DOI:10.32604/biocell.2021.017227
| BIOCELL DOI:10.32604/biocell.2021.017227 | |
| Article |
Viral integration detection strategies and a technical update on Virus-Clip
1Department of Pathology, The University of Hong Kong, Hong Kong, China
2State Key Laboratory of Liver Research, The University of Hong Kong, Hong Kong, China
*Address correspondence to: Irene Oi-lin Ng, iolng@hku.hk
#Authors contributed equally to this study
Received: 23 April 2021; Accepted: 31 May 2021
Abstract: Oncovirus infection is crucial in human malignancies. Certain oncoviruses can lead to structural variations in the human genome known as viral genomic integration, which can contribute to tumorigenesis. Existing viral integration detection tools differ in their underlying algorithms pinpointing different aspects or features of viral integration phenomenon. We discuss about major procedures in performing viral integration detection. More importantly, we provide a technical update on Virus-Clip to facilitate its usage on the latest human genome builds (hg19 and hg38) and the adoption of multi-thread mode for faster initial read alignment. By comparing the execution of Virus-Clip using single-thread and multi-thread modes of read alignment on targeted-panel sequencing data of HBV-associated hepatocellular carcinoma patients, we demonstrate the marked improvement of multi-thread mode in terms of significantly reduced execution time, while there is negligible difference in memory usage. Taken together, with the current update of Virus-Clip, it will continue supporting the in silico detection of oncoviral integration for better understanding of various human malignancies.
Keywords: Oncovirus; Viral genomic integration; In silico detection; Tumorigenesis; Human malignancies
Oncovirus infection is a major risk factor for human cancers (Muller-Coan et al., 2018). Some of the viruses may integrate into the human genome, leading to structural variation known as viral genomic integration. Infections of common oncoviruses, e.g., hepatitis B virus (HBV), human papillomavirus (HPV) and Epstein-Barr virus (EBV) are known to cause viral integration events and they are involved in the carcinogenesis process of liver cancer, cervical cancer and nasopharyngeal carcinoma, respectively. Viral integration may result in genome instability, disruption of human genes, aberrant human gene expression, and/or expression of chimeric oncogenic proteins, which contribute to the consequence of tumorigenesis.
Using hepatocellular carcinoma (HCC, a major form of primary liver cancer) as an illustration, it is a prevalent cancer and one of the leading causes of cancer death worldwide (El-Serag, 2011; Villanueva and Llovet, 2014). It has poor prognosis and only few effective treatment options are available. Despite years of efforts in studying the molecular mechanism of HCC carcinogenesis, current understanding on this lethal disease is still limited, with high recurrence and metastasis being the major hurdles for disease cure. Among the identified etiological risk factors for HCC (Ho et al., 2016), which include chronic viral infections (HBV and hepatitis C virus), chronic alcohol consumption, non-alcoholic fatty liver disease and non-alcoholic steatohepatitis, chronic HBV infection accounts for around 50% of cases (Llovet et al., 2021). One of the distinctive features of HBV genome is that it can integrate into the human genome, which in turn disrupts the endogenous tumor suppressors and other regulatory genes, or enhances the activity of proto-oncogenes. The imbalance of the overall oncogenic and tumor suppressive signals may result in enhanced cell survival, proliferation and reduced apoptosis and lead to HCC development (Ho et al., 2016).
Given the prominent role of viral integration in certain oncovirus-driven human cancers, it is important to characterize their underlying oncogenic mechanisms. With the wide adoption of next-generation sequencing (NGS), it is possible to have more systematic and unbiased survey of viral integration events. Throughout the past decade, different computational tools have emerged to detect viral integration and determine the exact breakpoint position of the human-virus chimera. Existing tools differs in the underlying algorithms, pinpointing different unique aspects/features of viral integration.
Target-panel sequencing data on HBV-associated HCC patients
We executed Virus-Clip using the target-panel sequencing data (Sze et al., 2021) of two selected HBV-associated HCC cases. They were detected with HBV integration events at KMT2B and TERT genes, respectively, at the human genome.
Performance evaluation of Virus-Clip
Virus-Clip was executed using single-thread and multi-thread modes for the initial read alignment using BWA-MEM. We compared the performance of the two modes in terms of execution time, and the number of CPU and memory used.
The latest version of Virus-Clip is available at https://github.com/dwhho/Virus-Clip.
Overview of Viral Integration Detection Tools
Different bioinformatics tools have been developed to investigate viral integration (Tab. 1). In general, major steps of viral integration detection include: (1) read alignment and extract of chimeric pairs and/or soft-clipped reads; (2) quality control of aligned and extracted reads; (3) integration of candidate discovery and determination of integration breakpoints (Fig. 1). We examined computational tools for viral integration detection, including VirusSeq (Chen et al., 2013), ViralFusionSeq (Li et al., 2013), VirusFinder (Wang et al., 2013), SummonChimera (Katz and Pipas, 2014), VERSE (Wang et al., 2015), Vy-PER (Forster et al., 2015), Virus-Clip (Ho et al., 2015), BATVI (Tennakoon and Sung, 2017), ViFi (Nguyen et al., 2018), HGT-ID (Baheti et al., 2018), VirTect (Xia et al., 2019), VIcaller (Chen et al., 2019a), SurVirus (Rajaby et al., 2021).
Read alignment and extract of chimeric pairs and/or soft-clipped reads
Most viral integration detection tools begin with having input as FASTQ or BAM files. After filtering for low-quality sequencing data, reads are mapped to the human and/or HBV genome and they search for potentially useful reads that indicate viral integration events.
The most frequently used read aligners for detecting viral integration detection are different variants of BWA and BLASTN. BLASTN can map with greater accuracy and at shorter read length (with word size of 11 by default) (McGinnis and Madden, 2004), but it is relatively time consuming compared to other aligners. On the other hand, BWA can align with longer and pair-end reads with much faster speed. To shorten the time for read alignment, some algorithms, e.g., BATVI and SurVirus, apply initial k-mer candidate filtering strategy to narrow down the possible set of input reads.
For the alignment strategy, there are mainly three ways to extract chimeric pairs and/or soft-clipped reads (Chen et al., 2019b). Strategy 1 ‘Human-Virus’: Raw reads are first mapped to the human genome, with partially mapped or unmapped reads obtained and then aligned to the virus genome. Vy-PER, HGT-ID and VIcaller use this strategy. Strategy 2 ‘Virus-Human’: It is similar to strategy 1 but is performed in reverse order. Virus-Clip, ViralFusionSeq, and BATVI, employ this strategy. Strategy 3 ‘Human+Virus’: Raw reads are aligned to a hybrid genome concatenating human and virus genomes. Tools use this strategy include VirusSeq, ViFi and VirTect. The remaining ones adopt a combinatorial approach, like VirusFinder and VERSE combine strategy 1 and 3, while SurVirus and SummonChimera integrate strategy 2 and 3. Due to the huge difference between the size of human and virus genome, the choice of the initial reference genome (reads to be mapped to) will make significant difference on the execution time. Tools, e.g., Virus-Clip initially aligns reads to the virus reference genome can substantially speed up the alignment and minimize the required computational resources.
Due to the genetic variability of virus genome and the virus-induced host genome instability, they are the rate-limiting factors for detecting viral integration. To improve the mapping ability, VERSE is designed to use short reads to iteratively modify reference genomes by SNPs and indels, so as to build a customize reference genome. ViFi applies phylogenetic methods to derive evolutionary relationships by a collection of profile Hidden Markov Models (HMMs) between known viral strains, and novel or mutated viral strains to identify viral reads from any region of the virus family of interest.
Quality control of aligned and extracted reads
To eliminate potential false-positives and ambiguous viral integration events, quality control for supportive reads is crucially important. Tools are having procedures to clean up low quality reads before detecting integration breakpoints. More recent ones, e.g., SurVirus, VIcaller, and ViFi, pay much more attention to quality control as compared to the earlier tools such as VirusSeq, ViralFusionSeq, and VirusFinder. Read alignment in BAM/SAM format has a mapping quality (MAPQ) score suggesting the precision of a read that aligned to the reference genome. In order to improve accuracy for detecting viral integration, MAPQ score are applied to filter low quality alignments. ViFi uses a criterion to remove low quality reads with MAPQ score <10. HGT-ID and VIcaller discard reads with MAPQ score <20. Besides, PCR duplicates could also cause false positives. Detection tools like BATVI, SurVirus and VIcaller remove redundant reads before subsequent analysis.
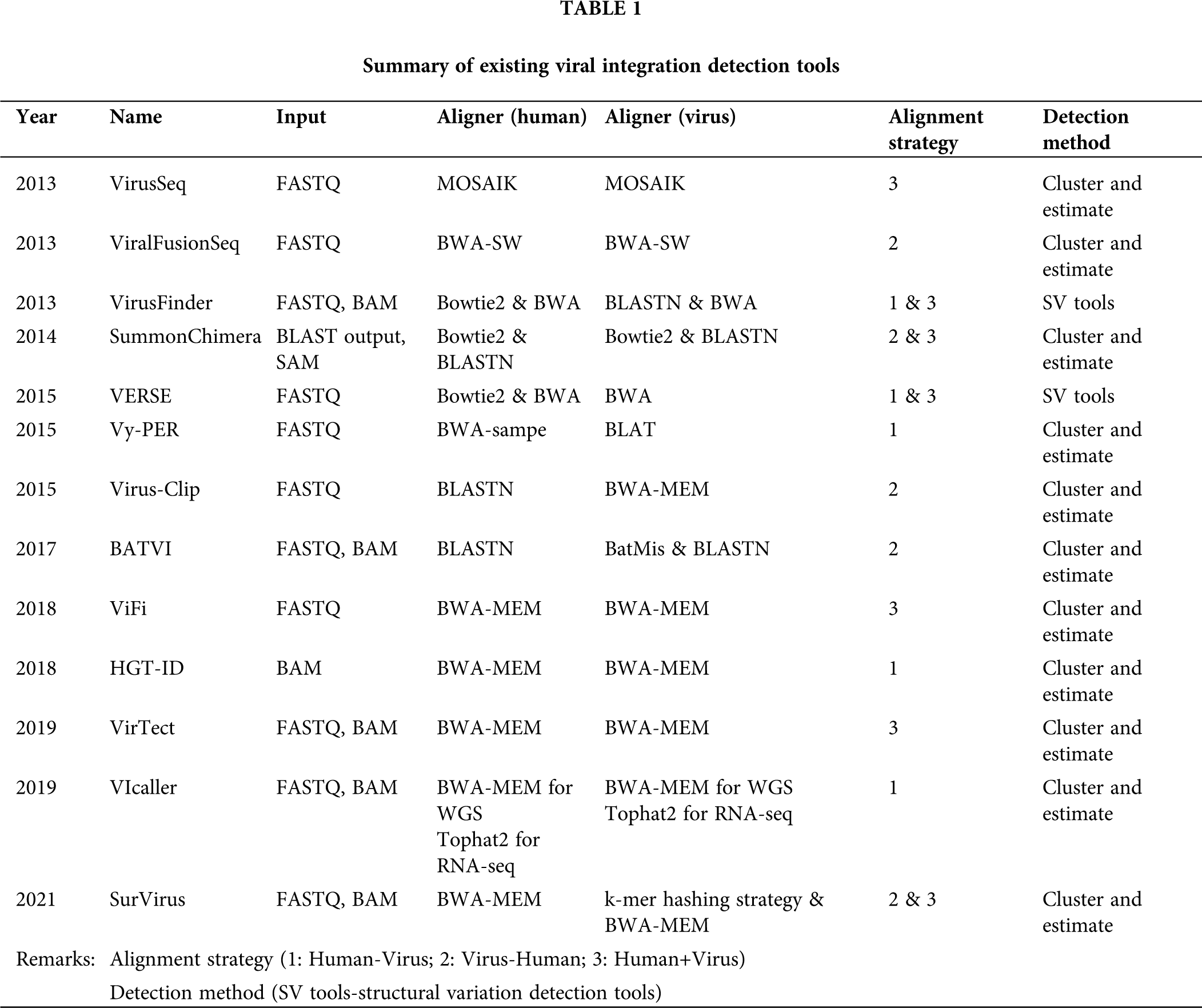

Figure 1: Major procedures in viral integration detection.
As human genome contains over 50% repetitive sequences (de Koning et al., 2011; Hannan, 2018; Lander et al., 2001), e.g., tandem repeats, satellite DNA and transposable elements. There may be high sequence similarities between human and virus repetitive sequences. Thus, detecting viral integration in repetitive regions can be challenging, since aligners usually fail to map reads correctly in repetitive regions. It tends to identify more false positives in those regions. Therefore, Vy-PER, SurVirus, ViFi, BATVI and VIcaller have dedicated strategies to reduce such artefacts.
Soft-clipped reads that have chimeric human-virus sequences (one end can be mapped to the reference genome while the other one cannot) are critical to provide information for indicating viral integration and suggesting the exact breakpoint positions. However, some soft-clipped sequence portions are too short to be unambiguously aligned. Hence, ViFi, VirTect, and Vicaller only extract soft-clipped sequence more than a certain threshold, while Virus-Clip only retains events that have soft-clipped sequence portions specifically realigned to reduce false positives. Soft-clipped sequence portions that unmapped to neither human nor virus genome, are not always invalid. The unmapped soft-clipped portions might be caused by the limited length of sequence, or the possibility of a short random sequence insertion. With the above consideration, BATVI has a dedicated strategy to rescue for putative soft-clipped reads that are either having soft-clipped sequence portions too short to be re-aligned or due to short random sequence inserted within viral integration site. Alternatively, ViFi attempts to rescue reads that are viral but might be unmapped due to evolutionarily divergent from the virus reference genome by using ensemble of HMMs. Taken together, different tools differ in their underlying consideration of viral integration features and they result in variable performance (efficiency and accuracy) in viral integration detection.
Integration of candidate discovery and determination of integration breakpoints
With the identification of high-quality read pairs or chimeric reads, they are used for deriving the exact integration breakpoint positions. Tools either cluster reads to determine breakpoints or rely on additional structural variation detection programs. Most of the existing tools follow the first approach. Concerning about the issue of tumor heterogeneity (Chan et al., 2021), some viral integration sites are shared among samples and they are believed to carry a higher degree of accuracy than the singleton ones. Nevertheless, directly pooling all sequencing data from different samples can be computationally intensive and time consuming for data analyses. Particularly, soft-clipped reads are pivotal to locate the exact breakpoint positions of the viral integration events and this strategy is using in tools, e.g., Virus-Clip. Alternatively, if soft-clipped reads are absent, tools, e.g., BATVI will assemble sequences to determine the breakpoints. Given that there could be mismatches and gaps around the breakpoints, tools, e.g., VirTect applies local HMM realignment to improve accuracy.
In summary, the procedures for identifying viral integration breakpoint are similar among the tools. They mainly differ in the threshold or refining strategy used to improve the precision of candidate breakpoints.
Technical Update of Virus-Clip
Virus-Clip was developed as a fast and memory efficient computational tool for detecting viral integration and determining the breakpoint position at single-base resolution. It takes raw reads in FASTQ format as input and can handle both single- and paired-end reads from ordinary NGS sequencers. Unlike most of the other tools that was developed at that time, Virus-Clip adopted ‘Virus-Human’ mapping strategy i.e., initially performing read alignment to virus reference genome. Due to this simple but yet important optimization, the efficiency of the entire viral integration detection process can be greatly enhanced. Besides, the installation of Virus-Clip is relatively easy, and we have provided necessary setup instructions. This is important because some tools have been reported to fail the installation due to complex compilation and/or execution errors (Chen et al., 2019b). Furthermore, BLASTN is employed to map the candidate soft-clipped sequence portions that are putatively of human origin. With the default minimal length of 11bp as input, it can effectively reduce false positives by discarding short candidate soft-clipped sequence portions (low discriminative power or high chance of random match due to short length). Another useful feature of Virus-Clip is the integrated annotation function that can determine the affected human genes without the need for additional annotation by another tool.
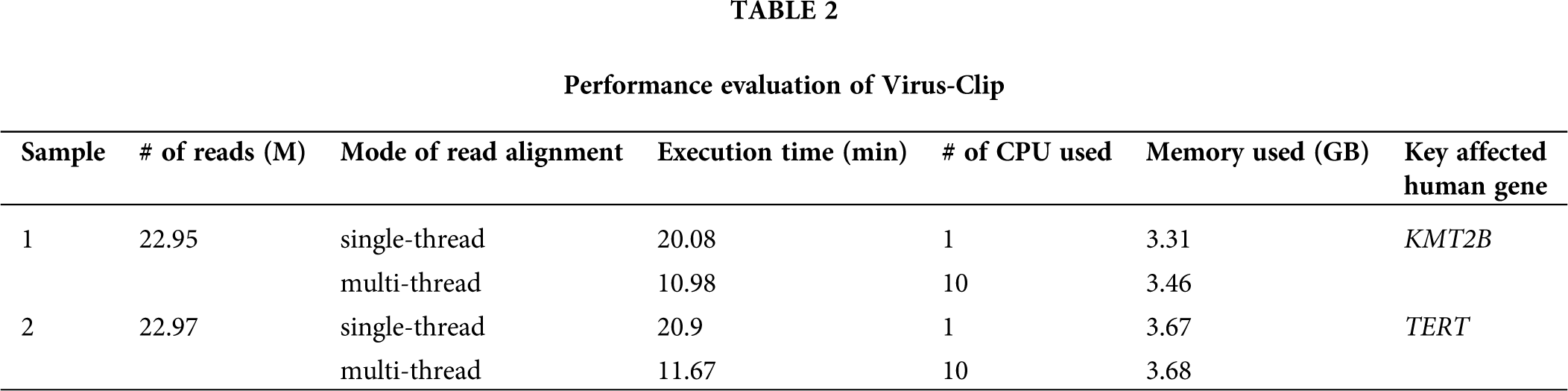
In the previous version of Virus-Clip, it was developed solely for the genome build hg19 and it is assumed to be run in single-thread mode. Therefore, in the current updated version (Fig. 2), we have revised Virus-Clip to allow for using either genome build hg19 or hg38 as human reference. Although Virus-Clip running in single-thread mode can already achieve good efficiency, with the increasing large size of NGS data, we have provided instructions to allow for using multi-thread mode in the initial read alignment. Indeed, Virus-Clip was tested to analyze the target-panel sequencing data of two HBV-associated HCC patients and the empirical performance of single-thread and multi-thread modes demonstrated significant improvement in executive time, while there was negligible difference in memory requirement (Tab. 2). Results justified the adoption of multi-thread mode in the initial read alignment of Virus-Clip, when computational resources are available. Regarding the precision of the viral integration events identified by Virus-Clip, as exemplified by our previous reports using empirical statistics on analyzing whole-transcriptome (Ho et al., 2015) and targeted sequencing (Sze et al., 2021) data, it can achieve good success rate of experimental confirmation using a threshold of at least 3 supporting reads.

Figure 2: Workflow of Virus-Clip. With the current technical update, users are provided with instructions to choose single-thread or multi-thread mode in the initial alignment step. With the availability of necessary computation resources, utilization of multi-thread mode can achieve better efficiency in execution time. Besides, the current version of Virus-Clip can support the analysis using the latest human genome builds (hg38 and hg19).
Taken together, with the current update of Virus-Clip, we hope to continue delivering a simple and useful bioinformatics tool that have all-rounded performance in terms of simplicity of installation, execution efficiency, low requirement of computational resources, and good experimentally validated accuracy of detection. We believe Virus-Clip will continue facilitating the detection of oncoviral integration and the studies of their related human malignancies.
Availability of Data and Materials: The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Author Contribution: Study conception and design: DWH, ION; Data collection and analysis: DWH, XL; All authors reviewed the results and approved the final version of the manuscript.
Funding Statement: The study was supported by the National Natural Science Foundation of China (81872222), Hong Kong Research Grants Council Theme-based Research Scheme (T12-704/16-R), Innovation and Technology Commission grant for State Key Laboratory of Liver Research, and University Development Fund of The University of Hong Kong. I.O.L. Ng is Loke Yew Professor in Pathology.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
Baheti S, Tang X, O’Brien DR, Chia N, Roberts LR, Nelson H, Boughey JC, Wang L, Goetz MP, Kocher JA, Kalari KR (2018). HGT-ID: An efficient and sensitive workflow to detect human-viral insertion sites using next-generation sequencing data. BMC Bioinformatics 19: 271. DOI 10.1186/s12859-018-2260-9. [Google Scholar] [CrossRef]
Chan LK, Tsui YM, Ho DW, Ng IO (2021). Cellular heterogeneity and plasticity in liver cancer. Seminars in Cancer Biology. DOI 10.1016/j.semcancer.2021.02.015. [Google Scholar] [CrossRef]
Chen X, Kost J, Sulovari A, Wong N, Liang WS, Cao J, Li D (2019a). A virome-wide clonal integration analysis platform for discovering cancer viral etiology. Genome Research 29: 819–830. DOI 10.1101/gr.242529.118. [Google Scholar] [CrossRef]
Chen X, Kost J, Li D (2019b). Comprehensive comparative analysis of methods and software for identifying viral integrations. Briefings in Bioinformatics 20: 2088–2097. DOI 10.1093/bib/bby070. [Google Scholar] [CrossRef]
Chen Y, Yao H, Thompson EJ, Tannir NM, Weinstein JN, Su X (2013). VirusSeq: Software to identify viruses and their integration sites using next-generation sequencing of human cancer tissue. Bioinformatics 29: 266–267. DOI 10.1093/bioinformatics/bts665. [Google Scholar] [CrossRef]
de Koning AP, Gu W, Castoe TA, Batzer MA, Pollock DD (2011). Repetitive elements may comprise over two-thirds of the human genome. PLoS Genetics 7: e1002384. DOI 10.1371/journal.pgen.1002384. [Google Scholar] [CrossRef]
El-Serag HB (2011). Hepatocellular carcinoma. New England Journal of Medicine 365: 1118–1127. DOI 10.1056/NEJMra1001683. [Google Scholar] [CrossRef]
Forster M, Szymczak S, Ellinghaus D, Hemmrich G, Ruhlemann M, Kraemer L, Mucha S, Wienbrandt L, Stanulla M (2015). UFO Sequencing Consortium within I-BFM Study Group, Franke A Vy-PER: Eliminating false positive detection of virus integration events in next generation sequencing data. Scientific Reports 5: 11534. DOI 10.1038/srep11534. [Google Scholar] [CrossRef]
Hannan AJ (2018). Tandem repeats mediating genetic plasticity in health and disease. Nature Reviews Genetics 19: 286–298. DOI 10.1038/nrg.2017.115. [Google Scholar] [CrossRef]
Ho DW, Lo RC, Chan LK, Ng IO (2016). Molecular pathogenesis of hepatocellular carcinoma. Liver Cancer 5: 290–302. DOI 10.1159/000449340. [Google Scholar] [CrossRef]
Ho DW, Sze KM, Ng IO (2015). Virus-Clip: A fast and memory-efficient viral integration site detection tool at single-base resolution with annotation capability. Oncotarget 6: 20959–20963. DOI 10.18632/oncotarget.4187. [Google Scholar] [CrossRef]
Katz JP, Pipas JM (2014). SummonChimera infers integrated viral genomes with nucleotide precision from NGS data. BMC Bioinformatics 15: 348. DOI 10.1186/s12859-014-0348-4. [Google Scholar] [CrossRef]
Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC et al. (2001). International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature 409: 860–921. DOI 10.1038/35057062. [Google Scholar] [CrossRef]
Li JW, Wan R, Yu CS, Co NN, Wong N, Chan TF (2013). ViralFusionSeq: Accurately discover viral integration events and reconstruct fusion transcripts at single-base resolution. Bioinformatics 29: 649–651. DOI 10.1093/bioinformatics/btt011. [Google Scholar] [CrossRef]
Llovet JM, Kelley RK, Villanueva A, Singal AG, Pikarsky E, Roayaie S, Lencioni R, Koike J, Zucman-Rossi J, Finn RS (2021). Hepatocellular carcinoma. Nature Reviews Disease Primers 7: 6. DOI 10.1038/s41572-020-00240-3. [Google Scholar] [CrossRef]
McGinnis S, Madden TL (2004). BLAST: At the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Research 32: W20–25. DOI 10.1093/nar/gkh435. [Google Scholar] [CrossRef]
Muller-Coan BG, Caetano BFR, Pagano JS, Elgui de Oliveira D (2018). Cancer progression goes viral: The role of oncoviruses in aggressiveness of Malignancies. Trends Cancer 4: 485–498. DOI 10.1016/j.trecan.2018.04.006. [Google Scholar] [CrossRef]
Nguyen ND, Deshpande V, Luebeck J, Mischel PS, Bafna V (2018). ViFi: Accurate detection of viral integration and mRNA fusion reveals indiscriminate and unregulated transcription in proximal genomic regions in cervical cancer. Nucleic Acids Research 46: 3309–3325. DOI 10.1093/nar/gky180. [Google Scholar] [CrossRef]
Rajaby R, Zhou Y, Meng Y, Zeng X, Li G, Wu P, Sung WK (2021). SurVirus: A repeat-aware virus integration caller. Nucleic Acids Research 49: e33. DOI 10.1093/nar/gkaa1237. [Google Scholar] [CrossRef]
Sze KM, Ho DW, Chiu YT, Tsui YM, Chan LK, Lee JM, Chok KS, Chan AC, Tang CN, Tang VW, Lo IL, Yau DT, Cheung TT, Ng IO (2021). Hepatitis B Virus-Telomerase Reverse Transcriptase Promoter Integration Harnesses Host ELF4. Resulting in Telomerase Reverse Transcriptase Gene Transcription in Hepatocellular Carcinoma. Hepatology 73: 23–40. DOI 10.1002/hep.31231. [Google Scholar] [CrossRef]
Tennakoon C, Sung WK (2017). BATVI: Fast, sensitive and accurate detection of virus integrations. BMC Bioinformatics 18: 101–111. [Google Scholar]
Villanueva A, Llovet JM (2014). Liver cancer in 2013: Mutational landscape of HCC--the end of the beginning. Nature Reviews Clinical Oncology 11: 73–74. DOI 10.1038/nrclinonc.2013.243. [Google Scholar] [CrossRef]
Wang Q, Jia P, Zhao Z (2013). VirusFinder: software for efficient and accurate detection of viruses and their integration sites in host genomes through next generation sequencing data. PLoS One 8: e64465. DOI 10.1371/journal.pone.0064465. [Google Scholar] [CrossRef]
Wang Q, Jia P, Zhao Z (2015). VERSE: A novel approach to detect virus integration in host genomes through reference genome customization. Genome Medicine 7: 2. DOI 10.1186/s13073-015-0126-6. [Google Scholar] [CrossRef]
Xia Y, Liu Y, Deng M, Xi R (2019). Detecting virus integration sites based on multiple related sequencing data by VirTect. BMC Medical Genomics 12: 19. DOI 10.1186/s12920-018-0461-8. [Google Scholar] [CrossRef]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |