DOI:10.32604/biocell.2022.016718
| BIOCELL DOI:10.32604/biocell.2022.016718 | |
| Article |
Phylogenetic analysis and target gene prediction of miR477 gene family in grape
1College of Horticulture and Plant Protection, Henan University of Science and Technology, Luoyang, 471023, China
2Henan Engineering Technology Research Center of Quality Regulation of Horticultural Plants, Luoyang, 471023, China
*Address correspondence to: Da-Long Guo, guodalong@haust.edu.cn
Received: 19 March 2021; Accepted: 06 May 2021
Abstract: To understand the molecular characteristics of the miR477 gene family of grape (Vvi-miR477) and to predict its target genes, the Vvi-miR477 genes were identified from previous small RNA sequencing data, then phylogenetic analysis and prediction of target gene were conducted. The Vvi-miR477 family consists of two precursor sequences and three mature sequences. The miR477 family members were mostly 19-22nt in length. The sequence is relatively conservative. Vvi-MIR477a and Vvi-MIR477b are located on chromosomes 1 and 2, respectively. These precursor sequences can form the typical stable stem-loop structure. Their minimum folding free energy is −39.10 kcal/mol and −50.90 kcal/mol, respectively. The MIR477 family can be divided into three groups. The prediction of target genes showed that Vvi-miR477 targets 26S proteasome, DEAD-box, GRAS family protein, Protein Phosphatase 2C, etc. The GO function of target genes was mainly enriched to six categories. The catabolic process, carboxylic ester hydrolase activity is shown to be high. This study provided a theoretical basis for further exploration of the molecular mechanism of miR477 in grape berry ripening.
Keywords: Vitis vinifera; miRNA; Fruit ripening; Phylogenetic characteristics; Target genes
MicroRNAs (miRNAs) are endogenous, non-coding single-stranded small RNA molecules with about 21–23 nucleotides, which are widely distributed in eukaryotes (Ye et al., 2019). A large number of studies have shown that miRNAs play a crucial role in regulating various metabolic and biological processes, including growth (Ding et al., 2020; Lelandais-Brière et al., 2010), flowering time (Yang et al., 2019), sex determination (Sun, 2012), and phytohormone signaling (Liu and Chen, 2009). For example, miR172 regulates the development of Brassica napus flower organs by targeting the euAP2 gene (Wang et al., 2019). Auxin response factors of miR167 plays an important role in flower development and fertility of rice (Li et al., 2020b). The overexpression of miR319a reduced the transcription level of its targeted gene family TCP, significantly improved the trichome density of transgenic Populus tomentosa leaves, and thus relieved the harm of insect herbivores (Fan et al., 2020). In strawberry, overexpression of miR399a could increase the content of soluble sugar, soluble solids, and vitamin C, indicating that the expression of miR399a is positively correlated with the content of sugar (Wang et al., 2017). The regulation of anthocyanin biosynthesis by miR858 has been revealed in grape (Tirumalai et al., 2019), tomato (Jia et al., 2015) and kiwifruit (Li et al., 2020a). By means of base complementary pairing, miRNA cleaves the mRNA or inhibits the translation of target genes to participate in the regulation of plant growth and development (Chipman and Pasquinelli, 2019).
The miR477 family is conserved and present in a number of plant species (Wang et al., 2020). Studies have shown that miR477 plays an important role in the biological processes of plants (Wang et al., 2020). For example, the downregulation of miR477 resulted in the accumulation of lipids and the transformation of nutrients during the fruit ripening process of Camellia oleifera (Liu et al., 2019). In cotton, ghr-miR477 directly lyses the mRNA of GhCBP60A during the post-transcriptional process. While miR477-CBP60A module participates in the resistance response of plants to V. dahliae. The silencing of ghr-miR477 and the knockout of GhCBP60A increased the susceptibility and the resistance of plants to V. dahlia, respectively (Hu et al., 2020). In addition, miR477 was differently expressed in the roots and leaves of bread wheat under drought stress (Lu et al., 2008) and up-regulated under salt stress in maize (Ding et al., 2009).
Grape is an important fruit crop in the world and has high nutritional value. In China, grapes are mainly used for natural consumption, and there are also many processed products, such as wine, raisins, grape juice, grape seeds, etc., Guo et al. (2020). Fruit development is a sequential process, which involves fruit set, fruit growth and fruit ripening (Osorio et al., 2013). During fruit development, each step is strictly regulated by complex molecular mechanisms, post-transcriptional regulation by microRNAs is one of the important ways (Farinati et al., 2020). At present, most of our knowledge about the genetic and molecular mechanisms of fruit ripening comes from the model plant, tomato (Gao et al., 2015). The detailed information of grape berry ripening is relatively few.
Previous reports showed that Fengzao is an early-ripening bud mutant of Kyoho, which matures nearly 30 days earlier (Guo and Zhang, 2015). The whole transcriptome characteristics of Kyoho and Fengzao were determined by high-throughput sequencing, which demonstrated that Vvi-miR477b-3p was one of the major differentially expressed miRNAs between Kyoho and Fengzao and up-regulated in Fengzao (Guo et al., 2018).
In this study, two precursor sequences and three mature sequences of Vvi-miR477 gene family were identified based on the high-throughput sequencing of small RNAs in the previous study (Guo et al., 2018). Sequence alignment, phylogenetic analysis, prediction and GO enrichment analysis of the target genes were performed for miR477 family members. This study provided the bases for future research on the regulatory mechanism of miR477 in grape early ripening process.
Acquisition of precursor and mature sequences of miR477 family members
The family members, precursor and mature sequences of Vvi-miR477 were identified from previous small RNA sequencing data (Guo et al., 2018) through homologs searches of blast using already known miRNAs in other species according to the method of Xie et al. (2010). The precursor and mature sequences of miR477 for the investigated plants were downloaded from the miRBase database (http://www.mirbase.org/index.shtml).
Analysis of the mature sequence and precursor sequence of the miR477 family
The miR477 mature sequences of Vitis vinifera, Nicotiana tabacum, Malus domestica, Citrus sinensis, Manihot esculenta, Populus trichocarpa, Fragaria vesca, Physcomitrella patens, Lotus japonicus, Prunus persica, Solanum tuberosum, Aquilegia caerulea, Solanum lycopersicum, Asparagus officinalis, Gossypium raimondii, Amborella trichopoda, Carica papaya, Cucumis melo were subjected to multiple sequence alignment using CLUSTALW online software with the default parameters (https://www.genome.jp/tools-bin/clustalw). Meanwhile, the grape miR477 precursor sequence (Vvi-MIR477) and the mature sequence (Vvi-miR477) were separately aligned. The phylogenetic tree of miR477 gene family was constructed using the Maximum Likelihood method of MEGA version 7.0.26. Bootstrap confidence values were obtained by applying 1000 replications. Kimura 2-parameter model was chosen as the substitution model. Other parameters were set at their default value. RNAfold (http://rna.tbi.univie.ac.at/) of the ViennaRNA package was used to analyze the secondary structure of the Vvi-MIR477. The default values of the software were used.
Gene location of Vvi-MIR477 family member
The localization of Vvi-MIR477a and Vvi-MIR477b on chromosomes was analyzed by local blast method. MapGene2Chrom V1.0 (http://mg2c.IASK.in/mg2C_v1.0/) online software was employed to map the chromosome location.
Target gene prediction of grape miR477 family members
PsRNATarget (http://plantgrn.noble.org/psRNATarget/) online software was used to predict the target genes of Vvi-miR477 family members. The grape transcript from University of Padua CRIBI Genomics V2.1 (http://genomes.cribi.unipd.it) was used as target prediction database. The maximum expectation value was set to 3 and the remaining parameters were as the default values.
Expression of miR477 and functional enrichment analysis of its target genes
A heatmap was drawn using the R pheatmap package to demonstrate the expression of Vvi-miR477 family members in Kyoho and Fengzao. The predicted miR477 target genes were then for Gene Ontology (GO) enrichment analysis with ClusterProfiler 3.8.0. GO terms that had a P-value less than 0.05 after Bonferroni correction were scored as significant.
Analysis of plant miR477 family mature sequence
Seventy-four mature miR477 sequences belonging to 17 species from the miRBase database were obtained. The number of members of the miR477 family varies greatly among the species. For example, there are 11 miR477 mature sequences for Manihot esculenta which is the largest. Citrus sinensis and Physcomitrella patens have 10 miR477 mature sequences. There are 9 miR477 mature sequences in Populus trichocarpa, 7 in Aquilegia caerulea, 6 in Prunus persica, 4 in Solanum tuberosum. There are two miR477 mature sequences in the Nicotiana tabacum, Malus domestica, Fragaria vesca, Lotus japonicus, Solanum lycopersicum, Asparagus officinalis, Cucumis melo. There is only one miR477 mature sequence in Gossypium raimondii, Amborella trichopoda, and Carica papaya. Three mature miR477 sequences of Vitis vinifera were identified from small RNA sequencing data, including Vvi-miR477a, Vvi-miR477b-3p and Vvi-miR477b-5p (Guo et al., 2018).
The mature sequences of miR477 from investigated plants were compared using CLUSTALW online software. The results showed that the length of most miR477 family members in different species was 20–21 nt, while few of them were 19 nt and 22 nt long (Fig. 1). Members of the same family from different species and different members of the same species have the different sequence length. For example, the sequences of miR477a-3p, miR477a-5p, miR477b-3p, miR477b-5p were different between Prunus persica and Citrus sinensis. The mature sequences of fve-miR477a and fve-miR477b were also different even if both of them are from Fragaria vesca miR477 family. The mature sequences of three members of the miR477 family of grapes are also not identical (Fig. 2). But their sequence is relatively conservative.
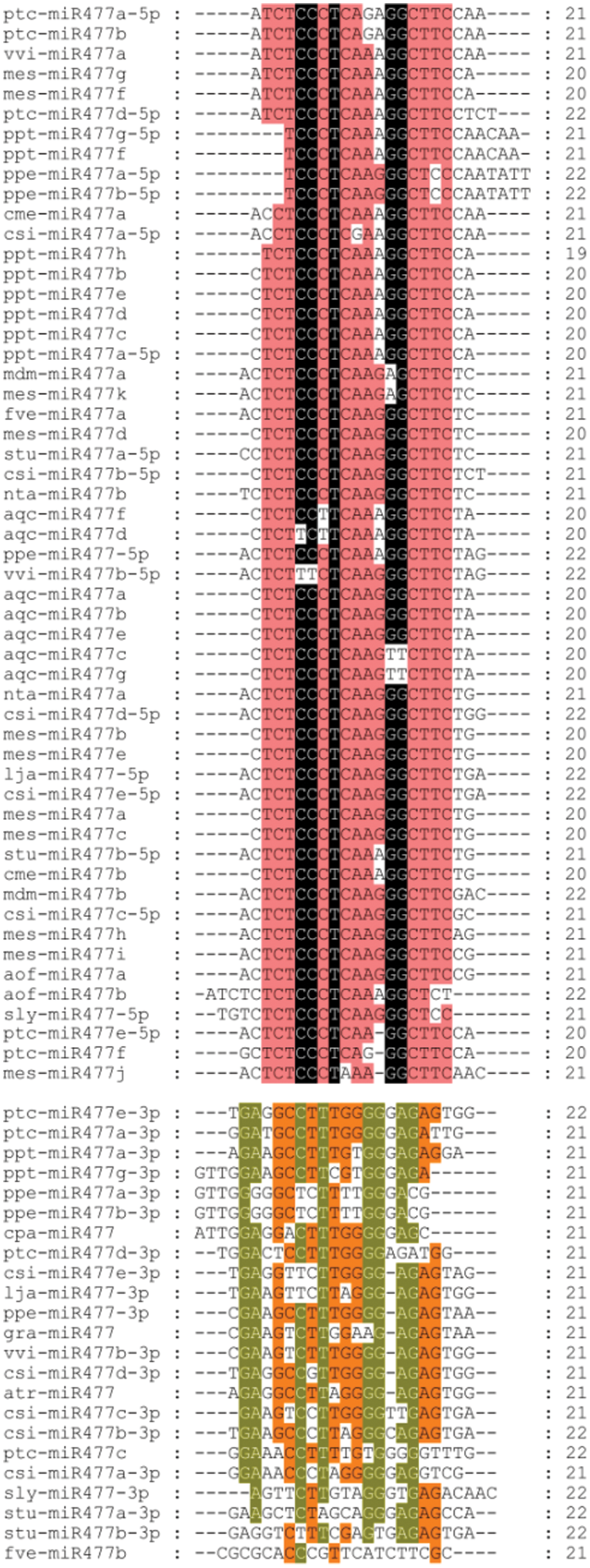
Figure 1: Sequence alignment of the mature miR477 from different plants.
Note: ptc, Populus trichocarpa; vvi, Vitis vinifera; mes, Manihot esculenta; ppt, Physcomitrella patens; ppe, Prunus persica; cme, Cucumis melo; csi, Citrus sinensis; mdm, Malus domestica; fve, Fragaria vesca; stu, Solanum tuberosum; nta, Nicotiana tabacum; aqc, Aquilegia caerulea; lja, Lotus japonicus; aof, Asparagus officinalis; sly, Solanum lycopersicum; gra, Gossypium raimondii; atr, Amborella trichopoda; cpa, Carica papaya.
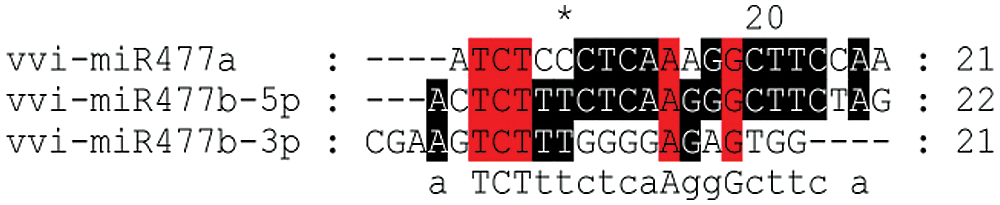
Figure 2: Sequence alignment of miR477 mature sequences in grape.
Note: Red indicates the highest level of conservatism, followed by black.
Localization of the Vvi-MIR477 gene family in the genome
The chromosome distribution of the Vvi-miR477 precursors is shown in Fig. 3. Vvi-MIR477 was unevenly distributed in the 19 chromosomes of grape, which was restricted to chromosomes 1 and 2. The Vvi-MIR477a was located on chromosome 1, and Vvi-MIR477b was located on chromosome 2.
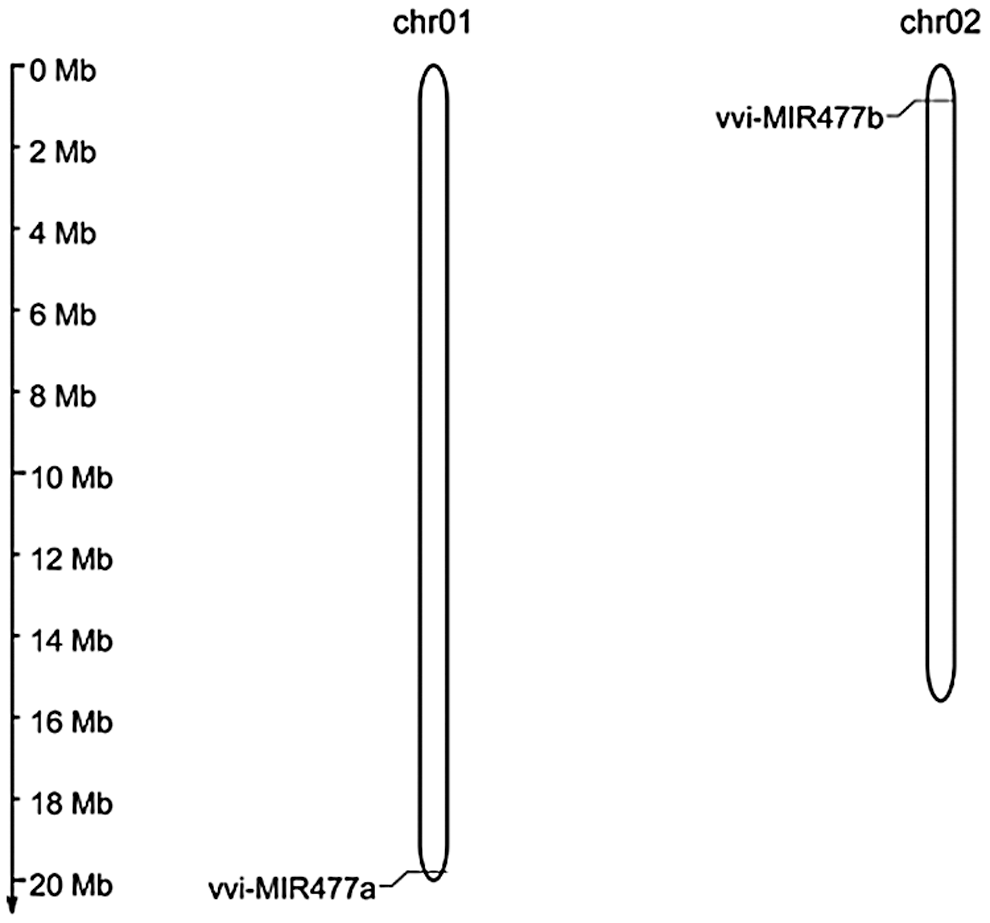
Figure 3: Localization of the Vvi-miR477 gene family in the grape genome.
Sequence analysis and secondary structure prediction of precursors of MIR477 family in grape
Previous studies have shown that the precursor miRNAs of miR477 family members in grape are Vvi-MIR477a and Vvi-MIR477b. Their lengths are 94 nt and 117 nt, respectively (Fig. 4). There is a 23-nt difference between these two precursors, and the sequences had a certain diversity. The RNAfold Server (http://rna.tbi.univie.ac.at/) in ViennaRNA Web Services is used to predict the secondary structure of the Vvi-MIR477 members. The minimum folding free energy (dG) of Vvi-MIR477a and Vvi-MIR477b is −39.10 kcal/mol, −50.90 kcal/mol, respectively (Fig. 5). It showed the high reliability forecast of miR477 family members. The red part indicated the mature sequence of miR477 (Fig. 5).
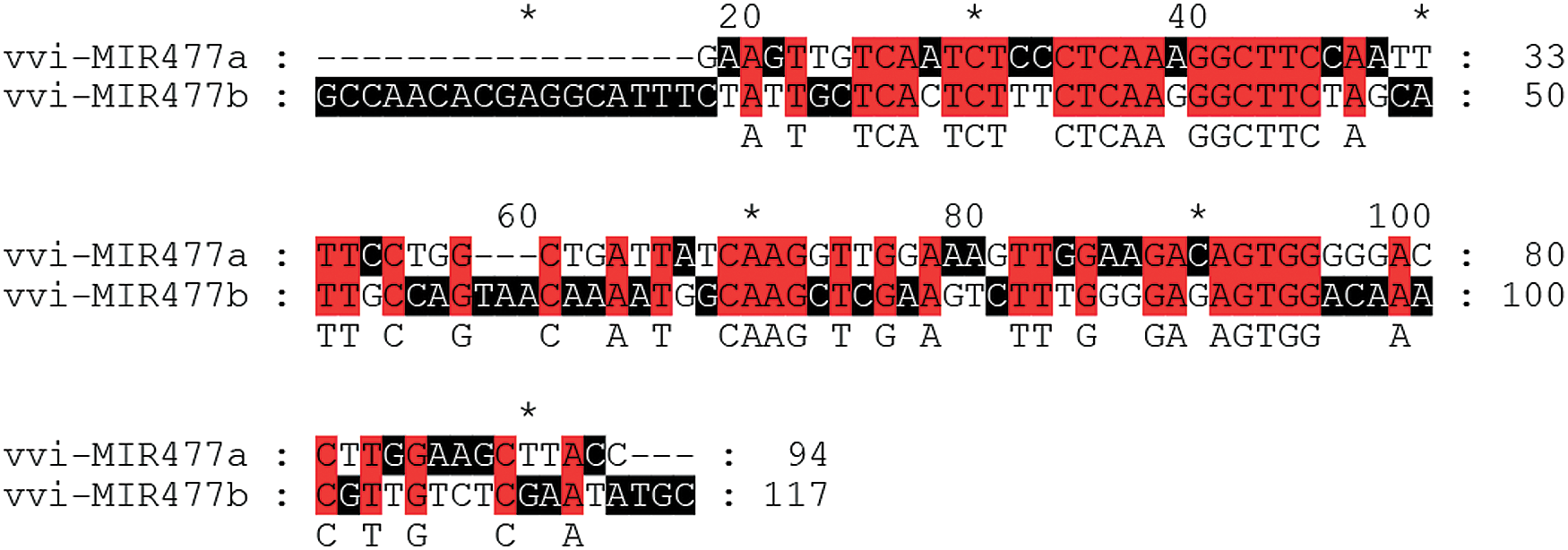
Figure 4: Sequence alignment of miR477 precursor sequences in grape.
Note: Red indicates the highest level of conservatism, followed by black.
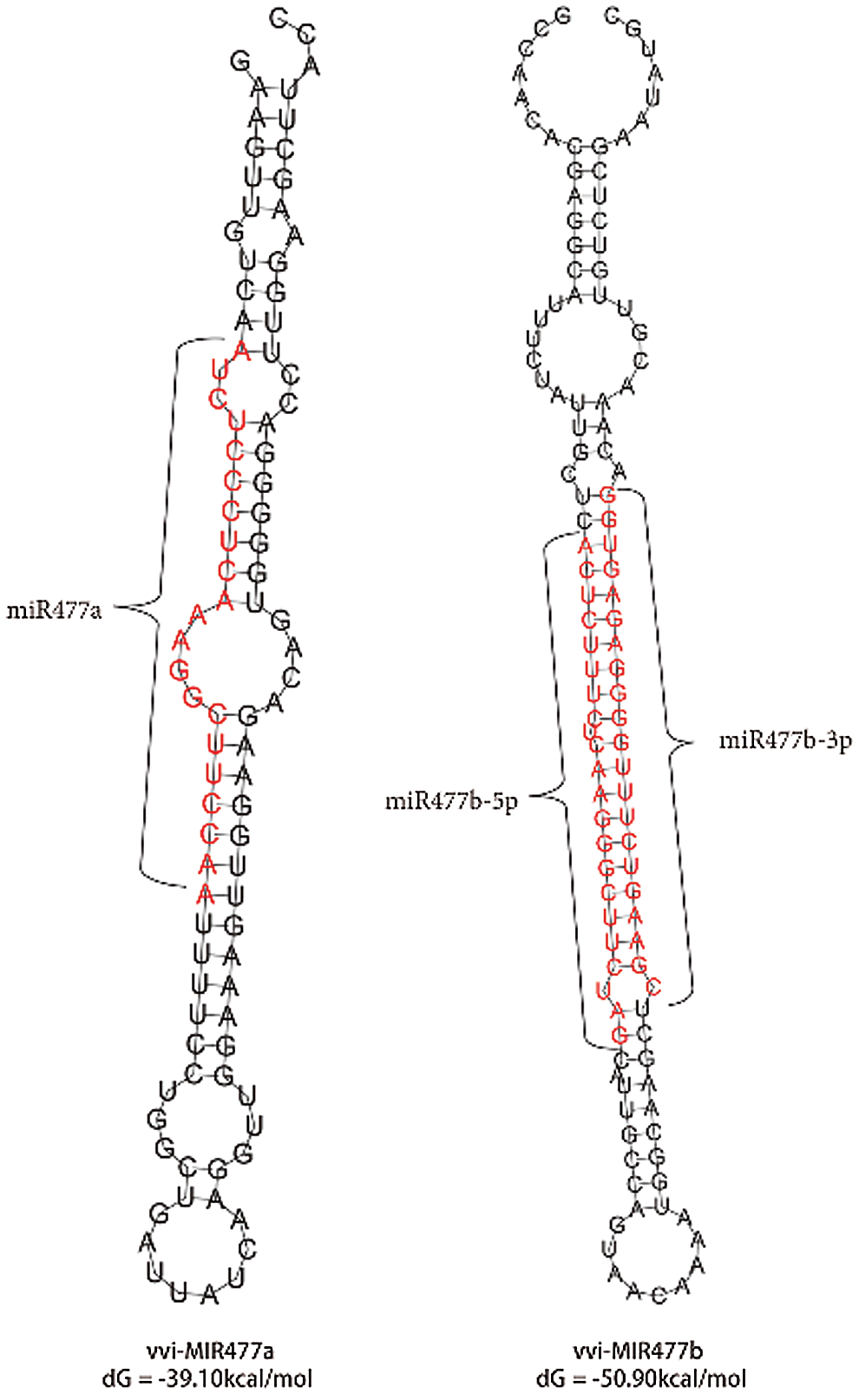
Figure 5: The stem-loop structure of Vvi-MIR477 genes.
Note: The mature miRNA sequences or putative exons encoding the precursors are in red. The junction of the red and black region are the canonical splice sites.
Phylogenetic tree of the miR477 family
To explore the phylogenetic relationships among members of the MIR477 family. The precursor sequences of MIR477 for the investigated plants from the miRBase database were downloaded. The maximum likelihood method in MEGA Version 7.0.26. Software was used to construct precursor evolutionary trees of MIR477. According to phylogenetic tree analysis, the MIR477 family can be divided into three major groups (Fig. 6). MIR477b from strawberry and MIR477h from cassava are clustered in individual group, and the rest of the precursors were clustered into another group. The MIR477 family of grape were clustered with apple and Populus trichocarpa MIR477. MIR477 members were found to be relatively dispersed among different species, indicating that the phylogenetic relationship of MIR477 family members is less related to species.
Different MIR477 members of the same species clustered into different branches, which also reflected the phylogenetic diversity of the plant MIR477 family. It is speculated that MIR477 genes may evolve at different rates and in different ways, indicating the complexity of plant miRNA precursor evolution.
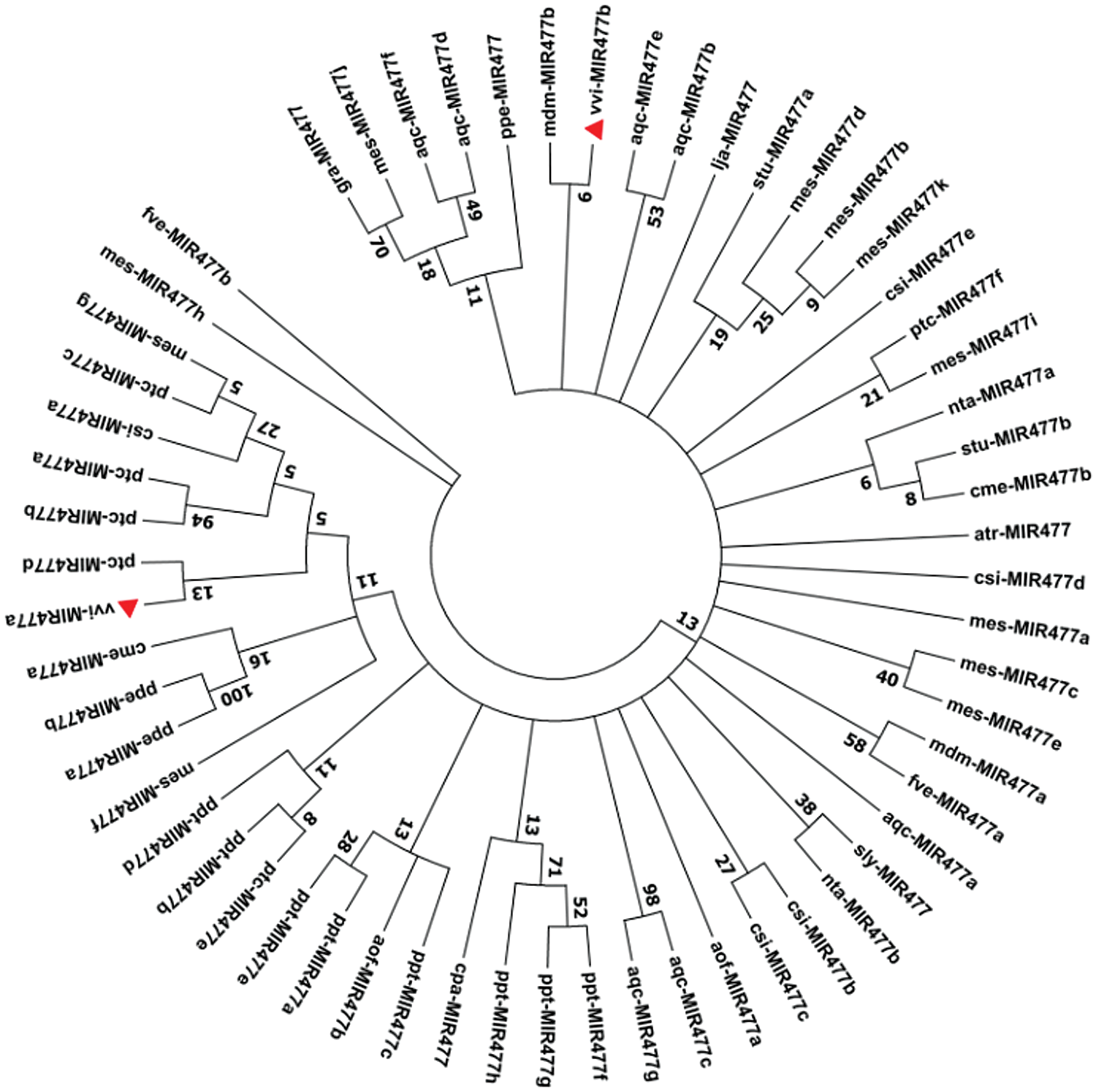
Figure 6: Phylogenetic tree of plants MIR477 precursor sequences based on the Maximum Likelihood method of MEGA version 7.0.26.
Note: ptc, Populus trichocarpa; Vvi, Vitis vinifera; mes, Manihot esculenta; ppt, Physcomitrella patens; ppe, Prunus persica; cme, Cucumis melo; csi, Citrus sinensis; mdm, Malus domestica; fve, Fragaria vesca; stu, Solanum tuberosum; nta, Nicotiana tabacum; aqc, Aquilegia caerulea; lja, Lotus japonicus; aof, Asparagus officinalis; sly, Solanum lycopersicum; gra, Gossypium raimondii; atr, Amborella trichopoda; cpa, Carica papaya.
Prediction of Vvi-miR477 target gene
The target genes of miR477 in grape were predicted with PsRNATarget software. The prediction results showed that different members of miR477 gene family in grape have the same target genes (Table 1). For example, both Vvi-miR477a and Vvi-miR477b-3p target 26S Proteasome components. Both Vvi-miR477a and Vvi-miR477b-5p target DEAD-box ATP-dependent RNA helicase. In addition, all Vvi-miR477 family members inhibit the transcription of target genes by cleavage. The target genes of different members of the miR477 family are not the same.
Expression of miR477 and functional enrichment analysis of its target genes
In order to specifically reflect the expression levels of Vvi-miR477 in Kyoho and Fengzao, a heat map of gene expression at different berry developmental stages were made from the previous data (Guo et al., 2018). As can be seen from the diagram (Fig. 7a), the expression levels of miR477 family members are different at different developmental stages. Vvi-miR477b-5p and Vvi-miR477b-3p were mostly up-regulated in Fengzao, and miR477b-5p was the highest in FZ3 period.
To reflect a global overview of the regulatory functions of miRNAs, the GO terms of all target genes were analyzed through a GO annotation. Target prediction analysis showed that the target genes regulate a wide range of biological processes, cellular components, and molecular functions. The GO enrichment analysis showed that the predicted target genes are enriched in catabolic process; carboxylic ester hydrolase activity; water homeostasis; regulation of root morphogenesis; water transport; hydrolase activity; Cellular response to hypoxia; regulation of root development (Fig. 7b).

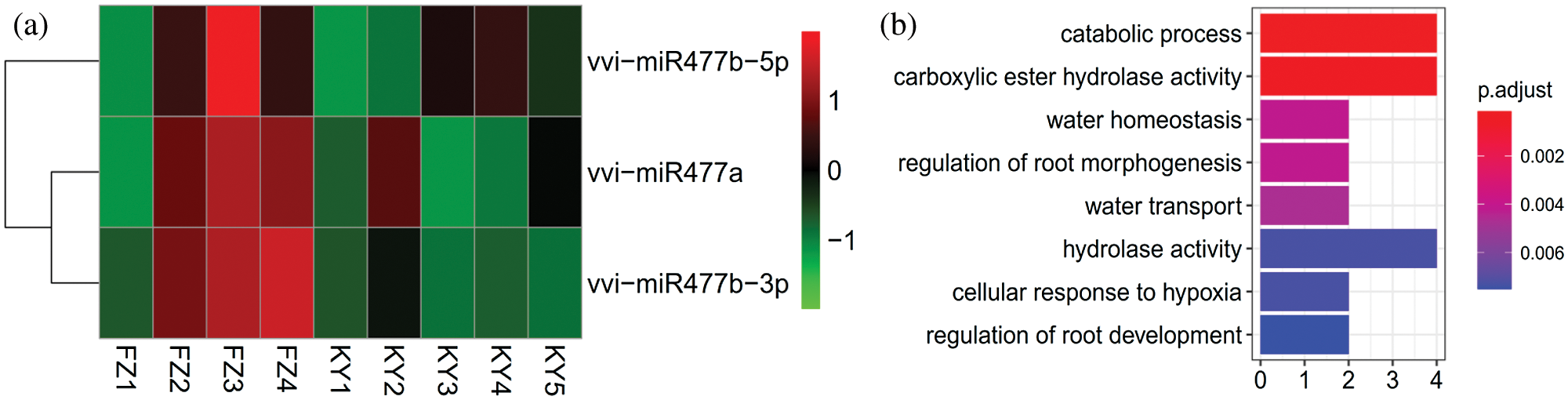
Figure 7: The expression level of Vvi-miR477 at different berry developmental stages and the GO enrichment analysis of their target genes.
(a) Expression analysis of miR477 family members at different stages of Kyoho and Fengzao. The abscissa represents the different developmental stages of Kyoho and Fengzao (data from the previous results (Guo et al., 2018)). FZ represents Fengzao and KY represents Kyoho. Red represents up-regulated expression and green represents down-regulated expression. (b) Scatter gram of overrepresented GO terms (P < 0.05) in molecular function categories from GO enrichment analysis of miRNA targets gene of all the miRNAs identified in this study using ClusterProfiler. Enrichment term is represented by colored dots (red indicates high enrichment and blue indicates low enrichment).
Molecular characteristics of miR477 family members
Many studies have shown that miRNAs play the important role in fruit development and ripening (Gao et al., 2015; Karlova et al., 2013). In this study, the molecular characteristics of miR477 were analyzed. The miR477 mature sequences of the investigated plants were obtained from the miRBase database, and the results showed that miR477 is widely distributed in angiosperms and mosses (Wang et al., 2020). Three mature miR477 members were revealed in grape. Sequence alignment analysis showed that miR477 had multiple conserved base regions, and MIR477 in grape could form a stable secondary structure. Wang et al. (2020) also indicated in tea plant (Camellia sinensis) that miR477 family members are conservative. The phylogenetic analysis showed that Vvi-miR477a and Vvi-miR477b were clustered into different branches. It can also be seen from the larger branches that different members of the same family are distributed in different branches. The similar cases were documented in tomato and Korla fragrant Pear (Liu et al., 2017; Ma et al., 2020b). The conservation of miR477 was also reflected in the results of target gene prediction.
In plants, miRNA genes are mostly discrete, independent transcription units, and about 50% of miRNAs have multiple sites in the genome, similar to protein-coding genes (Liang et al., 2012). Sequence alignment revealed that the length of the mature sequence of miR477 was inconsistent, with a difference of 2–3 nt. In addition, the secondary structure of MIR477 has a different minimum folding free energy (dG). The target genes of miR477 are not all the same. Tan et al. (2020) also discovered this by studying the function of miRNAs in plants. This is due to evolutionary processes which leads to the diversity of miRNA sequences with nucleotide substitutions, insertions, or deletions (Liang et al., 2012). These results suggested that miR477 is conserved in different species but is also diverse. Similar results have been reported for members of the MIR156 family in Korla fragrant pears (Ma et al., 2020b).
Target genes prediction of miR477 members
Fruit development is a highly coordinated and complex process. A range of physiological and biochemical changes influence the flavor, color, and texture of the berry (Ma et al., 2020a). Fengzao is an early-ripening bud mutant of Kyoho, which matures nearly 30 days earlier (Guo and Zhang, 2015). In addition, previous studies have shown that miR477 is differentially expressed in Kyoho and Fengzao and upregulated in Fengzao (Guo et al., 2018).
miRNAs exert their functions by regulating target genes (Alptekin et al., 2017). The prediction results of target genes showed that miRNAs targeted 26S proteasome, GRAS family protein, Protein Phosphatase 2C, DEAD-box ATP-dependent RNA helicase and other target genes. A growing number of genetic analyses have shown that the Ub/26S proteasome pathway plays a role in plant physiological processes, such as embryonic development, hormonal response, flower homeosis, cell differentiation and senescence (Gu and Ma, 2018). GRAS family protein regulates plant growth and development (Hakoshima, 2018). The negative regulator of type 2C protein phosphatase ABI1 functioned in strawberry fruit ripening (Jia et al., 2013). DEAD-box RNA helicase not only involved in plant normal growth and development, cell proliferation, differentiation, miRNA biological process, but also participating in plant abiotic stress responses (Wang et al., 2020). Therefore, we speculate that the precocity of Fengzao may regulated by miR477 through the regulation of the above target genes. However, the authenticity of target gene prediction and the function of target gene still need to be verified by further experiments.
GO enrichment analysis showed that Vvi-miR477 was involved in Cellular response to hypoxia. Hypoxic conditions can lead to hypoxic respiration and accumulation of toxic end products of reactive oxygen species (ROS) (Yang, 2014). Hydrogen peroxide (H2O2) is a type of reactive oxygen species (ROS). Under oxygen-deficient conditions, production of both hydrogen peroxide (H2O2) is triggered by hypoxia signaling (Pucciariello et al., 2012). Vergara et al. (2012) showed that hypoxia and respiratory inhibitors, such as potassium cyanide (KCN) and sodium nitroprusside (SNP), triggered the production of H2O2 in grape buds. While hydrogen peroxide (H2O2) treatment could promote the ripening of Kyoho grape (Guo et al., 2020). In addition, Hydrogen peroxide is also involved in ripening tomatoes (Jimenez et al., 2002) and pears (Chiriboga et al., 2013). Therefore, miR477 may involve in the regulation of grape ripening.
In this study, three mature sequences and two precursor sequences of the miR477 family of grape were obtained by bioinformatic analyses, Vvi-miR477a, Vvi-miR477b-3p, Vvi-miR477b-5p, Vvi-MIR477a, Vvi-MIR477b. The phylogenetic analysis showed that the members of Vvi-miR477 were clustered in different clades. Multiple sequence alignment results showed that most miR477 family members were 20 nt and 21 nt in length. The secondary structure diagrams of the two MIR477 members in grape are relatively stable. The results of target gene prediction showed that Vvi-miR477 targeted different genes, including 26S proteasome, DEAD-box ATP-dependent RNA helicase, Protein phosphatase 2C and GRAS family protein, etc. It showed the diversity of target genes of miR477. The GO function of target genes were enriched, mainly in six functions. The catabolic process, Carboxylic ester hydrolase activity is shown to be high. The expression levels, and the target gene functions of miR477, suggested that Vvi-miR477 may involve grape fruit ripening by regulating its target genes.
Availability of Data and Materials: The datasets generated during and analysed during the current study are available from the corresponding author on reasonable request.
Author Contribution: D-LG conceived and designed the experiment. M-SP analyzed and interpreted the data. H-YJ wrote the manuscript. All authors read and approved the final manuscript.
Funding Statement: This work was financially supported by National Key Research and Development Program of China (2018YFD1000105), Natural Science Foundation of China (NSFC: U1904113), and Program for Innovative Research Team (in Science and Technology) in University of Henan Province (21IRTSTHN021), China.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
Alptekin B, Akpinar BA, Budak H (2017). A comprehensive prescription for plant miRNA identification. Frontiers in Plant Science 7: 2058. [Google Scholar]
Chipman LB, Pasquinelli AE (2019). miRNA targeting: Growing beyond the seed. Trends in Genetics 35: 215–222. [Google Scholar]
Chiriboga MA, Bordonaba JG, Schotsmans WC, Larrigaudière C, Recasens I (2013). Antioxidant potential of ‘Conference’ pears during cold storage and shelf life in response to 1-methylcyclopropene. LWT-Food Science and Technology 51: 170–176. [Google Scholar]
Ding D, Zhang L, Wang H, Liu Z, Zhang Z, Zheng Y (2009). Differential expression of miRNAs in response to salt stress in maize roots. Annals of Botany 103: 29–38. [Google Scholar]
Ding Y, Huang L, Jiang Q, Zhu C (2020). MicroRNAs as important regulators of heat stress responses in plants. Journal of Agricultural and Food Chemistry 68: 11320–11326. [Google Scholar]
Fan D, Ran L, Hu J, Ye X, Xu D, Li J, Su H, Wang X, Ren S, Luo K (2020). miR319a/TCP module and DELLA protein regulate trichome initiation synergistically and improve insect defenses in Populus tomentosa. New Phytologist 227: 867–883. [Google Scholar]
Farinati S, Forestan C, Canton M, Varotto S, Bonghi C (2020). microRNA regulation of fruit development. Plant microRNAs, pp. 75–98. Cham: Springer. [Google Scholar]
Gao C, Ju Z, Cao D, Zhai B, Qin G, Zhu H, Fu D, Luo Y, Zhu B (2015). Micro RNA profiling analysis throughout tomato fruit development and ripening reveals potential regulatory role of RIN on micro RNA s accumulation. Plant Biotechnology Journal 13: 370–382. [Google Scholar]
Gu X, Ma S (2018). Recent advances in the discovery of novel peptide inhibitors targeting 26S proteasome. Anti-Cancer Agents in Medicinal Chemistry (Formerly Current Medicinal Chemistry-Anti-Cancer Agents) 18: 1656–1673. [Google Scholar]
Guo DL, Li Q, Lv WQ, Zhang GH, Yu YH (2018). MicroRNA profiling analysis of developing berries for ‘Kyoho’and its early-ripening mutant during berry ripening. BMC Plant Biology 18: 285. [Google Scholar]
Guo DL, Wang ZG, Pei MS, Guo LL, Yu YH (2020). Transcriptome analysis reveals mechanism of early ripening in Kyoho grape with hydrogen peroxide treatment. BMC Genomics 21: 784. [Google Scholar]
Guo DL, Zhang GH (2015). A new early-ripening grape cultivar–‘Fengzao’. Acta Horticulturae 1082: 153–156. [Google Scholar]
Hakoshima T (2018). Structural basis of the specific interactions of GRAS family proteins. FEBS Letters 592: 489–501. [Google Scholar]
Hu G, Hao M, Wang L, Liu J, Zhang Z, Tang Y, Peng Q, Yang Z, Wu J (2020). The cotton miR477-CBP60A module participates in plant defense against Verticillium dahlia. Molecular Plant-Microbe Interactions 33: 624–636. [Google Scholar]
Jia HF, Lu D, Sun JH, Li CL, Xing Y, Qin L, Shen YY (2013). Type 2C protein phosphatase ABI1 is a negative regulator of strawberry fruit ripening. Journal of Experimental Botany 64: 1677–1687. [Google Scholar]
Jia X, Shen J, Liu H, Li F, Ding N, Gao C, Pattanaik S, Patra B, Li R, Yuan L (2015). Small tandem target mimic-mediated blockage of microRNA858 induces anthocyanin accumulation in tomato. Planta 242: 283–293. [Google Scholar]
Jimenez A, Creissen G, Kular B, Firmin J, Robinson S, Verhoeyen M, Mullineaux P (2002). Changes in oxidative processes and components of the antioxidant system during tomato fruit ripening. Planta 214: 751–758. [Google Scholar]
Karlova R, van Haarst JC, Maliepaard C, van de Geest H, Bovy AG, Lammers M, Angenent GC, de Maagd RA (2013). Identification of microRNA targets in tomato fruit development using high-throughput sequencing and degradome analysis. Journal of Experimental Botany 64: 1863–1878. [Google Scholar]
Lelandais-Brière C, Sorin C, Declerck M, Benslimane A, Crespi M, Hartmann C (2010). Small RNA diversity in plants and its impact in development. Current Genomics 11: 14–23. [Google Scholar]
Li Y, Cui W, Qi X, Lin M, Qiao C, Zhong Y, Hu C, Fang J (2020a). MicroRNA858 negatively regulates anthocyanin biosynthesis by repressing AaMYBC1 expression in kiwifruit (Actinidia arguta). Plant Science 296: 110476. [Google Scholar]
Li Y, Li J, Chen Z, Wei Y, Qi Y et al. (2020b). OsmiR167a-targeted auxin response factors modulate tiller angle via fine-tuning auxin distribution in rice. Plant Biotechnology Journal 18: 2015–2026. [Google Scholar]
Liang T, Guo L, Liu C (2012). Genome-wide analysis of mir-548 gene family reveals evolutionary and functional implications. Journal of Biomedicine and Biotechnology 2012: 679563. [Google Scholar]
Liu M, Yu H, Zhao G, Huang Q, Lu Y, Ouyang B (2017). Profiling of drought-responsive microRNA and mRNA in tomato using high-throughput sequencing. BMC Genomics 18: 481. [Google Scholar]
Liu Q, Chen YQ (2009). Insights into the mechanism of plant development: Interactions of miRNAs pathway with phytohormone response. Biochemical and Biophysical Research Communications 384: 1–5. [Google Scholar]
Liu XX, Luo XF, Luo KX, Liu YL, Pan T, Li ZZ, Duns GJ, He FL, Qin ZD (2019). Small RNA sequencing reveals dynamic microRNA expression of important nutrient metabolism during development of Camellia oleifera fruit. International Journal of Biological Sciences 15: 416. [Google Scholar]
Lu S, Sun YH, Chiang VL (2008). Stress-responsive microRNAs in Populus. Plant Journal 55: 131–151. [Google Scholar]
Ma L, Mu J, Grierson D, Wang Y, Gao L et al. (2020a). Noncoding RNAs: Functional regulatory factors in tomato fruit ripening. Theoretical and Applied Genetics 133: 1753–1762. [Google Scholar]
Ma L, Zhou L, Quan S, Xu H, Yang J, Niu J (2020b). Evolutionary characteristics and expression patterns of miR156 gene family in Korla fragrant pear (Pyrus sinkiangensis Yu). Journal of Horticultural Science and Biotechnology 95: 303–315. [Google Scholar]
Osorio S, Scossa F, Fernie A (2013). Molecular regulation of fruit ripening. Frontiers in Plant Science 4: 198. [Google Scholar]
Pucciariello C, Parlanti S, Banti V, Novi G, Perata P (2012). Reactive oxygen species-driven transcription in Arabidopsis under oxygen deprivation. Plant Physiology 159: 184–196. [Google Scholar]
Sun G (2012). MicroRNAs and their diverse functions in plants. Plant Molecular Biology 80: 17–36. [Google Scholar]
Tan H, Li B, Guo H (2020). The diversity of post-transcriptional gene silencing mediated by small silencing RNAs in plants. Essays in Biochemistry 64: 919–930. [Google Scholar]
Tirumalai V, Swetha C, Nair A, Pandit A, Shivaprasad PV (2019). miR828 and miR858 regulate VvMYB114 to promote anthocyanin and flavonol accumulation in grapes. Journal of Experimental Botany 70: 4775–4792. [Google Scholar]
Vergara R, Parada F, Rubio S, Pérez FJ (2012). Hypoxia induces H2O2 production and activates antioxidant defence system in grapevine buds through mediation of H2O2 and ethylene. Journal of Experimental Botany 63: 4123–4131. [Google Scholar]
Wang XM, Kong RR, Zhang T, Gao YY, Xu JL et al. (2020). A DEAD-box RNA helicase TCD33 that confers chloroplast development in rice at seedling stage under cold stress. Journal of Plant Physiology 248: 153138. [Google Scholar]
Wang S, Liu S, Liu L, Li R, Guo R, Xia X, Wei C (2020). miR477 targets the phenylalanine ammonia-lyase gene and enhances the susceptibility of the tea plant (Camellia sinensis) to disease during Pseudopestalotiopsis species infection. Planta 251: 59. [Google Scholar]
Wang T, Ping X, Cao Y, Jian H, Gao Y et al. (2019). Genome-wide exploration and characterization of miR172/euAP2 genes in Brassica napus L. for likely role in flower organ development. BMC Plant Biology 19: 336. [Google Scholar]
Wang Y, Zhang J, Cui W, Guan C, Mao W, Zhang Z (2017). Improvement in fruit quality by overexpressing miR399a in woodland strawberry. Journal of Agricultural and Food Chemistry 65: 7361–7370. [Google Scholar]
Xie F, Frazier TP, Zhang B (2010). Identification and characterization of microRNAs and their targets in the bioenergy plant switchgrass (Panicum virgatum). Planta 232: 417–434. [Google Scholar]
Yang CY (2014). Ethylene and hydrogen peroxide are involved in hypoxia signaling that modulates AtERF73/HRE1 expression. Plant Signaling & Behavior 9: 877–885. [Google Scholar]
Yang R, Li P, Mei H, Wang D, Sun J et al. (2019). Fine-tuning of MiR528 accumulation modulates flowering time in rice. Molecular Plant 12: 1103–1113. [Google Scholar]
Ye J, Xu M, Tian X, Cai S, Zeng S (2019). Research advances in the detection of miRNA. Journal of Pharmaceutical Analysis 9: 217–226. [Google Scholar]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |