DOI:10.32604/biocell.2022.015390
| BIOCELL DOI:10.32604/biocell.2022.015390 | |
| Article |
Identification of tumorigenic risk genes in human placenta-derived mesenchymal stem cells treated with 3-methylcholanthrene
Institute of Human Stem Cell Research, General Hospital of Ningxia Medical University, Yinchuan, 750004, China
*Address correspondence to: Xueyun Liang, liangxy2104@hotmail.com; Xiaoming Liu, erc1080@163.com
Received: 17 December 2020; Accepted: 04 March 2021
Abstract: Mesenchymal stem cells (MSCs) capable of tumour topotaxis have been served as cellular vehicles to deliver anti-tumour agents. As cellular components of the tumour microenvironment, MSCs also affect tumour progression. However, the tumour transformation-related genes of MSCs remain unclear since either tumorigenic or tumour suppressor effects within these cells have been researched. Hence, we aimed to identify potential biomarkers indicative of tumorigenic risk by RNA-seq analysis of human placenta tissue-derived MSCs (hPTMSCs) exposed to the carcinogenic agent, 3-methylcholanthrene (3-MC). Twenty-nine tumour transformation-related genes and three pluripotency-related genes were appraised as differentially expressed genes (DEGs) in hPTMSCs. Overexpression of sfrp1 led to reduced cell viability, migration, and colony formation in A549. In contrast, the overexpression of ptgs2 exerted the opposite effect. These results indicate that A549 cells with high ptgs2 expression but low sfrp1 expression may have a more potential tumorigenic capacity. Taken together, this study suggests that ptgs2 and sfrp1 may be tumorigenic risk genes.
Keywords: Human placenta-derived mesenchymal stem cells; Transcriptional profile; Tumorigenicity
Mesenchymal stem cells (MSCs) are worthy candidates for many clinical applications, such as tissue regeneration, immunomodulation, and cell therapy (Park et al., 2016). Their characteristics include reproductive and multi-directional differentiation ability in vitro and expression of MSC-characteristic antigens CD90, CD73, and CD105 (Dominici et al., 2006). MSCs can be isolated from bone marrow, cord blood, adipose tissue, and placentas (Tian et al., 2011). Of which, human placental tissue is available and plentiful for the acquisition of stem cells. Therefore, human placenta tissue-derived MSCs (hPTMSCs) have become an attractive source for cell therapy. The safety of human adipose tissue-derived MSCs (hATMSCs) for intravenous infusion in animals and humans was reported by Ra et al. (2011). It has been also reported that no oncogenicity was detected in mice transplanted with adipose stromal/stem cells by MacIsaac et al. (2012). However, in tumour models, MSCs play a role in the progression of tumorigenesis, tumour growth, and metastasis. Some investigators reported that MSCs accelerate tumour growth, while others found that MSCs suppress tumour growth (Klopp et al., 2011, Akimoto et al., 2013, Keramidas et al., 2013). Zhang et al. (2013) found that bone marrow-derived MSCs (BM-MSCs) promote the growth and angiogenesis of breast and prostate tumours. Rodini et al. (2018) also found that MSCs enhanced the tumorigenicity of human glioblastoma by cell-cell communication. Nevertheless, Qiao et al. (2008) reported that DKK-1 produced by MSCs suppressed the growth of breast cancer cells through the inhibition of the WNT signalling pathway. Some mechanism research of MSC-related tumour enhancing effects have been found, including vascular (Zhang et al., 2013, Wang et al., 2017, Yang et al., 2017) and tumour microenvironment support, immunosuppressive effects, and metastatic support (Huang et al., 2015, He et al., 2016, Gonzalez et al., 2017). On the other hand, the tumour inhibition mechanisms of MSCs could be related to cell cycle arrest (Lu et al., 2008, Allahverdi et al., 2020), angiogenesis inhibition (Otsu et al., 2009), and regulatory of some soluble factors, such as DKK-1, that may interdict tumour-related signalling pathways (Han et al., 2018). Still and all, the tumour regulation mechanisms of MSCs are especially complicated and stiff to illustrate. Therefore, the potential tumorigenicity of MSCs should be further explored and monitored to clarify the risk related to MSC-based therapies (Lee and Hong, 2017).
The accumulation of mutations, genetic susceptibility of certain factors, exposure to environmental agents, and lifestyle all play important roles in the tumorigenesis process. 3-Methylcholanthrene (3-MC), a potent polycyclic aromatic hydrocarbon (PAH), is a common environmental pollutant that can be obtained as a by-product of combustion of various organic substances, such as coal, garbage, gas, and oil (Chang et al., 2017). 3-MC and other PAHs have many adverse effects, including immune suppression, endocrine disruption, birth defects, and tumorigenesis (Kwon et al., 2002; Bhattacharya and Haldar, 2012). These compounds suppress the proliferation of human umbilical vascular endothelial cells and induce tumours in rats and mice (Pang et al., 2008).
Here, we aimed to identify potential biomarkers indicative of tumorigenic risk by RNA-seq analysis of hPTMSCs exposed to the carcinogenic agent 3-MC and provide markers for detecting tumorigenicity related to quality and safety control of clinical-grade MSCs in cell therapy.
The collection of human placentas was authorized by the Ethics Committee of Human Research of Ningxia Medical University. Informed consent was signed with each mother prior to delivery. Three human full-term placentas were obtained from healthy mothers after elective caesarean section in the General Hospital of Ningxia Medical University. Human PTMSCs were isolated as described previously (Wang et al., 2012, Zhu et al., 2014, Zhu et al., 2015). Cells from the three mothers were pool together and cultivated in MesenCult-XF Basal Medium added to MesenCult-XF Supplement (STEMCELL Technologies Inc., Grenoble, France). At about 90% confluence, cells were digested using the MesenCult-ACF Dissociation Kit (STEMCELL Technologies Inc., Grenoble, France) and passaged. The hPTMSCs at passage 3 were treated with 3-MC (SIGMA-ALDRICH, Saint Louis, USA)) at a concentration of 5 μg/mL for serial 7 days to induce carcinogenesis. All the studies were conducted with five passages of the isolated hPTMSCs. Human lung adenocarcinoma cell A549 were purchased from the Cell Resource Center of the Chinese Academy of Sciences (Shanghai, China) and cultured in RPMI-1640 medium added to 10% fetal bovine serum (Gibco, Grand Island, NY, USA). Cells were collected to spread assays when confluence reached about 90% using TrypLE™ Express (Invitrogen, USA). All cultures were maintained at 37°C in a sterile humidified incubator with 5% CO2.
Flow cytometry (Zhu et al., 2015)
The immunological characterization of hPTMSCs was identified by flow cytometry on a FACS Calibur flow cytometer (BD Biosciences, San Diego, CA, USA). All antibodies were purchased from BD Pharmingen (Franklin Lakes, NJ, USA). Briefly, cells (1 × 106) in 100 μL PBS were incubated with the antibody for 30 min on the ice: IgG1-PE, CD34-PE, CD73-PE, IgG2a-FITC, CD14-FITC, CD45-FITC, CD90-FITC, CD105-FITC. After extensive washing, cells were resuspended in 500 μL PBS and used for flow cytometry analysis.
Total RNA was extracted from DMSO control and 3-MC treated hPTMSCs using TRIzol Reagent (Ambion, America). Sequencing libraries were generated using NEBNext UltraTM RNA Library Prep Kit for Illumina (NEB, USA), and index codes were added to attribute sequences to each sample. The clustering of the index-coded samples was performed on a cBot Cluster Generation System using TruSeq PE Cluster Kit v4-cBot-HS (Illumia). After cluster generation, the library preparations were sequenced on an Illumina Hiseq 2500 platform, and paired-end reads were generated. Gene expression levels were estimated by FPKM (fragments per kilobase of transcript per million fragments mapped) using Cuffquant and Cuffnorm (Florea et al., 2013). Differential expression analysis of two samples was performed using the DEGseq R package. P-value was adjusted using q-value. q-value < 0.005 and |log2 (foldchange)| ≥ 1 was set as the threshold for significantly differential expression.
Human LV-CON, LV-sfrp1, LV-sfrp1-RNAi, LV-ptgs2, and LV-ptgs2-RNAi were purchased from Genechem (Shanghai, China). hPTMSCs and A549 cells were infected with LV-CON, LV-sfrp1, LV-sfrp1-RNAi, LV-ptgs2, or LV-ptgs2-RNAi, respectively. sfrp1-targeting small hairpin RNA (shRNA) (ACCTTTCAGTCCGTGTTTA) and ptgs2-targeting shRNA (TGAATTTAACACCCTCTAT) were cloned into the GV Lentivirus plasmid (Genechem, China). The scramble sequence (TTCTCCGAACGTGTCACGT) of LV-CON was a control shRNA that does not complement any human gene.
qRT-PCR (Wu et al., 2017)
Total RNA was separated using TRIzol Reagent (Ambion, USA). RNA degradation was monitored on 1% agarose gels. RNA purity and concentration were checked using a Nanodrop™ spectrophotometer (Thermo Fisher Scientific Inc., San Diego, CA, USA). Then, 1μg of total RNA was converted to cDNA with cDNA Synthesis SuperMix (TransGen Biotech, Beijing, China). Afterwards, real-time PCR was carried out using TransStart Tip Green qPCR SuperMix (TransGen Biotech, Beijing, China) on Bio-rad IQ5 (Bio-Rad Inc., USA). The expression levels of gene transcripts were estimated according to the respective standard curves and normalized to the amount of actb or gapdh endogenous control transcripts by the comparative Ct value. Specific primers sets were synthesized by Shenggong (Shanghai, China) and illustrated in Tab. 1.

Western Blot analysis (Yan et al., 2019)
Whole protein was isolated using Whole Cell Lysis Assay kit (KeyGEN, Nanjing, China). Equal protein from all samples was separated by 10% or 12% SDS-PAGE and transferred to polyvinylidene fluoride membranes (Millipore, Billerica, MA, USA), then probed with rabbit anti SFRP1, PTGS2, SOX2, CMYC, OCT4, NANOG, and ACTB antibody (Proteintech, Wuhan, China) at 4°C overnight followed by appropriate secondary antibodies (BOSTER, Wuhan, China). The protein bands were detected via Superstar ECL Plus Ready-to-use (BOSTER, Wuhan, China) and quantified using Gel Imaging System (Bio-Rad Inc., USA). By using ACTB as an internal control, the ratio of grey level between the target bands and the internal control was considered the relative expression of target proteins.
Cell counting kit-8 (CCK-8) cell viability assays
Human LV-CON, LV-sfrp1, LV-sfrp1-RNAi, LV-ptgs2, and LV-ptgs2-RNAi infected A549 cells (3 × 103) were seeded in 96-well plates and cultured overnight in a CO2 incubator. CCK-8 assay (Dojindo, Kumamoto, Japan) was carried out. The plates were incubated for 4 h and then the absorbance was read at 450 nm in a plate reader (Bio-Rad Laboratories Inc., Hercules, USA). The following formula was used to calculate the percentage of viable cells. Percentage of Cell Viability = OD sample at 450 nm/OD MOCK at 450 nm × 100%.
Human LV-CON, LV-sfrp1, LV-sfrp1-RNAi, LV-ptgs2, and LV-ptgs2-RNAi infected A549 cells (1 × 103) were seeded in 6-well plates for colony formation. The cells were incubated in a humidified CO2 incubator for 10 to 14 days. The colonies were fixed with 4% paraformaldehyde and stained with crystal violet. Colonies with more than 50 cells were considered clonogenic survivors.
Human LV-CON, LV-sfrp1, LV-sfrp1-RNAi, LV-ptgs2, and LV-ptgs2-RNAi infected A549 cells were made artificial wounds and cultured for another 24 h. Cell migration ability was represented by the wound gap distance in the microscopic field at the time points of 0 and 24 h.
All data collected in this study was obtained from at least three independent experiments for each condition. SPSS19.0 analysis software (USA) and PRISM5 (GraphPad, USA) were used for the statistical analysis. Statistical evaluation of the data was performed by one- way ANOVA when more than two groups were compared with a single control, and a t-test was employed for comparison of differences between two groups. Quantitative data were presented as the mean ± standard deviation (SD). The statistical significance was set at P < 0.05. *P < 0.05, **P < 0.01 denote significant differences compared with the MOCK or sh-NC group.
Sequencing data and quality control
The Illumina HiSeq 2500 RNA sequencing platform was used to sequence the whole transcriptome of hPTMSCs. After stringent data filtration and quality inspection, approximately 62 million high-quality clean reads were obtained with 87.11% and 86.44% Q30 bases (base quality was more than 30) for DMSO control and 3-MC treated hPTMSCs, respectively. 29,251,305 and 32,739,136 cleanpaired-end reads as well as 8,730,066,236 and 9,771,496,192 clean bases were generated from DMSO control and 3-MC treated hPTMSCs, respectively (Tab. 2). These clean reads were then mapped to the human reference genome sequence (GRCh38) using STAR (version 2.5.2b) (Dobin et al., 2013). Only reads with a perfect match or mismatch were further analysed and annotated based on the reference genome.

Differentially expressed unigene analysis
Unigene expression was calculated by FPKM (fragments per kilobase of transcript per million fragments mapped) (Florea et al., 2013) using Cuffquant and Cuffnorm. Differential expression analysis was determined by EBseq. The threshold of the false discovery rate (FDR) was set to less than 0.01 and with a fold change greater than or equal to 2. Through this calculation, the upregulation and downregulation of both DMSO control- and 3-MC-treated transcripts were determined. Ultimately, 1081 differentially expressed genes (DEGs) were identified, including 498 upregulated and 583 downregulated genes (Figs. 1A–B and Suppl. Tab. S1). Hierarchical cluster analysis of DEGs is presented in Fig. 1C.

Figure 1: Expression levels of differentially expressed genes (DEGs) in DMSO group and 3-MC treated hPTMSCs group.
(A and B) Up-regulated and down-regulated genes are represented by red and green spots respectively, while undifferentially expressed genes are represented by black spots. (C) A hierarchical cluster analysis of DEGs.
To further study how hPTMSCs regulate their biological functions in response to 3-MC at the molecular level, we focused on tumour transformation-related and pluripotency-related DEGs. A total of 268 tumour transformation-related genes were identified, among which 17 upregulated (c1qtnf1, cd14, chi3l1, cxcl8, cxcl16, gata3, gch1, irak3, mn1, pid1, plxdc2, ptgs2, ptk2b, tnfaip3, tnfaip6, tnfaip8, and tnfsf13b) and 12 downregulated (il18, krt18, mtus1, mtus2, prame, pttg1, sfrp1, tacstd2, tlr4, tnfaip8l1, tnfrsf19, and txnip) (Figs. 2A and 2C, Suppl. Tab. S2). In addition, 66 pluripotency-related genes were identified, including jag1, lfng, and maml3, which were downregulated in the NOTCH signalling pathway (Figs. 2B and 2C and Suppl. Tab. S3).
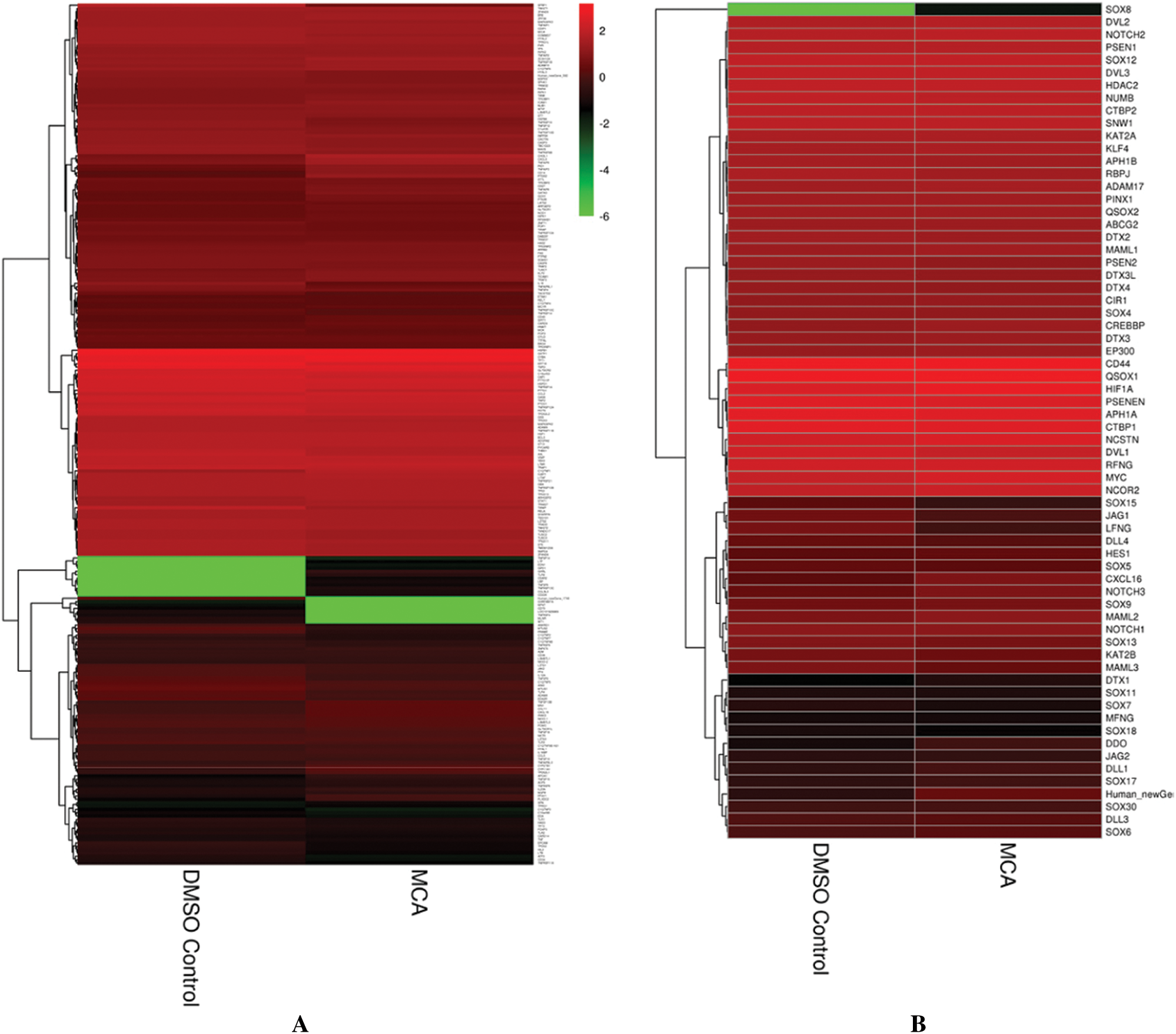

Figure 2: Hierarchical cluster analysis of tumor transformation-related and pluripotency-related genes. (A) The hierarchical cluster analysis of tumor transformation-related genes (268). (B) The hierarchical cluster analysis of pluripotency-related genes (66). (C) The hierarchical cluster analysis of tumor transformation-related and pluripotency-related DEGs (32).
Functional annotation, gene ontology classification and pathway analysis
Functional annotation provided information on protein function annotation, pathway annotation, COG annotation, and GO annotation. Unigenes were subjected to sequence alignment with protein databases, such as NR, Swiss-Prot, GO, KOG, Pfam, and KEGG, using BLAST software, which is based on sequence similarities to public databases. In total, 1077 DEGs were successfully annotated. All DEGs were annotated using the NR (1077) followed by eggNOG (1063), Swiss-Prot (1055), Pfam (996), GO (948), KEGG (709), KOG (695), and COG databases (325). The remaining DEGs had no matches. The GO database is an international standard biological annotation system with three ontologies: biological process, cellular component, and molecular function (Wang et al., 2010). The GO classification of DEGs is presented in Fig. 3A. The pathway-based analysis provides information and further understanding of how hPTMSCs regulate their biological functions in response to 3-MC at the molecular level. KEGG is a database resource for understanding the high-level functions and utilities of biological systems, especially large-scale molecular datasets generated by genome sequencing and other high-throughput experimental technologies. We used KOBAS software to test the statistical enrichment of DEGs in KEGG pathways. In total, 865 DEGs were mapped to the KEGG database. These DEGs were classified into 50 KEGG pathways of four main KEGG categories, including cellular processes (129 DEGs), environmental information processing (294 DEGs), human diseases (343 DEGs), and organismal systems (99 DEGs) (Fig. 3B). Out of 50 pathways, the first 20 pathways (q-value ≤ 1.00) are presented in Fig. 3C and Suppl. Tab. S4.
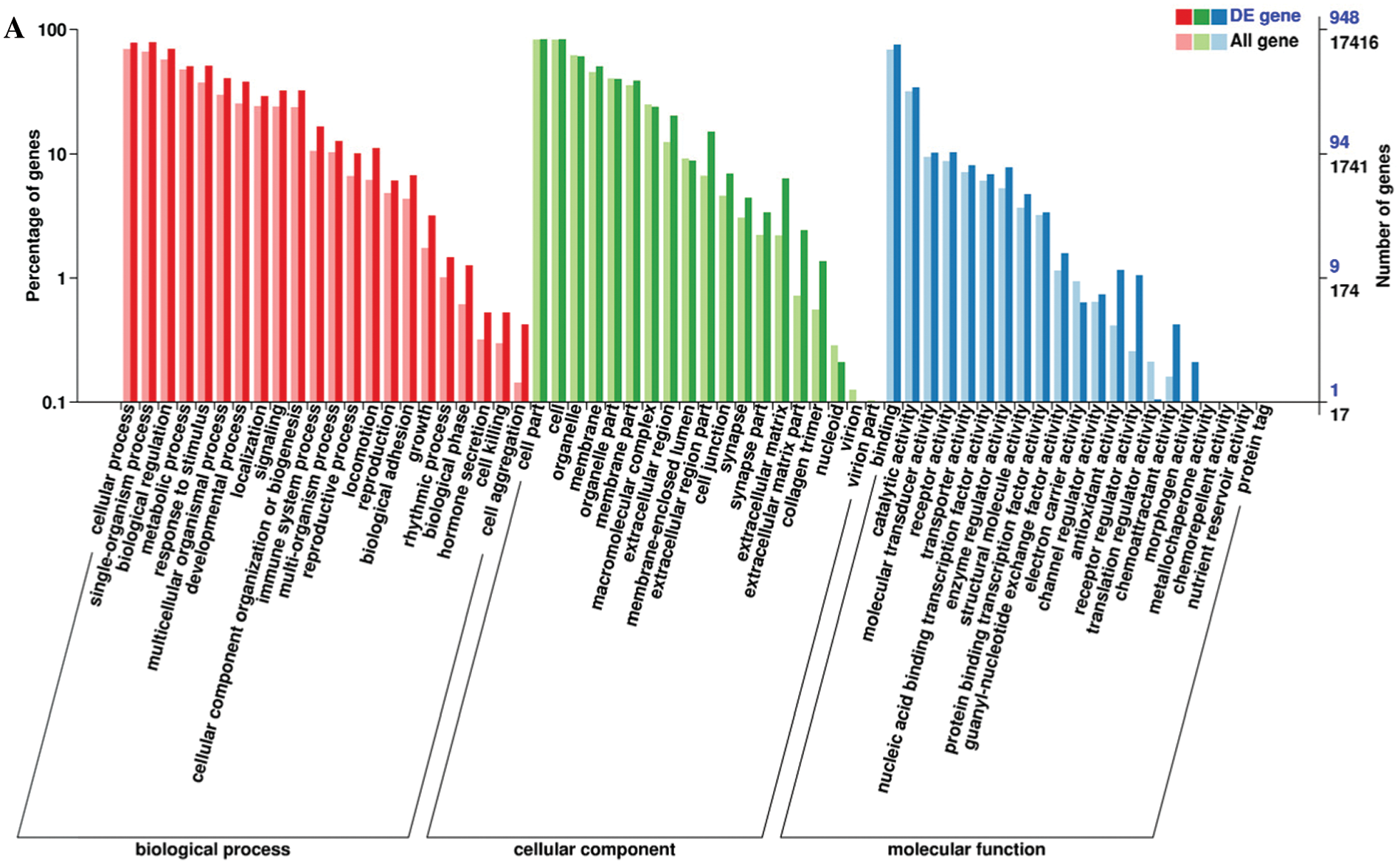

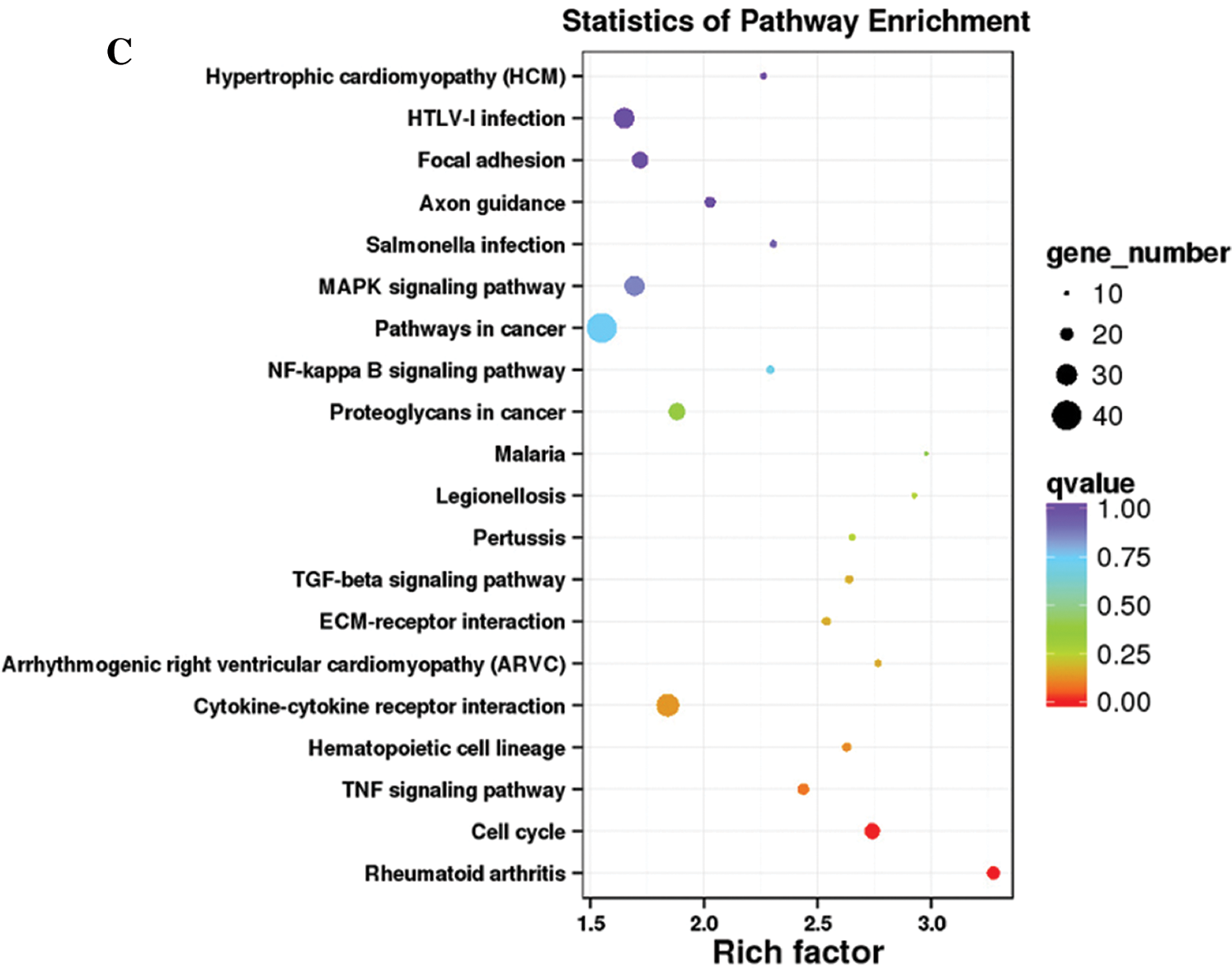
Figure 3: Histogram of GO classification and KEGG pathways of DEGs. (A) The GO classification of DEGs. (B) The percentages of DEGs that were mapped in 50 KEGG pathways of four main KEGG categories. (C) The first 20 KEGG pathways that are enriched with DEGs (q-value ≤ 1.00).
Verification of tumour transformation-related and pluripotency-related DEGs
To verify the results of tumour transformation-related and pluripotency-related DEGs in RNA sequencing, we characterized the expression of these DEGs by qPCR. The expression trend of 27 tumour transformation-related DEGs, as well as 3 pluripotency-related DEGs in the NOTCH signalling pathway, exhibited consistency with the RNA sequencing, whereas the other 2 tumour transformation-related DEGs did not change (Fig. 4A). Among these, sfrp1 and ptgs2 showed higher deregulation as seen by qPCR. To elucidate the possible roles of sfrp1 and ptgs2 in the 3-MC induced tumorigenesis of hPTMSCs, stable sfrp1 and ptgs2 overexpression or silencing in hPTMSCs and A549 cells were achieved using the respective lentiviruses. mRNA expression analysis of sox2, cmyc, oct4, and nanog in hPTMSCs and A549 cells with sfrp1 or ptgs2 overexpression and silencing showed that the cells express the four pluripotency gene markers (Fig. 4B). Western blot analysis of all samples showed that only CMYC and OCT4 were expressed at the protein level (Figs. 4C and 4D). Since these four genes have critical roles in the regulation of pluripotency, these results indicate that sfrp1 and ptgs2 alteration may affect the pluripotent state of hPTMSCs. To analyse the tumorigenic capacity of sfrp1 and ptgs2, CCK-8, colony formation, and wound healing assays were performed in A459 cells. Overexpression of sfrp1 led to reduced cell viability, colony formation, and migration of A549 cells. In contrast, the overexpression of ptgs2 resulted in significantly increased cell viability, colony formation, and migration. These results suggest that A549 cells with high ptgs2 expression but low sfrp1 expression may have more potential tumorigenic capacity, low ptgs2 expression, and high sfrp1 expression may exhibit less tumorigenic capacity (Fig. 5).


Figure 4: The verification of tumor transformation-related and pluripotency-related DEGs.(A) Detection of mRNA of transformation and pluripotency related DEGs in 3-MC treated hPTMSCs. (B) mRNA of pluripotent markers sox2, cmyc, oct4, and nanog measured by qPCR and normalized against gapdh or actb. MOCK was arbitrarily set to 1.0. (C and D) western blotting of OCT4 and CMYC in hPTMSCs and A549 cells with sfrp1 and ptgs2 overexpression and knockdown. *P < 0.05, **P < 0.01 denote significant differences compared with the MOCK or sh-NC group.

Figure 5: The inhibitory or promotion effects of SFRP1 and PTGS2 overexpression on the cell viability, colony formation, and migration of A549 cells.
(A) Detections of mRNA of sfrp1 and ptgs2 overexpression and knockdown efficiencies in stable A549 subcell lines constructed with indicated viruses. (B) Cell viability of A549 cells was analyzed by CCK-8 assay at 24 h. sfrp1 silence significantly facilitated the cell viability compared with the sh-NC group. ptgs2 silence significantly suppressed the cell viability compared with the sh-NC group. (C) Representative images of the indicated A549 subcell lines assessed by colony formation assay. sfrp1 overexpression significantly suppressed the colony formation compared with the MOCK or sh-NC group. ptgs2 overexpression significantly facilitated the colony formation compared with the MOCK or sh-NC group. (D) Representative images and quantitative data of migration of the indicated A549 subcell lines assessed by wound-healing assay. sfrp1 silence significantly facilitated the migration compared with the MOCK or sh-NC group. ptgs2 overexpression significantly facilitated the migration compared with the MOCK or sh-NC group. All the experiments were performed in triplicate, and the data are presented as the mean ± SD. *P < 0.05, **P < 0.01 denote significant differences compared with the MOCK or sh-NC group.
Clinically, MSCs may contribute to tissue reconstitution and maintenance of tissue homeostasis, which brings about some concern regarding the potential tumorigenicity of MSC-based therapies (Fekete et al., 2015). Although some studies suggest that MSCs have inhibitory effects on tumour development, an overwhelming number of studies demonstrate that MSCs exert stimulatory effects on tumour pathogenesis (Lee and Hong, 2017).
In the present study, we aimed to identify potential tumorigenic risk genes by RNA-seq analysis of hPTMSCs exposed to 3-MC. In total, 268 tumour transformation-related genes of 3-MC-induced MSCs were identified by RNA-seq, including 17 upregulated (c1qtnf1, cd14, chi3l1, cxcl8, cxcl16, gata3, gch1, irak3, mn1, pid1, plxdc2, ptgs2, ptk2b, tnfaip3, tnfaip6, tnfaip8, and tnfsf13b) and 12 downregulated (il18, krt18, mtus1, mtus2, prame, pttg1, sfrp1, tacstd2, tlr4, tnfaip8l1, tnfrsf19, and txnip) genes. In addition, 66 pluripotency-related genes were identified, including 3 downregulated NOTCH signalling pathway genes (jag1, lfng, and maml3). We then characterized the expression of these DEGs by qPCR and found that the expression trends of 27 tumour transformation-related DEGs, as well as 3 pluripotency-related DEGs in the NOTCH signalling pathway, were consistent with the RNA sequencing results; the other 2 tumour transformation-related DEGs did not change.
Secreted Frizzled-related protein 1 (sfrp1), which is located within the 8p11-12 region, is homologous to the extracellular cysteine-rich domain of the wingless and integration site growth factor (WNT) receptor Frizzled but lacks the intracellular and transmembrane domains (Yang et al., 2009). It has been reported that the downregulation of sfrp1 can lead to the over activation of WNT signalling pathways, which promotes tumorigenesis in human breast tissues (Veeck et al., 2006, Schlange et al., 2007). A recent focus in cancer prevention involves the modulation of molecular crosstalk between inflammation and cancer growth signalling networks converging on the redox-sensitive transcription factor nuclear factor-kB (NF-kB). NF-kB is directly associated with inflammatory responses with further links to the promotion of carcinogenesis (Yamamoto and Gaynor, 2001, Coussens and Werb, 2002, Pikarsky et al., 2004, Wang et al., 2009). NF-kB-mediated inflammation-driven carcinogenesis involves transactivation of a major molecular target, prostaglandin-endoperoxide synthase 2 (ptgs2) (Wang et al., 2007). ptgs2 is involved in prostaglandin (PG) biosynthesis and inflammation and is upregulated in various premalignant and malignant tissues (Wang et al., 2007). Abnormally high levels of ptgs2 contribute to tumorigenesis in various animal models of cancer, and downregulation of ptgs2 is spontaneously preventive (Boolbol et al., 1996, Takahashi et al., 2006, Lau et al., 2010). Various stimuli and mediators are involved in transient induction of NF-kB and transactivation of ptgs2, thus eliciting inflammation and consequent tumorigenesis (Boolbol et al., 1996, Yamamoto and Gaynor, 2001, Takahashi et al., 2006, Wang et al., 2007, Lau et al., 2010). Thus, targeted inhibition of NF-kB and ptgs2 has become a promising and practical tool in cancer prevention. Reports suggest that diminishing NF-kB-ptgs2 crosstalk may prevent tumorigenesis. More recently, genomic studies demonstrate that MSCs express the pluripotent markers oct4, nanog, and sox2, expounding their undifferentiated state (Patterson et al., 2012). sox2, cmyc, oct4, and nanog are four important transcription factors essential for self-renewal of ESCs (Nichols et al., 1998) and required for maintenance of pluripotency state.
We analysed the effects of the over-expression or inhibition of sfrp1 and ptgs2 upon the expression of sox2, cmyc, oct4, and nanog. By transfection experiments, we could demonstrate that the deregulation of sfrp1 and ptgs2 alter the expression of the four markers at the mRNA level and of OCT4 and CMYC at the protein level in hPTMSCs and human lung adenocarcinoma cells line A549. Since these marker genes have critical roles in the regulation of pluripotency, these results indicate that sfrp1 and ptgs2 alteration may affect the pluripotent state of hPTMSCs. The molecular mechanisms of sfrp1 and ptgs2 for pluripotency regulation are not clear. Previous studies show changes in DNA methylation and histone modification status of oct4 and nanog promoters following treatment with ESC extracts (Cho et al., 2010). Other studies also found that high cellular proliferation rates promote the generation of induced pluripotent stem cells at least partially by inducing passive DNA demethylation and up-regulating pluripotency-related genes (He et al., 2017). Promoter methylation of the sfrp1 gene is associated with lymph node metastasis and poor mean overall survival and it can be a prognostic marker in colorectal carcinoma (Kumar et al., 2019). L-securinine was a potential agent in the treatment of lung cancer by upregulation of the sfrp1 gene expression and changing the sfrp1 gene methylation (Yang et al., 2018). Moreover, enhanced ptgs2 expression in hepatocytes is sufficient to induce hepatocellular carcinoma through inducing promoter hypermethylation by reducing tet1, silencing tumor-suppressive genes, and activating key oncogenic pathways. Inhibition of ptgs2 represents a mechanism-based target for hepatocellular carcinoma prevention (Chen et al., 2017).
To determine the effects of overexpression or downregulation, gain and loss of function experiments were carried out. CCK-8, colony-formation, and wound healing assays showed that overexpression of sfrp1 led to reduced cell viability, colony formation, and migration of A549 cells. In contrast, overexpression of ptgs2 exerted the opposite effect. So, we inferred that our results might from different mechanisms.
In conclusion, 29 tumour transformation-related genes and 3 pluripotency-related genes were identified as differentially expressed genes (DEGs) in hPTMSCs exposed to 3-MC based on RNA-seq transcriptional profile analysis using functional annotation, Gene Ontology classification, and pathway analysis. Overexpression of sfrp1 led to reduced cell viability, colony formation, and migration of A549 cells. In contrast, the overexpression of ptgs2 exerted the opposite effect. These results indicate that A549 cells with high ptgs2 expression but low sfrp1 expression may have more potential tumorigenic capacity. Taken together, this study suggests that ptgs2 and sfrp1 may be tumorigenic risk genes, but their role in hPTMSCs need more gain- and loss-of-function experiment to verify for future studies.
Availability of Data and Materials: All data generated or analysed during this study are included in this manuscript.
Supplementary Material: The supplementary material is available online at DOI: https://10.32604/biocell.2022.015390.
Author Contribution: Study conception and design: Xueyun Liang and Xiaoming Liu: data collection: Xiaona Ma, Xiurui Yan and Jing Xue; analysis and interpretation of results: Yuanyuan Jia and Tingting Yang; draft manuscript preparation: Yuanyuan Jia. All authors reviewed the results and approved the final version of the manuscript.
Ethics Approval: The collection of human placentas was approved by the Ethics Committee of Human Research at General Hospital of Ningxia Medical University (2020-289).
Funding Statement: This work was supported by grants from the Ningxia Science and Technological Supporting Project(2015KJHM38) and the Ningxia Natural Science Foundation (2019AAC03231).
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
Akimoto K, Kimura K, Nagano M, Takano S, To’a Salazar G, Yamashita T, Ohneda O (2013). Umbilical cord blood-derived mesenchymal stem cells inhibit, but adipose tissue-derived mesenchymal stem cells promote, glioblastoma multiforme proliferation. Stem Cells and Development 22: 1370–1386. DOI 10.1089/scd.2012.0486. [Google Scholar] [CrossRef]
Allahverdi A, Arefian E, Soleimani M, Ai J, Nahanmoghaddam N, Yousefi-Ahmadipour A, Ebrahimi-Barough S (2020). MicroRNA-4731-5p delivered by AD-mesenchymal stem cells induces cell cycle arrest and apoptosis in glioblastoma. Journal of Cellular Physiology 235: 8167–8175. DOI 10.1002/jcp.29472. [Google Scholar] [CrossRef]
Bhattacharya S, Haldar PK (2012). Chemopreventive property of Trichosanthes dioica root against 3-methylcholanthrene-induced carcinogenesis in albino mice. Journal of Environmental Pathology, Toxicology and Oncology 31: 109–119. DOI 10.1615/JEnvironPatholToxicolOncol.v31.i2.30. [Google Scholar] [CrossRef]
Boolbol SK, Dannenberg AJ, Chadburn A, Martucci C, Guo XJ, Ramonetti JT, Abreu-Goris M, Newmark HL, Lipkin ML, DeCosse JJ, Bertagnolli MM (1996). Cyclooxygenase-2 overexpression and tumor formation are blocked by sulindac in a murine model of familial adenomatous polyposis. Cancer Research 56: 2556–2560. [Google Scholar]
Chang CC, Hsu YH, Chou HC, Lee YG, Juan SH (2017). 3-Methylcholanthrene/aryl-hydrocarbon receptor-mediated hypertension through eNOS inactivation. Journal of Cellular Physiology 232: 1020–1029. DOI 10.1002/jcp.25497. [Google Scholar] [CrossRef]
Chen H, Cai W, Chu ESH, Tang J, Wong CC, Wong SH, Sun W, Liang Q, Fang J, Sun Z, Yu J (2017). Hepatic cyclooxygenase-2 overexpression induced spontaneous hepatocellular carcinoma formation in mice. Oncogene 36: 4415–4426. DOI 10.1038/onc.2017.73. [Google Scholar] [CrossRef]
Cho HJ, Lee CS, Kwon YW, Paek JS, Lee SH, Hur J, Lee EJ, Roh TY, Chu IS, Leem SH, Kim Y, Kang HJ, Park YB, Kim HS (2010). Induction of pluripotent stem cells from adult somatic cells by protein-based reprogramming without genetic manipulation. Blood 116: 386–395. DOI 10.1182/blood-2010-02-269589. [Google Scholar] [CrossRef]
Coussens LM, Werb Z (2002). Inflammation and cancer. Nature 420: 860–867. DOI 10.1038/nature01322. [Google Scholar] [CrossRef]
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR (2013). STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. DOI 10.1093/bioinformatics/bts635. [Google Scholar] [CrossRef]
Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, Deans R, Keating A, Prockop D, Horwitz E (2006). Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 8: 315–317. DOI 10.1080/14653240600855905. [Google Scholar] [CrossRef]
Fekete N, Erle A, Amann EM, Furst D, Rojewski MT, Langonne A, Sensebe L, Schrezenmeier H, Schmidtke-Schrezenmeier G (2015). Effect of high-dose irradiation on human bone-marrow-derived mesenchymal stromal cells. Tissue Engineering Part C: Methods 21: 112–122. DOI 10.1089/ten.tec.2013.0766. [Google Scholar] [CrossRef]
Florea L, Song L, Salzberg SL (2013). Thousands of exon skipping events differentiate among splicing patterns in sixteen human tissues. F1000Research 2: 188. DOI 10.12688/f1000research.2-188.v1. [Google Scholar] [CrossRef]
Gonzalez ME, Martin EE, Anwar T, Arellano-Garcia C, Medhora N, Lama A, Chen YC, Tanager KS, Yoon E, Kidwell KM, Ge C, Franceschi RT, Kleer CG (2017). Mesenchymal stem cell-induced DDR2 mediates stromal-breast cancer interactions and metastasis growth. Cell Reports 18: 1215–1228. DOI 10.1016/j.celrep.2016.12.079. [Google Scholar] [CrossRef]
Han I, Yun M, Kim EO, Kim B, Jung MH, Kim SH (2018). Retraction Note: Umbilical cord tissue-derived mesenchymal stem cells induce apoptosis in PC-3 prostate cancer cells through activation of JNK and downregulation of PI3K/AKT signaling. Stem Cell Research & Therapy 9: 54. DOI 10.1186/s13287-018-1113-9. [Google Scholar] [CrossRef]
He L, Zhao F, Zheng Y, Wan Y, Song J (2016). Loss of interactions between p53 and survivin gene in mesenchymal stem cells after spontaneous transformation in vitro. The International Journal of Biochemistry & Cell Biology 75: 74–84. DOI 10.1016/j.biocel.2016.03.018. [Google Scholar] [CrossRef]
He S, Sun H, Lin L, Zhang Y, Chen J, Liang L, Li Y, Zhang M, Yang X, Wang X, Wang F, Zhu F, Chen J, Pei D, Zheng H (2017). Passive DNA demethylation preferentially up-regulates pluripotency-related genes and facilitates the generation of induced pluripotent stem cells. Journal of Biological Chemistry 292: 18542–18555. DOI 10.1074/jbc.M117.810457. [Google Scholar] [CrossRef]
Huang JC, Basu SK, Zhao X, Chien S, Fang M, Oehler VG, Appelbaum FR, Becker PS (2015). Mesenchymal stromal cells derived from acute myeloid leukemia bone marrow exhibit aberrant cytogenetics and cytokine elaboration. Blood Cancer Journal 5: e302. DOI 10.1038/bcj.2015.17. [Google Scholar] [CrossRef]
Keramidas M, de Fraipont F, Karageorgis A, Moisan A, Persoons V, Richard MJ, Coll JL, Rome C (2013). The dual effect of mesenchymal stem cells on tumour growth and tumour angiogenesis. Stem Cell Research & Therapy 4: 41. DOI 10.1186/scrt195. [Google Scholar] [CrossRef]
Klopp AH, Gupta A, Spaeth E, Andreeff M, Marini F3rd (2011). Concise review: Dissecting a discrepancy in the literature: do mesenchymal stem cells support or suppress tumor growth? Stem Cells 29: 11–19. DOI 10.1002/stem.559. [Google Scholar] [CrossRef]
Kumar A, Gosipatala SB, Pandey A, Singh P (2019). Prognostic relevance of SFRP1 gene promoter methylation in colorectal carcinoma. Asian Pacific Journal of Cancer Prevention 20: 1571–1577. DOI 10.31557/APJCP.2019.20.5.1571. [Google Scholar] [CrossRef]
Kwon YW, Ueda S, Ueno M, Yodoi J, Masutani H (2002). Mechanism of p53-dependent apoptosis induced by 3-methylcholanthrene: Involvement of p53 phosphorylation and p38 MAPK. Journal of Biological Chemistry 277: 1837–1844. DOI 10.1074/jbc.M105033200. [Google Scholar] [CrossRef]
Lau GT, Ye L, Leung LK (2010). The licorice flavonoid isoliquiritigenin suppresses phorbol ester-induced cyclooxygenase-2 expression in the non-tumorigenic MCF-10A breast cell line. Planta Medica 76: 780–785. DOI 10.1055/s-0029-1240699. [Google Scholar] [CrossRef]
Lee HY, Hong IS (2017). Double-edged sword of mesenchymal stem cells: Cancer-promoting versus therapeutic potential. Cancer Science 108: 1939–1946. DOI 10.1111/cas.13334. [Google Scholar] [CrossRef]
Lu YR, Yuan Y, Wang XJ, Wei LL, Chen YN, Cong C, Li SF, Long D, Tan WD, Mao YQ, Zhang J, Li YP, Cheng JQ (2008). The growth inhibitory effect of mesenchymal stem cells on tumor cells in vitro and in vivo. Cancer Biology & Therapy 7: 245–251. DOI 10.4161/cbt.7.2.5296. [Google Scholar] [CrossRef]
MacIsaac ZM, Shang H, Agrawal H, Yang N, Parker A, Katz AJ (2012). Long-term in-vivo tumorigenic assessment of human culture-expanded adipose stromal/stem cells. Experimental Cell Research 318: 416–423. DOI 10.1016/j.yexcr.2011.12.002. [Google Scholar] [CrossRef]
Nichols J, Zevnik B, Anastassiadis K, Niwa H, Klewe-Nebenius D, Chambers I, Scholer H, Smith A (1998). Formation of pluripotent stem cells in the mammalian embryo depends on the POU transcription factor Oct4. Cell 95: 379–391. DOI 10.1016/S0092-8674(00)81769-9. [Google Scholar] [CrossRef]
Otsu K, Das S, Houser SD, Quadri SK, Bhattacharya S, Bhattacharya J (2009). Concentration-dependent inhibition of angiogenesis by mesenchymal stem cells. Blood 113: 4197–4205. DOI 10.1182/blood-2008-09-176198. [Google Scholar] [CrossRef]
Pang PH, Lin YH, Lee YH, Hou HH, Hsu SP, Juan SH (2008). Molecular mechanisms of p21 and p27 induction by 3-methylcholanthrene, an aryl-hydrocarbon receptor agonist, involved in antiproliferation of human umbilical vascular endothelial cells. Journal of Cellular Physiology 215: 161–171. DOI 10.1002/jcp.21299. [Google Scholar] [CrossRef]
Park SJ, Kim HJ, Kim W, Kim OS, Lee S, Han SY, Jeong EJ, Park HS, Kim HW, Moon KS (2016). Tumorigenicity evaluation of umbilical cord blood-derived mesenchymal stem cells. Toxicological Research 32: 251–258. DOI 10.5487/TR.2016.32.3.251. [Google Scholar] [CrossRef]
Patterson M, Chan DN, Ha I, Case D, Cui Y, Van Handel B, Mikkola HK, Lowry WE (2012). Defining the nature of human pluripotent stem cell progeny. Cell Research 22: 178–193. DOI 10.1038/cr.2011.133. [Google Scholar] [CrossRef]
Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, Gutkovich-Pyest E, Urieli-Shoval S, Galun E, Ben-Neriah Y (2004). NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature 431: 461–466. DOI 10.1038/nature02924. [Google Scholar] [CrossRef]
Qiao L, Xu ZL, Zhao TJ, Ye LH, Zhang XD (2008). Dkk-1 secreted by mesenchymal stem cells inhibits growth of breast cancer cells via depression of Wnt signalling. Cancer Letters 269: 67–77. DOI 10.1016/j.canlet.2008.04.032. [Google Scholar] [CrossRef]
Ra JC, Shin IS, Kim SH, Kang SK, Kang BC, Lee HY, Kim YJ, Jo JY, Yoon EJ, Choi HJ, Kwon E (2011). Safety of intravenous infusion of human adipose tissue-derived mesenchymal stem cells in animals and humans. Stem Cells and Development 20: 1297–1308. DOI 10.1089/scd.2010.0466. [Google Scholar] [CrossRef]
Rodini CO, Goncalves da Silva PB, Assoni AF, Carvalho VM, Okamoto OK (2018). Mesenchymal stem cells enhance tumorigenic properties of human glioblastoma through independent cell-cell communication mechanisms. Oncotarget 9: 24766–24777. DOI 10.18632/oncotarget.25346. [Google Scholar] [CrossRef]
Schlange T, Matsuda Y, Lienhard S, Huber A, Hynes NE (2007). Autocrine WNT signaling contributes to breast cancer cell proliferation via the canonical WNT pathway and EGFR transactivation. Breast Cancer Research 9: 2000. DOI 10.1186/bcr1769. [Google Scholar] [CrossRef]
Takahashi T, Baba M, Nishino H, Okuyama T (2006). Cyclooxygenase-2 plays a suppressive role for induction of apoptosis in isoliquiritigenin-treated mouse colon cancer cells. Cancer Letters 231: 319–325. DOI 10.1016/j.canlet.2005.02.025. [Google Scholar] [CrossRef]
Tian LL, Yue W, Zhu F, Li S, Li W (2011). Human mesenchymal stem cells play a dual role on tumor cell growth in vitro and in vivo. Journal of Cellular Physiology 226: 1860–1867. DOI 10.1002/jcp.22700. [Google Scholar] [CrossRef]
Veeck J, Niederacher D, An H, Klopocki E, Wiesmann F, Betz B, Galm O, Camara O, Durst M, Kristiansen G, Huszka C, Knuchel R, Dahl E (2006). Aberrant methylation of the Wnt antagonist SFRP1 in breast cancer is associated with unfavourable prognosis. Oncogene 25: 3479–3488. DOI 10.1038/sj.onc.1209386. [Google Scholar] [CrossRef]
Wang L, Feng Z, Wang X, Wang X, Zhang X (2010). DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 26: 136–138. DOI 10.1093/bioinformatics/btp612. [Google Scholar] [CrossRef]
Wang L, Yang Y, Zhu Y, Ma X, Liu T, Zhang G, Fan H, Ma L, Jin Y, Yan X, Wei J, Li Y (2012). Characterization of placenta-derived mesenchymal stem cells cultured in autologous human cord blood serum. Molecular Medicine Reports 6: 760–766. DOI 10.3892/mmr.2012.1000. [Google Scholar] [CrossRef]
Wang MT, Honn KV, Nie D (2007). Cyclooxygenases, prostanoids, and tumor progression. Cancer and Metastasis Reviews 26: 525–534. DOI 10.1007/s10555-007-9096-5. [Google Scholar] [CrossRef]
Wang S, Liu Z, Wang L, Zhang X (2009). NF-kappaB signaling pathway, inflammation and colorectal cancer. Cellular & Molecular Immunology 6: 327–334. DOI 10.1038/cmi.2009.43. [Google Scholar] [CrossRef]
Wang Y, Liu J, Jiang Q, Deng J, Xu F, Chen X, Cheng F, Zhang Y, Yao Y, Xia Z, Xu X, Su X, Huang M, Dai L, Yang Y, Zhang S, Yu D, Zhao RC, Wei Y, Deng H (2017). Human adipose-derived mesenchymal stem cell-secreted CXCL1 and CXCL8 facilitate breast tumor growth by promoting angiogenesis. Stem Cells 35: 2060–2070. DOI 10.1002/stem.2643. [Google Scholar] [CrossRef]
Wu J, Wang Y, Liu G, Jia Y, Yang J, Shi J, Dong J, Wei J, Liu X (2017). Characterization of air-liquid interface culture of A549 alveolar epithelial cells. Brazilian Journal of Medical and Biological Research 51: 363. DOI 10.1590/1414-431x20176950. [Google Scholar] [CrossRef]
Yamamoto Y, Gaynor RB (2001). Therapeutic potential of inhibition of the NF-kappaB pathway in the treatment of inflammation and cancer. Journal of Clinical Investigation 107: 135–142. DOI 10.1172/JCI11914. [Google Scholar] [CrossRef]
Yan X, Fu X, Jia Y, Ma X, Tao J, Yang T, Ma H, Liang X, Liu X, Yang J, Wei J (2019). Nrf2/Keap1/ARE signaling mediated an antioxidative protection of human placental mesenchymal stem cells of fetal origin in alveolar epithelial cells. Oxidative Medicine and Cellular Longevity 2019: 1–12. DOI 10.1155/2019/2654910. [Google Scholar] [CrossRef]
Yang KQ, Liu Y, Huang QH, Mo N, Zhang QY, Meng QG, Cheng JW (2017). Bone marrow-derived mesenchymal stem cells induced by inflammatory cytokines produce angiogenetic factors and promote prostate cancer growth. BMC Cancer 17: 277. DOI 10.1186/s12885-017-3879-z. [Google Scholar] [CrossRef]
Yang X, Han SW, Liu H, Zhu L, Chen YX, Ji ZN (2018). Secreted frizzled-related protein 1 (SFRP1) gene methylation changes in the human lung adenocarcinoma cells treated with L-securinine. Journal of Asian Natural Products Research 20: 163–171. DOI 10.1080/10286020.2017.1329828. [Google Scholar] [CrossRef]
Yang ZQ, Liu G, Bollig-Fischer A, Haddad R, Tarca AL, Ethier SP (2009). Methylation-associated silencing of SFRP1 with an 8p11-12 amplification inhibits canonical and non-canonical WNT pathways in breast cancers. International Journal of Cancer 125: 1613–1621. DOI 10.1002/ijc.24518. [Google Scholar] [CrossRef]
Zhang T, Lee YW, Rui YF, Cheng TY, Jiang XH, Li G (2013). Bone marrow-derived mesenchymal stem cells promote growth and angiogenesis of breast and prostate tumors. Stem Cell Research & Therapy 4: 70. DOI 10.1186/scrt221. [Google Scholar] [CrossRef]
Zhu Y, Song X, Han F, Li Y, Wei J, Liu X (2015). Alteration of histone acetylation pattern during long-term serum-free culture conditions of human fetal placental mesenchymal stem cells. PLoS One 10: e0117068. DOI 10.1371/journal.pone.0117068. [Google Scholar] [CrossRef]
Zhu Y, Yang Y, Zhang Y, Hao G, Liu T, Wang L, Yang T, Wang Q, Zhang G, Wei J, Li Y (2014). Placental mesenchymal stem cells of fetal and maternal origins demonstrate different therapeutic potentials. Stem Cell Research & Therapy 5: 48. DOI 10.1186/scrt436. [Google Scholar] [CrossRef]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |