DOI:10.32604/biocell.2021.015250
| BIOCELL DOI:10.32604/biocell.2021.015250 | |
| Article |
Complete genome sequence of two strawberry vein banding virus isolates from China
1School of Plant Protection, Anhui Agricultural University, Hefei, 230036, China
2Anhui Province Key Laboratory of Integrated Pest Management on Crops, Hefei, 230036, China
3Key Laboratory of Biology and Sustainable Management of Plant Diseases and Pests of Anhui Higher Education Institutes, Anhui Agricultural University, Hefei, 230036, China
*Address correspondence to: Tong Jiang, jiangtong4650@sina.com
#These authors contributed equally to this paper
Received: 04 December 2020; Accepted: 08 February 2021
Abstract: It was rarely reported about strawberry vein banding virus (SVBV) genome sequence in China and most countries worldwide. In this work, we determined the complete genome sequences of two SVBV isolates in China, designated SVBV-AH and SVBV-BJ, that were obtained from naturally infected strawberry samples from Anhui province and Beijing city of China, respectively. The complete genomes of SVBV-AH and SVBV-BJ were 7,862 nucleotides (nts) and 7,863 nts long, respectively, and both constituted with seven genes typical of the caulimoviruses. Alignment of complete nucleotide sequences showed that SVBV-AH and SVBV-BJ shared a significant nucleotide sequence identity of 97.7% of each other and had 85.7% and 86.0% sequence identity related to SVBV from the United States (SVBV-US), respectively. Phylogenetic trees, based on the alignment of complete nucleotide sequences and amino acid sequences of Coat Protein (CP), both showed that SVBV-AH and SVBV-BJ clustered into one branch with all the other SVBV isolates, and other species of caulimoviruses clustered into another tree branch. It illustrated that all the SVBV isolates had an extremely high relationship but had a distant relationship with other species of caulimoviruses. We further confirmed that SVBV-AH infectious clone could cause similar symptoms to SVBV-infected in strawberry under natural conditions. Taken together, our study provided valuable information to elucidate the origin and dissemination of SVBV Chinese isolates, meanwhile providing the necessary vector for studying the gene functions of strawberry.
Keywords: Strawberry vein banding virus; Chinese isolate; Complete genome; Coat Protein (CP); Phylogenetic analysis; Infectious clone
Strawberry vein banding virus (SVBV) was originally described as a distinct virus infecting strawberry in 1955 (Frazier, 1955). Its presence has been reported in cultivated strawberries in many countries worldwide: America, Australia, Brazil, Asia, and European countries (Ratti et al., 2009; Honetšlegrová et al., 1995; Petrzik et al., 1998), recently, it is reported in Jilin, Hebei, Henan, and Liaoning provinces of China (Chen et al., 2016). SVBV has become a major virus infecting strawberry and causes severe damage to strawberry production, especially infections mixed with other strawberry viruses (Bolton, 1974; Mráz et al., 1997; Thompson et al., 2003). SVBV is transmitted by grafting or by several aphid species such as Chaetosiphon sp. in a semi-persistent manner (Vašková et al., 2004). The host range of SVBV is restricted to strawberry (Fragaria) (Mráz et al., 1997). Although SVBV virulence may be reduced in commercial strawberry (Fragaria × ananassa cv. Sachinoka) cultivars, partial inhibition in growth potential, accompanied by a decrease in the number of creeping stems still existed on strawberry plants (Vašková et al., 2004). When indicator plants wild strawberries (Fragaria vesca, Fragaria virginiana) were infected with SVBV, leaves of strawberry plants showed chlorotic and mild vein banding symptoms exhibited along the main leaf veins and failed to unfold properly (Vašková et al., 2004).
SVBV is a well-defined virus species and classified as a member of the genus Caulimovirus in the family Caulimoviridae (Pattanaik et al., 2004). SVBV has a double-stranded DNA (dsDNA) genome encapsulated in icosahedral particles with the size of approximately 45 nm diameter (Kitajima et al., 1973; Stenger et al., 1988). The genome of SVBV was reported to be nearly 8 kb and comprises seven ORFs, potentially coding for seven proteins (Petrzik et al., 1998). So far, a large number of SVBV detection works have been conducted in the world, and the PCR method was used to amplify partial cp gene for SVBV detection (Mráz et al., 1998; Hanzliková et al., 2006). The variability in genomes of SVBV was also determined in many countries previously. Comparison of the sequences of partial cp genes was used to reflect the variation of the SVBV complete genome due to the lack of sufficient complete genomes information of SVBV (Vašková et al., 2004). The first attempt to compare sequence homology of cp genes of SVBV was carried out among the samples from American and European countries. Variability of SVBV was determined only by sequencing a fragment less than 1/3 of the full ORF IV, 431 nts of the middle part of cp gene (Mráz et al., 1998). Thus, the variability of partial cp genes seemed to be unreliable and insufficient to reflect the variation of complete genomes of SVBV. Up to now, only a few complete genomes of SVBV was determined. The first complete genome sequence of SVBV was reported in the United States (Accession No. X97304) (Frazier, 1955), and the other isolates were all from China (Feng et al., 2016).
In our research, we reported two complete nucleotide sequences of SVBV isolates in naturally infected strawberry plants from China. Phylogenetic relationships and nucleotide sequence identities between the two SVBV China isolates (SVBV-AH and SVBV-BJ) and other previously characterized isolates of caulimoviruses were analyzed. Agrobacterium tumefaciens cultures containing SVBV-AH infectious clone were infiltrated into Fragaria vesca leaves and resulted in systemic infection with obvious symptoms of yellowing bands along the main leaf veins, indicated that the SVBV-AH isolate poses the ability to cause disease in natural strawberry, and probable the causal agent of the vein banding disease in strawberries. Collectively, our study will provide useful information on SVBV from China to elucidate its origin and evolutionary status.
Plant sample collections and detection
Field samples were collected in cultivated strawberry (Fragaria × ananassa cv. Sachinoka) planting gardens, twenty-six samples from Changfeng county, Anhui province, and nine samples from Daxing district, Beijing city, China. Samples showed suspected disease symptoms of growth decline with fewer creeping stems and manifest leaf vein chlorotic. Firstly, SVBV was detected by ELISA with an antibody raised against SVBV CP protein, which antiserum was prepared using the purity recombination protein as antigen to immunize rabbits by our laboratory. Two positive samples were obtained, SVBV-AH was from Anhui province, and SVBV-BJ was from Beijing city.
DNA extraction and PCR amplification
The total DNA of the two samples was extracted from 100 mg of leaf tissues using the CTAB method (Stein et al., 2001). The DNA quality was assessed by using a NanoDrop 2000 Spectrophotometer (Thermoscientific, Waltham, MA, USA) and by electrophoresis on 0.8% agarose TAE gels. To amplify the complete genome sequences of two SVBV isolates, 2 pairs of overlapping PCR primers (Tab. 1) were designed manually based on the complete sequences of SVBV and other caulimoviruses in GenBank (Accession Nos. KX249735, KX249736, KX249737, KX249738, MH894295, KT250632, KP311681, MF197916, LC315804, HE681085, X97304, KX950836, JX272320, AF454635, X06166, AJ853858, JQ926983, EU554423, JX429923, X79465, KJ716236, AB863169, JX912267, AF140604). The amplified fragments could be generated along the overall length of the genome. PCR was performed by using the total DNA (the concentration between 100–500 ng/µL) as a template, the 50 µL PCR mixture including 1 µL DNA, 1 µM each specific primer, 1 µL KOD FX Neo DNA Polymerase, 25 µL 2×PCR Buffer for KOD FX Neo, 11 µL 2 mM dNTPs (TOYOBO, Japan) and 11 µL distilled water. The PCR reaction was conducted under the following conditions: 2 min at 94°C; 30 cycles of 10 s at 94°C, 30 s at 68°C and 120 s at 68°C, with a final extension at 68°C for 5 min and the amplified PCR products were purified by a TaKaRa PCR purification Kit (TaKaRa, Dalian, China).
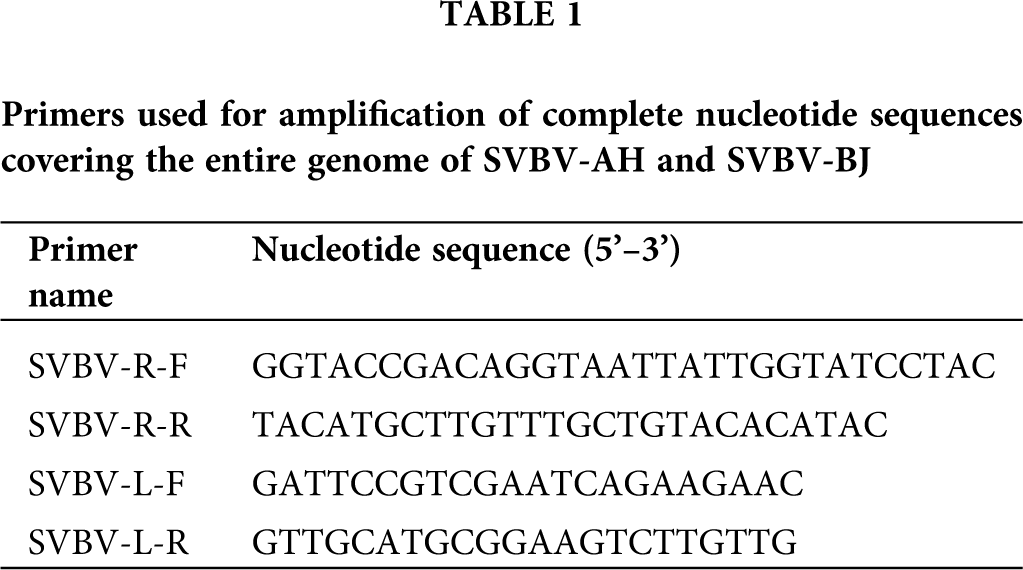
The 3’-A added DNA products were cloned into a pMD18-T vector (TaKaRa, Dalian, China) using TA-cloning strategy followed by transformation of chemically competent Escherichia coli DH5α cells according to standard protocols (Hanahan, 1983); then, the positive clones were obtained and sequenced used the corresponding primer pairs of the amplification (Tab. 1) by Invitrogen (Shanghai, China).
Sequence analysis and alignment
Each sequence was assembled into the full-length SVBV genome by using the sequence assembly program SeqMan (Lasergene 7.1.0, DNASTAR Inc, USA). Snap Gene Viewer (GSL Biotech, Chicago, IL) was used to search for potential ORFs in the genome. Sequence analyses comparison of the SVBV-AH and SVBV-BJ to the reference sequences were performed using the DNAStar 5.01 package (DNASTAR, Madison, USA). Complete nucleotide sequences of other isolates of Caulimovidae used for comparison were retrieved from the National Center for Biotechnology Information (NCBI) databases (http://www.ncbi.nlm.nih.gov/). Conserved domains of the putative proteins were identified by Conserved Domain Search (CD-Search) in NCBI (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) (Marchler et al., 2015).
Molecular phylogenetic tree analysis was performed by using MEGA5 software, and the phylogenetic trees were constructed by the neighbor-joining method with 1,000 bootstrap replicates. Detailed information of caulimoviruses used for comparisons (including GenBank accession number, the abbreviations, and the region where it was isolated) are shown in phylogenetic trees.
Construction and infiltration of SVBV infectious clone
The detailed construction procedures of the SVBV infectious clone (pBin-1.25SVBV-AH) were described previously (Feng et al., 2016). SVBV-AH infectious clone and the pBinPLUS empty vector were transferred into Agrobacterium tumefaciens (GV3101), and then the transformed A. tumefaciens suspensions were infiltrated into Fragaria vesca leaves (a seedling of 6–8 leaves) respectively using vacuum infiltration method according to our previous reports (Feng et al., 2016). The pBin-1.25SVBV-AH infectious clone was infiltrated into 30 strawberry samples (F. vesca), and the empty vector (pBinPLUS) was infiltrated into 10 strawberry samples, respectively. The infiltrated leaves were photographed with a Canon EOS 700 D digital camera (Canon, Taiwan, China) with a Y48 yellow filter. Mock-inoculated and SVBV-infected strawberry leaves were harvested 25 dpi. Southern blot hybridization was performed to detect SVBV DNA accumulation by digoxigenin-labeled SVBV probes using DIG High prime DNA labeling and detection starter kit II (Roche) according to the manufacturer’s instructions.
Features of ORFs and encoded proteins of SVBV isolates
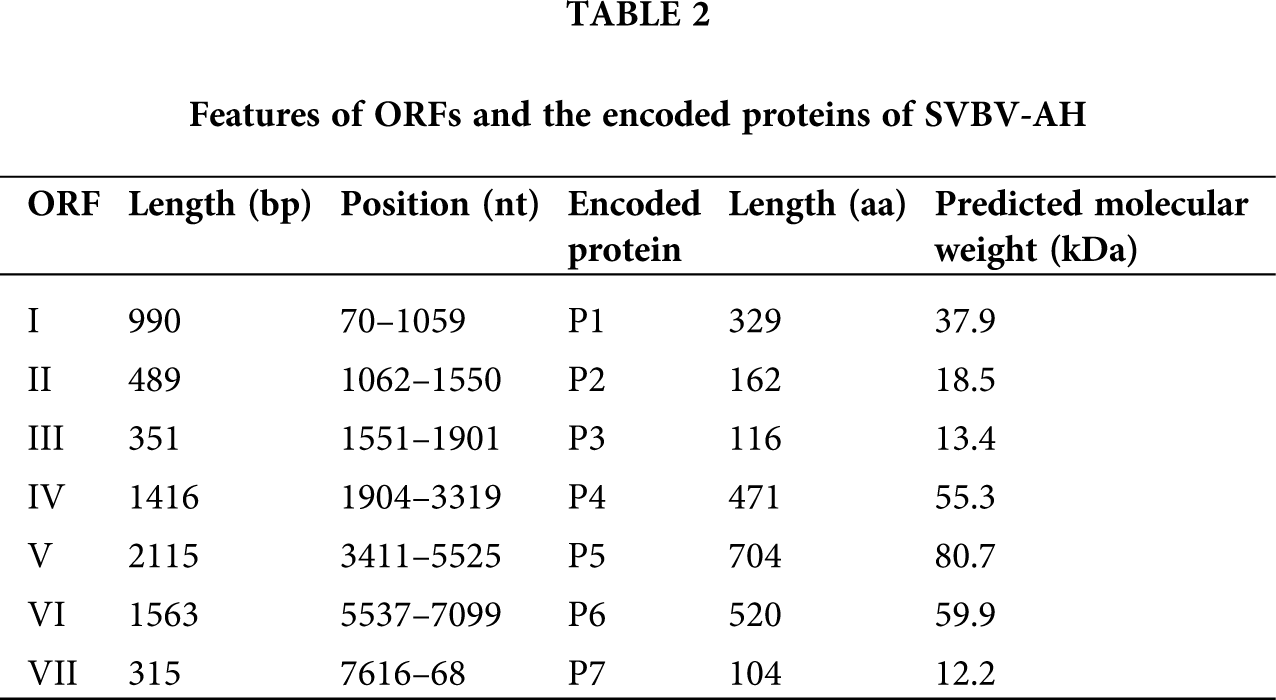
The complete genomes of SVBV-AH (GenBank Accession No. KX787430) and SVBV-BJ (GenBank Accession No. KR080547) are 7,862 nts and 7,863 nts in length, respectively. The complete genome sequences of SVBV-AH and SVBV-BJ both existed in a large untranslated region (UTR) and some small intergenic regions (IRs) between ORFs. The length of the large untranslated region between ORF VI and ORF VII of SVBV-AH and SVBV-BJ is 516 nts and 515 nts, respectively. Additionally, the complete genomes of two SVBV isolates also present a relatively small intergenic region of 91 nts between ORF VI and ORF VII and 2 nts intergenic space between ORF I and ORF II, ORF III, and ORF IV. The interval between ORF V and ORF VI of SVBV-AH is 11 nts, and that of SVBV-BJ is 12 nts. Moreover, ORF II and ORF III of two SVBV isolates are continuous.
Sequence analysis revealed that the genomes of SVBV-AH and SVBV-BJ contain seven open reading frames (ORFs) that encode seven proteins, respectively. The speculated functions of the corresponding putative proteins of SVBV-AH are as follows: ORF I encodes a 37.9 kDa viral movement protein (MP) P1 with a main function domain between 37 to 228 aa which is conserved in caulimoviruses. It was known that viral MP could facilitate intracellular trafficking of the viral genomes and assist the spread of the viral replication complexes between plant cells. Beyond that, P1 of CaMV could interact with the plasmodesmata and possessed the ability to bind viral RNA to achieve its cell-to-cell movement (Carluccio et al., 2014).
ORF II encodes an 18.5 kD protein P2. P2 is acquired by aphids to associate with aphid transmission of the virus that is found in various caulimoviruses; P2 was also known as the aphid transmission factor (ATF) and could assist plant–plant transmission of a non-transmissible CaMV isolate from crude extracts of infected plants (Bouchery, 1990). P2 of SVBV contains two domains, one domain near the N-terminus is necessary for the aphid transmission, the other one between 103 aa and 145 aa is a Crotonase/Enoyl-Coenzyme A (CoA) hydratase. CoA hydratase superfamily contains a diverse set of enzymes, which play important roles in fatty acid metabolism (Woolston et al., 1983).
ORF III encodes a 13.4 kD protein, and no conserved motif could be found in P3 of SVBV. However, P3 of CaMV contains a C-terminal basic domain located at 112–126 aa, which possesses non-sequence-specific DNA binding activity. P3 is also essential for the infection cycle. Interaction of P3 with P2 could promote the efficiency of aphid transmission, and it is a second ‘helper’ factor required for CaMV transmission by aphids (Véronique et al., 1999).
ORF IV encodes a 55.3 kDa protein P4, P4 contains a C2HC type zinc-finger conserved domain at amino acid position 396–412, which is a typical component of the coat protein of all caulimoviruses. P4 plays an important role in the encapsulation of viral DNA and is frequently used for serological detection of caulinoviruses (Singh et al., 2014).
ORF V encodes an 80.7 kDa multifunctional protein P5. P5 of CaMV has been proved to be a polyprotein precursor, and it possesses activities of proteinase, reverse transcriptase, aspartate proteinase, and ribonuclease H. There are four predicted conserved domains present in P5 of SVBV. A reverse transcriptases (RTs) domain located at 301–481 aa and an RNA-dependent DNA polymerase domain located at 325–481 aa were reported to be conserved in caulimoviruses. In addition, P5 also contains a Ribonuclease H (RNase H) domain located at 576–698 aa and a peptidase A3 domain close to the N-terminus (Muriel et al., 2002).
ORF VI encodes a 59.9 kDa protein P6. The function of P6 is predicted as the viroplasmin proteins of caulimoviruses. Viroplasmin protein is reported as the main components of viral inclusion bodies and responsible for viral assembly and accumulation. P6 of CaMV is a multifunctional protein and is probably involved in controlling the specificity of virus-host interaction, the severity of host symptom, and the translational transactivation of other ORFs of CaMV (Schoelz et al., 1986; Johannes et al., 1989).
No conserved motif could be detected in protein encoded by ORF VII of SVBV, and also no related research works about P7 of CaMV were reported till now.
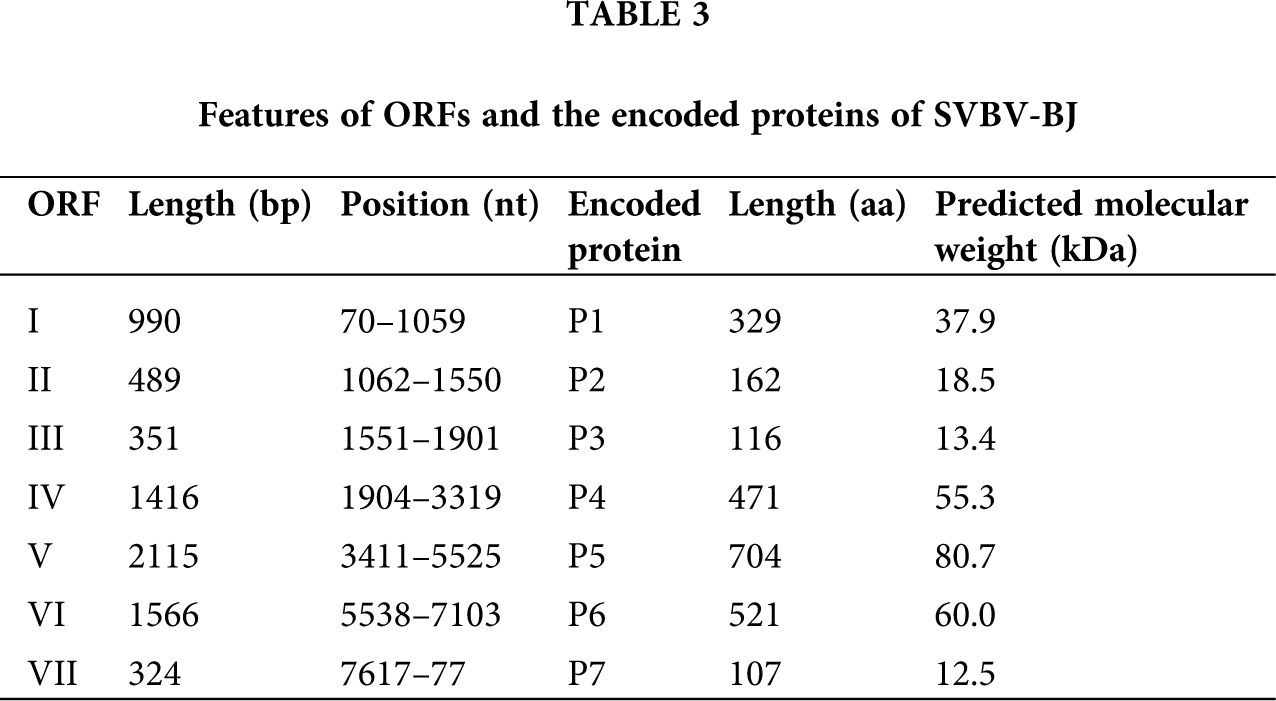
Although the size of each ORF of SVBV-BJ has some differences from that of SVBV-AH, the putative functions of proteins of SVBV-BJ are identical to the corresponding proteins of SVBV-AH. More information on the encoded proteins of the two SVBV isolates is present in Tabs. 2 and 3.
Nucleotide and amino acid sequence identities between SVBV and those of other representative caulimoviruses
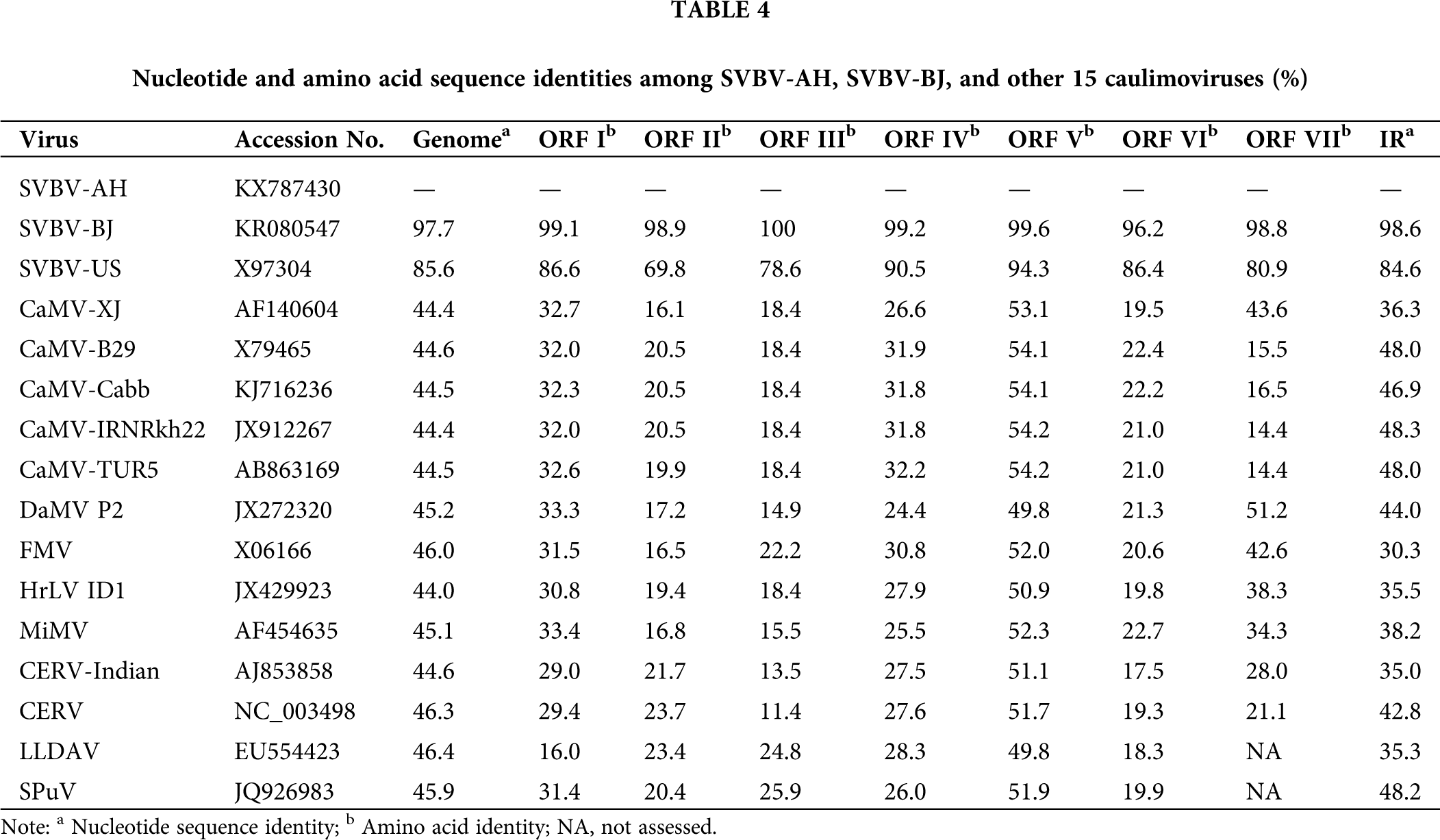
The complete genome sequences comparison showed that all 14 SVBV isolates shared relative high sequences homology (85.7%–97.7%) with each other. Among them, SVBV-AH and SVBV-BJ shared very high nucleotide sequence identity (97.7%) with each other, and 96.5%–98.6% sequence identity with other SVBV China isolates. Besides, they shared 97.7% and 97.4% sequence identity with the Japan isolate, while they had relatively lower sequence identity with Canada isolate (92.3% and 92.9%) and only 85.7% and 86.0% sequences identity with SVBV-US, respectively. SVBV-AH and SVBV-BJ shared 43.9%–46.1% sequence identity with other 12 members of caulimoviruses and had only 44.4%–44.7% nucleotide sequence identity with 5 isolates of CaMV. Although the genome structure of SVBV and CaMV was very similar, the sequence identity of the complete nucleotide sequences of SVBV and CaMV was very low. It was probably due to SVBV and CaMV infecting different families of hosts, SVBV infects Rosaceae plants, and CaMV infects Brassicaceae plants (Farzadfar and Pourrahim, 2013). In the long term of evolution, both SVBV and CaMV produced very strong host specificity, and coadaptation and coevolution of SVBV and CaMV with their hosts resulted in great genome variation. However, SVBV isolates derived from different countries, even isolated with the vast Pacific Ocean, still shared a relatively higher sequence identity. This illustrated that the genome variation of SVBV isolates had little relationship with their geographical origins.
Despite the remarkable identity of the arrangement of the ORFs of SVBV-AH and SVBV-BJ isolates to that of other caulimoviruses, the ORFs of our two SVBV isolates share relatively lower amino acid sequences identities (13.5%–58.2%) with the corresponding ORFs of other species of caulimoviruses. Among them, ORF II, ORF III, and ORF VI of our two SVBV isolates had very low amino acid sequence identity (13.5%–23.0%) with the corresponding ORFs of other species of caulimoviruses, whereas ORF V of two SVBV shared relatively higher amino acid sequence identity (55.9%–58.2%) with that of other species of caulimoviruses. Both the full length and ORFs amino acid sequences of the SVBV-AH and SVBV-BJ isolates share extremely high identity (97.7% and 96.2%–100%) with each other. It could be suspected that the two SVBV had distant evolutionary relationships with other species of caulimoviruses in ORF II, ORF III, and ORF VI, and ORF V of two SVBV had a relatively closer evolutionary relationship with that of other species of caulimoviruses (Tab. 4).
Phylogenetic trees based on sequence alignments between SVBV and those of other representative caulimoviruses obtained by BLASTp search
A phylogenetic dendrogram was performed to determine the evolutionary relationship based on the complete nucleotide sequences of 14 SVBV isolates aligned with other species of caulimoviruses derived from different geographic areas. As shown in Fig. 1A, all the isolates of caulimoviruses were distributed into two major clades. All of the SVBV isolates clustered into one branch, while other species of caulimoviruses clustered into another branch. The 13 SVBV isolates from China, Japan, and Canada clustered into a sub-branch, and the SVBV-US isolate formed a sub-branch alone. Besides, DaMV, MiMV, and FMV formed a sub-branch, and 5 CaMV, CERVs, SpuV, LLDAV, and HrLV formed another sub-branch. It was illustrated that SVBV-AH, SVBV-BJ, and other SVBV isolates had a very close relationship with each other but had an extremely distant relationship with other species of caulimoviruses (Fig. 1A).
A phylogenetic tree of the amino acid sequences of ORF IV of SVBV was constructed based on 14 SVBV isolates and other representative species of caulimoviruses. From the phylogenetic tree, it was found that all caulimoviruses were distributed into two major branches. SVBV-AH and SVBV-BJ clustered into a separate branch together with other 10 SVBV isolates except for SVBV-NS8 (Canada) and SVBV-USA, indicating that 12 SVBV isolates from China and Japan had extremely close evolutionary relationships with each other but had a relatively distant relationship with SVBV isolates from Canada and USA. Besides, DaMV, MiMV and FMV clustered into a sub-branch, and LLDAV, SpuV, CERV, HrLV and 5 CaMV isolates clustered into another sub-branch. We could find that the structures of the two phylogenetic trees were extremely similar. It could be illustrated that the phylogenetic relationships of amino acid sequences of ORF IV could reflect the relationships of caulimoviruses to some extent, and the evolutionary relationship is almost consistent with the geographical distribution of all the viruses (Fig. 1B).
In our previous study, the first complete genome sequence of SVBV from China (SVBV-CN) was reported. Now, we determined two novel SVBV isolates derived from other regions of China and described the complete nucleotide sequences and the genetic characterization of the two SVBV isolates. Although further and more detailed research is desirable, the results of the present study help us deeply understand the genetic diversity of SVBV isolates in China and benefit us to reveal the regulation of epidemiology and evolution of SVBV throughout the world.

Figure 1: Phylogenetic trees based on sequence alignments between SVBV and other 14 caulimoviruses.
(A) Phylogenetic tree based on the complete nucleotide sequences of two SVBV China isolates and other 14 caulimoviruses. (B) Phylogenetic tree based on the amino acid sequences of ORF IV of two SVBV China isolates and other 14 caulimoviruses. The trees were constructed using MEGA version 4.1 by the observed-divergency method with 1000 bootstrap replicates, and the bootstrap values (>90%) are shown. The length of the branches and the percentage of bootstrap frequency are indicated. (A) Complete genome (B) ORF IV.
Inoculation with SVBV-AH infectious clone
The A. tumefaciens suspension containing SVBV-AH infectious clone and the empty vector were infiltrated into F. vesca leaves, respectively. When F. vesca was inoculated with SVBV-AH infectious clone for 25 days, yellow banding symptoms on the newly developed leaves could be observed. While the newly developed leaves inoculated with the empty vector showed no symptoms, which similar to non-infection leaves (Fig. 2A).
We collected the samples from newly developed leaf tissues 25 days after SVBV-AH infection to prevent possible cross-contamination. Southern blot analysis confirmed the SVBV DNA accumulation in SVBV-AH infected F. vesca, whereas no viral DNA could be detected in control plants (Fig. 2B). Therefore, we conclude that the SVBV-AH infectious clone is the causal agent of the tested strawberry and poses pathogenicity, causing a similar symptom as SVBV-strawberry infected under natural conditions. So far, there are few available vectors infecting strawberry, hampering the ability for independent research for studying the gene functions of strawberry. It is expected that SVBV-AH infectious clone would contribute to this field.
Anhui province has a large strawberry planting area, where virus diseases seriously damage the yield and quality of strawberry fruits. Therefore, it is urgent to breed some new resistant varieties suitable for cultivating in Anhui province. Inoculation with local SVBV isolate could reflect the actual resistance level of the new strawberry varieties in Anhui province more accurately.

Figure 2: Symptoms and viruses accumulation in F. vesca plants infected by SVBV-AH infectious clone and empty vector.
(A) A. tumefaciens suspension containing SVBV-AH infectious clone and the empty vector (Mock) were infiltrated into F. vesca leaves respectively using vacuum infiltration method. (B) Southern blot analysis of SVBV DNA accumulation in SVBV-infected plants and the negative control plants, SVBV plasmids was used as the positive control.
Acknowledgement: The undergraduates Xuechun Kou and BingJin Luo of Anhui agricultural university also participated in sample collection and data processing in this study.
Availability of Data and Materials: All data generated or analysed during this study are included in this article.
Author Contributions: Lei Jiang and Shuai Li developed the experiments; Lei Jiang, Shuai Li, and Xizi Jiang participated in the study design and wrote the manuscript. Tong Jiang conceived the study and finalized the manuscript. All authors have read and approved the final manuscript.
Funding Statement: This work was supported by grants from the National Natural Science Foundation of China (Nos. 32072386 and 31801700), the Key Research and Development Project of Anhui Province (202004a06020013), and the Anhui Postdoctoral Fund (2019B360).
Conflicts of Interest: All authors declare that they have no conflict of interest.
Bolton AT (1974). Effects of three virus diseases and their combinations on fruit yield of strawberries. Canadian Journal of Plant Science 54: 271–275. DOI 10.4141/cjps74-043. [Google Scholar] [CrossRef]
Bouchery Y (1990). Comparison of short- and long-feed transmission of the cauliflower mosaic virus Cabb-S strain and SΔII hybrid by two species of aphid: Myzus persicae (Sulzer) and Brevicoryne brassicae (L.). Research in Virology 141: 677–683. DOI 10.1016/0923-2516(90)90040-P. [Google Scholar] [CrossRef]
Carluccio AV, Zicca S, Stavolone L (2014). Hitching a ride on vesicles: Cauliflower mosaic virus movement protein trafficking in the endomembrane system. Plant Physiology 164: 1261–1270. DOI 10.1104/pp.113.234534. [Google Scholar] [CrossRef]
Chen J, Zhang HP, Feng MF, Zuo DP, Hu YH, Jiang T (2016). Transcriptome analysis of woodland strawberry (Fragaria vesca) response to the infection by strawberry vein banding virus (SVBV). Virology Journal 13: 19. DOI 10.1186/s12985-016-0584-5. [Google Scholar] [CrossRef]
Feng MF, Zhang HP, Pan Y, Hu YH, Chen J, Zuo DP, Jiang T (2016). Complete nucleotide sequence of strawberry vein banding virus Chinese isolate and infectivity of its full-length DNA clone. Virology Journal 13: 128. DOI 10.1186/s12985-016-0624-1. [Google Scholar] [CrossRef]
Frazier NW (1955). Strawberry vein banding virus. Phytopathology 45: 307–312. [Google Scholar]
Farzadfar S, Pourrahim R (2013). Biological and molecular variation of Iranian Cauliflower mosaic virus (CaMV) isolates. Virus Genes 47: 347–356. DOI 10.1007/s11262-013-0948-5. [Google Scholar] [CrossRef]
Hanahan D (1983). Studies on transformation of Escherichia coli with plasmids. Journal of Molecular Biology 166: 557–580. DOI 10.1016/S0022-2836(83)80284-8. [Google Scholar] [CrossRef]
Kitajima EW, Betti JA, Costa AS (1973). Strawberry vein-banding virus, a member of the cauliflower mosaic virus group. Journal General of Virology 20: 117–119. [Google Scholar]
Honetšlegrová J, Mráz I, Špak J (1995). Detection and isolation of strawberry vein banding virus in the Czech Republic. Acta Horticulturae 385: 29–32. [Google Scholar]
Hanzliková VD, Špak J, Petrzik K (2006). Variability in sequence of strawberry vein banding virus. Biologia Plantarum 50: 660–666. DOI 10.1007/s10535-006-0103-4. [Google Scholar] [CrossRef]
Johannes F, Karl G, Pierre P, Hélèlene S, Barbara P, Jean-Marc B, Thomas H (1989). Differential inhibition of downstream gene expression by the cauliflower mosaic virus 35S RNA leader. Virus Genes 3: 45–55. DOI 10.1007/BF00301986. [Google Scholar] [CrossRef]
Marchler BA, Derbyshire MK, Gonzales NR, Lu S, Chitsaz F, Geer LY, Geer RC, He J, Gwadz M, Hurwitz DI, Lanczycki CJ, Lu F, Marchler GH, Song JS, Thanki N, Wang Z, Yamashita RA, Zhang D, Zheng C, Bryant SH (2015). CDD: NCBI’s conserved domain database. Nucleic Acids Research 43: 222–226. DOI 10.1093/nar/gku1221. [Google Scholar] [CrossRef]
Muriel H, Marina B, Angèle G, Pierre Y, Mario K (2002). Cauliflower mosaic virus: Still in the news. Molecular Plant Pathology 3: 419–429. DOI 10.1046/j.1364-3703.2002.00136.x. [Google Scholar] [CrossRef]
Mráz I, Petrzik K, Šíp M, Fránová HJ (1998). Variability in coat protein sequence homology among American and European sources of strawberry vein banding virus. Plant Disease 82: 544–546. DOI 10.1094/PDIS.1998.82.5.544. [Google Scholar] [CrossRef]
Mráz I, Petrzik K, Fránová HJ, Síp M (1997). Detection of strawberry vein banding virus by polymerase chain reaction and dot blot hybridization. Acta Virologica 44: 241–242. [Google Scholar]
Petrzik K, Benes V, Mraz I, Honetslegrova FJ, Ansorge W, Spak J (1998). Strawberry vein banding virus-definitive member of the genus Caulimovirus. Virus Genes 16: 303–305. DOI 10.1023/A:1008039024963. [Google Scholar] [CrossRef]
Ratti C, Pisi A, Autonell CR (2009). First report of strawberry vein banding virus on strawberry in Italy. Plant Disease 93: 675. DOI 10.1094/PDIS-93-6-0675A. [Google Scholar] [CrossRef]
Pattanaik S, Dey N, Bhattacharyya S, Maiti IB (2004). Isolation of full-length transcript promoter from the Strawberry vein banding virus (SVBV) and expression analysis by protoplasts transient assays and in transgenic plants. Plant Science 167: 427–438. DOI 10.1016/j.plantsci.2004.04.011. [Google Scholar] [CrossRef]
Stenger DC, Mullin RH, Morris TJ (1988). Isolation, molecular cloning, and detection of strawberry vein banding virus DNA. Phytopathology 78: 154–159. DOI 10.1094/Phyto-78-154. [Google Scholar] [CrossRef]
Schoelz J, Shepherd RJ, Daubert S (1986). Region VI of cauliflower mosaic virus encodes a host range determinant. Molecular and Cellular Biology 6: 2632–2637. DOI 10.1128/MCB.6.7.2632. [Google Scholar] [CrossRef]
Singh P, Prabha K, Rakesh KJ, Baranwal VK (2014). N-terminal in coat protein of Garlic virus X is indispensible for its serological detection. Virus Genes 48: 128–132. DOI 10.1007/s11262-013-0990-3. [Google Scholar] [CrossRef]
Stein N, Herren G, Keller B (2001). A new DNA extraction method for high-throughput marker analysis in a large-genome species such as Triticum aestivum. Plant Breeding 120: 354–356. DOI 10.1046/j.1439-0523.2001.00615.x. [Google Scholar] [CrossRef]
Thompson JR, Wetzel S, Klerks MM, Vaskova D, Schoen CD, Spak J, Jelkmann W (2003). Multiplex RT-PCR detection of four aphid-borne strawberry viruses in Fragaria spp. in combination with a plant mRNA specific internal control. Journal of Virological Methods 111: 85–93. DOI 10.1016/S0166-0934(03)00164-2. [Google Scholar] [CrossRef]
Vašková D, Špak J, Klerks MM, Schoen CD, Thompson JP, Jelkmann W (2004). Real-time NASBA for detection of strawberry vein banding virus. European Journal of Plant Pathology 110: 213–221. DOI 10.1023/B:EJPP.0000015378.27255.12. [Google Scholar] [CrossRef]
Véronique L, Emmanuel J, Angèle G, Thomas H, Denis L, Martine C, Pierre Y, Mario K, Stéphane B (1999). Aphid transmission of cauliflower mosaic virus requires the viral PIII protein. EMBO Journal 18: 7077–7085. DOI 10.1093/emboj/18.24.7077. [Google Scholar] [CrossRef]
Woolston CJ, Covey SN, Penswick JR, Davies JW (1983). Aphid transmission and a polypeptide are specified by a defined region of the cauliflower mosaic virus genome. Gene 23: 15–23. DOI 10.1016/0378-1119(83)90212-3. [Google Scholar] [CrossRef]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |