DOI:10.32604/biocell.2022.016807
| BIOCELL DOI:10.32604/biocell.2022.016807 | |
| Article |
Hyperbaric oxygen protects against PC12 and H9C2 cell damage caused by oxygen–glucose deprivation/reperfusion via the inhibition of cell apoptosis and autophagy
1Hepatobiliary Surgery, Guangxi Academy of Medical Sciences, The People’s Hospital of Guangxi Zhuang Autonomous Region, Nanning, 530021, China
2Department of Emergency, The People’s Hospital of Guangxi Zhuang Autonomous Region, Nanning, 530021, China
3Department of Endocrinology, The People’s Hospital of Guangxi Zhuang Autonomous Region, Nanning, 530021, China
4Department of Neurology, The People’s Hospital of Guangxi Zhuang Autonomous Region, Nanning, 530021, China
5Department of Pharmacy, The People’s Hospital of Guangxi Zhuang Autonomous Region, Nanning, 530021, China
6Department of Hyperbaric Oxygen, The People’s Hospital of Guangxi Zhuang Autonomous Region, Nanning, 530021, China
7Department of Research Center of Medical Sciences, The People’s Hospital of Guangxi Zhuang Autonomous Region, Nanning, 530021, China
*Address correspondence to: Chunxia Chen, chunxia251401@126.com; Wensheng Lu, Lwswxqz@163.com
#These authors contributed equally to this work
Received: 28 March 2021; Accepted: 14 May 2021
Abstract: In this study, we investigated the protective effect of hyperbaric oxygen (HBO) on PC12 and H9C2 cell damage caused by oxygen-glucose deprivation/reperfusion and its possible mechanism. PC12 and H9C2 cell oxygen-glucose deprivation/reperfusion model were established. Cells were divided into a control group, model group, hyperbaric air (HBA) group and HBO group. The cell viability was detected by the CCK8 assay. Hoechst 33342 and PI staining assays and mitochondrial membrane potential (MMP) assays were used to detect cell apoptosis. The ultrastructure of cells, including autophagosomes, lysosomes, and apoptosis, were examined using a transmission electron microscope. The expression of autophagy-related proteins was detected by cellular immunofluorescence and immunocytochemistry. Our results showed that HBO can significantly improve the vitality of damaged PC12 and H9C2 cells caused by oxygen–glucose deprivation/reperfusion. HBO can significantly inhibit apoptosis of PC12 and H9C2 cells caused by oxygen-glucose deprivation/reperfusion. Importantly, we found that the protective mechanism of PC12 and H9C2 cell damage caused by oxygen-glucose deprivation/reperfusion may be related to the inhibition of the autophagy pathway. In this study, the results of cellular immunofluorescence and immunocytochemistry experiments showed that the 4E-BP1, p-AKt and mTOR levels of PC12 and H9C2 cells in the model group decreased, while the levels of LC3B, Atg5 and p53 increased. However, after HBO treatment, these autophagy-related indexes were reversed. In addition, observation of the cell ultrastructure with transmission electron microscopy found that in the model group, a significant increase in the number of autophagic vesicles was observed. In the HBO group, a decrease in autophagic vesicles was observed. The study demonstrated that hyperbaric oxygen protects against PC12 and H9C2 cell damage caused by oxygen-glucose deprivation/reperfusion via the inhibition of cell apoptosis and autophagy.
Keywords: Hyperbaric oxygen; PC12 cells; H9C2 cells; Celoxygen-glucose deprivation/reperfusion; Apoptosis; Autophagy
Vascular dementia (VD) is a common dementia disease worldwide (Harper et al., 2014; Kivimäki et al., 2015). In Asia and many developing countries, the incidence of VD even surpasses that of Alzheimer’s disease, causing intense concern among clinical and scientific researchers. Research on VD has found that arteriosclerosis and hypertension, among others, cause vascular damage or carotid artery stenosis or occlusion, which makes the important parts of the brain involved in cognitive function in a state of ischemia and hypoxia for a long time; important brain areas such as the hippocampus and prefrontal cortex neurons are damaged, and cognitive dysfunction gradually appears (Ferro et al., 2019). Coronary ischemic disease is widespread worldwide and affects more than 15 million adults in the United States (Nabel and Braunwald, 2012; Mozaffarian et al., 2016). Insufficient blood supply to the coronary arteries caused by thrombosis or acute changes in coronary atherosclerotic plaques can lead to myocardial ischemia (MI) (Buja, 2005). How to restore the blood supply to the ischemic area as soon as possible and to save dying neurons, glial cells, and vascular endothelial cells, is the key to the treatment of ischemic cardiovascular and cerebrovascular diseases. Currently, early restoration of blood flow is a common treatment strategy for cerebral ischemia and myocardial ischemia (Levisman and Price, 2015). However, disappointingly, reperfusion can easily cause further serious brain and heart damage, called cerebral ischemia reperfusion injury (CIRI) and myocardial ischemia reperfusion injury (MIRI), respectively (Yellon and Hausenloy, 2007; Ibáñez et al., 2007). Therefore, finding ways to limit or to improve ischemia-reperfusion injury (IRI) is urgent and has important clinical significance.
In recent years, the role of autophagy and autophagy–lysosomal pathways, in cardiovascular and cerebrovascular diseases, has attracted increasing attention. Studies have confirmed that the mammalian target of rapamycin (mTOR)-mediated autophagy signaling pathway participates in and regulates cell biological processes such as apoptosis and proliferation and plays an important role in the occurrence and development of CIRI and MIRI (Hu et al., 2017; Jia et al., 2015; Hao et al., 2017; Xuan and Jian, 2016; Wang et al., 2015). Autophagy is an evolutionarily conserved lysosome-dependent degradation process and an intracellular self-protection mechanism that can degrade aged misfolded proteins and damaged organelles. The factors that induce autophagy after cerebral ischemia and myocardial ischemia–reperfusion include (1) oxidative stress caused by the release of a large amount of oxygen free radicals that can induce autophagy by upregulating the expression of autophagy-related proteins LC3 and Beclin1 (Qin et al., 2010); (2) endoplasmic reticulum stress, i.e., after endoplasmic reticulum homeostasis is unbalanced, unfolded or misfolded proteins accumulate in the endoplasmic reticulum and induce autophagy in cells by inhibiting the activation of mTOR protein, a key factor in the autophagy pathway (Sheng et al., 2012); and (3) decreased levels of apoptosis-related genes such as Bcl-2 protein can cause autophagy activation (Luo and Rubinsztein, 2010). Among them, the autophagy pathway mediated by mTOR is a common pathway for cerebral ischemia and myocardial ischemia-reperfusion to induce autophagy, and it is at the center of CIRI and MIRI molecular mechanisms.
A large amount of evidence indicates that neuronal death caused by excessive autophagy occurs during cerebral ischemia reperfusion injury (IR) (Zhang et al., 2019; Yin et al., 2013). Current research reports that autophagy plays a dual role in MIRI, including being slightly induced during ischemia to promote cell survival and increasing sharply during reperfusion, thereby triggering cardiomyocyte death (Hariharan et al., 2011; Wei et al., 2012). Mammalian target of rapamycin (mTOR) kinase is an important regulator of the classic autophagy pathway (Kang et al., 2011; Yu et al., 2010). The coupling system of autophagy-related small molecule 5 (Atg5) and microtubule-associated protein 1A/1B light chain 3 (LC3) is necessary for the formation of autophagic vesicles (Han et al., 2018). Eukaryotic initiation factor 4E binding protein 1 (4E-BP1) binds to eukaryotic initiation factor 4E (eIF4E) (Pópulo et al., 2012). Phosphorylation of 4E-BP1 disrupts the assembly of the eIF4E/4E-BP1 complex, which initiates eIF4E-dependent translation, thereby activating cap- dependent mRNA translation (Bramham et al., 2016). In addition to regulating cell cycle checkpoints and apoptosis (Foster et al., 2012), p53 has also been shown to mediate the transactivation of autophagy inducers (Tasdemir et al., 2008; Crighton et al., 2006). Serine/threonine protein kinase (Akt), also known as protein kinase B, is a multifunctional adaptor protein that can be used in a variety of signaling pathways, including mTOR-mediated autophagy, nuclear factor-κB (NF-κB) and apoptosis (Yao and Han, 2014). The study of the mTOR-mediated autophagy pathway not only helps to understand the molecular mechanism of CIRI and MIRI pathogenesis but also provides new intervention targets for the prevention and treatment of CIRI and MIRI.
Hyperbaric oxygen therapy (HBO) refers to the clinical treatment of breathing 100% O2 in a room at a higher pressure (usually 2–3 times the atmospheric pressure). Clinically, HBO has a therapeutic effect on a variety of ischemic diseases, especially diseases such as cerebral infarction and myocardial infarction (Dekleva et al., 2004; Bennett et al., 2015; Rusyniak et al., 2003; Liu et al., 2016). Our previous studies have proven (Chen et al., 2017; Chen et al., 2016b; Pan et al., 2015) that HBO treatment has a strong effect of reducing oxidative stress and a strong anti-inflammatory effect, protecting cholinergic nerves, and delaying nerve cell apoptosis. In an experiment of HBO treatment of myocardial ischemia-reperfusion injury (Chen et al., 2016a), it was found that HBO treatment can improve the heart function of MIRI rats and reduce the area of myocardial infarction. Further studies have demonstrated the increase of p-mTOR protein and the reduction of LC3-II protein in the myocardial cells of MIRI rats after treatment with HBO. This finding suggests that HBO’s anti-IRI protection of nerve cells and cardiomyocytes may be related to the regulation of the mTOR-mediated autophagy signaling pathway. PC12 cells are rat adrenal pheochromocytoma cells derived from Rattus norvegicus adrenal pheochromocytoma (Meiri et al., 1998; Greene and Tischler, 1976). PC12 cells do not synthesize epinephrine, and after cell differentiation is induced by nerve growth factor, PC12 cells can have neural cell characteristics and are a commonly used as a cell line for studying neural development and function. H9C2 is a rat cardiomyocyte, a subclonal cell line derived from embryonic BD1X rat heart tissue and is often used in the study of cardiomyopathy (Vaudry et al., 2002). The method of nerve cell or cardiomyocyte damage caused by oxygen–glucose deprivation/reperfusion is often used to simulate ischemia–reperfusion injury of nerve cells or cardiomyocytes in vitro experiments. This method is ideal for studying central nerve cell and cardiomyocyte ischemia-reperfusion injury in an in vitro model (Globus et al., 1988).
Therefore, this project intends to use PC12 cells and H9C2 hypoxia–reoxygenation injury models to explore the regulatory mechanism of HBO on the mTOR-mediated autophagy signaling pathway.
Dulbecco’s modified Eagle’s medium (DMEM) and fetal bovine serum (FBS) were purchased from Gibco (Grand Island, NY, USA). Rapamycin, an inhibitor of autophagy, was provided by Meilunbio (Meilunbio, Dalian, China). A Cell Counting Kit (CCK-8) was obtained from Dojindo (Dojindo, Shanghai, China). Hoechst 33342, propidium iodide (PI), and a mitochondrial membrane potential (MMP) assay kit were supplied by Nanjing Jiancheng Bioengineering Institute (Nanjing Jiancheng, Nanjing, China). Glutaraldehyde (2.5%) and DAPI solution were purchased from Solarbio Life Sciences (Solarbio, Beijing, China). Antibodies against 4E-BP1, Atg5, LC3B, m-TOR, p53, p-Akt and Caspase-3 were purchased from Abcam Inc. (Cambridge, UK).
Rat pheochromocytoma PC12 cells and rat cardiomyocyte H9C2 cells were purchased from American Type Culture Collection (ATCC, Maryland, USA). PC12 cells and H9C2 cells were cultured in medium consisting of DMEM, 10% fetal bovine serum (FBS) and 1% penicillin–streptomycin. The conditions of the cell incubator were 37°C with 5% CO2. Cells were divided into a control group, model group, hyperbaric air (HBA) group and HBO group. Cells in the control group were untreated. Cells in the model group were cultured in medium without sugar and serum and put into a 37°C incubator with a mixture of 95% N2 and 5% CO2 gas for 0.5 h for oxygen sugar deprivation. Then, the cells were taken out from the incubator. Sugar-free and serum-free medium was removed. Cells were cultured in complete medium and placed in a 37°C incubator with a mixture of 95% O2 and 5% CO2 for 24 h. Cells in the HBO group were placed in a hyperbaric chamber (Yantai Hongyuan Co., Ltd., China) sterilized by ultraviolet rays and purged with pure oxygen. Then, the pressure was increased to 0.25 MPa at a rate of 100 kPa/min and received treatment with pure oxygen for 90 min/day. After treatment for 3 days, cells were collected for assays. Cells in the rapamycin (Rap) group were treated with rapamycin (300 nM). Cells in the Rap + HBO group were simultaneously treated with rapamycin (300 nM) and pure oxygen. The conditions of cells in the HBA group were the same as those in the HBO group in addition to hyperbaric air. The cells of HBO, HBA, Rap, Rap + HBO groups have the same treatment as model group before treating with HBO, HBA and Rap.
Cell viability was measured by using the CCK8 assay. After cells in each group received treatment accordingly, 10 μL of CCK-8 solution was added to each well followed by 2 h of incubation in a 37°C incubator. The absorbance of the plate was then measured at 450 nm by using a microplate reader. Cell viability was expressed as the percentage of control (nontreated) cells.
Hoechst 33342 and PI staining assay
Hoechst 33342 is a nuclear dye that can penetrate cell membranes and insert nuclear DNA. It can enter normal cells in a small amount, making normal cells appear light blue under a fluorescence microscope and can enter apoptotic cells in a large amount, making apoptotic cells appear bright blue. PI can only stain the nucleus through cells with damaged cell membranes. Cells were cultured in 6-well plates (5 × 104 cells per well) for treatment accordingly. Then, the cells were washed twice with PBS, and 1 mL of Hoechst 33342 staining solution was added to each well and incubated in a 37°C incubator for 25 min. Next, the cells were washed twice with PBS, and PI (50 μg/mL) was added to each well and incubated in the dark for 15 min at room temperature. After washing twice with PBS, the cells were observed, and pictures were taken under an inverted fluorescence microscope.
Mitochondrial membrane potential (MMP) assay
The decrease in mitochondrial membrane potential is a landmark event in the early stage of cell apoptosis. The decrease in cell membrane potential can be easily detected by the transition from red fluorescence to green fluorescence of JC-1. At the same time, the transition from red fluorescence to green fluorescence of JC-1 can also be used as an indicator of early cell apoptosis. Cells were cultured in a 6-well plate for treatment accordingly. After washing once with PBS, 1 ml cell culture solution and 1 mL JC-1 staining working solution were added to each well. After incubating for 20 min at 37°C in a cell culture incubator, the supernatant was aspirated, and the cells were washed twice with JC-1 staining buffer (1×). Two milliliters of cell culture medium were added to each well, and the cells were observed under an inverted fluorescence microscope.
Observation of the ultrastructure by transmission electron microscopy (TEM)
Treated accordingly, cells were fixed in cold 3% glutaraldehyde for 2 h and then washed with phosphate-buffered saline (pH = 7.2). Next, the cells were postfixed in 1% osmium tetroxide for 2 h and then dehydrated step by step with ethanol and acetone. Subsequently, they were soaked with acetone and embedding agent overnight. Finally, they were stained with uranium acetate and lead citrate. The ultrastructure of cells, including autophagosomes, lysosomes, and apoptosis, were examined using a transmission electron microscope (Hitachi H-7650, Japan).
The expression of autophagy-related proteins was detected by cellular immunofluorescence. Then, the cells were washed three times with PBS, and goat serum was used for blocking for 30 min. Next, the cell slides were incubated with eIF4E-binding protein 1 (4E-BP1, 1:400), Atg5 (1:400), LC3B (1:400), and p53 (1:400) primary antibodies overnight at 4°C in a humid chamber. After washing three times with PBS, the cell slides were incubated with fluorescent secondary antibodies for 30 min at 37°C. Finally, the cell slides were washed three times with PBS and then stained with DAPI. Fluorescent films were photographed using a fluorescence microscope (Olympus, Germany).
The expression of autophagy-related proteins was detected by immunocytometry. The paraformaldehyde-fixed cell slides were washed three times with PBS for 5 min and then immersed in the permeabilization solution on ice for 2 min. Goat serum was added dropwise after rinsing three times with PBS, and the cell slides were incubated at room temperature in a humidified box for 35 min. Then, the cell slides were incubated with eIF4E-binding protein 1 (4E-BP1, 1:400), Atg5 (1:400), LC3B (1:400), p-Akt (1:400), p53 (1:400), m-TOR (1:400), and Caspase-3 (1:400) primary antibodies overnight at 4°C in a humid chamber. After washing three times with PBS (0.1 mol/L), the cell slides were incubated with secondary antibodies for 30 min at 37°C and then incubated with DAB. Finally, the cell slides were dehydrated, cleared, and fixed. Positive cells were stained brown and counted in five different fields of view for each slide by a pathological image analyzer system (Leica DM6000, Germany).
SPSS 19.0 software was used for statistical analysis. The data are expressed as the mean ± SEM. One-way analysis of variance (ANOVA) was performed for differences between groups. The significant level was set at P < 0.05.
Effects of HBO on PC12 and H9C2 cell viability
As shown in Fig. 1, compared with the control group, the PC12 and H9C2 cell viability of the model groups was significantly reduced (both P < 0.01). The PC12 and H9C2 cell activities of the HBO groups were significantly higher than those of the model groups (both P < 0.01). As shown in Figs. 1B and 1D, like the HBO group, the PC12 and H9C2 cell activities of the rapamycin (Rap) groups were significantly higher than those of the model groups (both P < 0.01). Particularly, PC12 and H9C2 cell activities were higher than those of the HBO groups or the rapamycin groups after being treated with rapamycin and HBO at the same time. These results indicated that HBO protect cells from damage by inhibiting autophagy.
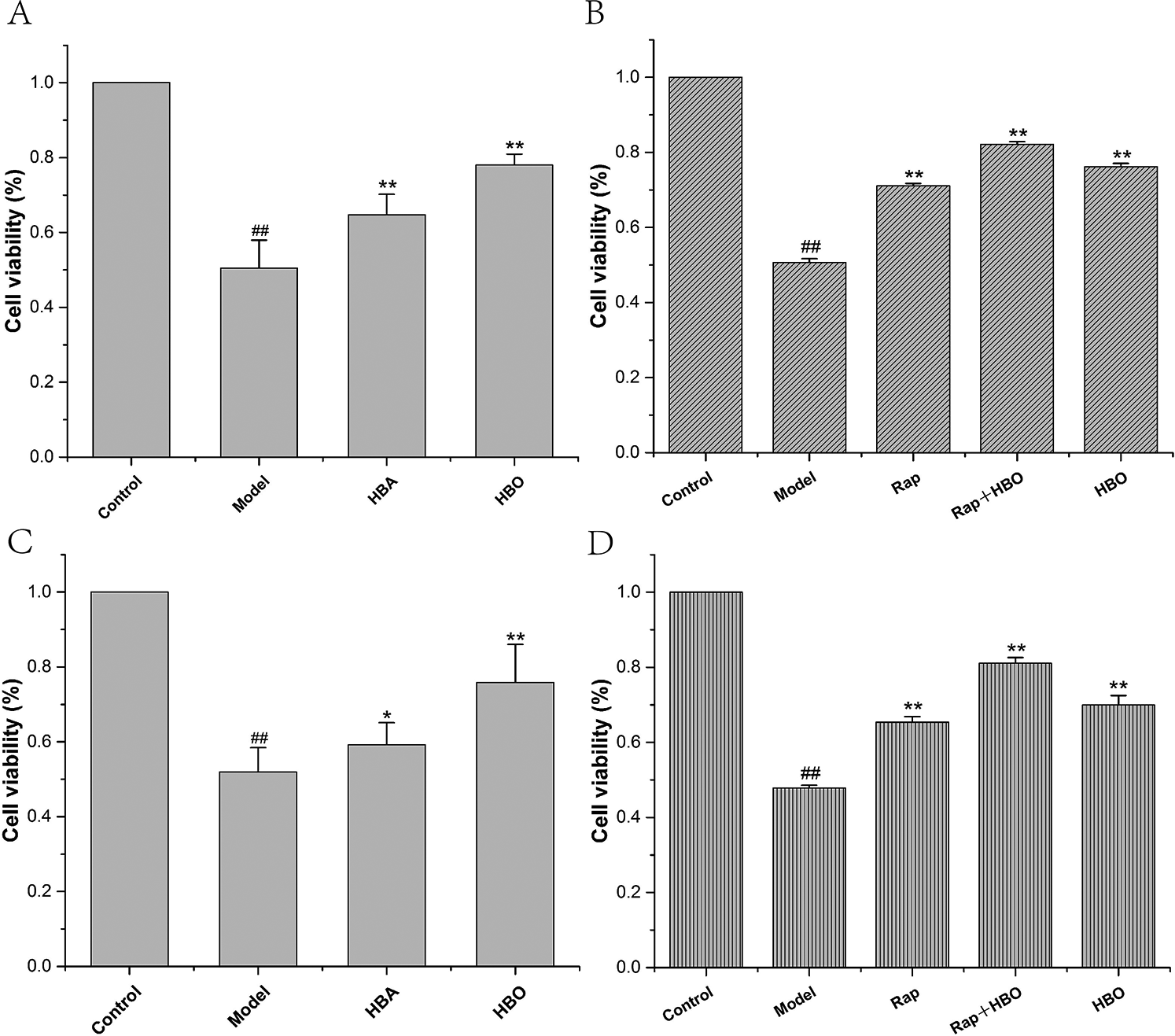
Figure 1: Effects of HBO on PC12 (A, B) and H9C2 (C, D) cell viability. The results are presented as the mean ± SEM (N = 5). ##P < 0.01 compared with the control group, *P < 0.05, **P < 0.01 compared with the model group.
Effect of HBO on oxygen-glucose deprivation/reperfusion-induced apoptosis in PC12 and H9C2 cells
The PC12 and H9C2 cells in the model group showed typical characteristics of apoptosis using Hoechst 33342 staining and PI staining. Under a fluorescence microscope, apoptotic cells stained with Hoechst 33342 showed bright blue fluorescence. As shown in Fig. 2, the apoptotic cells of PC12 and H9C2 in the model groups increased significantly compared with the control group and the apoptotic cells of PC12 and H9C2 in HBO groups reduced significantly. These results suggested HBO effectively inhibited oxygen-glucose deprivation/reperfusion-induced apoptosis in PC12 and H9C2 cells.
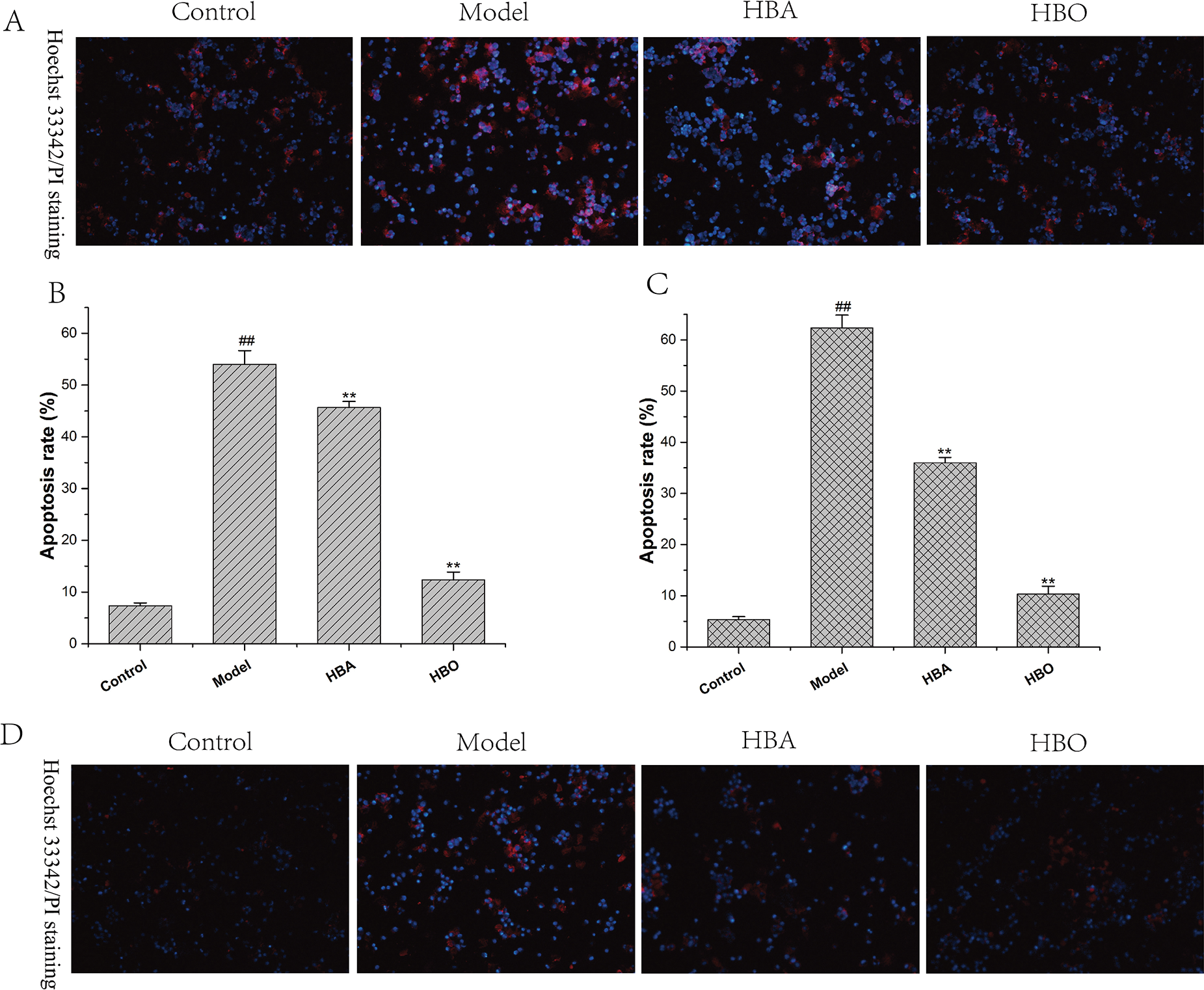
Figure 2: Effect of HBO on ischemia-reperfusion injury-induced apoptosis in PC12 (A, B) and H9C2 (C, D) cells by Hoechst 33342 and PI staining (×200). The results are presented as the mean ± SEM (N = 5). ##P < 0.01 compared with the control group, **P < 0.01 compared with the model group.
Effect of HBO on oxygen-glucose deprivation/reperfusion-induced MMP in PC12 and H9C2 cells
In normal cells, the mitochondrial membrane potential is higher, and JC-1 aggregates in the matrix of the mitochondria to form J-aggregates, which can produce orange-red fluorescence. However, in apoptotic cells, the mitochondrial membrane potential is lower, and JC-1 cannot accumulate in the matrix of the mitochondria. At this time, JC-1 is a monomer and can produce green fluorescence. As shown in Figs. 3A and 3B, PC12 and H9C2 cells in the model groups exhibited a marked decrease in the red/green fluorescence ratio compared with the control group. In contrast, PC12 and H9C2 cells in the HBO groups showed an increase in the red/green fluorescence ratio compared with the model groups. This showed HBO can inhibit ischemia-reperfusion injury-induced apoptosis in PC12 and H9C2 cells.
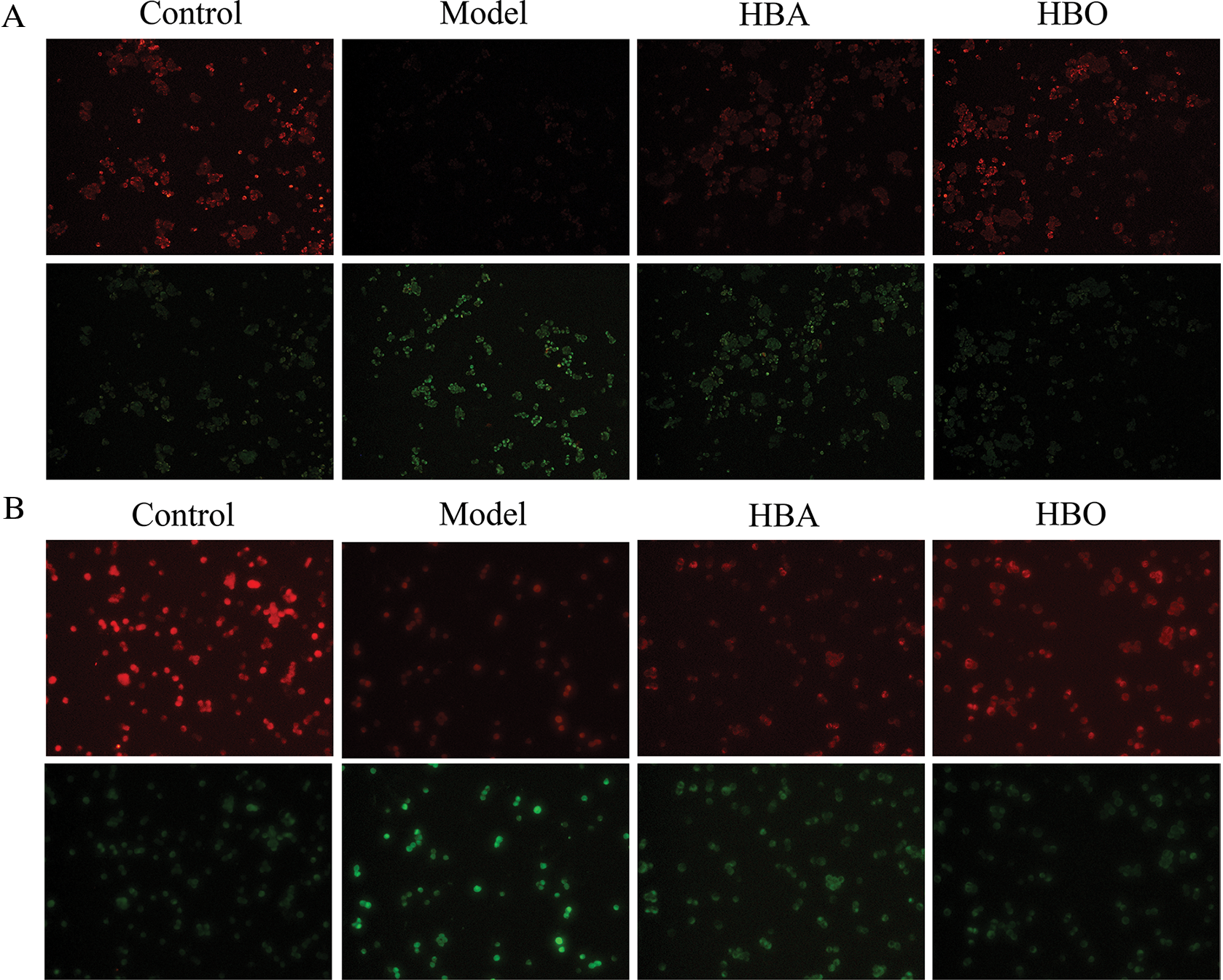
Figure 3: Effect of HBO on ischemia-reperfusion injury-induced MMP in PC12 (A) and H9C2 (B) cells by JC-1 (×200).
Effects of HBO on the ultrastructure of PC12 and H9C2 cells
The purpose of observing the ultrastructure of cells by transmission electron microscopy was to explore the effect of HBO on cell apoptosis and autophagy. As shown in Figs. 4 and 5, in the control group, the cell membrane was intact, and a normal number and structure of organelles, including lysosomes, mitochondria, and endoplasmic reticulum, were observed. In the model group, typical characteristics of apoptosis were observed, including cell membrane rupture and cell necrosis and vacuolization. In addition, the number of mitochondria, lysosomes, and autophagic vesicles increased significantly. Moreover, in the HBO group, fewer autophagic vacuoles, restoration of organelle rupture and reduction of apoptosis were observed. These results suggested HBO effectively inhibited oxygen-glucose deprivation/reperfusion-induced apoptosis and autophagy in PC12 and H9C2 cells.
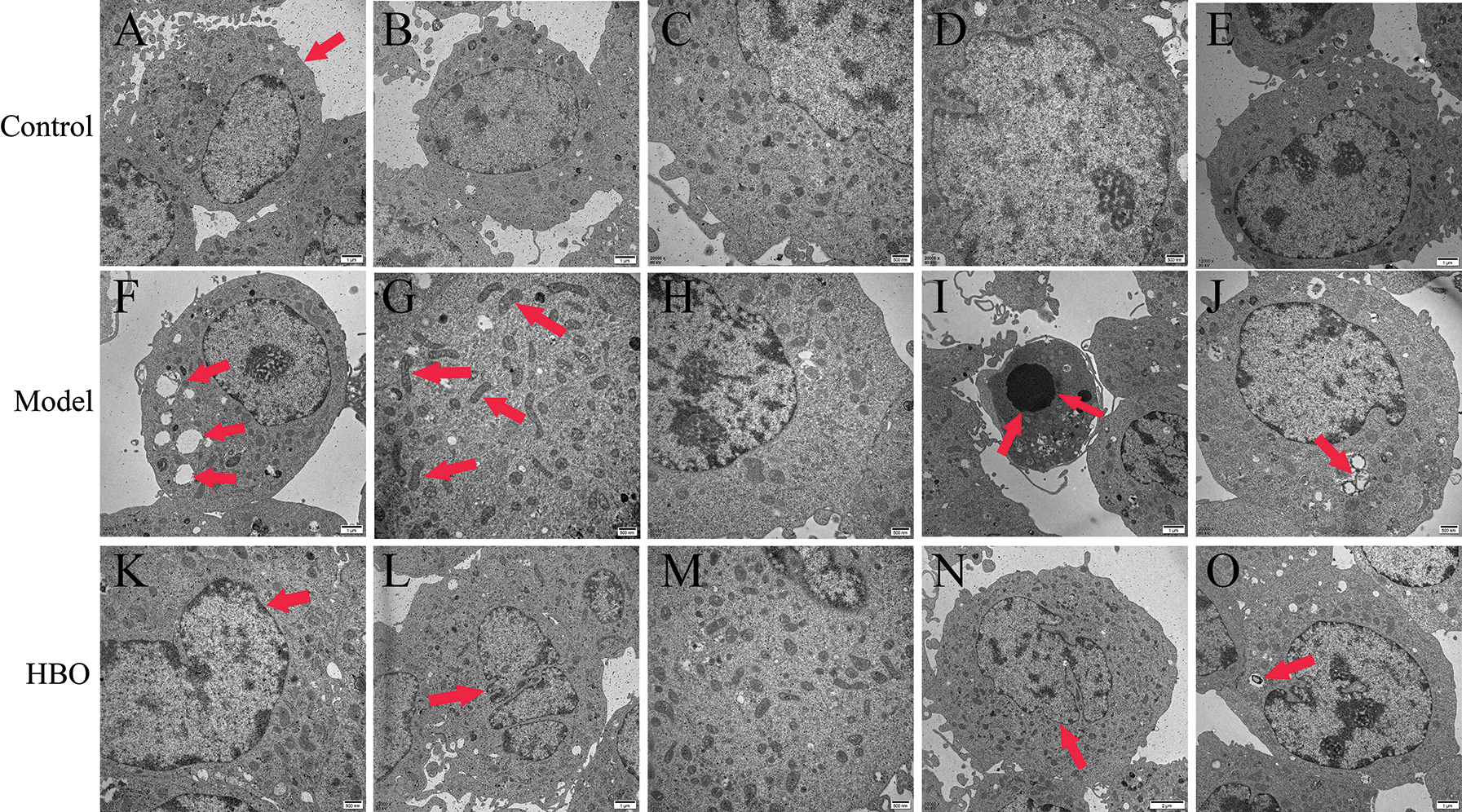
Figure 4: Effects of HBO on the ultrastructure of PC12 cells by transmission electron microscopy. (A) Cell membrane was normal. (B, C, D, and E) Mitochondria, lysosomes, endoplasmic reticulum, and other organelles were normal. (F) Cells were necrotic and vacuolated. (G) Mitochondria were increased. (H) Endoplasmic reticulum was reduced. (I) Cells were apoptotic. (J) Autophagic vesicles were massively increased. (K) The cell membrane was basically normal. (L and M) The cells were roughly normal. (N) The nucleus is slightly degenerated. (O) Autophagic vesicles were gradually reduced.
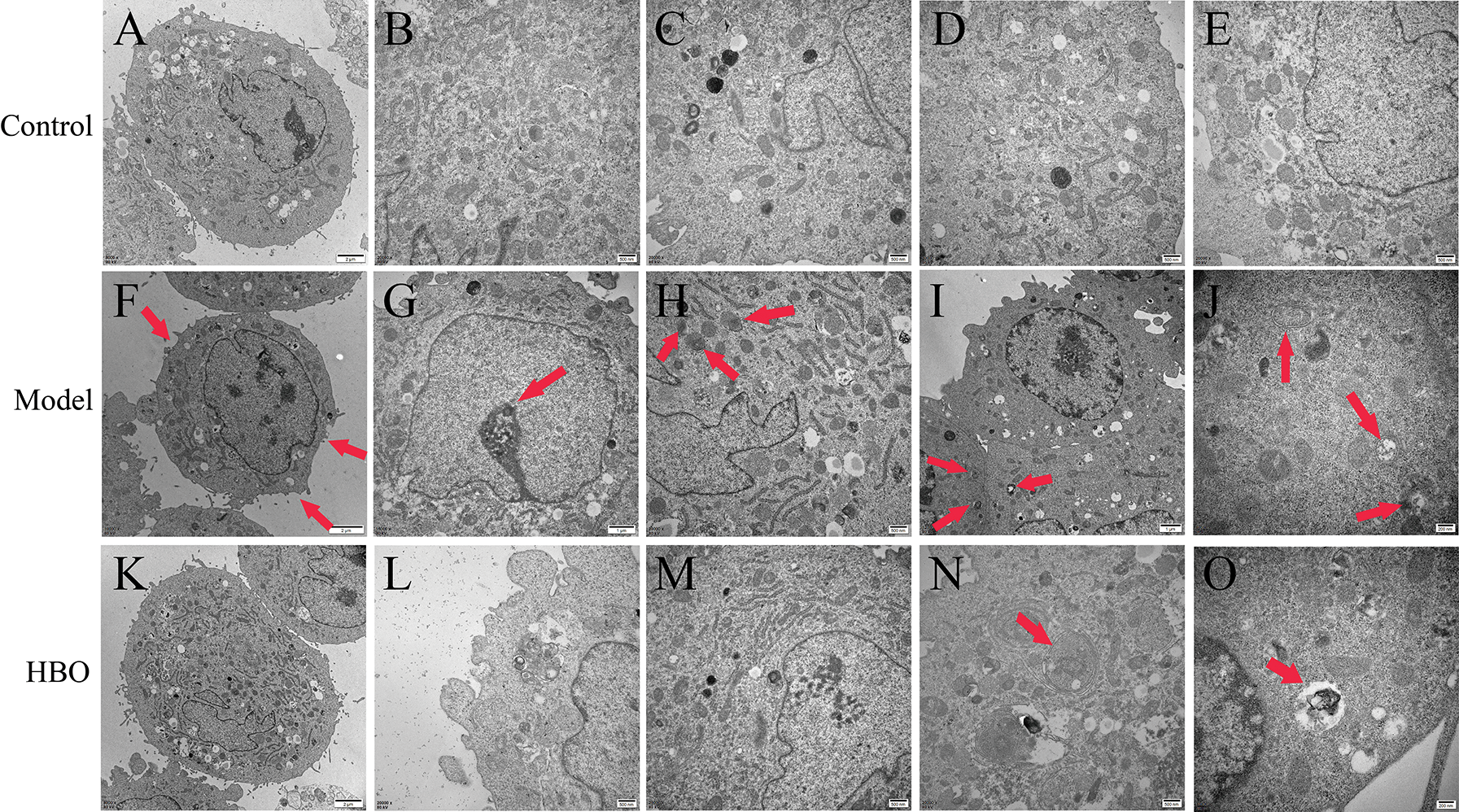
Figure 5: Effects of HBO on the ultrastructure of H9C2 cells by transmission electron microscopy. (A) The cell membrane was intact. (B, C) Mitochondria and lysosomes were normal. (D and E) The endoplasmic reticulum and lysosome were normal. (F) The cell membrane was ruptured. (G) The nucleus was apoptotic. (H) Lysosomes were autophagy. (I and J) Autophagic vesicles were greatly increased. (K) The cell membrane was basically normal. (L) Lysosomes were normal. (M) The endoplasmic reticulum and mitochondria were basically normal. (N and O) Autophagic vesicles were gradually reduced.
Effects of HBO on the expression of autophagy-related proteins by cellular immunofluorescence
The 4E-BP1, Atg5, LC3B and p53 proteins were stained green via cellular immunofluorescence, and the intensity of fluorescence was used to indicate the expression of protein. As shown in Figs. 6A and 6B, compared with the control group, the fluorescence intensity of 4E-BP1 significantly decreased and the fluorescence intensity of Atg5, LC3B and p53 significantly increased in the model group. In contrast, the fluorescence intensity of 4E-BP1 significantly increased, and the fluorescence intensity of Atg5, LC3B and p53 significantly decreased in the HBO group compared with the model group. These results indicated that HBO can reversely regulate the changes in key autophagy factors caused by oxygen-glucose deprivation/reperfusion in PC12 and H9C2 cells.
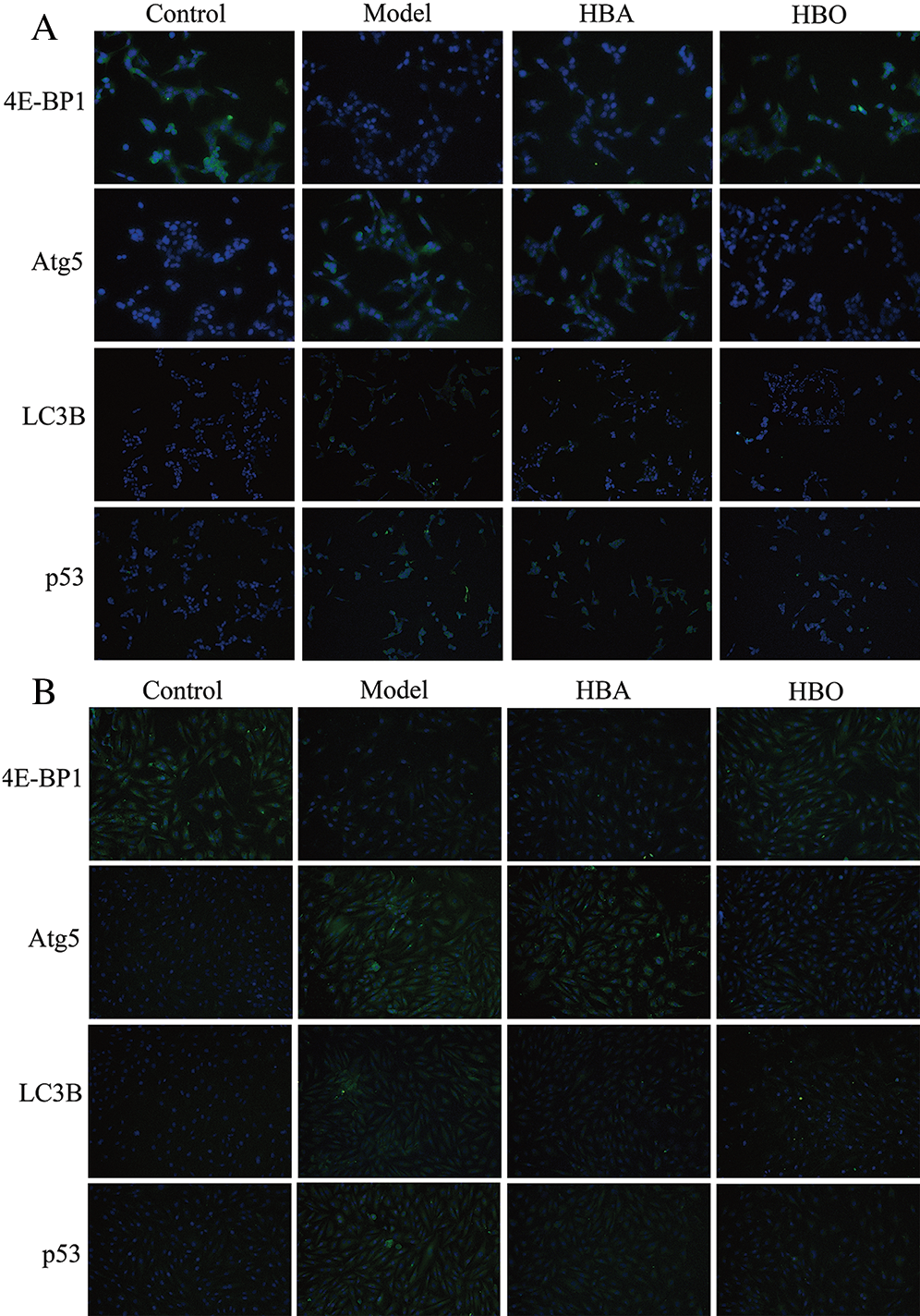
Figure 6: Effects of HBO on the expression of autophagy-related proteins in PC12 (A) and H9C2 (B) cells by cellular immunofluorescence (×400).
Effects of HBO on the expression of autophagy- related proteins by immunocytochemistry
The 4E-BP1, Atg5, LC3B, m-TOR, p53, p-Akt and Caspase-3 proteins were mainly present in the cytoplasm and stained brown or yellow via immunohistochemical staining. As shown in Figs. 7 and 8, compared with the control group, the expressions of 4E-BP1, p-Akt and m-TOR were significantly downregulated (P < 0.01 or P < 0.05), and the expressions of Atg5, LC3B, p53 and Caspase-3 were significantly upregulated (P < 0.01 or P < 0.05) in the model group. In contrast, the expressions of 4E-BP1, p-Akt and m-TOR were significantly upregulated (P < 0.01 or P < 0.05), and the expression of Atg5, LC3B, p53 and Caspase-3 (P < 0.01 or P < 0.05) were significantly downregulated in the HBO group compared with the model group. These results suggested HBO protected PC12 and H9C2 cells from damage and apoptosis by regulating the mTOR-mediated autophagy signaling pathway.
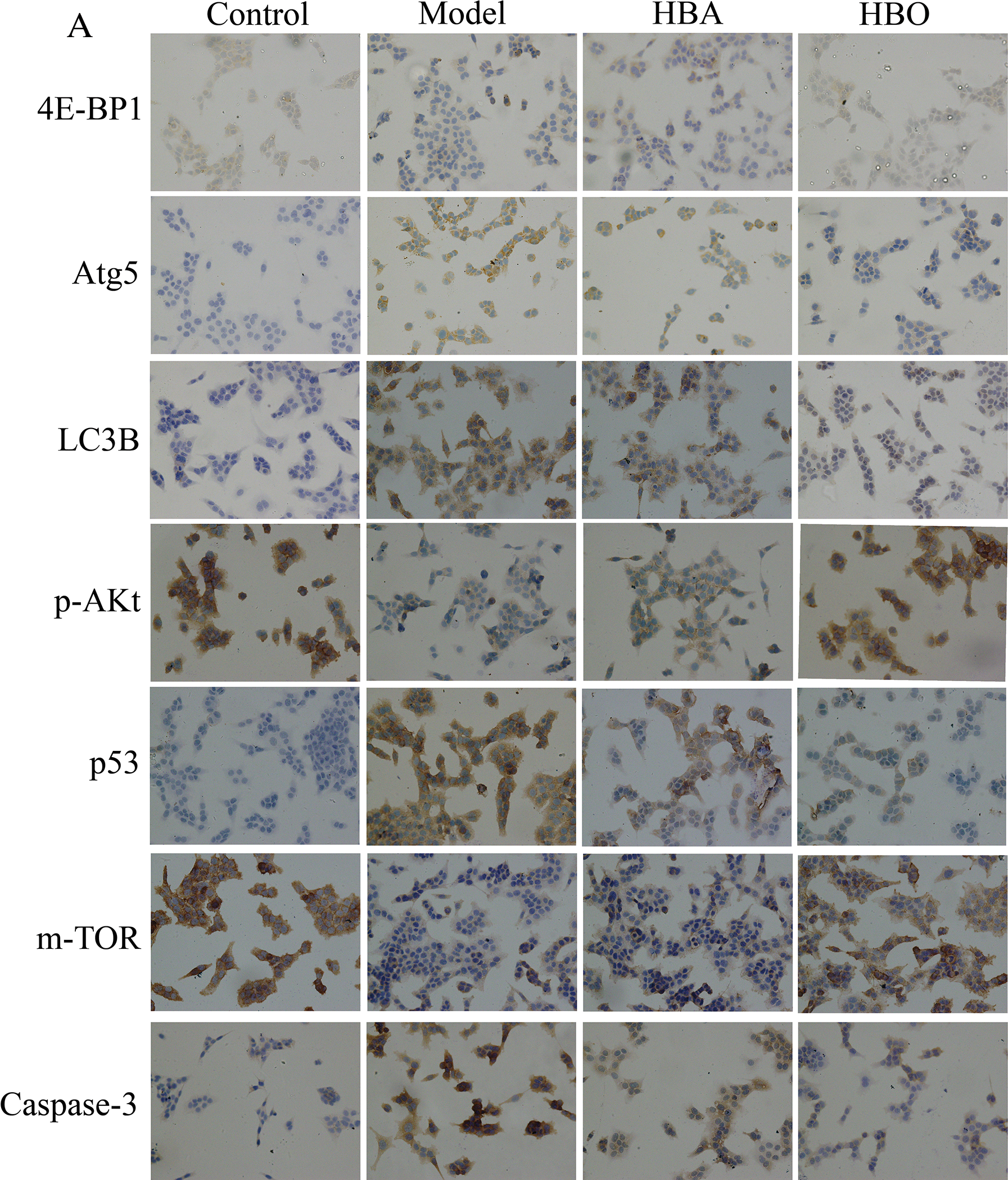
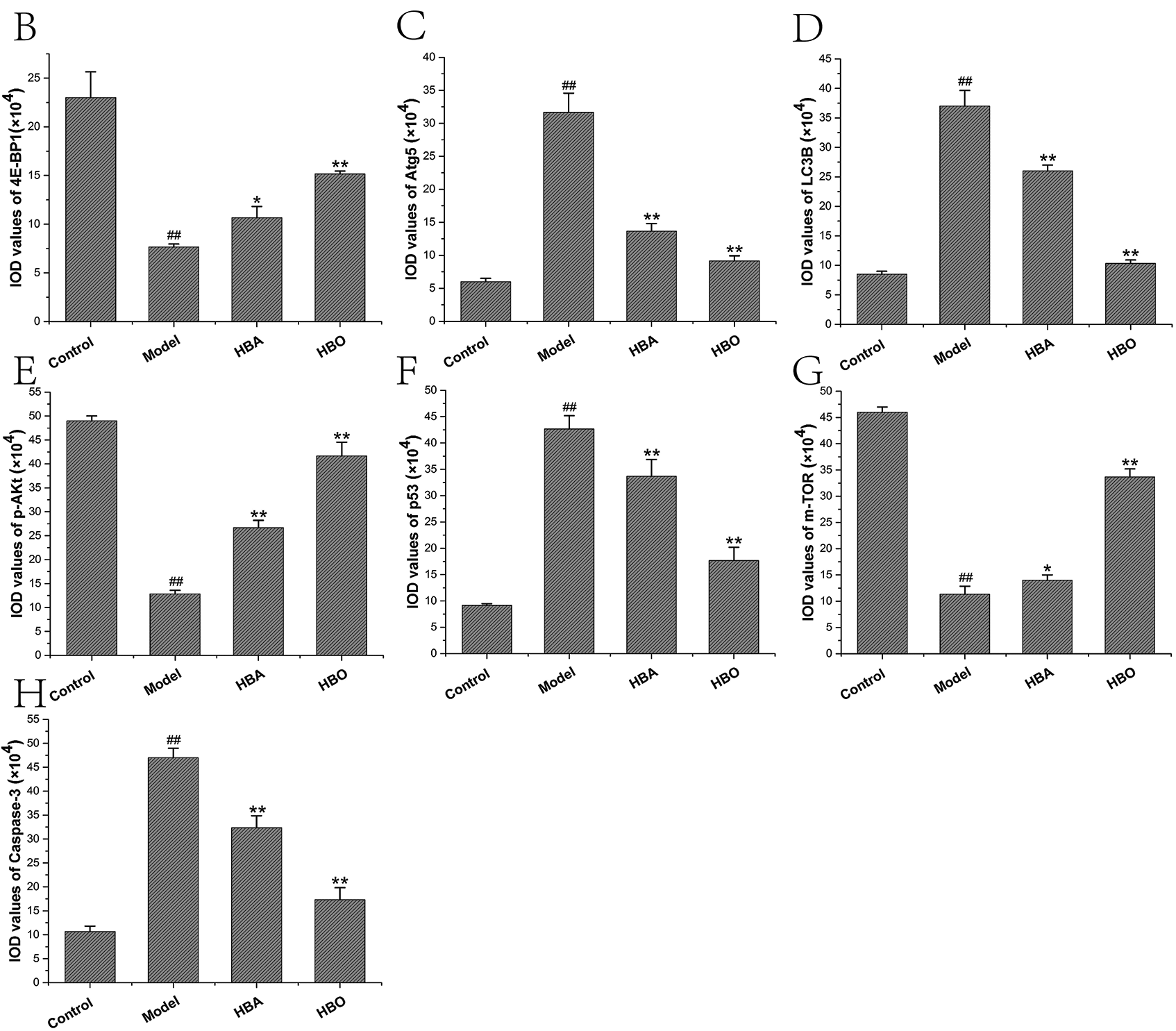
Figure 7: Effects of HBO on the expression of autophagy-related proteins in PC12 cells by immunocytochemistry. The results are presented as the mean ± SEM (N = 5). ##P < 0.01 compared with the control group, *P < 0.05, **P < 0.01 compared with the model group.
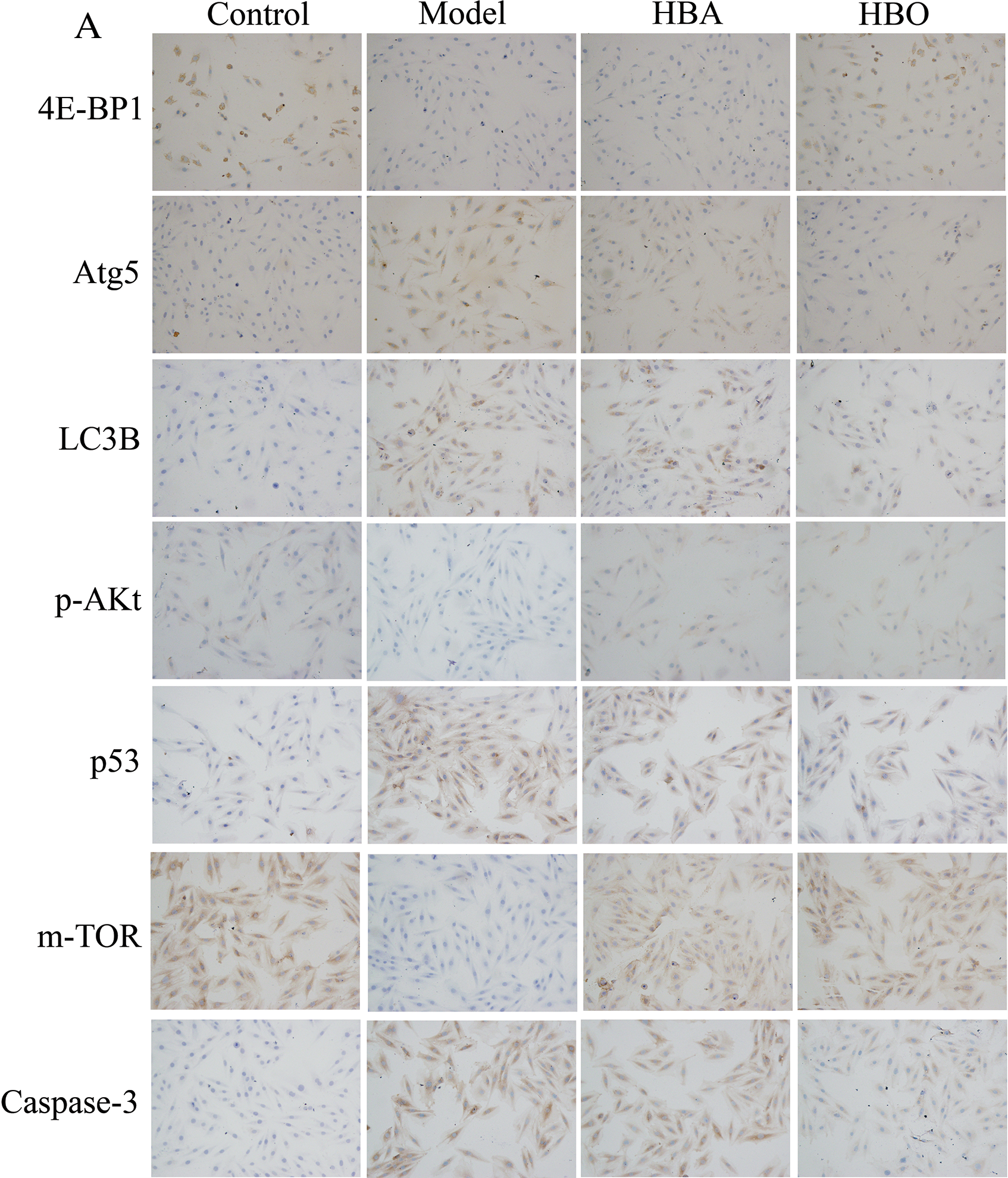
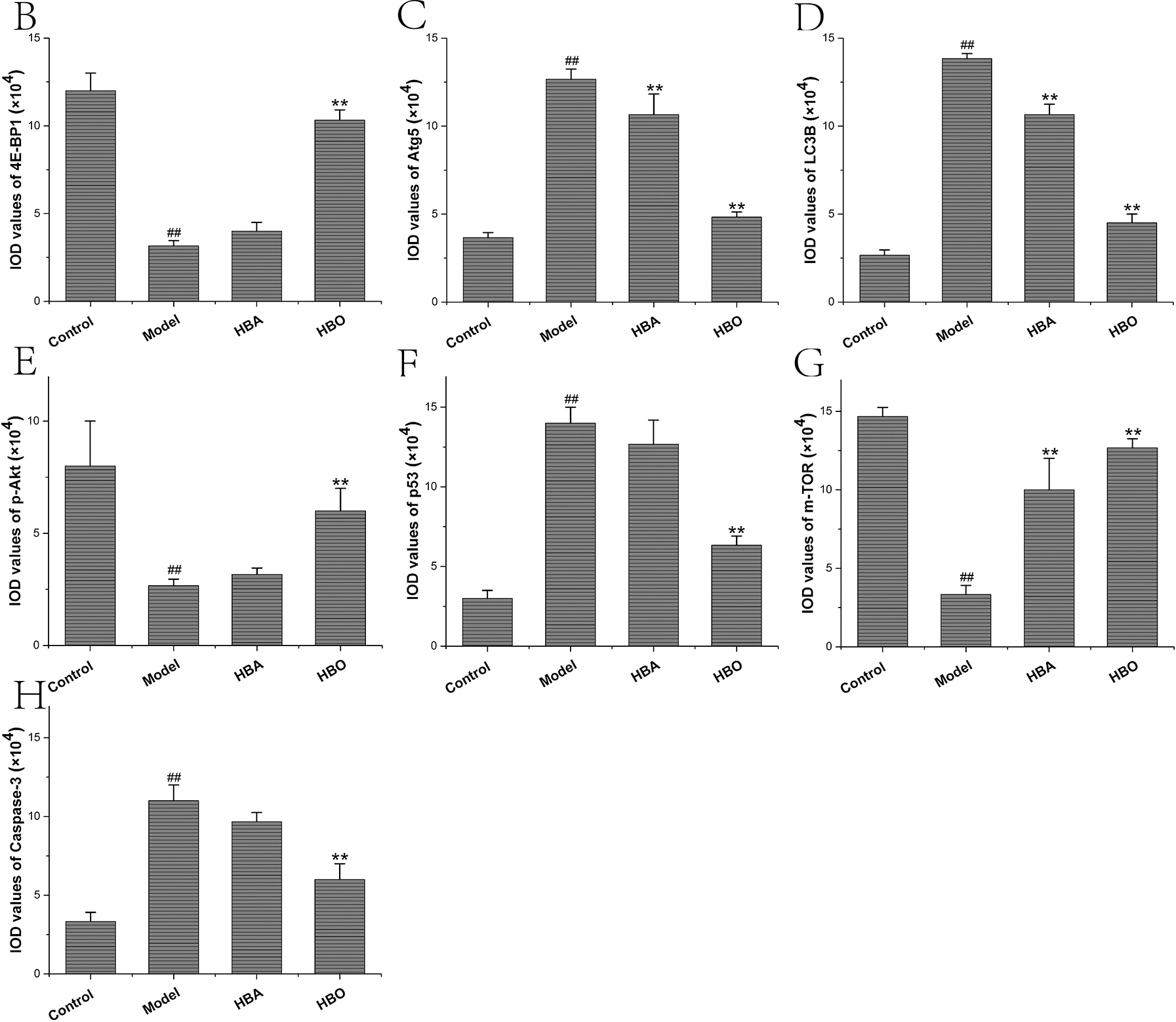
Figure 8: Effects of HBO on the expression of autophagy-related proteins in H9C2 cells by immunocytochemistry. The results are presented as the mean ± SEM (N = 5). ##P < 0.01 compared with the control group, *P < 0.05, **P < 0.01 compared with the model group.
In this study, oxygen-glucose deprivation/reperfusion model in PC12 cells and H9C2 cells were constructed and divided into a control group, oxygen-glucose deprivation/reperfusion injury model group, HBA group and HBO group. In this experiment, obvious damage to PC12 cells and H9C2 cells caused by oxygen glucose deprivation/reperfusion was observed. Under the microscope, the cells were swollen, some cells lost their original shape, and adhesion was significantly reduced. Some cells were lysed, and some cells gradually underwent apoptosis or even died. Cell viability was detected by the CCK-8 assay, and the results showed that the viability of PC12 cells and H9C2 cells in the model group was significantly decreased. Compared with the model group, the cell viability of the HBA group and HBO group was significantly increased, especially the HBO group. Like the HBO group, the PC12 and H9C2 cell activities of the rapamycin (Rap) groups were significantly higher than those of the model groups (both P < 0.01). Particularly, PC12 and H9C2 cell activities were higher than those of the HBO groups or the rapamycin groups after being treated with rapamycin and HBO at the same time. These results indicated that HBO may protect cells from damage by inhibiting autophagy. The Hoechst 33324-PI and JC-1 experimental results showed that the number of apoptotic PC12 and H9C2 cells in the model group increased significantly, while the number of apoptotic PC12 and H9C2 cells in the HBO group decreased significantly. Observation of the cell ultrastructure with transmission electron microscopy found that in the model group, typical characteristics of cell apoptosis were observed, including cell membrane ruptures, cell necrosis and vacuolization. In addition, the number of mitochondria and lysosomes increased significantly. In addition, in the HBO group, restoration of organelle rupture and reduction of apoptosis were observed. These results suggest that HBO intervention can inhibit the apoptosis of PC12 cells and H9C2 cells caused by oxygen-glucose deprivation/reperfusion injury.
Apoptosis, also called programmed cell death, is characterized by typical morphology, including chromatin condensation, the formation of apoptotic bodies, and the controlled degradation of chromosomal DNA. The importance of caspases in the process of neuronal death has been recognized. Caspase-3 and caspase-9 are the two main types of caspases involved in apoptosis, and some caspases, such as caspase-11 and caspase-12, are only activated under pathological conditions (Baturcam et al., 2017; Kang et al., 2000; Zhang et al., 2016). There is much evidence that nerve ischemia can activate caspases. In animal models of focal and global cerebral ischemia, caspase-3 activity can be upregulated before neuronal death. Within a few hours of stroke caused by atherosclerosis, caspase-3 precursor levels can be seen to increase. In transient and permanent neuroischemia, caspase-9 activates caspase-3, all of which strongly suggest that after cerebral ischemia, the mechanism of the mitochondrial-mediated apoptosis pathway involves caspases. Caspase-3 is considered to be the key executor of nuclear degradation, and it may be caused by DNA fragmentation caused by DNase activated by caspases (Sakurai et al., 2003). The immunohistochemistry results showed that caspase-3 in the normal group was basically not expressed in the cells, and caspase-3 protein expression in PC12 cells and H9C2 cells in the model group was significantly increased. Compared with the model group, the caspase-3 protein expression of PC12 cells and H9C2 cells in the HBO group was significantly reduced. Autophagy plays an important role in the regulation of cell survival and cell death, especially in the apoptosis signaling pathway. Evidence that autophagy regulates apoptosis includes mitochondrial phagocytosis. Autophagy can selectively degrade damaged mitochondria and maintain mitochondrial homeostasis. It is well known that the mitochondrial-dependent (endogenous) pathway is an important mechanism of apoptotic cell death (Bursch, 2001; Shimizu et al., 2004; Yu et al., 2004). p53 can co-regulate autophagy and apoptosis (Luo and Rubinsztein, 2010). p53 regulates the expression of apoptosis-related genes (Bcl-2 and Apaf1) and autophagy-related pathways (AMPK/mTOR and Bmf/Beclin-1) (Guo et al., 2014; Feng et al., 2005). In addition, p53 targets the expression of DRAM and can stimulate the interaction between apoptosis and autophagy (Crighton et al., 2006), which is critical to cell fate, but is complicated by their contradictory effects under certain conditions. Autophagy and apoptosis both affect each other as partners.
In recent years, the role of autophagy and autophagy–lysosomal pathways, in neurological diseases, has attracted increasing attention. Studies have confirmed that in the occurrence of VD, the mammalian target of rapamycin (mTOR)-mediated autophagy signaling pathway is involved in the pathological process of hippocampal neuronal apoptosis and necrosis (Hu et al., 2017; Jia et al., 2015). Previous studies have found that HBO has significant preventive effects in improving cognitive function and resisting myocardial ischemia reperfusion injury. Moreover, 4E-BP1 is regulated by the mTOR complex and participates in stimulating ribosomal proteins and encoding elongation factors (Tang et al., 2001). The ULK1 complex recruits LC3B and ATG5 to the surface of phagocytic vesicles to form autophagic vesicles (Tanida et al., 2004). Phosphate acyl-3-inositol kinase- protein kinase B (PI3K-Akt) and AMPK have been shown to be the main ways to initiate the mTOR pathway (Chen et al., 2019). In addition, Akt plays a role in regulating synaptic plasticity through its phosphorylation at Ser473 (Horwood et al., 2006; Noshita et al., 2003). In another AMPK pathway, nuclear p53 activates AMPK to accelerate mTOR-induced autophagy. In this study, the results of cellular immunofluorescence and immunocytochemistry experiments showed that the 4E-BP1, p-AKt and mTOR levels of PC12 cells and H9C2 cells in the model group decreased, while the levels of LC3B, Atg5 and p53 increased. However, after HBO treatment, these autophagy-related indexes were reversed. Similar results were observed in the HBA group, but the efficiency was lower than that of HBO treatment. In addition, observation of the cell ultrastructure with transmission electron microscopy found that in the model group, a significant increase in the number of autophagic vesicles was observed. In the HBO group, a decrease in autophagic vesicles was observed. These results indicate that the protective mechanism of PC12 and H9C2 cell damage caused by oxygen-glucose deprivation/reperfusion may be related to the inhibition of the autophagy pathway.
In summary, our findings indicate that hyperbaric oxygen can protect PC12 cells and H9C2 cells from damage caused by oxygen-glucose deprivation/reperfusion by inhibiting apoptosis and autophagy. Our results provide laboratory evidence to support that HBO may prevent clinical IRI. There are still some limitations in this study. Western blot results are easier to quantify than immunofluorescence or immunocytochemistry. However, because of a lack of time and funds, WB experiments cannot be carried out in this study. In the future, we will continue to conduct in-depth research and improve the experiment.
Acknowledgement: The authors would like to thank Dr. Jianquan Li from Guangxi Medical University, China, for his technical support in immunohistochemistry.
Availability of Data and Materials: The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Author Contributions: The authors confirm contribution to the paper as follows: study conception and design: Chunxia Chen, Wensheng Lu; data collection: Jianrong Yang, Wan Chen, Yaoxuan Li, Xiaorong Pan; analysis and interpretation of results: Xing Zhou, Yaoxuan Li, Zhihuang Nong, Liyuan Zhou, Xuan Wei; draft manuscript preparation: Jianrong Yang, Wan Chen, Xing Zhou. All authors reviewed the results and approved the final version of the manuscript.
Funding Statement: This study was supported by the National Natural Science Foundation of China (81960246, 81701089, 81560044 and 30860113), the Guangxi Natural Science Foundation (2020GXNSFAA238003 and 2017GXNSFBA198010), the Guangxi Medical and Health Appropriate Technology Research and Development Project (S2020076, S201422-01 and S2019087), and the Shanxi Health Research Project (2019165).
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
Baturcam E, Snape N, Yeo TH, Schagen J, Thomas E et al. (2017). Human metapneumovirus impairs apoptosis of nasal epithelial cells in asthma via HSP70. Journal of Innate Immunity 9: 52–64. [Google Scholar]
Bennett MH, Lehm JP, Jepson N (2015). Hyperbaric oxygen therapy for acute coronary syndrome. Cochrane Database Systematic Reviews 7: CD004818. [Google Scholar]
Bramham CR, Jensen KB, Proud CG (2016). Tuning specific translation in cancer metastasis and synaptic memory: Control at the MNK-eIF4E axis. Trends in Biochemical Sciences 41: 847–858. [Google Scholar]
Buja LM (2005). Myocardial ischemia and reperfusion injury. Cardiovascular Pathology 14: 170–175. [Google Scholar]
Bursch W (2001). The autophagosomal-lysosomal compartment in programmed cell death. Cell Death & Differentiation 8: 569–581. [Google Scholar]
Chen CX, Chen W, Nong ZH, Ma Y, Qiu SL, Wu GW (2016a). Cardioprotective effects of combined therapy with hyperbaric oxygen and diltiazem pretreatment on myocardial ischemia-reperfusion injury in rats. Cellular Physiology and Biochemistry 38: 2015–2029. [Google Scholar]
Chen CX, Huang LY, Nong ZH, Li YX, Chen W et al. (2017). Hyperbaric oxygen prevents cognitive impairments in mice induced by D-galactose by improving cholinergic and anti- apoptotic functions. Neurochemical Research 42: 1240–1253. [Google Scholar]
Chen XW, Li C, Chen Y, Ni C, Chen XX et al. (2019). Aflatoxin B1 impairs Leydig cells through inhibiting AMPK/mTOR mediated autophagy flux pathway. Chemosphere 233: 261–272. [Google Scholar]
Chen XY, Li YX, Chen W, Nong ZH, Huang JP, Chen CX (2016b). Protective effect of hyperbaric oxygen on cognitive impairment induced by D-Galactose in mice. Neurochemical Research 41: 3032–3041. [Google Scholar]
Crighton D, Wilkinson S, O’Prey J, Syed N, Smith P et al. (2006). DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 126: 121–134. [Google Scholar]
Dekleva M, Neskovic A, Vlahovic A, Putnikovic B, Beleslin B et al. (2004). Adjunctive effect of hyperbaric oxygen treatment after thrombolysis on left ventricular function in patients with acute myocardial infarction. American Heart Journal 148: 031. [Google Scholar]
Feng Z, Zhang H, Levine AJ, Jin S (2005). The coordinate regulation of the p53 and mTOR pathways in cells. Proceedings of the National Academy of Sciences of the United States of America 102: 8204–8209. [Google Scholar]
Ferro DA, van den Brink H, Exalto LG, Boomsma JMF, Barkhof F et al. (2019). Clinical relevance of acute cerebral microinfarcts in vascular cognitive impairment. Neurology 92: e1558–e1566. [Google Scholar]
Foster SS, de S, Johnson LK, Petrini JH, Stracker TH (2012). Cell cycle- and DNA repair pathway-specific effects of apoptosis on tumor suppression. Proceedings of the National Academy of Sciences of the United States of America 109: 9953–9958. [Google Scholar]
Globus MY, Busto R, Dietrich WD, Martinez E, Valdes I et al. (1988). Effect of ischemia on the in vivo release of striatal dopamine, glutamate, and γ-aminobutyric acid studied by intracerebral microdialysis. Journal of Neurochemistry 51: 1455–1464. [Google Scholar]
Greene LA, Tischler AS (1976). Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proceedings of the National Academy of Sciences of the United States of America 73: 2424–2428. [Google Scholar]
Guo W, Yan L, Yang L, Liu X, Qiukai E et al. (2014). Targeting GRP75 improves HSP90 inhibitor efficacy by enhancing p53-mediated apoptosis in hepatocellular carcinoma. PLoS One 9: e85766. [Google Scholar]
Han YF, Zhao YB, Li J, Li L, Li YG et al. (2018). Stat3-Atg5 signal axis inducing autophagy to alleviate hepatic ischemia-reperfusion injury. Journal of Cellular Biochemistry 119: 3440–3450. [Google Scholar]
Hao M, Zhu S, Hu L, Zhu H, Wu X, Li Q (2017). Myocardial ischemic postconditioning promotes autophagy against ischemia reperfusion injury via the activation of the nNOS/AMPK/mTOR pathway. International Journal of Molecular Sciences 18: 614. [Google Scholar]
Hariharan N, Zhai P, Sadoshima J (2011). Oxidative stress stimulates autophagic flux during ischemia/reperfusion. Antioxidants & Redox Signaling 14: 2179–2190. [Google Scholar]
Harper L, Barkhof F, Scheltens P, Schott JM, Fox NC (2014). An algorithmic approach to structural imaging in dementia. Journal of Neurology, Neurosurgery & Psychiatry 85: 692–698. [Google Scholar]
Horwood JM, Dufour F, Laroche S, Davis S (2006). Signalling mechanisms mediated by the phosphoinositide 3-kinase/Akt cascade in synaptic plasticity and memory in the rat. European Journal of Neuroscience 23: 3375–3384. [Google Scholar]
Hu M, Liu ZJ, Lv PY, Wang HB, Zhu YF et al. (2017). Autophagy and Akt/CREB signalling play an important role in the neuroprotective effect of nimodipine in a rat model of vascular dementia. Behavioural Brain Research 4328: 31178. [Google Scholar]
Ibáñez B, Heusch G, Ovize M, van de Werf F (2007). Evolving therapies for myocardial ischemia/reperfusion injury. Journal of the American College of Cardiology 65: 1454–1471. [Google Scholar]
Jia YQ, Jin W, Xiao YN, Dong YH, Wang TJ et al. (2015). Lipoxin A4 methyl ester alleviates vascular cognition impairment by regulating the expression of proteins related to autophagy and ER stress in the rat hippocampus. Cellular and Molecular Biology Letters 20: 475–487. [Google Scholar]
Kang R, Zeh HJ, Lotze MT, Tang D (2011). The Beclin 1 network regulates autophagy and apoptosis. Cell Death & Differentiation 18: 571–580. [Google Scholar]
Kang SJ, Wang S, Hara H, Peterson EP, Namura S et al. (2000). Dual role of caspase-11 in mediating activation of caspase-1 and caspase-3 under pathological conditions. Journal of Cell Biology 149: 613–622. [Google Scholar]
Kivimäki M, Jokela M, Nyberg ST, Singh-Manoux A, Fransson EI et al. (2015). Long working hours and risk of coronary heart disease and stroke: A systematic review and meta-analysis of published and Unpublished data for 603,838 individuals. Lancet 386: 1739–1746. [Google Scholar]
Levisman J, Price MJ (2015). Update on the guidelines for the management of ST-elevation myocardial infarction. American Journal of Cardiology 115: A3–A9. [Google Scholar]
Liu WC, Yang SN, Wu CW, Chen LW, Chan JY (2016). Hyperbaric oxygen therapy alleviates carbon monoxide poisoning-induced delayed memory impairment by preserving brain-derived neurotrophic factor-dependent hippocampal neurogenesis. Critical Care Medicine 44: e25–39. [Google Scholar]
Luo S, Rubinsztein DC (2010). Apoptosis blocks Beclin 1- dependent autophagosome synthesis: An effect rescued by Bcl-xL. Cell Death & Differentiation 17: 268–277. [Google Scholar]
Meiri KF, Saffell JL, Walsh FS, Doherty P (1998). Neurite outgrowth stimulated by neural cell adhesion molecules requires growth-associated protein-43 (GAP-43) function and is associated with GAP-43 phosphorylation in growth cones. Journal of Neuroscience 18: 10429–10437. [Google Scholar]
Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ et al. (2016). Executive summary: Heart disease and stroke statistics-2016 update: A report from the American Heart Association. Circulation 133: 447–454. [Google Scholar]
Nabel EG, Braunwald E (2012). A tale of coronary artery disease and myocardial infarction. New England Journal of Medicine 366: 54–63. [Google Scholar]
Noshita N, Sugawara T, Lewen A, Hayashi T, Chan PH (2003). Copper-zinc superoxide dismutase affects Akt activation after transient focal cerebral ischemia in mice. Stroke 34: 1513–1518. [Google Scholar]
Pan XR, Chen CX, Huang JP, Wei HM, Fan QP (2015). Neuroprotective effect of combined therapy with hyperbaric oxygen and madopar on 6-hydroxydopamine-induced Parkinson’s disease in rats. Neuroscience Letters 600: 220–225. [Google Scholar]
Pópulo H, Lopes JM, Soares P (2012). The mTOR signalling pathway in human cancer. International Journal of Molecular Sciences 13: 1886–1918. [Google Scholar]
Qin L, Wang Z, Tao L, Wang Y (2010). ER stress negatively regulates AKT/TSC/mTOR pathway to enhance autophagy. Autophagy 6: 239–247. [Google Scholar]
Rusyniak DE, Kirk MA, May JD, Kao LW, Brizendine EJ et al. (2003). Hyperbaric oxygen therapy in acute ischemic stroke: Results of the hyperbaric oxygen in acute ischemic stroke trial pilot study. Stroke 34: 571–574. [Google Scholar]
Sakurai M, Nagata T, Abe K, Horinouchi T, Itoyama Y, Tabayashi K (2003). Survival and death-promoting events after transient spinal cord ischemia in rabbits: Induction of Akt and caspase3 in motor neurons. Journal of Thoracic and Cardiovascular Surgery 125: 370–377. [Google Scholar]
Sheng R, Liu XQ, Zhang LS, Gao B, Han R et al. (2012). Autophagy regulates endoplasmic reticulum stress in ischemic preconditioning. Autophagy 8: 310–325. [Google Scholar]
Shimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawa-Kobayashi S et al. (2004). Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nature Cell Biology 6: 1221–1228. [Google Scholar]
Tang H, Hornstein E, Stolovich M, Levy G, Templeton D et al. (2001). Amino acid- induced translation of TOP mRNAs is fully dependent on phosphatidylinositol 3-kinase-mediated signaling, is partially inhibited by rapamycin, and is independent of S6K1 and rpS6 phosphorylation. Molecular and Cellular Biology 21: 8671–8683. [Google Scholar]
Tanida I, Ueno T, Kominami E (2004). LC3 conjugation system in mammalian autophagy. International Journal of Biochemistry & Cell Biology 36: 2503–2518. [Google Scholar]
Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri-Mergny M et al. (2008). Regulation of autophagy by cytoplasmic p53. Nature Cell Biology 10: 676–687. [Google Scholar]
Vaudry D, Stork PJ, Lazarovici P, Eiden LE (2002). Signaling pathways for PC12 cell differentiation: Making the right connections. Science 296: 1648–1649. [Google Scholar]
Wang B, Zhong S, Zheng F, Zhang Y, Gao F et al. (2015). N-n-butyl haloperidol iodide protects cardiomyocytes against hypoxia/reoxygenation injury by inhibiting autophagy. Oncotarget 6: 24709–24721. [Google Scholar]
Wei K, Wang P, Miao CY (2012). A double-edged sword with therapeutic potential: An updated role of autophagy in ischemic cerebral injury. CNS Neuroscience & Therapeutics 18: 879–886. [Google Scholar]
Xuan F, Jian J (2016). Epigallocatechin gallate exerts protective effects against myocardial ischemia/reperfusion injury through the PI3K/Akt pathway-mediated inhibition of apoptosis and the restoration of the autophagic flux. International Journal of Molecular Medicine 38: 328–336. [Google Scholar]
Yao H, Han X (2014). The cardioprotection of the insulin- mediated PI3K/Akt/mTOR signaling pathway. American Journal of Cardiovascular Drugs 14: 433–442. [Google Scholar]
Yellon DM, Hausenloy DJ (2007). Myocardial reperfusion injury. New England Journal of Medicine 357: 1121–1135. [Google Scholar]
Yin B, Liang H, Chen YG, Chu KT, Huang L et al. (2013). EGB1212 post-treatment ameliorates hippocampal CA1 neuronal death and memory impairment induced by transient global cerebral ischemia/reperfusion. American Journal of Chinese Medicine 41: 1329–1341. [Google Scholar]
Yu L, Alva A, Su H, Dutt P, Freundt E et al. (2004). Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science 304: 1500–1502. [Google Scholar]
Yu L, McPhee CK, Zheng LX, Mardones GA, Rong YG et al. (2010). Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 465: 942–946. [Google Scholar]
Zhang DM, Zhang T, Wang MM, Wang QinXX, Wu YY et al. (2019). TIGAR alleviates ischemia/reperfusion-induced autophagy and ischemic brain injury. Free Radical Biology and Medicine 137: 1323. [Google Scholar]
Zhang Q, Liu JN, Chen SL, Liu J, Liu LJ et al. (2016). Caspase-12 is involved in stretch-induced apoptosis mediated endoplasmic reticulum stress. Apoptosis 21: 432–442. [Google Scholar]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |