 sgRNA SMN2 was successfully constructed and analyzed by sequencing (Fig. 1D).
The CRISPR/Case 9 sgRNA precisely target edit ISS intron 7 sequence of SMN2 in HEK293 cell line.
To detect the target editing of the ISS sequence, the primers were designed on both wings of intron 7 in the genome sequence. The product covers part of intron 6, whole exon 7, whole intron 7, and part of exon 8. Using normal HEK293 cells as a control, the results of PCR showed that the size of the products of the sgRNA recombinant vector edited was similar to that of normal HEK293 cells (Fig. 2A), suggesting that there was no deletion or insertion of large fragments.
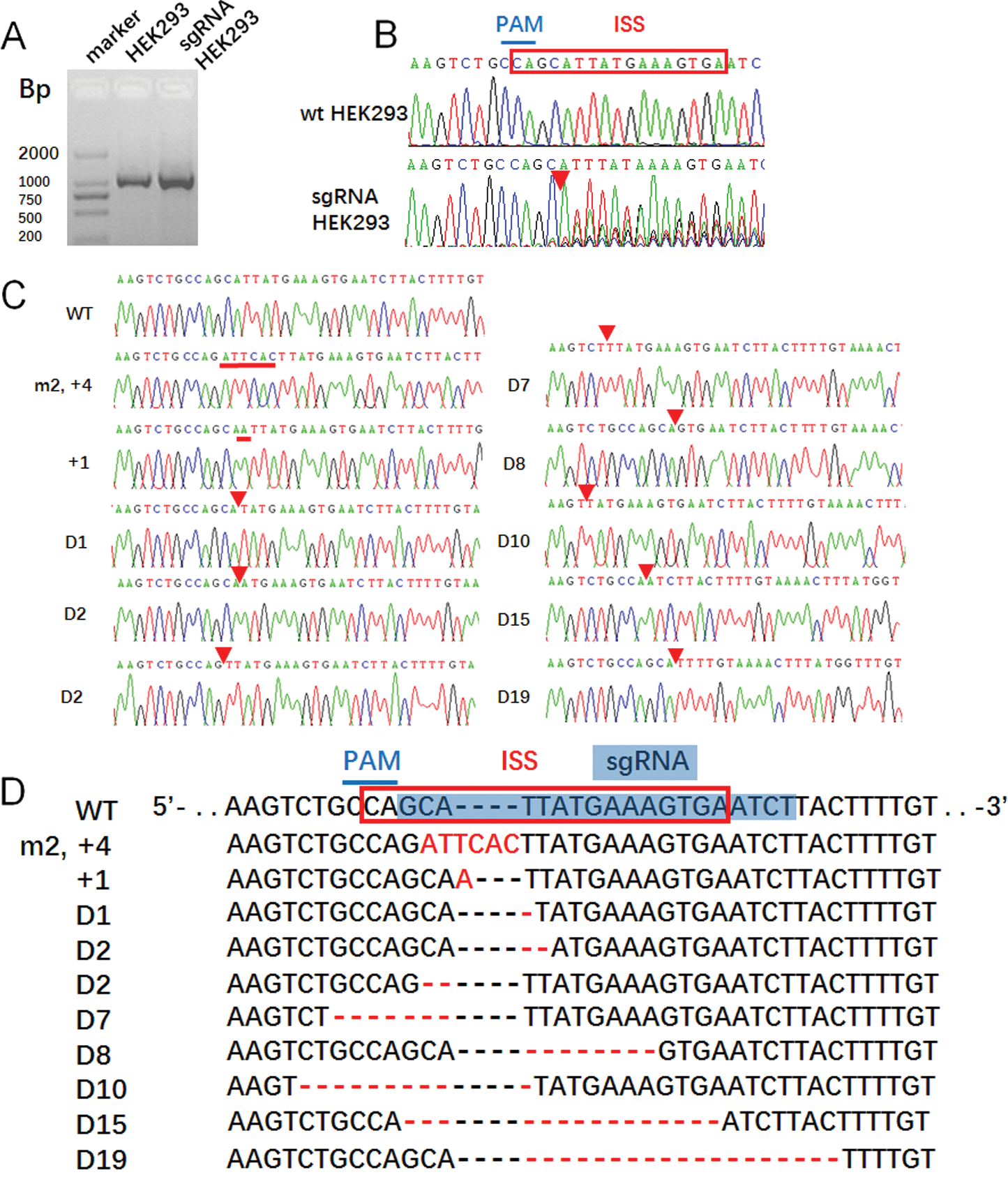
Figure 2: The CRISPR/Case 9 sgRNA precisely target editing the ISS sequence of SMN2 in HEK293 cell line.
(A) PCR product of HEK293 and transfection HEK293 cells were sequenced, and (B) there were overlapping peaks behind the position PAM. (C) Mutation PCR products linked to cloning vector pGEM-T easy to obtain recombinant plasmids pGEM-TEasy-G7, and then single cloning sequencing, and sequence alignment to HEK293. (D) The mutation positions were in the ISS sequence, which agreed with the theory.
Direct sequencing of PCR products showed that from the PAM downstream side, the NGG complementary sequence CCA has set peaks and continued downstream (Fig. 2B), suggesting that there was gene editing in the PCR products from CAA, but the specific editing situation required monoclonal sequencing by connecting vectors. After connecting the PCR products with T vectors, 10 monoclones were selected for sequencing (Fig. 2C). The result showed that one clone had two base mutations and four base insertions downstream of CCA. The second clone had one base insertion, and the other eight clones were as deleted edited, in which the longest deletion being 19 bases and the least deletion for one base, except two clones deleted CCA sequence and one and three bases to the 5’ end, the other six clones were deleted from the 3’-end from CCA (Fig. 2D).
The editing of the ISS sequence enhances Exon 7 inclusion of SMN2
The six pore plates of a cell line stably expressing sgRNA CRISPR/Case 9 screened by puromycin were culture for 48 h, with normal HEK293 cell line as control. After 48 hours of culture, the cells were harvest for extracted total RNA, RT-PCR, the PCR products digestion by restriction enzyme of DdeI, and 2% agarose gel electrophoresis (Fig. 3A). By calculating the gray value quantitatively, the proportion of SMN2 gene FL was analyzed according to the calculation formula. The results showed that SMN2 FL increased by 8.03% on average in HEK293 cells after editing the ISS sequence (Fig. 3B).
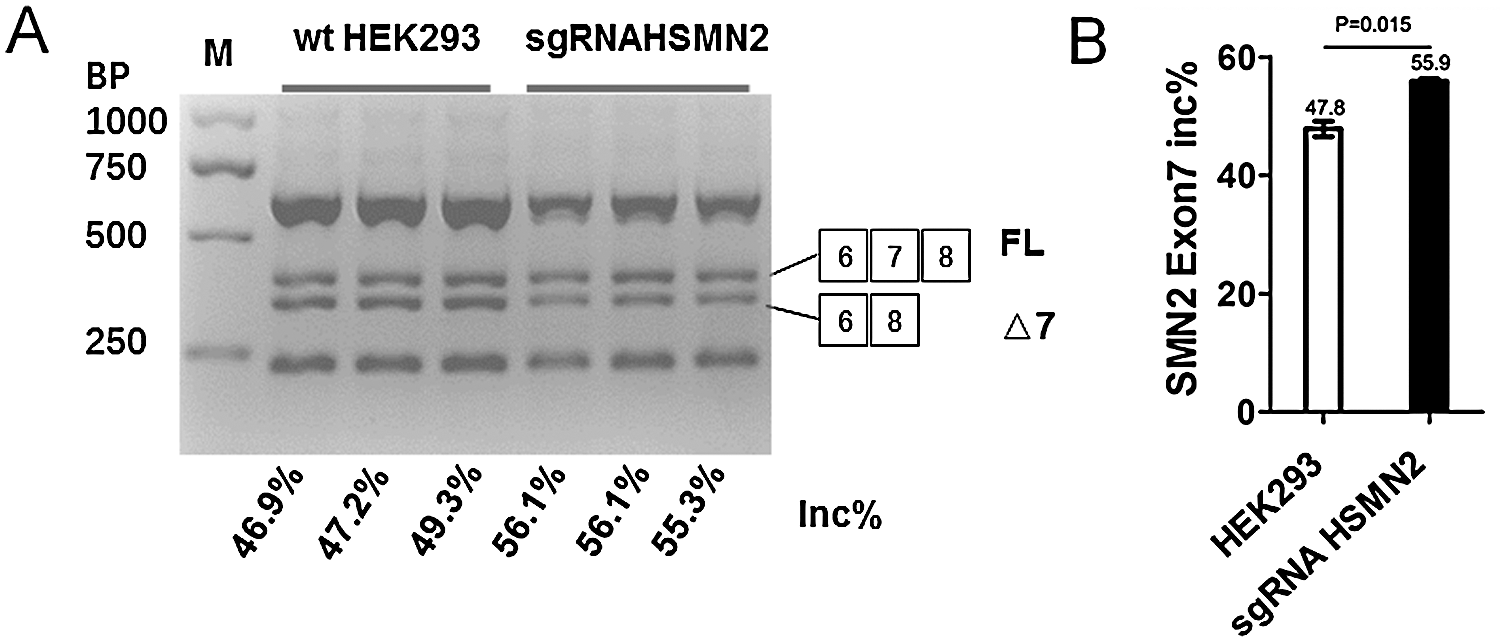
Figure 3: The editing of the ISS sequence enhances Exon 7 inclusion of SMN2.
(A) HEK293 and puromycin selection sgRNA HEK293 were cultured in six-well plates, after 48 h, harvested cell and extraction total RNA, using SMN1/2 primer radioactive RT-PCR analysis of SMN2 exon 7 inclusion, FL, full-length transcript; Δ7, exon 7-skipped transcript; Inc%, 100 × FL/(FL + Δ7). (B) Statistical analysis.
The target editing of ISS effectively corrects SMN2 splicing and restores SMN expression in vitro cultured primary cells from SMA type I neonatal mice
Because the sgRNA recombinant vector was ultimately designed to promote the inclusion of FL in the SMN2 gene, while only the SMN2 gene was transfected in type I mice of Taiwan model mouse, the primary culture of chondrocyte of type I mice (Fig. 4A) was established. Six-well plates were used to transfect the sgRNA recombinant vector with an empty vector as control, and cells were harvested 48 h later. Total RNA extracted and the expression of the SMN2 FL gene was detected by real-time PCR. The results showed that the expression of the SMN2 FL gene increased by 23%. The difference was statistically significant (Fig. 4B). The total protein extracted was used for the detection of the expression of SMN protein by Western blot. The results showed that the level of sgRNA targeted editing protein was significantly higher than that of the control group (Figs. 4C and 4D).
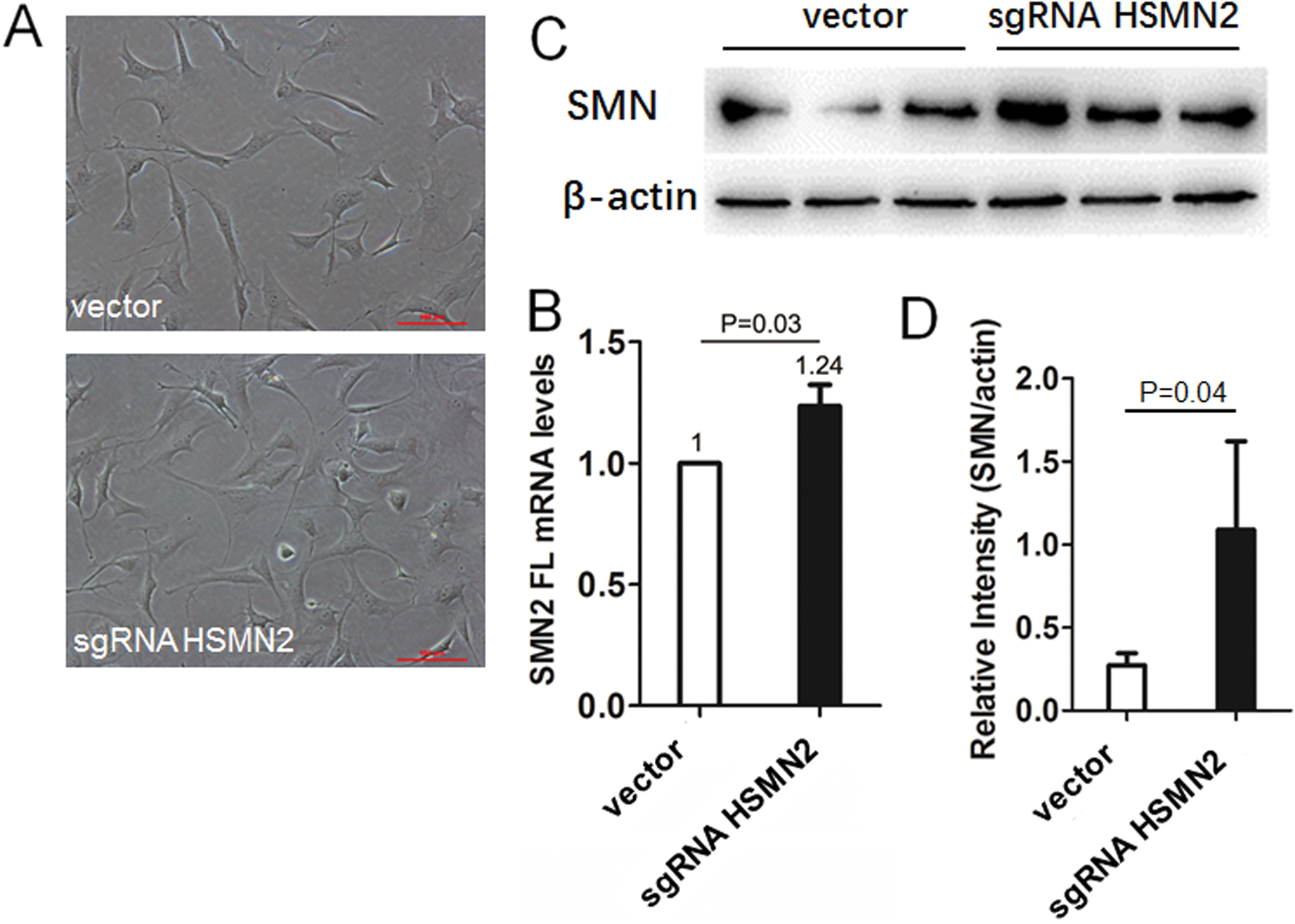
Figure 4: The targeted editing of ISS effectively corrects SMN2 splicing and restores SMN expression in vitro cultured primary chondrocyte from SMA type I neonatal mice.
(A) The chondrocyte culture system and transfection with sgRNAHSMN2 CRISPR case9 in six-well cluster dishes, harvested cell, and extraction of total RNA, using SMN1/2 FL primer radioactive. (B) RT-qPCR analysis of SMN2 FL mRNA levels. (C) and (D) Extracted protein, using Western blot analysis of SMN protein levels.
Previous studies have shown that the splicing repressor can inhibit the normal inclusion of exons by preventing a series of splicing factors from combining with ISS sequences (Fig. 5A) (Cartegni et al., 2002). The inclusion of exon 7 can be promoted by target blocking the ISS sequence with ASO (Fig. 5B) (Hua et al., 2011). Here, sgRNA was used to delete the ISS sequence, so as to promote the inclusion of exon 7 (Fig. 5C).
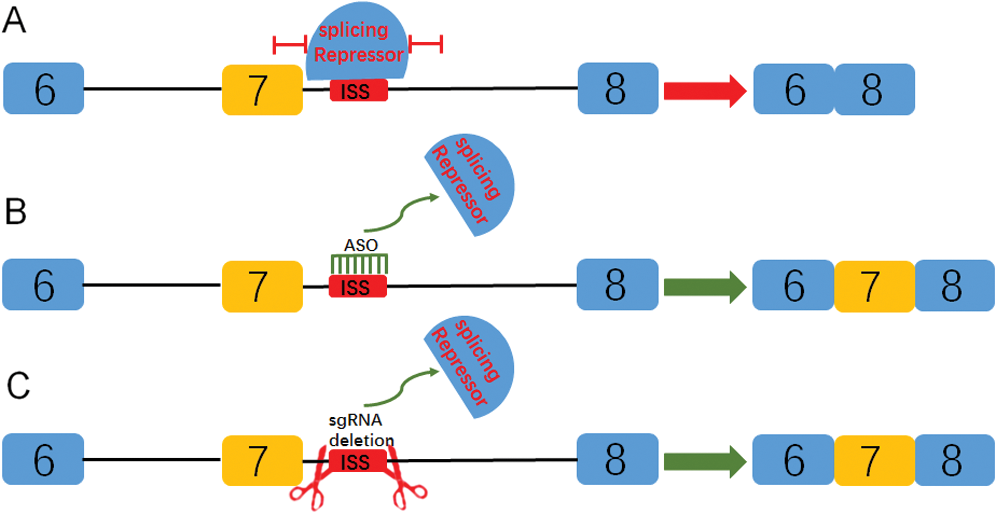
Figure 5: Diagram of targeted blocking or deletion of the ISS sequence promotes the inclusion of exon 7 of SMN2 gene.
(A) The splicing repressor can combine ISS sequence and inhibit the binding of splicing factors through a series of complex mechanisms, so as to promote the inclusion of exon 7. (B) By targeting the blocking ISS sequence with ASO, the splicing repressor could not combine the sequence so that the splicing can proceed normally. (C) In this study, sgRNA was used to target the deletion of the ISS sequence, which also prevented the binding of splicing repressor and promoted the inclusion of exon 7.
As SMN1 mutation causes SMA disease, targeting the SMN2 gene and promoting the inclusion of SMN2 gene FL were critical for the treatment of SMA disease (Alías et al., 2018).
Pre-mRNA splicing is a dynamic process of eliminating introns and connecting exons (Singh et al., 2018; Wang et al., 2002). In which it involves splicing trans-acting factors and cis-acting elements that important effects on splicing in exons or their flanking introns, such as exonic-splicing enhancers (ESEs), exonic-splicing silencers (ESSs), intronic-splicing enhancers (ISEs), and intronic-splicing silencers (ISSs) (Cartegni et al., 2002; Skordis et al., 2003). There was an ISS sequence in SMN2 intron 7, using targeted blocking by ASO can promote the increase of FL gene and protein of SMN2 gene, more importantly, that could rescue the type I mice from 10 days to about 500 days (Hua et al., 2008, 2011). For ASO with a certain half-life, if the ISS sequence was edited in genomic levels, which could be sustained promote the expression increase of SMN2 FL.
In recent years, CRISPR/Cas9 gene-editing technology and gene therapy have become a research hotspot. There was the genetic therapy of Duchenne muscular dystrophy (DMD), in which the animal model of DMD was used for in vivo experiments therapy. By constructing target edited sgRNA, CRISPR/Cas9 was guided to precisely target delete downstream PAM sequences in vivo. Exon 23 with a nonsense mutation was deleted to form a DMD gene, and the muscle pathology was improved, and the muscle strength of mice was increased (Nelson et al., 2016; Tabebordbar et al., 2016). Another study showed to convert the SMN2 gene 840 T to C in iPSC induced from SMA patient by two crRNAs and one sgRNA that recognized the SMN2 Exon 7 locus (Zhou et al., 2018). Interestingly, there happened to be the reverse sequence of PAM near the end of exon 7 in the ISS sequence of SMN2 intron 7, in which the sgRNA could guide CRISPR/Cas9 to edit to the 3’ end of ISS from 5’ end. If the editing effect was with expected, the ISS sequence will be deleted along the predetermined direction, but not the reverse direction near exon 7, which direction deletion results in loss of conserved sequence of exon 7. The editing effect with expected that was verification (Fig. 2), Two of the 10 clones are inserted mutation and the others are deleted mutation, which can edit the ISS sequence effectively. According to the analysis of the FL inclusion ratio (Fig. 3), the editing effect effectively improved the inclusion and increase of the FL gene by 8%.
SMN1 gene is highly homologous to SMN2 in human cells, which dilutes sgRNA to target the editing of ISS sequences in SMN2. In order to further verification of the proportion of sgRNA recombinant vector in SMA mice to promote FL after SMN2 editing, the SMA mice chondrocyte culture system was established, which was used for target editing by the sgRNA recombinant vector. Because only the SMN2 gene was knocked in the mice without smn1, so only real-time qPCR was needed to detect FL expression for SMN2, and it does not need to be digestion by restriction enzyme of DdeI.
In conclusion, the construction of sgRNA HSMN2 CRISPR/Case 9 recombinant plasmid could precisely target editing the ISS sequence of intron 7 of SMN2, and significantly promote the mRNA and protein levels of SMN2 FL in HEK293 or primary cell of SMA mice. Moreover, it is worth constructing a virus to act on SMA mice in the later stage, to study the life-saving effect of SMA mice, and to study the expression of SMN2 gene FL in SMA mice.
Acknowledgement: We are grateful to Professor Hua Yimin for his constructive advice in the edit target of recombination sgRNA CRISPR/Case9 plasmid.
Funding Statement: This work was funded by grants from Nantong Science and Technology Program, grant number (JC2018090), URL: http://kjj.nantong.gov.cn/, the Practice Innovation Training Program Projects for the Jiangsu College Students, grant number (201810304028z), URL: https://jsgjc.jse.edu.cn/cxcy/, and the Scientific Innovation Research of College Graduates in Jiangsu Province, grant number (KYCX18-2415), URL: http://jyt.jiangsu.gov.cn/. The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
Alías L, Bernal S, Calucho M, Martínez E, March F, Gallano P, Fuentes-Prior P, Abuli A, Serra-Juhe C, Tizzano EF (2018). Utility of two SMN1 variants to improve spinal muscular atrophy carrier diagnosis and genetic counselling. European Journal of Human Genetics 26: 1554–1557. DOI 10.1038/s41431-018-0193-4. [Google Scholar] [CrossRef]
Arnold ES, Fischbeck KH (2018). Spinal muscular atrophy. Handbook of Clinical Neurology 148: 591–601. [Google Scholar]
Baker CR, Hanson-Smith V, Johnson AD (2013). Following gene duplication, paralog interference constrains transcriptional circuit evolution. Science 342: 104–108. DOI 10.1126/science.1240810. [Google Scholar] [CrossRef]
Benchaouir R, Robin V, Goyenvalle A (2015). Gene and splicing therapies for neuromuscular diseases. Frontiers in Bioscience 20: 1190–1233. DOI 10.2741/4367. [Google Scholar] [CrossRef]
Brzustowicz LM, Lehner T, Castilla LH, Penchaszadeh GK, Wilhelmsen KC, Daniels R, Davies KE, Leppert M, Ziter F, Wood D, Dubowitz V, Zerres K, Hausmanowa-Petrusewicz I, Ott J, Munsat TL, Gilliam TC (1990). Genetic mapping of chronic childhood-onset spinal muscular atrophy to chromosome 5q11.2-13.3. Nature 344: 540–541. DOI 10.1038/344540a0. [Google Scholar] [CrossRef]
Burghes AH, Beattie CE (2009). Spinal muscular atrophy: why do low levels of survival motor neuron protein make motor neurons sick? Nature Reviews Neuroscience 10: 597–609. DOI 10.1038/nrn2670. [Google Scholar] [CrossRef]
Burnight ER, Gupta M, Wiley LA, Anfinson KR, Tran A, Triboulet R, Hoffmann JM, Klaahsen DL, Andorf JL, Jiao C, Sohn EH, Adur MK, Ross JW, Mullins RF, Daley GQ, Schlaeger TM, Stone EM, Tucker BA (2017). Using CRISPR-Cas9 to generate gene-corrected autologous iPSCs for the treatment of inherited retinal degeneration. Molecular Therapy 25: 1999–2013. DOI 10.1016/j.ymthe.2017.05.015. [Google Scholar] [CrossRef]
Cartegni L, Chew SL, Krainer AR (2002). Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nature Reviews Genetics 3: 285–298. DOI 10.1038/nrg775. [Google Scholar] [CrossRef]
Crawford TO, Pardo CA (1996). The neurobiology of childhood spinal muscular atrophy. Neurobiology of Disease 3: 97–110. DOI 10.1006/nbdi.1996.0010. [Google Scholar] [CrossRef]
Demirci S, Leonard A, Haro-Mora JJ, Uchida N, Tisdale JF (2019). CRISPR/Cas9 for sickle cell disease: applications, future possibilities, and challenges. Advances in Experimental Medicine and Biology 1144: 37–52. DOI 10.1007/5584_2018_331. [Google Scholar] [CrossRef]
García-Cabezas MA, García-Alix A, Martín Y, Gutiérrez M, Hernández C, Rodríguez JI, Morales C (2004). Neonatal spinal muscular atrophy with multiple contractures, bone fractures, respiratory insufficiency and 5q13 deletion. Acta Neuropathologica 107: 475–478. DOI 10.1007/s00401-004-0825-3. [Google Scholar] [CrossRef]
Greensmith L, Vrbová G (1997). Disturbances of neuromuscular interaction may contribute to muscle weakness in spinal muscular atrophy. Neuromuscular Disorders 7: 369–372. DOI 10.1016/S0960-8966(97)00047-3. [Google Scholar] [CrossRef]
Hsieh-Li HM, Chang JG, Jong YJ, Wu MH, Wang NM, Tsai CH, Li H (2000). A mouse model for spinal muscular atrophy. Nature Genetics 24: 66–70. DOI 10.1038/71709. [Google Scholar] [CrossRef]
Hua Y, Sahashi K, Hung G, Rigo F, Passini MA, Bennett CF, Krainer AR (2010). Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes & Development 24: 1634–1644. DOI 10.1101/gad.1941310. [Google Scholar] [CrossRef]
Hua Y, Sahashi K, Rigo F, Hung G, Horev G, Bennett CF, Krainer AR (2011). Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature 478: 123–126. DOI 10.1038/nature10485. [Google Scholar] [CrossRef]
Hua Y, Vickers TA, Okunola HL, Bennett CF, Krainer AR (2008). Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. American Journal of Human Genetics 82: 834–848. DOI 10.1016/j.ajhg.2008.01.014. [Google Scholar] [CrossRef]
Kolb SJ, Kissel JT (2011). Spinal muscular atrophy: a timely review. Archives of Neurology 68: 979–984. DOI 10.1001/archneurol.2011.74. [Google Scholar] [CrossRef]
Lefebvre S, Burglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M, Paslier D, Frézal J, Cohen D, Weissenbach J, Munnich A, Melki J (1995). Identification and characterization of a spinal muscular atrophy-determining gene. Cell 80: 155–165. DOI 10.1016/0092-8674(95)90460-3. [Google Scholar] [CrossRef]
Long C, Amoasii L, Mireault AA, Mcanally JR, Li H, Sanchez-Ortiz E, Bhattacharyya S, Shelton JM, Bassel-Duby R, Olson EN (2016). Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 351: 400–403. DOI 10.1126/science.aad5725. [Google Scholar] [CrossRef]
Miao K, Zhang X, Su SM, Zeng J, Huang Z, Chan UI, Xu X, Deng CX (2019). Optimizing CRISPR/Cas9 technology for precise correction of the Fgfr3-G374R mutation in achondroplasia in mice. Journal of Biological Chemistry 294: 1142–1151. DOI 10.1074/jbc.RA118.006496. [Google Scholar] [CrossRef]
Miskovic M, Lalic T, Radivojevic D, Cirkovic S, Vlahovic G, Zamurovic D, Guc-Scekic M (2011). Lower incidence of deletions in the survival of motor neuron gene and the neuronal apoptosis inhibitory protein gene in children with spinal muscular atrophy from Serbia. Tohoku Journal of Experimental Medicine 225: 153–159. DOI 10.1620/tjem.225.153. [Google Scholar] [CrossRef]
Nelson CE, Hakim CH, Ousterout DG, Thakore PI, Moreb EA, Castellanos Rivera RM, Madhavan S, Pan X, Ran FA, Yan WX, Asokan A, Zhang F, Duan D, Gersbach CA (2016). In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science 351: 403–407. DOI 10.1126/science.aad5143. [Google Scholar] [CrossRef]
Passini MA, Bu J, Richards AM, Kinnecom C, Sardi SP, Stanek LM, Hua Y, Rigo F, Matson J, Hung G, Kaye EM, Shihabuddin LS, Krainer AR, Bennett CF, Cheng SH (2011). Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Science Translational Medicine 3: 72ra18. DOI 10.1126/scitranslmed.3001777. [Google Scholar] [CrossRef]
Pearn J (1978). Incidence, prevalence, and gene frequency studies of chronic childhood spinal muscular atrophy. Journal of Medical Genetics 15: 409–413. DOI 10.1136/jmg.15.6.409. [Google Scholar] [CrossRef]
Prior TW, Krainer AR, Hua Y, Swoboda KJ, Snyder PC, Bridgeman SJ, Burghes AH, Kissel JT (2009). A positive modifier of spinal muscular atrophy in the SMN2 gene. American Journal of Human Genetics 85: 408–413. DOI 10.1016/j.ajhg.2009.08.002. [Google Scholar] [CrossRef]
Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG, Zhang F (2014). Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343: 84–87. DOI 10.1126/science.1247005. [Google Scholar] [CrossRef]
Sheng L, Wan B, Feng P, Sun J, Rigo F, Bennett CF, Akerman M, Krainer AR, Hua Y (2018). Downregulation of Survivin contributes to cell-cycle arrest during postnatal cardiac development in a severe spinal muscular atrophy mouse model. Human Molecular Genetics 27: 486–498. DOI 10.1093/hmg/ddx418. [Google Scholar] [CrossRef]
Singh NK, Singh NN, Androphy EJ, Singh RN (2006). Splicing of a critical exon of human survival motor neuron is regulated by a unique silencer element located in the last intron. Molecular and Cellular Biology 26: 1333–1346. DOI 10.1128/MCB.26.4.1333-1346.2006. [Google Scholar] [CrossRef]
Singh NN, Del Rio-Malewski JB, Luo D, Ottesen EW, Howell MD, Singh RN (2017). Activation of a cryptic 5’ splice site reverses the impact of pathogenic splice site mutations in the spinal muscular atrophy gene. Nucleic Acids Research 45: 12214–12240. DOI 10.1093/nar/gkx824. [Google Scholar] [CrossRef]
Singh NN, Luo D, Singh RN (2018). Pre-mRNA splicing modulation by antisense oligonucleotides. In: Yokota T, Maruyama R, eds. Exon Skipping and Inclusion Therapies. Methods in Molecular Biology. 1828: 415–437. New York, NY: Humana Press. [Google Scholar]
Skordis LA, Dunckley MG, Yue B, Eperon IC, Muntoni F (2003). Bifunctional antisense oligonucleotides provide a trans-acting splicing enhancer that stimulates SMN2 gene expression in patient fibroblasts. Proceedings of the National Academy of Sciences of the United States of America 100: 4114–4119. DOI 10.1073/pnas.0633863100. [Google Scholar] [CrossRef]
Tabebordbar M, Zhu K, Cheng JKW, Chew WL, Widrick JJ, Yan WX, Maesner C, Wu EY, Xiao R, Ran FA, Cong L, Zhang F, Vandenberghe LH, Church GM, Wagers AJ (2016). In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science 351: 407–411. DOI 10.1126/science.aad5177. [Google Scholar] [CrossRef]
Tisdale S, Pellizzoni L (2015). Disease mechanisms and therapeutic approaches in spinal muscular atrophy. Journal of Neuroscience 35: 8691–8700. DOI 10.1523/JNEUROSCI.0417-15.2015. [Google Scholar] [CrossRef]
Wan L, Dreyfuss G (2017). Splicing-correcting therapy for SMA. Cell 170: 5. DOI 10.1016/j.cell.2017.06.028. [Google Scholar] [CrossRef]
Wang P, Lou PJ, Leu S, Ouyang P (2002). Modulation of alternative pre-mRNA splicing in vivo by pinin. Biochemical and Biophysical Research Communications 294: 448–455. DOI 10.1016/S0006-291X(02)00495-3. [Google Scholar] [CrossRef]
Wu X, Wang SH, Sun J, Krainer AR, Hua Y, Prior TW (2017). A-44G transition in SMN2 intron 6 protects patients with spinal muscular atrophy. Human Molecular Genetics 26: 2768–2780. DOI 10.1093/hmg/ddx166. [Google Scholar] [CrossRef]
Zhou M, Hu Z, Qiu L, Zhou T, Feng M, Hu Q, Zeng B, Li Z, Sun Q, Wu Y, Liu X, Wu L, Liang D (2018). Seamless genetic conversion of SMN2 to SMN1 via CRISPR/Cpf1 and single-stranded oligodeoxynucleotides in spinal muscular atrophy patient-specific induced pluripotent stem cells. Human Gene Therapy 29: 1252–1263. DOI 10.1089/hum.2017.255. [Google Scholar] [CrossRef]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |