DOI:10.32604/biocell.2022.018798
| BIOCELL DOI:10.32604/biocell.2022.018798 | |
| Viewpoint |
Cell extrusion in development and cancer, what MARCKS the difference for epithelial integrity?
1Sección Biología Celular, Facultad de Ciencias, Universidad de la República, Montevideo, CP 11400, Uruguay
2Institut Pasteur de Montevideo, Montevideo, CP 11400, Uruguay
3Sección Biofísica y Biología de Sistemas, Facultad de Ciencias, Universidad de la República, Montevideo, CP 11400, Uruguay
*Address correspondence to: Gonzalo Aparicio, gaparicio@fcien.edu.uy; Flavio R. Zolessi, fzolessi@fcien.edu.uy
Received: 18 August 2021; Accepted: 24 September 2021
Abstract: Cell extrusion is an active mechanism to eliminate non-viable or supernumerary cells in healthy epithelia. It also plays a role in carcinogenesis, both in tumor growth (apical extrusion) and metastasis (basal extrusion). Embryonic tissues like the neuroepithelium, on the other hand, present rates of proliferation comparable to that of carcinomas, without the occurrence of cell extrusion. However, the downregulation or phosphorylation of actin-modulating proteins like MARCKS, causes extensive neuroepithelial apical cell extrusion. As changes in MARCKS proteins phosphorylation and expression have also been correlated to carcinogenesis, we propose here an integrated model for their functions in epithelial integrity.
Keywords: Epithelial-mesenchymal plasticity; Carcinoma; Neurulation; Neuroepithelium; Phosphorylation
Introduction: Cell Extrusion as an Active Mechanism
The function of epithelia as barriers is achieved by the tight mechanical coupling of their participating cells. This poses a major challenge: tissue integrity must be preserved despite the constant cell turnover caused by proliferation and cell death, or the eventual occurrence of aberrant cell behavior, such as in cancerous transformation (Villars and Levayer, 2020; Eisenhoffer et al., 2012). One active mechanism for maintaining epithelial density and integrity is cell extrusion, a set of processes involving biochemical and mechanical signaling concerted to achieve the expulsion of a cell from the epithelial layer, either through its apical or basal side (Nanavati et al., 2020; Eisenhoffer and Rosenblatt, 2013). In this section we will introduce the mechanisms seen in physiological conditions, and in the next, those in pathological or experimental situations.
Apical extrusion can expel apoptotic or live cells, with each having its own particular mechanisms. Extrusion of apoptotic cells is accompanied by medium-scale epithelial modifications, such as topological irregularities, pulsatile contractions and loss of tissue tension (Atieh et al., 2021; Saw et al., 2017). Individual apoptotic cells seem to become hypercontractile prior to extrusion (Nanavati et al., 2020) and secrete sphingosine-1-phosphate (S1P) to activate RhoA in neighboring cells (Villars and Levayer, 2020; Eisenhoffer et al., 2012), eliciting active lamellipodial protrusion and the assembly of a basally-localized contractile actomyosin ring, in a cadherin-dependent manner (Duszyc et al., 2021; Lubkov and Bar-Sagi, 2014; Rosenblatt et al., 2001).
Conversely, “live apical cell extrusion” appears as the main counterbalancing mechanism to maintain constant cell densities against epithelial proliferation, and it occurs regularly in many homeostatic epithelia, like the tips of intestinal microvilli, where it was first identified (Madara, 1990). In this type of extrusion, “live” means that the cell will remain alive and playing an active part in the process, to eventually die by anoikis after the extrusion has been completed (Slattum and Rosenblatt, 2014). It requires stretch-activated ion channels (Piezo), S1P and the Rho-myosin pathway, and also produces regular topological changes in the organization of the epithelium, based on the collective action of surrounding cells (Nanavati et al., 2020; Eisenhoffer et al., 2012).
Basal extrusion is not a common phenomenon in physiological conditions, except during embryogenesis. For example, in vertebrates, neural crest cells acquire the ability to traverse the basal lamina and migrate away from the ectoderm during neurulation, through a process of epithelial-mesenchymal transition (EMT), modulated by an increase in cell density and stiffness in the surrounding tissues (Nanavati et al., 2020; Barriga et al., 2018; Acloque et al., 2009). Epithelial-mesenchymal transition is now considered part of a bidirectional process, highly involved in both development and cancer, known as epithelial-mesenchymal plasticity (EMP; Yang et al., 2020).
Cell Proliferation vs. Cell Extrusion in Carcinogenesis
Responsible for nearly 90% of all cancer cases worldwide (http://www.iarc.fr), carcinomas (malignant cancers originated in epithelia) present an extreme case of proliferative activity. Cell extrusion appears as a pivotal cellular process, both in their control and pathogenesis. For example, a proposed cell competition mechanism termed Epithelial Defense Against Cancer (EDAC; Tanimura and Fujita, 2020), implies the live apical extrusion, followed by anoikis, of transformed cells. This mechanism has been extensively studied in mammalian epithelial (MDCK) cultures and zebrafish embryo epidermis, by experimentally transforming a subset of cells through the mosaic expression of oncogenic proteins, like H-RasV12 or v-Src. The extrusion mechanism involves the activity of the complexes paxillin/plectin/EPLIN and myosin-II/spectrin in these oncogenic-transformed cells (Kasai et al., 2018; Takagi et al., 2018) and polarized changes in tensile forces involving RhoA, filamin and vimentin in the surrounding cells (Kajita et al., 2014).
Excessive or maintained tension in the epithelium, however, can prevent apical detachment of the extruded cells, leading to the formation of multilayered cell accumulations. This phenomenon has been observed when depleting caveolae in non-transformed cells surrounding transformed ones, in a process involving the membrane phospholipid phosphatidylinositol(4,5)bisphosphate (PIP2) and the formin FML2 (Teo et al., 2020). Transformed cells resistant to anoikis can thus generate protrusive tumors, such as the polyps seen in the premalignant stages of colorectal adenocarcinoma. Furthermore, if cells from a growing tumor undergo EMT and basal extrusion, they would eventually cause metastasis (Ortiz et al., 2021; Slattum and Rosenblatt, 2014). Adenomatous Polyposis Coli (APC) was indicated as a switch in the decision between apical and basal extrusion. Normally, APC acts by favoring a basal accumulation of contractile F-actin, leading to apical extrusion, but either gene disruption or the expression of an oncogenic mutant APC, causes cells to extrude basally (Marshall et al., 2011). Basal cell extrusion was also promoted upon the expression of a different Ras protein, the more commonly found in cancers K-RasV12 (Slattum et al., 2014), as well as after down-regulation of E-cadherin in H-RasV12-expressing cells (Hogan et al., 2009). An interesting recent report linked autophagy, which has been associated with bad prognosis in carcinomas, to the downregulation of E-cadherin in breast cancer cells (Damiano et al., 2020).
Cell Proliferation and Morphogenesis vs. Cell Extrusion in Development
In embryonic development, epithelia must maintain their integrity while simultaneously enduring high proliferation rates and morphogenetic forces. An extreme case is represented by the neuroepithelium, from which the millions of neurons and glial cells in the central nervous system originate. In this naturally-overcrowded epithelium, cells accommodate by means of a pseudostratified organization maintained by interkinetic nuclear migration, a cell-cycle synchronic, cytoskeleton-driven movement of nuclei between the apical and basal poles of cells (Strzyz et al., 2016; Sauer, 1935). In addition, neuroepithelial cells have to confront the forces generated by the morphogenetic movements of neurulation, which include cell shape changes (elongation and apical constriction) and convergent extension cell rearrangements (Nikolopoulou et al., 2017; Wallingford, 2012). Despite all this, apical cell extrusion is not normally seen in the neuroepithelium, and the reason might again lie in tissue tension generated by the cytoskeleton.
The flexion of the neural folds has been shown to partly depend on the modulation of two properties of the neuroepithelium: cell density, associated to cell proliferation, and tissue stiffness, associated to actomyosin-based cell contractility (Zhang et al., 2019; McShane et al., 2015). For example, an experimental upregulation of the Hippo-YAP/TAZ pathway, which modulates cell proliferation in response to mechanical stress (Aragona et al., 2013), caused hyperproliferation in the chick neuroepithelium, eventually leading to the generation of apical cell protrusions (Cao et al., 2008). Conversely, YAP (Yes-Associated Protein) deletion in neural crest prospective cells, bordering the neural and non-neural ectoderm, resulted in neurulation defects (Wang et al., 2015). On the other hand, the apical constriction in primary neurulation relies on the association of actomyosin and Rho GTPases with the cadherin-based adhesion complexes of neuroepithelial cells (Escuin et al., 2015; Kinoshita et al., 2008). Severe neurulation defects, accompanied by massive apical cell extrusion, were evident upon the perturbation of Rho (Kinoshita et al., 2008) and another protein that is normally localized around the apical actomyosin ring: the Myristoylated Alanine Rich C-Kinase Substrate (MARCKS; Aparicio et al., 2018).
MARCKS Proteins: Bidirectional Modulators of Epithelial-Mesenchymal Plasticity and Cell Extrusion?
The nervous system-enriched, naturally-unfolded MARCKS proteins comprise a small family encoded by two genes in most vertebrates (MARCKS and MARCKS-Like 1), duplicated in teleosts (Prieto and Zolessi, 2017; Brudvig and Weimer, 2015; Wu et al., 1982). Three domains characterize these proteins: the N-terminal myristoylation domain, the MH2 domain, and the central effector domain, or ED (Toledo et al., 2013; Tapp et al., 2005). Through the 25 residue-long ED, they selectively bind and sequester PIP2 at the plasma membrane, bind Ca-calmodulin, and crosslink actin filaments. Their multiple cellular roles are usually related to cell shape changes or membrane dynamics, which they regulate through the modulation of actin cytoskeleton dynamics and/or PIP2 availability (El Amri et al., 2018; Iioka et al., 2004; Laux et al., 2000).
Normally, MARCKS accumulates apically in chick neural plate cells (Zolessi and Arruti, 2001) and the functional inactivation of both genes caused severe defects in neurulation and neural tube morphogenesis in mice (Chen et al., 1996; Stumpo et al., 1995) and zebrafish (Prieto and Zolessi, 2017). In the chick, either MARCKS knockdown or its protein kinase C (PKC)-induced phosphorylation (leading to a loss in membrane attachment and apical localization), caused a failure in neural tube closure, with the neural plate remaining flat and wider than normal (Aparicio et al., 2018). The most remarkable effect at the cellular level was a general reduction in apicobasal polarity, accompanied by massive apical cell extrusion. Supra-apically localized cells did not detach immediately and remained viable for some time, even dividing in that ectopic position. These observations indicated a live apical cell extrusion mechanism, as described above for oncogenic cells (Fig. 1).
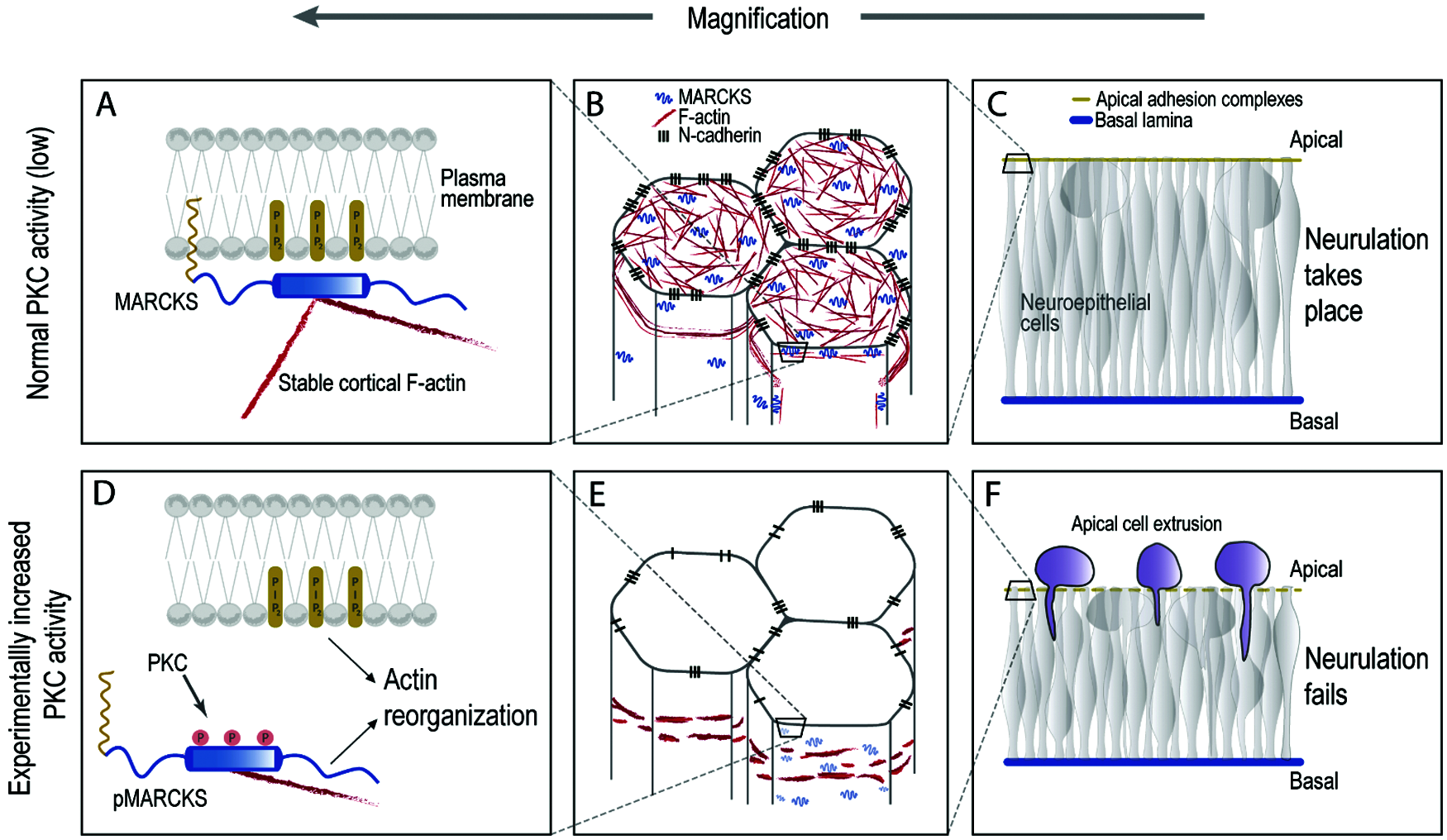
Figure 1: MARCKS hyper-phosphorylation at the ED causes apical extrusion in the neuroepithelium. Partly speculative model to explain the role of MARCKS in neurulation. In the normal chick neural plate cells (A–C), MARCKS is largely present in a non-phosphorylated form, hence attached to PIP2 at the plasma membrane, while simultaneously binding two actin filaments (A and B). This configuration would promote apico-basal cell polarity and cell adhesion, strengthening the epithelial phenotype of the neural plate cells, which is necessary for neurulation (C). If MARCKS is extensively phosphorylated upon experimental PKC activation (or if it is down-regulated, not shown in the figure), it detaches from the plasma membrane, releasing PIP2 and one actin filament, with an effect on cortical F-actin stability (D–E). This would cause a reduction in apico-basal cell polarity and adhesion, accompanied by apical cell extrusion and a failure to close the neural tube (F) (Aparicio et al., 2018).
On the other hand, PKC activity is usually deregulated in cancer (Newton, 2018), and different studies have indicated that MARCKS expression is downregulated in transformed and cancerous cells (Bickeböller et al., 2015; Rohrbach et al., 2015; Jarboe et al., 2012; Michel et al., 2009; Brooks et al., 1996; Joseph et al., 1992; Wolfman et al., 1987). In in vitro-transformed cells, this downregulation was at the transcriptional level, and was related to either v-Src tyrosine kinase activity or to an initially increased PKC activity, while deleterious mutations in the MARCKS-encoding gene were detected in human colorectal cancer cells displaying microsatellite instability (Michel et al., 2009). Some studies, however, have indicated a correlation between an increase in MARCKS expression and bad prognosis in different cancers (Chen et al., 2017; Browne et al., 2013; Micallef et al., 2009). This apparently paradoxical observation might have a very simple explanation on the observation of a higher-than-normal MARCKS ED phosphorylation in several highly-expressing cancers (Chen et al., 2017; Chen and Rotenberg, 2010). In lung cancer cells, the overexpression of a non-phosphorylatable MARCKS or the competitive inhibition of endogenous MARCKS phosphorylation with an ED-mimetic peptide, reduced EMT and cell invasiveness, and increased radiation sensitivity (Rohrbach et al., 2017, 2015; Chen et al., 2014). Altogether, these observations have led to the idea that MARCKS is a bonafide tumor suppressor, whose membrane-binding and actin crosslinking activities are necessary to prevent cells from acquiring a malignant phenotype, keeping at least three processes at bay: cell proliferation, cell invasiveness and resistance to apoptosis.
We can consider the epithelial phenotype as one endpoint of a continuous line that leads to the mesenchymal phenotype in the other, with several intermediate stages along the line, dynamically connected by EMP. We propose that MARCKS-family proteins act as dynamic switches continuously influencing the cell’s position along this line (Fig. 2). A well-regulated proportion of unphosphorylated MARCKS, enough to simultaneously bind most of the free PIP2 while anchoring crosslinked actin filaments to the membrane, will make cells firmly attach to each other and acquire an epithelial phenotype, getting closer to an endpoint. Any physiological, pathological or experimental changes in this balance (lower or too high MARCKS expression; higher PKC-type phosphorylation), would drive cells towards the mesenchymal endpoint of the EMP line. One of the resulting events in this epithelial-mesenchymal transition would be cell extrusion, either apical or basal, with different outcomes regarding the fate of the extruded cells depending on both cell-intrinsic and cell-extrinsic factors.
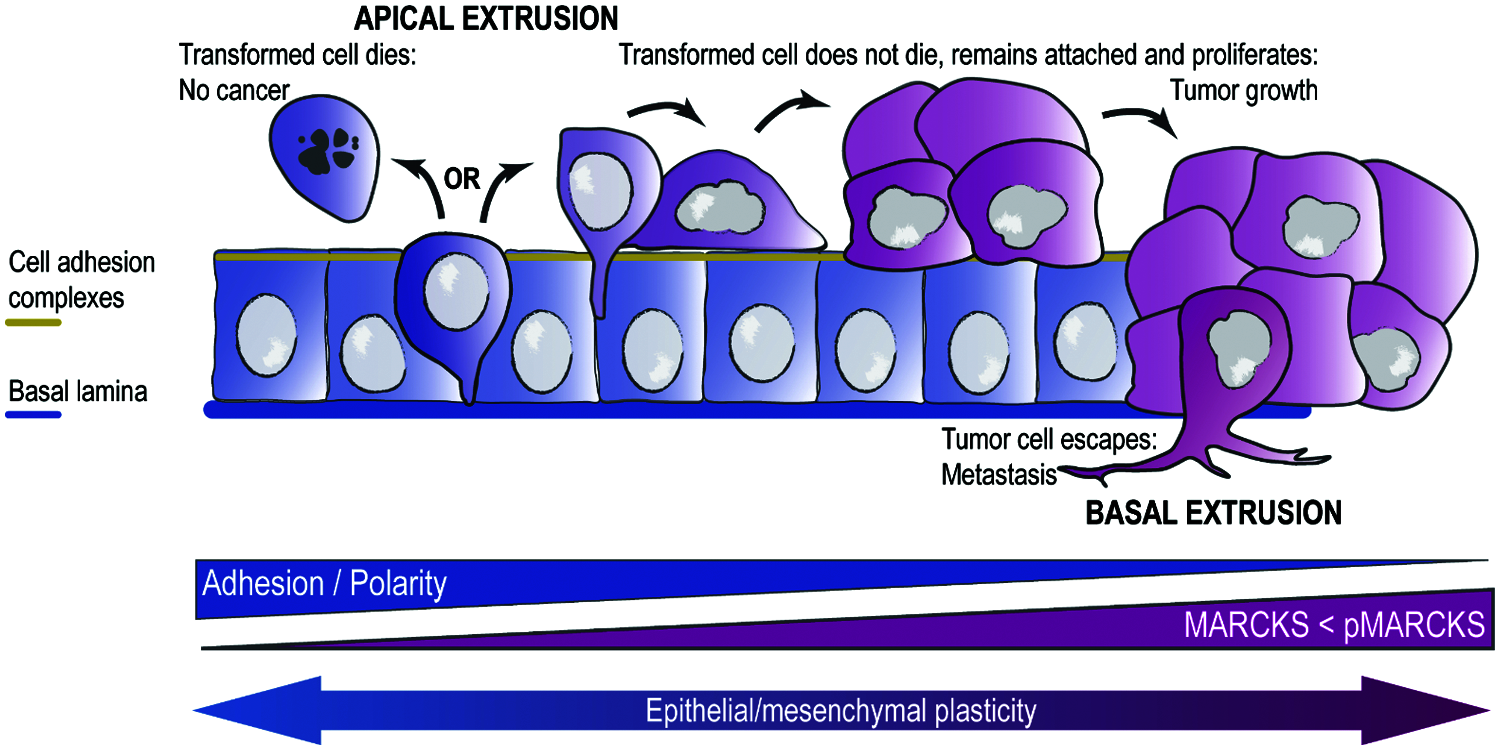
Figure 2: A model for the role of cell extrusion and MARCKS protein in carcinogenesis. In a healthy epithelium, transformed cells tend to be expelled by apical cell extrusion followed by cell death (anoikis). Some transformed cells may however gain the ability to stay attached to the apical side of the epithelium after extrusion. If these cells are at the same time resistant to cell death and proliferative (both hallmarks of cancer cells), they will generate a primary tumor, which is initially “benign”. Eventually, some cells in this tumor may gain the ability to basally extrude, escaping from the epithelium and invading other tissues and organs, where they form secondary tumors (metastasis). This implies a process known as “epithelial-mesenchymal plasticity”, which is associated with the degree of cell adhesion and apico-basal polarity. The balance between MARCKS expression and its phosphorylation at the ED would modulate this process.
Authors’ Contribution:The authors confirm contribution to the paper as follows: conception, bibliographic search, draft manuscript preparation: L. Veloz, S. A. Bosch, G. Aparicio, F. R. Zolessi. All authors reviewed the results and approved the final version of the manuscript.
Funding Statement: This work was partially funded by CSIC-UdelaR Grant C125-347 to FRZ; CAP-UdelaR Master’s fellowship to SAB; Dedicación Total-UdelaR, to FRZ; PEDECIBA.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present work.
Acloque H, Adams MS, Fishwick K, Bronner-Fraser M, Nieto MA (2009). Epithelial-mesenchymal transitions: The importance of changing cell state in development and disease. Journal of Clinical Investigation 119: 1438–1449. DOI 10.1172/JCI38019. [Google Scholar] [CrossRef]
Aparicio G, Arruti C, Zolessi FR (2018). MARCKS phosphorylation by PKC strongly impairs cell polarity in the chick neural plate. Genesis 56: e23104. DOI 10.1002/dvg.23104. [Google Scholar] [CrossRef]
Aragona M, Panciera T, Manfrin A, Giulitti S, Michielin F, Elvassore N, Dupont S, Piccolo S (2013). A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell 154: 1047–1059. DOI 10.1016/j.cell.2013.07.042. [Google Scholar] [CrossRef]
Atieh Y, Wyatt T, Zaske AM, Eisenhoffer GT (2021). Pulsatile contractions promote apoptotic cell extrusion in epithelial tissues. Current Biology 31: 1129–1140. DOI 10.1016/j.cub.2020.12.005. [Google Scholar] [CrossRef]
Barriga EH, Franze K, Charras G, Mayor R (2018). Tissue stiffening coordinates morphogenesis by triggering collective cell migration in vivo. Nature 554: 523–527. DOI 10.1038/nature25742. [Google Scholar] [CrossRef]
Bickeböller M, Tagscherer KE, Kloor M, Jansen L, Chang-Claude J et al. (2015). Functional characterization of the tumor-suppressor MARCKS in colorectal cancer and its association with survival. Oncogene 34: 1150–1159. DOI 10.1038/onc.2014.40. [Google Scholar] [CrossRef]
Brooks G, Brooks SF, Goss MW (1996). MARCKS functions as a novel growth suppressor in cells of melanocyte origin. Carcinogenesis 17: 683–689. DOI 10.1093/carcin/17.4.683. [Google Scholar] [CrossRef]
Browne BC, Hochgräfe F, Wu J, Millar EKA, Barraclough J et al. (2013). Global characterization of signalling networks associated with tamoxifen resistance in breast cancer. FEBS Journal 280: 5237–5257. DOI 10.1111/febs.12441. [Google Scholar] [CrossRef]
Brudvig JJ, Weimer JM (2015). X MARCKS the spot: Myristoylated alanine-rich C kinase substrate in neuronal function and disease. Frontiers in Cellular Neuroscience 9: 1–10. DOI 10.3389/fncel.2015.00407. [Google Scholar] [CrossRef]
Cao X, Pfaff SL, Gage FH (2008). YAP regulates neural progenitor cell number via the TEA domain transcription factor. Genes and Development 22: 3320–3334. DOI 10.1101/gad.1726608. [Google Scholar] [CrossRef]
Chen CH, Fong LWR, Yu E, Wu R, Trott JF, Weiss RH (2017). Upregulation of MARCKS in kidney cancer and its potential as a therapeutic target. Oncogene 36: 3588–3598. DOI 10.1038/onc.2016.510. [Google Scholar] [CrossRef]
Chen CH, Thai P, Yoneda K, Adler KB, Yang PC, Wu R (2014). A peptide that inhibits function of Myristoylated Alanine-Rich C Kinase Substrate (MARCKS) reduces lung cancer metastasis. Oncogene 33: 3696–3706. DOI 10.1038/onc.2013.336. [Google Scholar] [CrossRef]
Chen J, Chang S, Duncan SA, Okano HJ, Fishell G, Aderem A (1996). Disruption of the MacMARCKS gene prevents cranial neural tube closure and results in anencephaly. Proceedings of the National Academy of Sciences of the United States of America 93: 6275–6279. DOI 10.1073/pnas.93.13.6275. [Google Scholar] [CrossRef]
Chen X, Rotenberg SA (2010). PhosphoMARCKS drives motility of mouse melanoma cells. Cellular Signalling 22: 1097–1103. DOI 10.1016/j.cellsig.2010.03.003. [Google Scholar] [CrossRef]
Damiano V, Spessotto P, Vanin G, Perin T, Maestro R, Santarosa M (2020). The autophagy machinery contributes to E-cadherin turnover in breast cancer. Frontiers in Cell and Developmental Biology 8: 545. DOI 10.3389/fcell.2020.00545. [Google Scholar] [CrossRef]
Duszyc K, Gomez GA, Lagendijk AK, Yau MK, Nanavati BN et al. (2021). Mechanotransduction activates RhoA in the neighbors of apoptotic epithelial cells to engage apical extrusion. Current Biology 31: 1326–1336. DOI 10.1016/j.cub.2021.01.003. [Google Scholar] [CrossRef]
Eisenhoffer GT, Loftus PD, Yoshigi M, Otsuna H, Chien CB, Morcos PA, Rosenblatt J (2012). Crowding induces live cell extrusion to maintain homeostatic cell numbers in epithelia. Nature 484: 546–549. DOI 10.1038/nature10999. [Google Scholar] [CrossRef]
Eisenhoffer GT, Rosenblatt J (2013). Bringing balance by force: Live cell extrusion controls epithelial cell numbers. Trends in Cell Biology 23: 185–192. DOI 10.1016/j.tcb.2012.11.006. [Google Scholar] [CrossRef]
El Amri M, Fitzgerald U, Schlosser G (2018). MARCKS and MARCKS-like proteins in development and regeneration. Journal of Biomedical Science 25: 1–12. DOI 10.1186/s12929-018-0445-1. [Google Scholar] [CrossRef]
Escuin S, Vernay B, Savery D, Gurniak CB, Witke W, Greene NDE, Copp AJ (2015). Rho kinase-dependent actin turnover and actomyosin disassembly are necessary for mouse spinal neural tube closure. Journal of Cell Science 128: 2468–2481. DOI 10.1242/jcs.164574. [Google Scholar] [CrossRef]
Hogan C, Dupré-Crochet S, Norman M, Kajita M, Zimmermann C et al. (2009). Characterization of the interface between normal and transformed epithelial cells. Nature Cell Biology 11: 460–467. DOI 10.1038/ncb1853. [Google Scholar] [CrossRef]
Iioka H, Ueno N, Kinoshita N (2004). Essential role of MARCKS in cortical actin dynamics during gastrulation movements. Journal of Cell Biology 164: 169–174. DOI 10.1083/jcb.200310027. [Google Scholar] [CrossRef]
Jarboe JS, Anderson JC, Duarte CW, Mehta T, Nowsheen S et al. (2012). MARCKS regulates growth and radiation sensitivity and is a novel prognostic factor for glioma. Clinical Cancer Research 18: 3030–3041. DOI 10.1158/1078-0432.CCR-11-3091. [Google Scholar] [CrossRef]
Joseph CK, Qureshi SA, Wallace DJ, Foster DA (1992). MARCKS protein is transcriptionally down-regulated in v-Src-transformed BALB/c 3T3 cells. Journal of Biological Chemistry 267: 1327–1330. DOI 10.1016/S0021-9258(18)48433-5. [Google Scholar] [CrossRef]
Kajita M, Sugimura K, Ohoka A, Burden J, Suganuma H et al. (2014). Filamin acts as a key regulator in epithelial defence against transformed cells. Nature Communications 5: 1–13. DOI 10.1038/ncomms5428. [Google Scholar] [CrossRef]
Kasai N, Kadeer A, Kajita M, Saitoh S, Ishikawa S, Maruyama T, Fujita Y (2018). The paxillin-plectin-EPLIN complex promotes apical elimination of RasV12-transformed cells by modulating HDAC6-regulated tubulin acetylation. Scientific Reports 8: 988. DOI 10.1038/s41598-018-20146-1. [Google Scholar] [CrossRef]
Kinoshita N, Sasai N, Misaki K, Yonemura S (2008). Apical accumulation of Rho in the neural plate is important for neural plate cell shape change and neural tube formation. Molecular Biology of the Cell 19: 2289–2299. DOI 10.1091/mbc.e07-12-1286. [Google Scholar] [CrossRef]
Laux T, Fukami K, Thelen M, Golub T, Frey D, Caroni P (2000). GAP43, MARCKS, and CAP23 modulate PI(4,5)P(2) at plasmalemmal rafts, and regulate cell cortex actin dynamics through a common mechanism. Journal of Cell Biology 149: 1455–1472. DOI 10.1083/jcb.149.7.1455. [Google Scholar] [CrossRef]
Lubkov V, Bar-Sagi D (2014). E-cadherin-mediated cell coupling is required for apoptotic cell extrusion. Current Biology 24: 868–874. DOI 10.1016/j.cub.2014.02.057. [Google Scholar] [CrossRef]
Madara JL (1990). Maintenance of the macromolecular barrier at cell extrusion sites in intestinal epithelium: Physiological rearrangement of tight junctions. Journal of Membrane Biology 116: 177–184. DOI 10.1007/BF01868675. [Google Scholar] [CrossRef]
Marshall TW, Lloyd IE, Delalande JM, Näthke I, Rosenblatt J (2011). The tumor suppressor adenomatous polyposis coli controls the direction in which a cell extrudes from an epithelium. Molecular Biology of the Cell 22: 3962–3970. DOI 10.1091/mbc.e11-05-0469. [Google Scholar] [CrossRef]
McShane SG, Molè MA, Savery D, Greene NDE, Tam PPL, Copp AJ (2015). Cellular basis of neuroepithelial bending during mouse spinal neural tube closure. Developmental Biology 404: 113–124. DOI 10.1016/j.ydbio.2015.06.003. [Google Scholar] [CrossRef]
Micallef J, Taccone M, Mukherjee J, Croul S, Busby J, Moran MF, Guha A (2009). Epidermal growth factor receptor variant III-induced glioma invasion is mediated through myristoylated alanine-rich protein kinase C substrate overexpression. Cancer Research 69: 7548–7556. DOI 10.1158/0008-5472.CAN-08-4783. [Google Scholar] [CrossRef]
Michel S, Kloor M, Singh S, Gdynia G, Roth W, von Knebel Doeberitz M, Schirmacher P, Bläker H (2009). Coding microsatellite instability analysis in microsatellite unstable small intestinal adenocarcinomas identifies MARCKS as a common target of inactivation. Molecular Carcinogenesis 49: 175–182. DOI 10.1002/mc.20587. [Google Scholar] [CrossRef]
Nanavati BN, Yap AS, Teo JL (2020). Symmetry breaking and epithelial cell extrusion. Cells 9: 1416. DOI 10.3390/cells9061416. [Google Scholar] [CrossRef]
Newton AC (2018). Protein kinase C: Perfectly balanced. Critical Reviews in Biochemistry and Molecular Biology 53: 208–230. DOI 10.1080/10409238.2018.1442408. [Google Scholar] [CrossRef]
Nikolopoulou E, Galea GL, Rolo A, Greene NDE, Copp AJ (2017). Neural tube closure: Cellular, molecular and biomechanical mechanisms. Development 144: 552–566. DOI 10.1242/dev.145904. [Google Scholar] [CrossRef]
Ortiz MA, Mikhailova T, Li X, Porter BA, Bah A, Kotula L (2021). Src family kinases, adaptor proteins and the actin cytoskeleton in epithelial-to-mesenchymal transition. Cell Communication and Signaling 19: 67. DOI 10.1186/s12964-021-00750-x. [Google Scholar] [CrossRef]
Prieto D, Zolessi FR (2017). Functional diversification of the Four MARCKS family members in zebrafish neural development. Journal of Experimental Zoology Part B: Molecular and Developmental Evolution 328: 119–138. DOI 10.1002/jez.b.22691. [Google Scholar] [CrossRef]
Rohrbach TD, Jarboe JS, Anderson JC, Trummell HQ, Hicks PH et al. (2015). Targeting the effector domain of the myristoylated alanine rich C-kinase substrate enhances lung cancer radiation sensitivity. International Journal of Oncology 46: 1079–1088. DOI 10.3892/ijo.2014.2799. [Google Scholar] [CrossRef]
Rohrbach TD, Jones RB, Hicks PH, Weaver AN, Cooper TS, Eustace NJ, Yang ES, Jarboe JS, Anderson JC, Willey CD (2017). MARCKS phosphorylation is modulated by a peptide mimetic of MARCKS effector domain leading to increased radiation sensitivity in lung cancer cell lines. Oncology Letters 13: 1216–1222. DOI 10.3892/ol.2016.5550. [Google Scholar] [CrossRef]
Rosenblatt J, Raff MC, Cramer LP (2001). An epithelial cell destined for apoptosis signals its neighbors to extrude it by an actin- and myosin-dependent mechanism. Current Biology 11: 1847–1857. DOI 10.1016/S0960-9822(01)00587-5. [Google Scholar] [CrossRef]
Sauer FC (1935). Mitosis in the neural tube. Journal of Comparative Neurology 62: 377–405. DOI 10.1002/(ISSN)1096-9861. [Google Scholar] [CrossRef]
Saw TB, Doostmohammadi A, Nier V, Kocgozlu L, Thampi S, Toyama Y, Marcq P, Lim CT, Yeomans JM, Ladoux B (2017). Topological defects in epithelia govern cell death and extrusion. Nature 544: 212–216. DOI 10.1038/nature21718. [Google Scholar] [CrossRef]
Slattum GM, Gu Y, Sabbadini R, Rosenblatt J (2014). Autophagy in oncogenic K-Ras promotes basal extrusion of epithelial cells by degrading S1P. Current Biology 24: 19–28. DOI 10.1016/j.cub.2013.11.029. [Google Scholar] [CrossRef]
Slattum GM, Rosenblatt J (2014). Tumour cell invasion: An emerging role for basal epithelial cell extrusion. Nature Reviews Cancer 14: 495–501. DOI 10.1038/nrc3767. [Google Scholar] [CrossRef]
Strzyz PJ, Matejcic M, Norden C (2016). Heterogeneity, cell biology and tissue mechanics of pseudostratified epithelia: Coordination of cell divisions and growth in tightly packed tissues. International Review of Cell and Molecular Biology 325: 89–118. [Google Scholar]
Stumpo DJ, Bock CB, Tutlle JS, Tuttle JS, Blackshear PJ (1995). MARCKS deficiency in mice leads to abnormal brain development and perinatal death. Proceedings of the National Academy of Sciences of the United States of America 92: 944–948. DOI 10.1073/pnas.92.4.944. [Google Scholar] [CrossRef]
Takagi M, Ikegawa M, Shimada T, Ishikawa S, Kajita M, Maruyama T, Kamasaki T, Fujita Y (2018). Accumulation of the myosin-II-spectrin complex plays a positive role in apical extrusion of Src-transformed epithelial cells. Genes to Cells 23: 974–981. DOI 10.1111/gtc.12643. [Google Scholar] [CrossRef]
Tanimura N, Fujita Y (2020). Epithelial defense against cancer (EDAC). Seminars in Cancer Biology 63: 44–48. DOI 10.1016/j.semcancer.2019.05.011. [Google Scholar] [CrossRef]
Tapp H, Al-Naggar IM, Yarmola EG, Harrison A, Shaw G, Edison AS, Bubb MR (2005). MARCKS is a natively unfolded protein with an inaccessible actin-binding site. Journal of Biological Chemistry 280: 9946–9956. DOI 10.1074/jbc.M414614200. [Google Scholar] [CrossRef]
Teo JL, Gomez GA, Weeratunga S, Davies EM, Noordstra I et al. (2020). Caveolae control contractile tension for epithelia to eliminate tumor cells. Developmental Cell 54: 75–91. DOI 10.1016/j.devcel.2020.05.002. [Google Scholar] [CrossRef]
Toledo A, Zolessi FR, Arruti C (2013). A novel effect of MARCKS phosphorylation by activated PKC: The dephosphorylation of its serine 25 in chick neuroblasts. PLoS One 8: e62863. DOI 10.1371/journal.pone.0062863. [Google Scholar] [CrossRef]
Villars A, Levayer R (2020). Cell extrusion: Crowd pushing and sticky neighbours. Current Biology 30: 168–171. DOI 10.1016/j.cub.2019.12.033. [Google Scholar] [CrossRef]
Wallingford JB (2012). Planar cell polarity and the developmental control of cell behavior in vertebrate embryos. Annual Review of Cell and Developmental Biology 28: 627–653. DOI 10.1146/annurev-cellbio-092910-154208. [Google Scholar] [CrossRef]
Wang J, Xiao Y, Hsu CW, Martinez-Traverso IM, Zhang M et al. (2015). Yap and Taz play a crucial role in neural crest-derived craniofacial development. Development 143: 504–515. [Google Scholar]
Wolfman A, Wingrove T, Blackshear PJ, Macara I (1987). Down-regulation of protein kinase C and of an endogenous 80-kDa substrate in transformed fibroblasts. Journal of Biological Chemistry 262: 16546–16552. DOI 10.1016/S0021-9258(18)49290-3. [Google Scholar] [CrossRef]
Wu WC, Walaas SI, Nairn AC, Greengard P (1982). Calcium/phospholipid regulates phosphorylation of a Mr “87” substrate protein in brain synaptosomes. Proceedings of the National Academy of Sciences of the United States of America 79: 5249–5253. DOI 10.1073/pnas.79.17.5249. [Google Scholar] [CrossRef]
Yang J, Antin P, Berx G, Blanpain C, Brabletz T et al. (2020). Guidelines and definitions for research on epithelial-mesenchymal transition. Nature Reviews Molecular Cell Biology 21: 341–352. DOI 10.1038/s41580-020-0237-9. [Google Scholar] [CrossRef]
Zhang J, Raghunathan R, Rippy J, Wu C, Finnell RH, Larin KV, Scarcelli G (2019). Tissue biomechanics during cranial neural tube closure measured by Brillouin microscopy and optical coherence tomography. Birth Defects Research 111: 991–998. DOI 10.1002/bdr2.1389. [Google Scholar] [CrossRef]
Zolessi FR, Arruti C (2001). Apical accumulation of MARCKS in neural plate cells during neurulation in the chick embryo. BMC Developmental Biology 1: 7. DOI 10.1186/1471-213X-1-7. [Google Scholar] [CrossRef]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |