DOI:10.32604/biocell.2022.017383
| BIOCELL DOI:10.32604/biocell.2022.017383 | |
| Article |
Nicotinic acid induces apoptosis of glioma cells via the calcium-dependent endoplasmic reticulum stress pathway
1Department of R&D, Biotech & Science Company of UP, Guangzhou Branch, Guangzhou, 51000, China
2State Key Laboratory of Brain and Cognitive Science, Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming, 650223, China
*Address correspondence to: Jiejing Li, xenjiejingli@hotmail.com
Received: 06 May 2021; Accepted: 17 June 2021
Abstract: Malignant glioma is one of the most common and deadly tumors in the central nervous system while developing effective treatments for this devastating disease remains a challenge. Previously, we demonstrated that the vitamin nicotinic acid (NA) inhibits glioma invasion. Here, we show that high-dose NA induces apoptosis of malignant glioma cells in vitro and in vivo. In cultured U251 glioma cells treated with NA, we detected ER stress that was likely caused by elevated intracellular calcium levels. The elevated calcium can be attributed to the activation of TRPV1, a cation channel that has been implicated in cutaneous flushing caused by NA administration. Our data further suggested that NA-induced apoptosis is mediated by the calcium-dependent proteases called calpains, whose activities are drastically upregulated by NA. NA-induced apoptosis of U251 cells can be attenuated by blocking calpain activity or knocking down TRPV1. These results reveal a novel function of NA in regulating glioma cell apoptosis via the calcium-dependent ER stress pathway and imply a potential application of NA for the treatment of malignant glioma.
Keywords: Nicotinic acid; Apoptosis; Calcium; Endoplasmic reticulum stress
Glioma, which accounts for nearly 30% of all primary brain tumors, is one of the most challenging solid tumors to treat in the central nervous system (Weller et al., 2015). Although advanced technologies, including surgery, radiation, and chemotherapy, have been customized to treat glioma, the overall survival (OS) of patients rarely exceeds 2 years and does not meet the desirable expectations (Lwin et al., 2013). Gliomas are classified into pathological grades I–IV (Rasmussen et al., 2017), and grade IV, also called glioblastoma (GBM), is the most aggressive type of primary brain tumors. The median OS for GBM with current standard treatments is mere 12–15 months (Saito et al., 2018). Hence, it is crucial to develop more effective drugs for treating glioma.
Targeting apoptotic pathways has drawn much interest in cancer therapy, as cancer cell death via apoptosis causes minimal inflammation (Jan, 2019). However, induction of apoptosis has shown a limited benefit in glioma treatment to date, probably due to our lack of knowledge on the cellular apoptotic pathways in these tumors (Valdés-Rives et al., 2017). Apoptosis is typically controlled by the cell-surface death receptor-mediated extrinsic pathway and the mitochondria-mediated intrinsic pathway (D’arcy, 2019). In the endoplasmic reticulum (ER), perturbations of ER function can trigger the unfolded protein response (UPR) to restore protein homeostasis. However, when the UPR fails to alleviate ER stress, both the intrinsic and extrinsic apoptotic pathways can be activated (Sano and Reed, 2013). Three key signaling proteins, protein kinase R-like ER kinase (PERK), inositol-requiring protein-1α (IRE1α), and activating transcription factor 6 (ATF6), are involved in ER stress-induced apoptosis (Hacker, 2014; Rizzuto et al., 1998; Sano and Reed, 2013). PERK is responsible for activating the pro-apoptotic transcription factor C/EBP homologous protein (CHOP) (Cullinan and Diehl, 2004). IRE1α stimulates apoptosis signal-regulating kinase 1 (ASK1) and subsequent activation of the stress kinases c-Jun N-terminal kinase (JNK) and p38 mitogen-activated protein kinase (MAPK), which promote apoptosis (Urano et al., 2000). ATF6-mediated events activate ER chaperones such as glucose-regulated protein 94 (GRP94), immunoglobulin heavy chain-binding protein (BiP/GRP78), and calreticulin, as well as CHOP (Bailey and O’hare, 2007; Belmont et al., 2012). All of these pathways eventually lead to the activation of the caspase-12, -9, and -3 cascades and subsequent apoptosis. In addition, Ca2+ released from the stressed ER can upregulate the activities of Ca2+-dependent proteases calpains, which, in turn, activate both caspase-12 and the ASK1-JNK cascade to induce apoptosis (Nakagawa and Yuan, 2000; Tan et al., 2006). ER-localized calpains can also cleave Ca2+-regulatory proteins to initiate ER stress (Samanta et al., 2013). It has been shown that thapsigargin (TG) and tunicamycin, two ER stress inducers, can promote the death of GBM cells (Quick and Faison, 2012). Moreover, many chemicals, including desipramine, valproate, flavonoids, and cannabinoid, exert their pro-apoptotic function via ER stress in glioma and other tumors (Das et al., 2010; Kim et al., 2005; Ma et al., 2011; Salazar et al., 2009). Therefore, developing drugs that target ER stress to treat malignant glioma is of great potential.
Nicotinic acid (NA), a subclass of the vitamin B family, has been widely used for decades to lower the levels of very-low and low-density lipoproteins, as well as raise the levels of high-density lipoprotein (Carlson, 2005). However, side effects, such as cutaneous flushing, which is partly caused by the activation of the capsaicin receptor transient receptor potential cation channel subfamily V member 1 (TRPV1), limit the clinical application of NA (Gille et al., 2008; Ma et al., 2014). Previous studies have shown that NA is able to induce transient intracellular calcium elevation in CHO-K1 cells and neutrophils (Kostylina et al., 2008; Tunaru et al., 2003), and we also reported that NA upregulates the intracellular calcium in NIH3T3 cells (Li et al., 2014). Additionally, we showed that NA treatment can inhibit the invasion of U251 GBM cells by promoting Snail1 degradation and impairing the assembly of leading edge (Li et al., 2017; Yang et al., 2017). In the current study, we further assessed the effects of NA on glioma development, and our data suggest that high doses of NA induce ER stress and apoptosis of glioma cells through TRPV1-mediated calcium release and calpain activation.
NA (Cat. #72309), TG (Cat. #T9033), and calpain Inhibitor I (Cat. #A6185) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Pan Caspase Inhibitor Z-VAD-FMK (FMK001) was obtained from R&D Systems (Minneapolis, MN, USA). Small interfering RNA (siRNA) of TRPV1 (Assay ID: 104707) was purchased from Thermo Fisher Scientific Systems (Minneapolis, MN, USA). U251 GBM cells (Serial: TCHu 58) and C6 cells (Serial: TCR 1) were obtained from the Cell Bank of Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). U251 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, C11965500BT, Gibco, Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (FBS, 10099141C, Gibco, Thermo Fisher Scientific), and C6 cells in F12K medium (N3520, Sigma-Aldrich) supplemented with 15% equine serum (26050088, Gibco, Thermo Fisher Scientific) and 2.5% FBS. Both cells were incubated at 37°C with 30% humidity and 5% CO2.
Time-lapse measurement of intracellular [Ca2+]
U251 cells were seeded in a 96-well cell culture plate (Greiner Bio-One, Monroe, NC, USA) at 2 × 104 cells/well, and incubated with Fluo-4 (Fluo-4 Direct™ Calcium Assay Kits, Invitrogen Molecular Probes, Carlsbad, CA, USA) for 1 h at 37°C. Ca2+ levels were assessed by measuring the fluorescence intensity with a Zeiss LSM 510 META confocal microscope (Oberkochen, Germany); and Zeiss LSM Image Examiner software (FV10-ASW 2.1 Viewer) was used for quantitative analyses.
Cells were lysed using a RIPA Lysis Buffer (P0013B; Beyotime Institute of Biotechnology, Haimen, China) in the presence of the complete™ proteinase inhibitor cocktail (Roche Diagnostics, Basel, Switzerland), and then centrifuged at 13,201 × g and 4°C for 15 min. The supernatant was collected, and protein concentration was assessed using the BCA protein assay kit (Applygen Technologies, Beijing, China). Proteins (50 μg) were separated on a 12% SDS polyacrylamide gel and transferred to a 0.20 μm polyvinylidene difluoride (PVDF) membrane (EMD Millipore, Burlington, MA, USA). Membranes were blocked at room temperature (RT, 25°C) with 5% bovine serum albumin (BSA; Sigma-Aldrich) for 2 h and incubated with one of the following primary antibodies: caspase-3 (#14220, 1:2,000), cleaved caspase-3 (#9664, 1:2,000), cleaved caspase-9 (#52873, 1:2,000), GRP94 (#20292, 1:2,000), calreticulin (#12238, 1:2,000), protein disulfide isomerase (PDI) (#3501, 1:2,000), CHOP (#2895, 1:2,000), phospho-c-Jun (#91952, 1:2,000,) and GAPDH (#5174, 1:5,000; all from Cell Signaling Technology, Burlington, MA, USA), as well as β-actin (1:5,000; Sigma-Aldrich A5316), at 4°C overnight. Detection was carried out using Horseradish Peroxidase (HRP)-conjugated secondary antibodies (anti-mouse IgG, #7076, 1:10,000 and anti-rabbit IgG, #7074, 1:10,000; both from Cell Signaling Technology) for 2 h at RT (25°C), and signals were detected using SuperSignalTM West Pico PLUS stable peroxide solution (Thermo Fisher Scientific) and X-ray film.
Flow cytometry analyses of apoptosis
Harvest cells at a concentration of 1 × 106 cells/mL using trypsin without EDTA (Gibco, 15050-065). After Centrifuging at 300 g for 3 min at RT (25°C), remove the supernatant. Cell apoptosis was assessed by using the Annexin-V Apoptosis Detection kit (556547; BD Biosciences, Franklin Lakes, NJ, USA) according to the manufacturer’s instructions: Wash cells with cold phosphate-buffered saline (PBS) and then resuspend cells in 100 µL 1 × Binding Buffer. Add 5 µL of fluorescein isothiocyanate (FITC)-conjugated annexin-V and 5 µL propidium iodide (PI). Incubate for 15 min at RT (25°C) in the dark. Add 400 µL of 1 × Binding Buffer to each tube. Analyze by flow cytometry within 1 h. Fluorescence intensity was measured using a BD Biosciences FACSVantageSE flow cytometer. Original data were analyzed using WinMDI 2.9 software and presented in the form of dot plots, with FITC-annexin-V as the x-axis and PI as the y-axis.
Fluorescent immunohistochemistry
U251 cells were cultured on Lab-Tek chamber slides (Sigma-Aldrich). After treatment with PBS or NA, the cells were fixed with 4% paraformaldehyde and permeabilized with 0.4% Triton X-100 at RT (25°C). The cells were then blocked with 5% BSA and incubated with primary antibodies at 4°C overnight. The primary antibodies used were cleaved caspase-3 (#9664, 1:200; Cell Signaling Technology) and TRPV1 (AF3066-SP, 1:200; R&D Systems). The cells were subsequently incubated with PE or FITC-conjugated secondary antibodies (Invitrogen P-2771MP and Sigma-Aldrich F0382, respectively; 1:100) at RT (25°C), and labeled with DAPI (D8417; Sigma-Aldrich) to identify cell nuclei. Fluorescence was detected using an Olympus IX81S1F-3 laser confocal scanning microscope (Tokyo, Japan).
In vitro assays of calpain activities
To measure calpain activities, cytosolic proteins were extracted and analyzed with a Calpain Activity Fluorometric Assay Kit (K240, BioVision, Milpitas, CA, USA) according to the manufacturer’s instructions, and fluorescence was measured at 400/505 nm using a microplate reader. Active calpain I (positive control) and calpain inhibitor Z-LLY-FMK were included in the kit.
SiRNA transfection was performed by using Lipofectamine™ 2000 Transfection Reagent (11668019, Thermo Fisher Scientific Systems, Minneapolis, MN, USA) according to the manufacturer’s instructions: Dilute 100 pmol siRNA oligomer and 10 µL Lipofectamine 2000 in 250 µL Gibco™ Opti-MEM™ I Reduced Serum Medium without serum respectively. After incubation for 5 min at RT (25°C), mix the diluted siRNA and Lipofectamine 2000. Mix gently and incubate for 20 min at RT (25°C). Add the oligomer-Lipofectamine 2000 complexes to each well containing cells and medium. Incubate the cells at 37°C in a CO2 incubator for 48–72 h with following analysis.
The mRNA levels of target genes were analyzed using semi-quantitative RT-PCR. Total RNA was extracted from U251 cells with an RNA Simple total RNA kit (DP419, Tiangen, Beijing, China), and reverse transcription was performed using an M-MLV First Strand Kit (K1621, Thermo Fisher Scientific). Reverse transcription products were then amplified by semi-quantitative PCR with a HotStart Taq Master Mix Kit (KT121221, Tiangen, Beijing, China) using the primers for TRPV1 and GAPDH as described (Spinsanti et al., 2008). Following a “hot start” at 95°C for 3 min, the samples were cycled at 95°C for 30 s, 55°C for 30 s, and 72°C for 20 s.
Microelectrode array (MEA) measurement
U251 cells were seeded on multiwell MEA plates, which had 59 electrodes (8 × 8 grid) with a diameter of 30 μm and spaced 200 μm apart. Data were recorded simultaneously from 59 channels at a sampling rate of 20 kHz/channel.
Evaluating the effects of NA on C6 cell apoptosis in rats
A rat glioma model was established to test the in vivo effects of NA on C6 cell apoptosis as described previously (Li et al., 2017). Animals were maintained at a constant RT (24°C) with 50% humidity and free access to food and water under 12 h light/dark conditions. They were anesthetized via intraperitoneal injection of sodium pentobarbital (Nembutal, 50 mg/kg) before surgery. Following administration of sodium pentobarbital, the rats became ataxic, lost their righting reflex, had no responses to pain, and eventually remained immobile. Next, 2–5 × 105 C6 cells were orthotopically injected into the right striatum of Sprague-Dawley rat brains through a pre-settled stainless steel tube, and NA and control (PBS) were also injected through the tube. NA (70 mM) was administered at a volume of 5 μL on the fourth day after tumor cell transplantation. Rats were sacrificed by anesthesia overdose with an intraperitoneal injection of sodium pentobarbital (200 mg/kg) after continuous administration of NA for 5 days, and their brain tissues were removed and embedded immediately in optimal cutting temperature compound. Brain tissues were subsequently cut into 7-μm slices using a Leica CM1850 cryostat microtome (Wetzlar, Germany). The slices were fixed with 95% ethanol and stained with hematoxylin and eosin (H&E; Sigma-Aldrich). After sealing with a neutral mounting medium (Jiangyuan, China), the slices were examined under an Olympus upright microscope.
All experiments were repeated at least three times prior to statistical analyses. All experimental data are presented as the mean ± SD. Differences between samples were analyzed using the two-tailed Student’s t-test and regarded as statistically significant at P < 0.05.
High-dose NA induces apoptosis of glioma cells in vitro and in vivo
We previously demonstrated that 7.0 mM NA inhibits glioma invasion by facilitating Snail1 degradation (Li et al., 2017). To further explore the effects of NA on malignant glioma, we treated cultured U251 GBM cells and primary mouse neurons with increasing concentrations of NA, and we carried out PI and annexin-V double staining to detect apoptotic cells. After 20 h of treatment, 14 mM but not 3.5 or 7.0 mM NA significantly increased apoptosis of U251 cells (Figs. 1E–1H and 1O). This effect was likely specific for tumor cells, as the viability of normal mouse neurons was not affected under the same conditions (Figs. 1A–1D and 1N). Time-course analyses revealed that treatment with 14 mM NA induced apoptosis of U251 cells within 15 min (Figs. 1I–1M and 1P). We also performed immunofluorescence using an antibody that recognizes cleaved caspase-3, a hallmark of apoptosis, which detected drastically increased levels of cleaved caspase-3 in U251 cells treated with 14 mM NA for 4 h (Figs. 2A–2D). Western blotting using antibodies for total and cleaved caspase-3 confirms upregulated caspase-3 cleavage within 1 h after treatment with 14 mM NA (Fig. 2E), suggesting that the apoptotic pathways were activated.
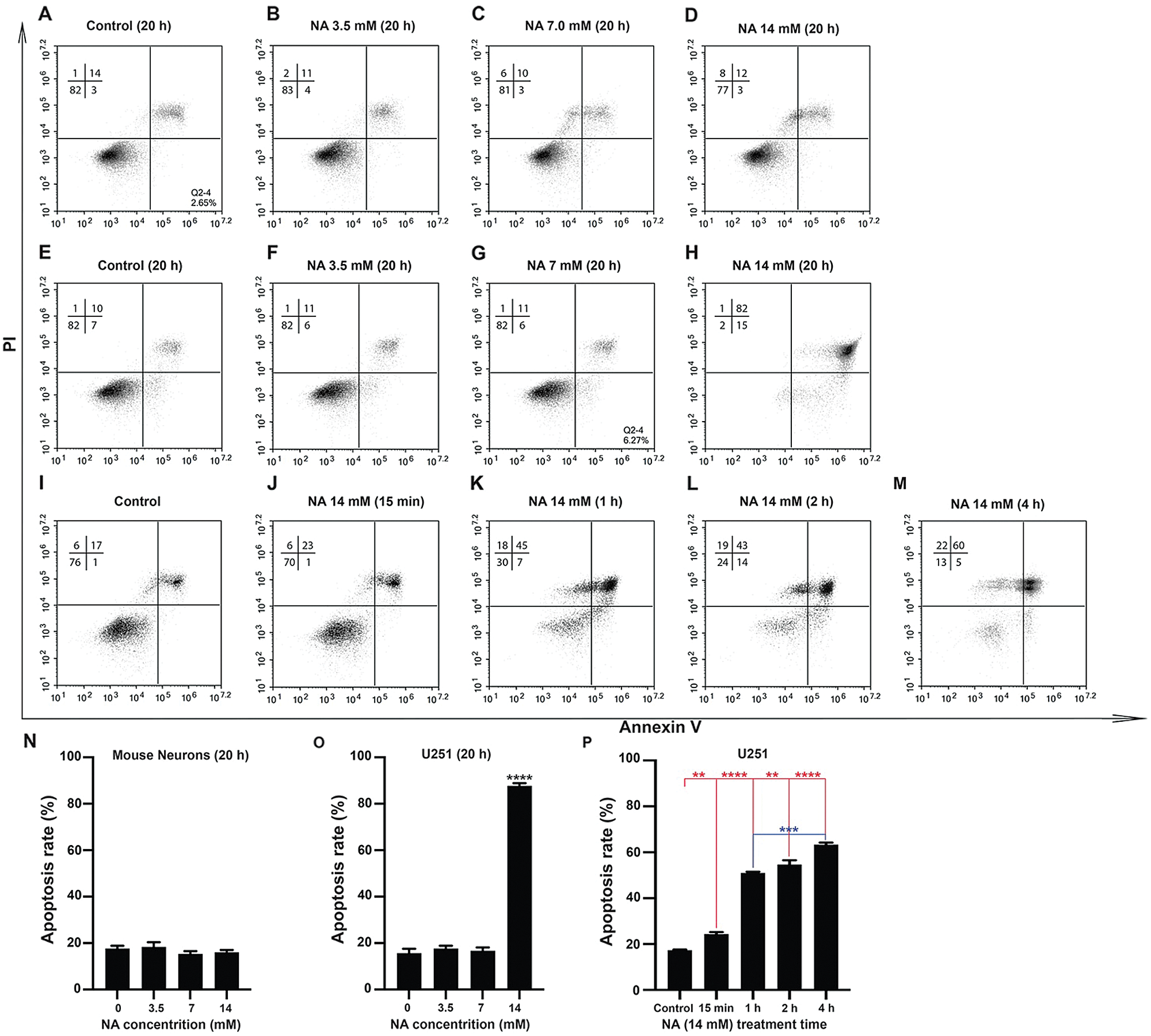
Figure 1: Apoptosis assays for U251 cells and mouse neurons with various concentrations of NA. U251 cells (E–M, O and P) and mouse neurons (A–D and N) were cultured in the indicated concentration of NA for different lengths of time. Cells were harvested and stained with annexin-V and PI, and cell sorting was carried out as described in the Materials and Methods section. Profiles of cells analyzed in a representative experiment are shown in A–M. The results of three independent experiments are summarized in N–P. The data are expressed as mean ± SD of three experiments (**P < 0.01, ***P < 0.001 and ****P < 0.0001 vs. untreated control).
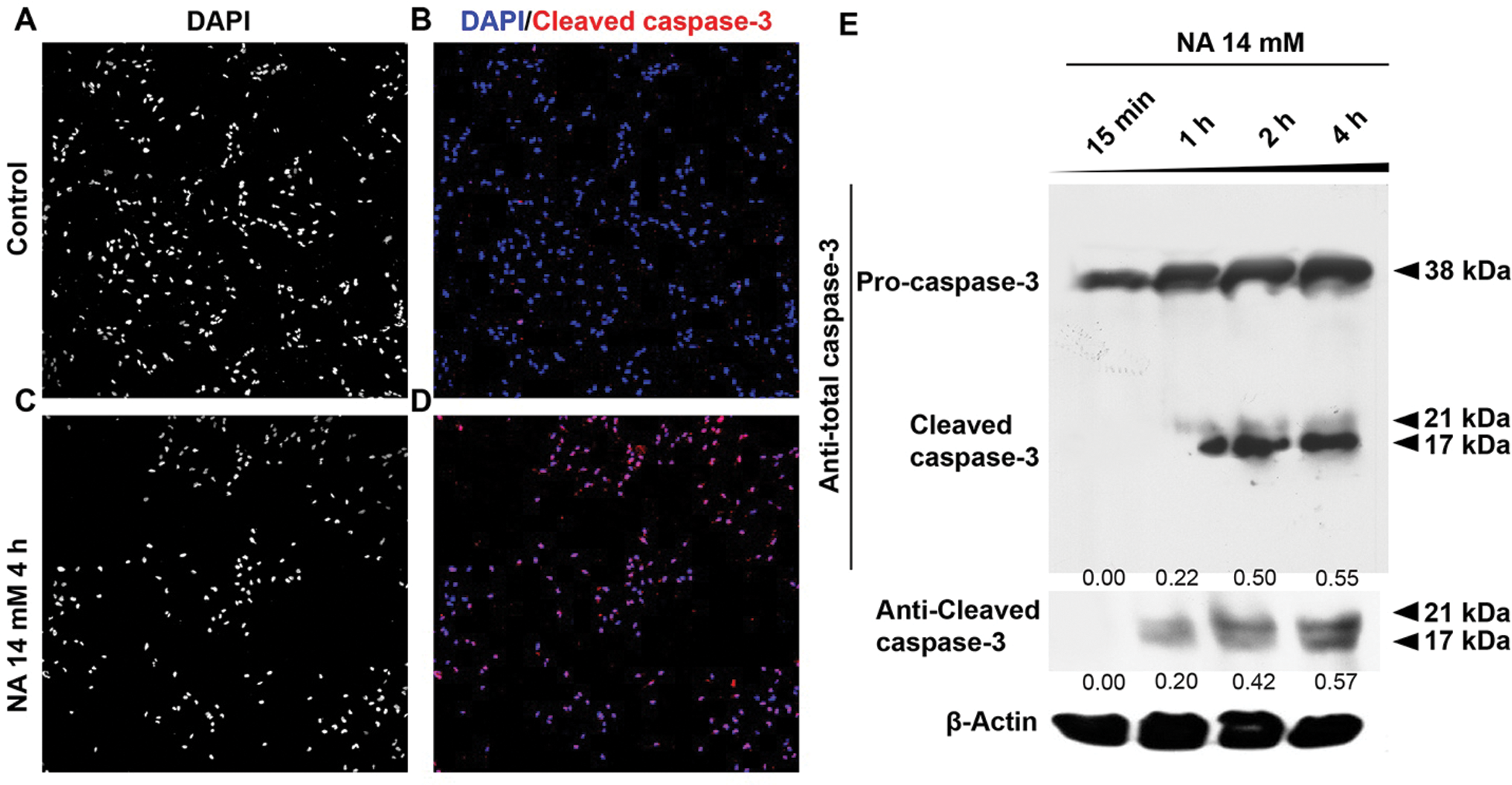
Figure 2: NA treatment promotes the cleavage of caspase-3. (A–D) Immunofluorescence was carried out to detect cleaved caspase-3 after treatment with 14 mM NA for 4 h (with a magnification of 10×). (E) The levels of pro-caspase-3 and cleaved caspase-3 were analyzed by western blotting after treatment with 14 mM NA for the indicated length of time. The relative expression of cleaved caspase-3 was quantified densitometrically and subsequently normalized against β-Actin.
Given the ability of NA to cause apoptosis of U251 cells in vitro, we further investigated the effect of NA on cell viability in vivo. We allografted rats with C6 glioma cells as described (Li et al., 2017; Xu et al., 2019), and administered NA orthotopically starting from day 4 after tumor cell transplantation. We previously detected a maximal NA tolerance of 120 mM in the allografted rats as well as a therapeutic effect when 5 μL of 70 mM NA was administered (Li et al., 2017) (data not shown). After 5 days of treatment with 5 μL of 70 mM NA, brain tissues were obtained, sectioned, and stained with H&E. In the core areas of NA-treated tumor grafts, we observed tissue condensation with nuclear fragmentation and cell shrinkage (Figs. 3A–3D). Immunofluorescence revealed a strikingly higher intensity of cleaved caspase-3 upon NA treatment (Figs. 3E and 3F). Based on these data, we conclude that NA induces apoptosis of glioma cells in vitro and in vivo.
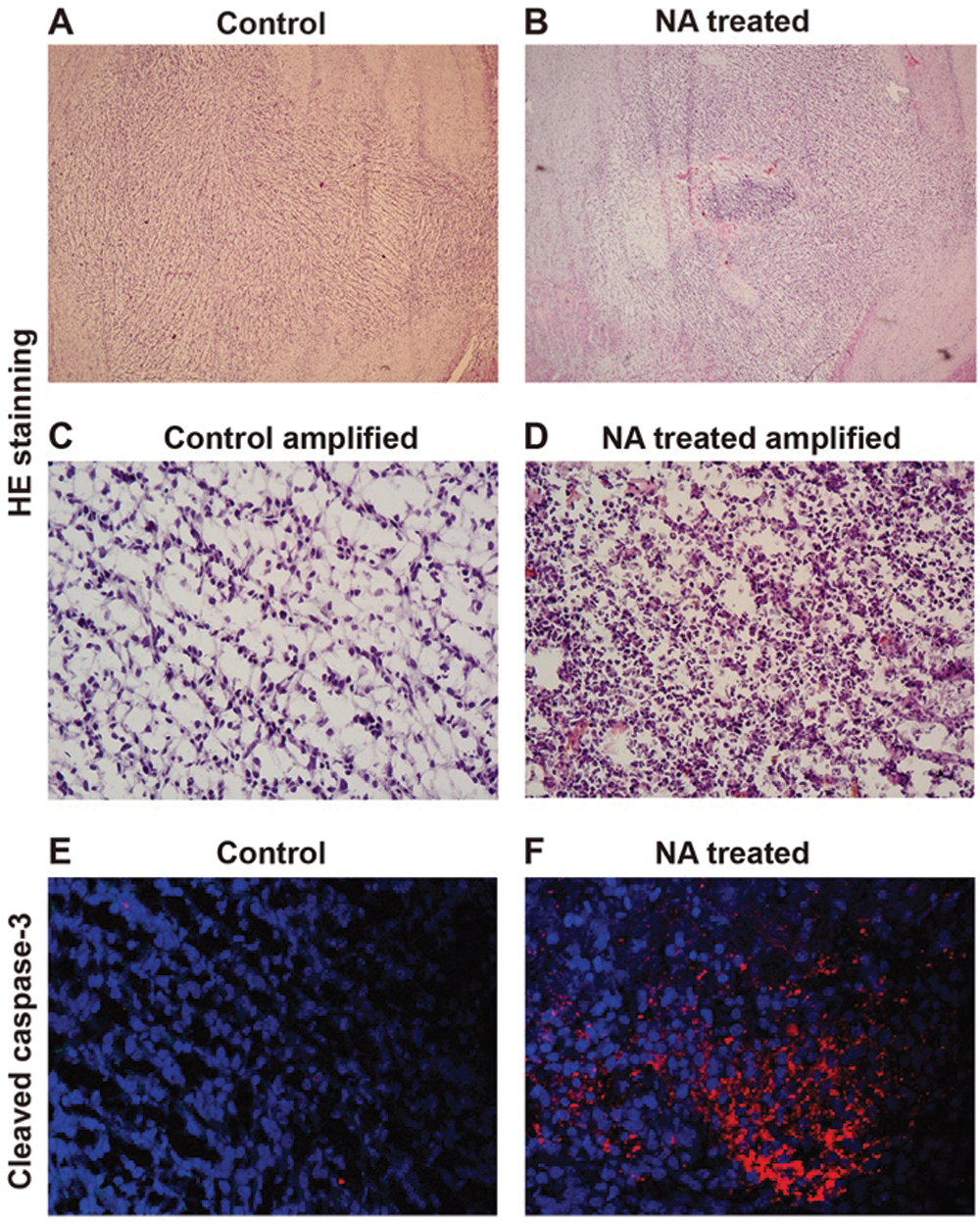
Figure 3: NA injection induces C6 glioma cell apoptosis in grafted rats. Rats allografted with C6 cells were injected with PBS (control) or NA as described in the Materials and Methods section. (A–D). Brain slices were collected, and H&E staining was carried out. Representative images of brain slices were shown with a magnification of 10× (A and B) and 50× (C and D). (E and F) Immunofluorescence of cleaved caspase-3 on the brain slices collected from rats injected with PBS or NA.
NA treatment leads to elevated intracellular [Ca2+] and ER stress
It has been demonstrated that ER plays a vital role in apoptosis by activating caspase-12 and CHOP. The activated caspase-12 subsequently triggers the activation of caspases-9, -7, and -3 (Masud et al., 2007; Szegezdi et al., 2003). Treatment of U251 cells with 14 mM NA elevated the levels of ER chaperones GRP94, calreticulin, and PDI in a time-dependent manner starting within 15 min (Fig. 4A), suggesting an enhancement in ER stress. A similar time-dependent increase of CHOP and cleaved caspase-9 was also observed, whereas the induction of JNK kinase activity lagged but was apparent after NA treatment for 2 h (Figs. 4A and 4B). Thus, it is possible that NA induces apoptosis by triggering ER stress.
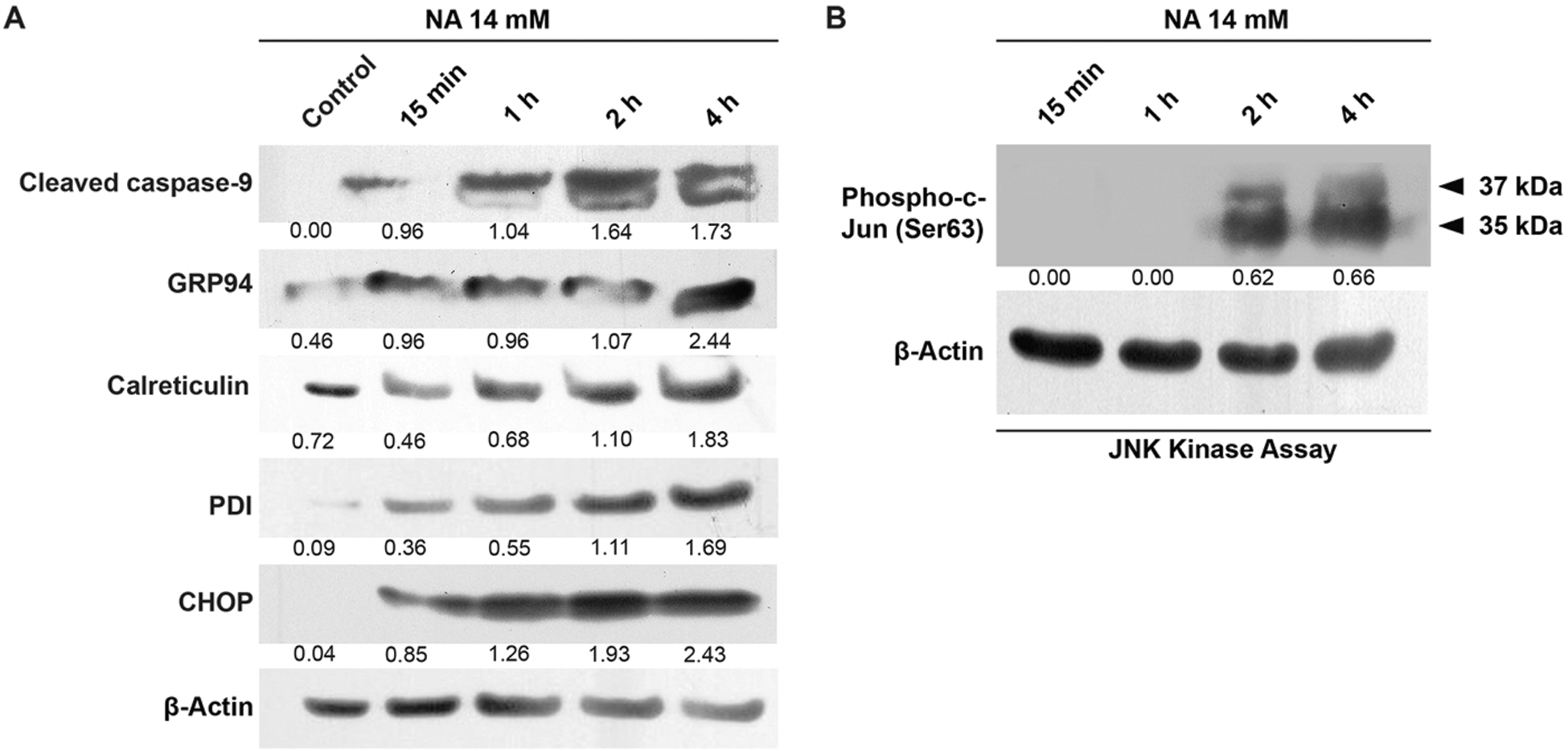
Figure 4: NA induces ER stress and apoptosis. The levels of activated caspase-9, GRP94, calreticulin, PDI and CHOP (A), and phospho-c-Jun (B) were analyzed by western blotting after treatment with 14 mM NA for the indicated length of time. The relative expression of indicated proteins was quantified densitometrically and subsequently normalized against β-actin.
One of the main functions of ER is to regulate Ca2+ homeostasis, and Ca2+-binding chaperones such as calreticulin facilitate protein folding in the ER. Accumulation of unfolded or misfolded proteins and perturbation of ER Ca2+ can both result in ER stress (Sano and Reed, 2013). Because we and others have shown that NA upregulates intracellular [Ca2+] in other types of cells (Kostylina et al., 2008; Li et al., 2014; Tunaru et al., 2003), we hypothesized that NA causes ER stress in U251 cells by interfering with Ca2+ homeostasis. To test this hypothesis, we used MEA to analyze the effects of NA on the cell action potential. As shown in Figs. 5A–5D, treatment of U251 cells with 14 mM NA induced action potential, which was blocked by the calcium ion chelator BAPTA. Visualization of intracellular Ca2+ using Fluo-4 showed that 7 mM NA initially elevated but later returned the intracellular [Ca2+] level to normal (Figs. 5E and 5H). In contrast, treatment with 14 mM NA led to the continuous elevation of intracellular [Ca2+], which could be suppressed by TG, an ER Ca2+-ATPase pump inhibitor (Figs. 5E and 5I). Elevated intracellular [Ca2+] was not observed in primary mouse or Macaca neurons treated with 14 mM NA (Figs. 5F, 5G, 5J, and 5K), suggesting that it is specific to glioma cells.
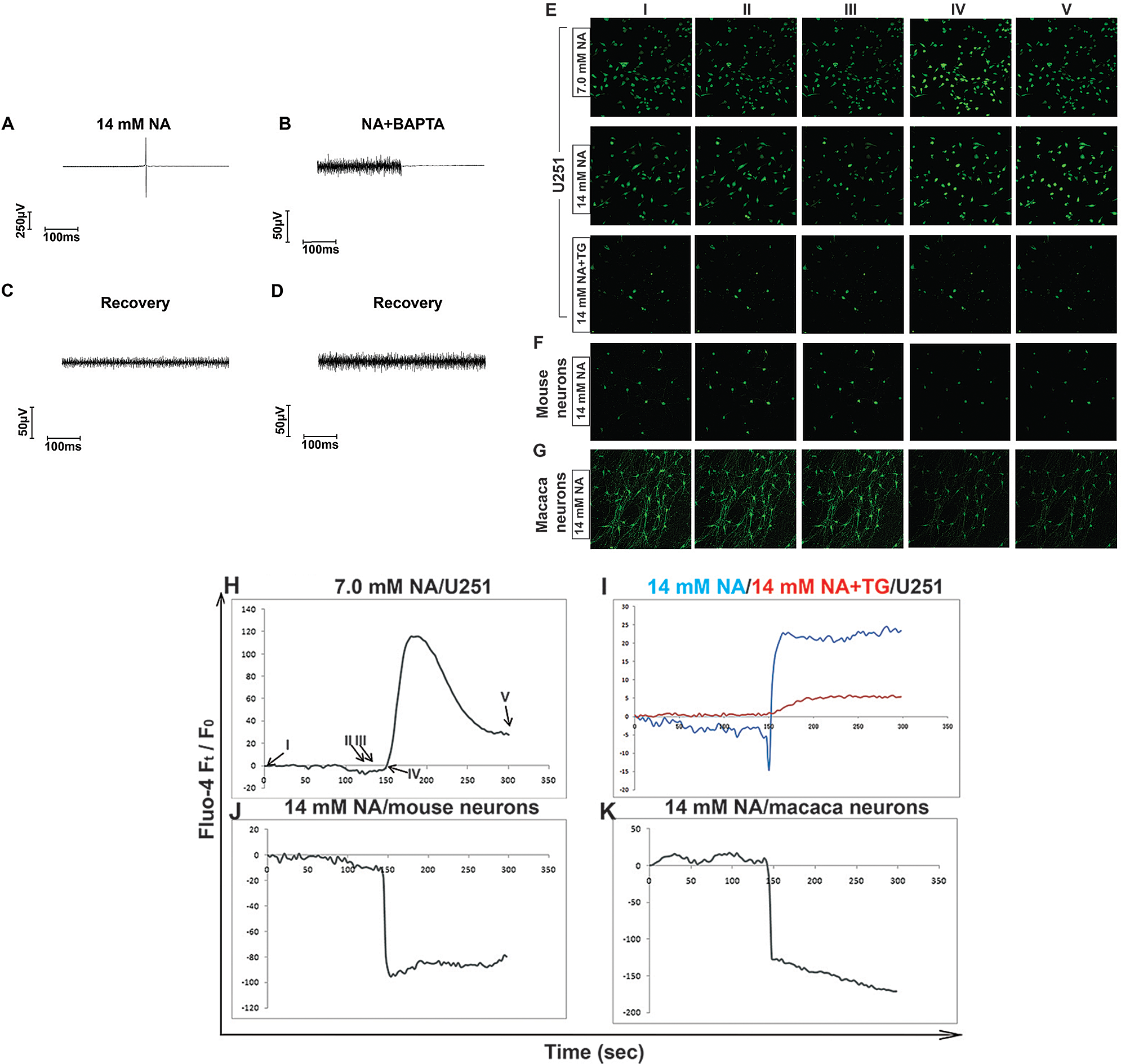
Figure 5: Assessment of intracellular [Ca2+] regulated by NA. (A–D) The influence of NA with or without the calcium chelator BAPTA on cellular action potentials in U251 cells was measured by MEA, as described in the Materials and Methods section. (A and C) NA caused action potentials in U251 cells. (B and D) BAPTA blocked cellular action potentials caused by NA. (E–G) Visualization of intracellular [Ca2+] using Fluo-4 in U251 cells (E) or mouse or Macaca neurons (F and G) after treatment with the indicated concentration of NA. (H–K) Time-lapse curves of intracellular [Ca2+] in U251 cells or mouse or Macaca neurons upon NA treatment. I, II, III, IV, and V are the representative time points captured during the assay (NA was added at time point IV (150 s)).
TRPV1 is responsible for [Ca2+] upregulation and apoptosis caused by NA
High doses of NA are known to cause flushing by activating the capsaicin receptor TRPV1 (Gille et al., 2008; Ma et al., 2014). TRPV1 is a cation channel whose activation can trigger an increase in cytosolic free Ca2+ and ER stress (Haustrate et al., 2020), and NA treatment is known to trigger an increase in intracellular [Ca2+] in keratinocytes and Langerhans cells, the main carriers of flushing that express high levels of TRPV1 (Benyo et al., 2006; Maciejewski-Lenoir et al., 2006). To test whether TRPV1 is involved in NA-induced [Ca2+] upregulation and apoptosis, we evaluated two siRNAs targeting TRPV1. As shown in Figs. 6A–6D, transfection of U251 cells with either siRNA1 depleted TRPV1 mRNA and protein, with siRNA2 being more effective. Unlike treatment with 14 mM NA alone, which caused sustained elevation of intracellular [Ca2+] (Figs. 5E and 5I), a combination of 14 mM NA and siRNA2 triggered a transient [Ca2+] spike that returned to near the baseline in approximately 90 s (Fig. 6E). Interestingly, no apparent apoptosis was observed in the first 2 h, and nearly 60% of the cells were still viable after 4 h of treatment (Figs. 6F–6J). This observation was in stark contrast with 14 mM NA alone, which induced detectable apoptosis within 15 min and induced cell apoptosis to >60% in 4 h (Figs. 1I–1M and 1P). Taken together, these results indicate that TRPV1 is at least partially responsible for NA-induced [Ca2+] elevation and subsequent apoptosis in U251 cells.
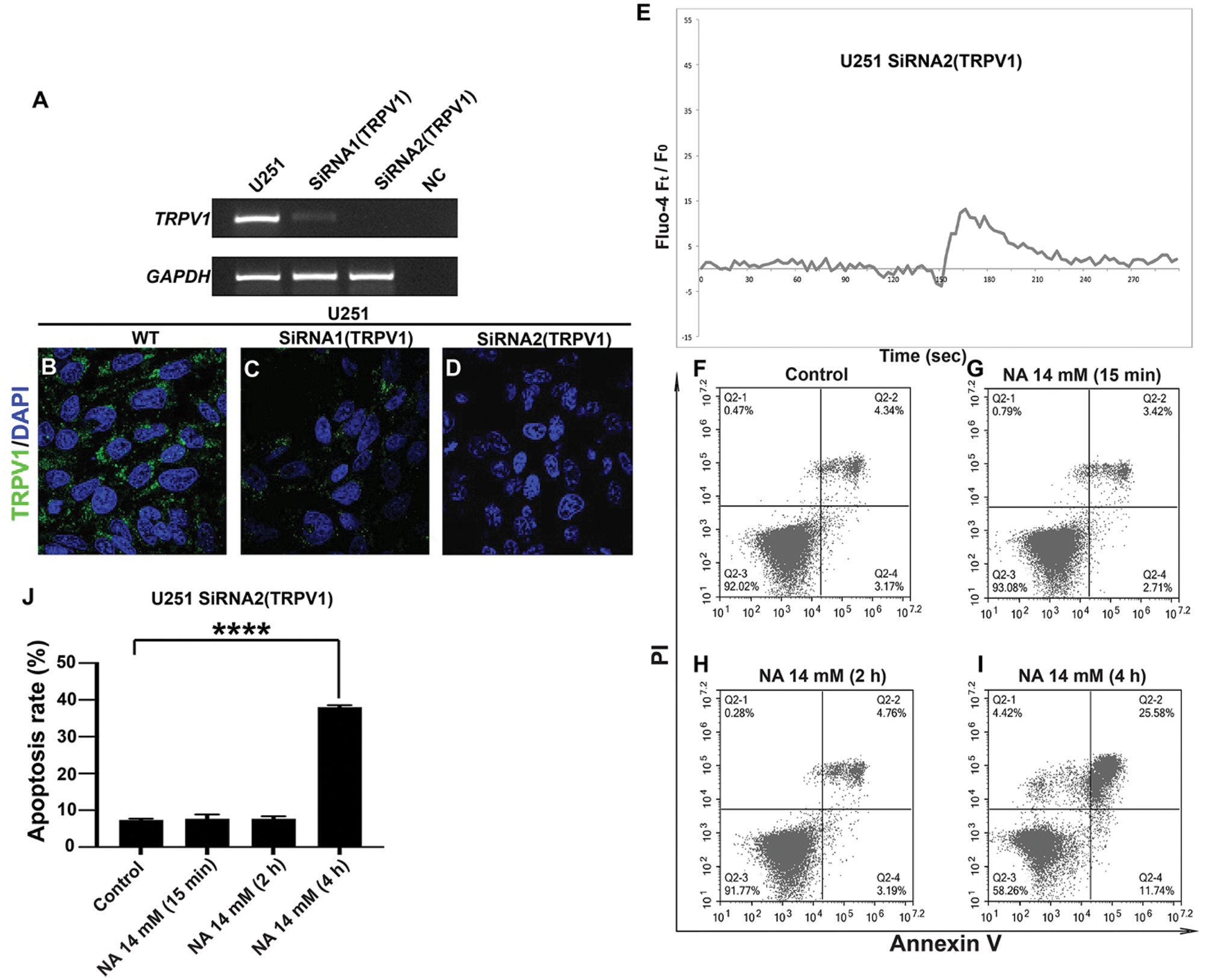
Figure 6: TRPV1 mediates NA function in U251 cells. (A–D) Evaluation of TRPV1 knockdown efficiency. U251 cells were transfected with two siRNAs targeting TRPV1, and RT-PCR (A) and immunofluorescence (B–D, pictures were taken with a magnification of 40×) were carried out to detect TRPV1 mRNA and protein levels, respectively. NC, negative control without reverse transcriptase. (E) Time-lapse curve of intracellular [Ca2+] after the transfection of TRPV1 siRNA2 and treatment with 14 mM NA. (F–I) Apoptosis of U251 cells after transfection with TRPV1 siRNA2 and treatment with 14 mM NA. Cells were harvested, stained with annexin-V and PI, and analyzed by flow cytometry. The percentage of U251 cells that underwent apoptosis with NA treatment from three independent experiments is summarized in J (****P < 0.0001).
NA induces apoptosis in U251 cells via calpain activation
Because the Ca2+-dependent proteases calpains are key regulators of ER-mediated cell death (Samanta et al., 2013), we examined calpain activities in U251 cells. Treatment with 14 mM but not lower concentrations of NA led to drastically enhanced cleavage of a fluorogenic peptide substrate for calpains, which was abrogated by a selective calpain inhibitor (Fig. 7A). These results point to a strong increase in calpain activity upon treatment with high-dose NA. We further tested if blocking calpain activities could “rescue” the apoptosis caused by NA treatment. Indeed, NA-induced apoptosis was significantly reduced by either the calpain inhibitor or a caspase inhibitor (Figs. 7B–7F), suggesting that NA induces apoptosis of U251 cells through calpains activation.
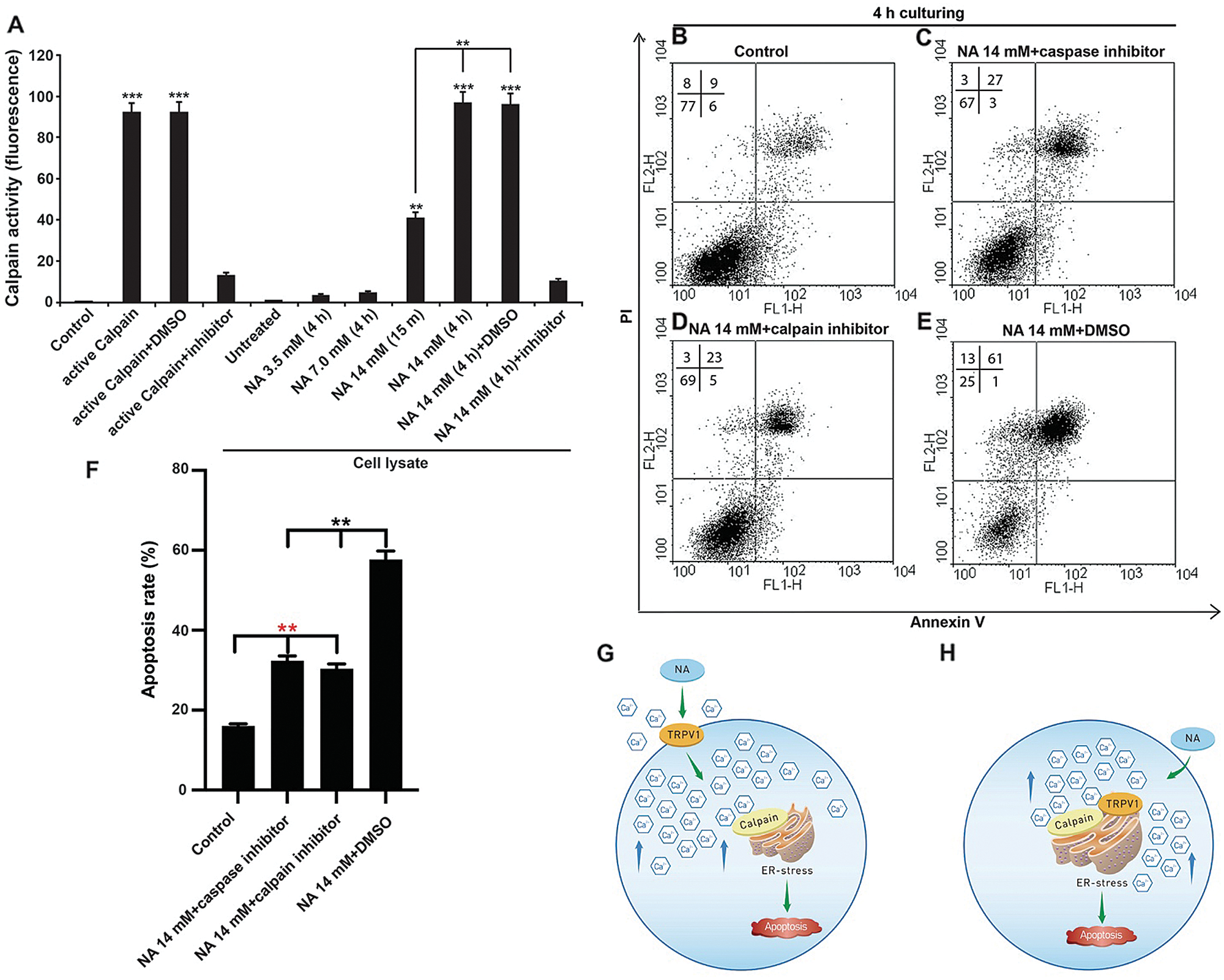
Figure 7: NA induces ER-mediated apoptosis by activating calpains. (A) NA activates calpains in U251 cells. After the indicated treatment, cellular calpain activities were assessed as described in the Materials and Methods section. (B–E) Apoptosis of U251 cells was evaluated by flow cytometry after the treatment with 14 mM NA and indicated inhibitors. The percentage of U251 cells that underwent apoptosis with NA treatment from three independent experiments is summarized in F (**P < 0.01 and ***P < 0.001). (G and H) Our models of NA function in inducing ER stress and apoptosis via TRPV1 and calpains. See the full text for a detailed explanation.
Although NA and its derivatives have been intensively used and studied for decades, the roles of this critical vitamin in cancer remain largely elusive. Previously, we showed that NA modulates intracellular calcium concentration in NIH3T3 cells and inhibits the invasion of glioma cells by promoting Snail1 degradation (Li et al., 2014; Li et al., 2017). In our following study, we observed that high doses of NA could also decrease the viability of U251 cells. Hence, we here focused on the effect of NA on glioma cell apoptosis. We demonstrated that 14 mM NA induces apoptosis of U251 cells in vitro and C6 cells in vivo in allografted rats. This novel function of NA is likely mediated by elevated intracellular [Ca2+] and ER stress via the cation channel TRPV1 and Ca2+-dependent proteases called calpains.
Disturbance of cellular energy production, redox, or calcium state can lead to ER stress, and sustained ER stress can initiate apoptosis involving PERK, ATF6, or IRE1α (Sano and Reed, 2013). Intracellular [Ca2+] homeostasis is well known to be involved in tumor initiation, angiogenesis, and metastasis (Cui et al., 2017), and targeting Ca2+ homeostasis is a promising direction in the development of anti-cancer drugs. In this report, we show that high dosages of NA can specifically disturb the Ca2+ homeostasis in U251 cells but not normal neurons, ultimately resulting in apoptosis. As far as we know, this is the first evidence that NA directly causes the death of cancer cells.
A major side effect of NA treatment is flushing, which has been shown to be caused by direct activation of the capsaicin receptor TRPV1 by NA from the intracellular side at physiological temperature (Ma et al., 2014). TRPV1 is a nonselective Ca2+-permeable cation channel, and it has been speculated that the NA-induced increase of intracellular [Ca2+] in keratinocytes and Langerhans cells, the main carriers of flushing, is mediated by TRPV1 (Ma et al., 2014). Here, we showed that in U251 GBM cells, knockdown of TRPV1 alleviates high-dose NA-induced sustained [Ca2+] elevation and apoptosis (Figs. 6E–6J), indicating that TRPV1 is a key effector downstream of NA in triggering increased intracellular [Ca2+]. We further demonstrated that NA treatment results in strongly upregulated activities of calpains (Fig. 7A), which are Ca2+-dependent proteases that can induce ER stress and apoptosis (Samanta et al., 2013). In addition to ER stress, calpains are also known to participate in the mitochondria-mediated apoptotic pathway by cleaving and releasing apoptosis-inducing factor (AIF) from the inner mitochondrial membrane (Jayanthi et al., 2004). Blocking calpain activities partially rescues NA-induced apoptosis (Figs. 7B–7F), providing a mechanistic connection between [Ca2+] elevation and glioma cell death (Fig. 7G).
Although TRP channels were previously thought to localize on the plasma membrane, research in the past decade has suggested that many of them also localize and function on intracellular membranes (Imler and Zinsmaier, 2014). In particular, TRPV1 has been found to be enriched on the ER membrane of neurons and other cells, where the activation of TRPV1 triggers Ca2+ release from the ER (Gallego Sandín et al., 2009; Wong et al., 2014). While the exact pool(s) of TRPV1 that are activated by NA remains unknown, it is possible that intracellular NA interacts with TRPV1 on the ER membrane to induce the release of Ca2+ from the ER, causing ER stress directly. In this scenario, calpains may not be required for initiating ER stress; instead, they are likely activated by Ca2+ released from the stressed ER and serve as downstream effectors to execute apoptosis and/or provide positive feedback to induce additional ER stress (Fig. 7H). Consistent with this model, we found that the calpain inhibitor only partially suppressed the apoptosis induced by NA (Figs. 7D and 7F), suggesting that there may be other pathways mediating NA-induced apoptosis.
Many small molecules can induce glioma cell death by causing ER malfunction (Kim et al., 2005; Ma et al., 2011; Salazar et al., 2009). Conversely, ER stress is also involved in the development of treatment resistance in glioma cells (Avril et al., 2017; Obacz et al., 2017). Here, we demonstrated that NA induces glioma cell apoptosis in vitro and in vivo, suggesting that it can be used as a potential therapy for glioma. Notably, 5-fluorouracil, an antimetabolite that is being actively tested for malignant glioma treatment (Cloughesy et al., 2016), has been reported to cause NA deficiency (Stevens et al., 1993). It has also been reported that patients receiving chemotherapy, especially those on complete parenteral nutrition, are prone to vitamin B3 deficiency (Inculet et al., 1987). These findings raise the intriguing possibility of using NA in combination with current treatments for glioma. Although the concentration of NA that induced apoptosis of U251 cells in our experiments (14 mM) was higher than the recommended daily dose as a vitamin, it is close to the dosages used clinically for lipid-lowering treatment and should be relatively safe (Goel and Dunbar, 2016). In conclusion, our results reveal a novel function of NA in regulating glioma apoptosis and support the possibility of applying NA to the treatment of this devastating disease. Further investigations are needed to thoroughly evaluate the effects of NA on malignant glioma and other cancers.
Authors’ Contribution: J.Q. and J.L. designed the experiments. X.Y., J.Q., J.L. performed the experiments. X.Y., J.Q. and J.L. analyzed the data and wrote the manuscript.
Availability of Data and Materials: The analyzed datasets generated during the present study are available from the corresponding author on reasonable request.
Ethics Statement: All experimental protocols of rats were approved by the Animal Care and Use Committee of Kunming Institute of Zoology, Chinese Academy of Sciences (Permit No. SYDW-20120110-66).
Funding Statement: This study was supported by the National Natural Science Foundation of China (Grant Nos. 81760503 and 81760660), the Applied Basic Research Key Project of Yunnan (Grant Nos. 2018FE001(-318) and 2018FE001(-123)), the Applied Basic Research projects of Yunnan Province (Grant No. 2019FA038), the Yunnan Health Science and Technology Plan Projects (Grant No. 2016NS207), and Yunnan health training project of high-level talents (Grant Nos. D2017026 and H-2018074).
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
Avril T, Vauleon E, Chevet E (2017). Endoplasmic reticulum stress signaling and chemotherapy resistance in solid cancers. Oncogenesis 6: e373. [Google Scholar]
Bailey D, O’hare P (2007). Transmembrane bZIP transcription factors in ER stress signaling and the unfolded protein response. Antioxidants & Redox Signaling 9: 2305–2322. [Google Scholar]
Belmont PJ, Chen WJ, Thuerauf DJ, Glembotski CC (2012). Regulation of microRNA expression in the heart by the ATF6 branch of the ER stress response. Journal of Molecular and Cellular Cardiology 52: 1176–1182. [Google Scholar]
Benyo Z, Gille A, Bennett CL, Clausen BE, Offermanns S (2006). Nicotinic acid-induced flushing is mediated by activation of epidermal langerhans cells. Molecular Pharmacology 70: 1844–1849. [Google Scholar]
Carlson LA (2005). Nicotinic acid: The broad-spectrum lipid drug. A 50th anniversary review. Journal of Internal Medicine 258: 94–114. [Google Scholar]
Cloughesy TF, Landolfi J, Hogan D, Bloomfield S, Carter BS et al. (2016). Phase 1 trial of vocimagene amiretrorepvec and 5-fluorocytosine for recurrent high-grade glioma. Science Translational Medicine 8: 341ra375. [Google Scholar]
Cui C, Merritt R, Fu L, Pan Z (2017). Targeting calcium signaling in cancer therapy. Acta Pharmaceutica Sinica B 7: 3–17. [Google Scholar]
Cullinan SB, Diehl JA (2004). PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. The Journal of Biological Chemistry 279: 20108–20117. [Google Scholar]
D’arcy MS (2019). Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biology International 43: 582–592. [Google Scholar]
Das A, Banik NL, Ray SK (2010). Flavonoids activated caspases for apoptosis in human glioblastoma T98G and U87MG cells but not in human normal astrocytes. Cancer 116: 164–176. [Google Scholar]
Gallego Sandín S, Rodríguez-García A, Alonso M, García-Sancho J (2009). The endoplasmic reticulum of dorsal root ganglion neurons contains functional TRPV1 channels. The Journal of Biological Chemistry 284: 32591–32601. [Google Scholar]
Gille A, Bodor ET, Ahmed K, Offermanns S (2008). Nicotinic acid: Pharmacological effects and mechanisms of action. Annual Review of Pharmacology and Toxicology 48: 79–106. [Google Scholar]
Goel H, Dunbar RL (2016). Niacin alternatives for dyslipidemia: Fool’s gold or gold mine? Part II: Novel niacin mimetics. Current Atherosclerosis Reports 18: 17. [Google Scholar]
Hacker G (2014). ER-stress and apoptosis: Molecular mechanisms and potential relevance in infection. Microbes and Infection 16: 805–810. [Google Scholar]
Haustrate A, Prevarskaya N, Lehen’kyi V (2020). Role of the TRPV channels in the endoplasmic reticulum calcium homeostasis. Cells 9: 317. [Google Scholar]
Imler E, Zinsmaier KE (2014). TRPV1 channels: Not so inactive on the ER. Neuron 84: 659–661. [Google Scholar]
Inculet RI, Norton JA, Nichoalds GE, Maher MM, White DE, Brennan MF (1987). Water-soluble vitamins in cancer patients on parenteral nutrition: A prospective study. Journal of Parenteral and Enteral Nutrition 11: 243–249. [Google Scholar]
Jan R (2019). Understanding apoptosis and apoptotic pathways targeted cancer therapeutics. Advanced Pharmaceutical Bulletin 9: 205. [Google Scholar]
Jayanthi S, Deng X, Noailles PA, Ladenheim B, Cadet JL (2004). Methamphetamine induces neuronal apoptosis via cross-talks between endoplasmic reticulum and mitochondria-dependent death cascades. FASEB Journal 18: 238–251. [Google Scholar]
Kim AJ, Shi Y, Austin RC, Werstuck GH (2005). Valproate protects cells from ER stress-induced lipid accumulation and apoptosis by inhibiting glycogen synthase kinase-3. Journal of Cell Science 118: 89–99. [Google Scholar]
Kostylina G, Simon D, Fey MF, Yousefi S, Simon HU (2008). Neutrophil apoptosis mediated by nicotinic acid receptors (GPR109A). Cell Death & Differentiation 15: 134–142. [Google Scholar]
Li J, Li Y, Zhang P, Niu H, Shi Y (2014). Nicotinic acid modulates intracellular calcium concentration and disassembles the cytoskeleton. Molecular Medicine Reports 10: 2805–2810. [Google Scholar]
Li J, Qu J, Shi Y, Perfetto M, Ping Z et al. (2017). Nicotinic acid inhibits glioma invasion by facilitating Snail1 degradation. Scientific Reports 7: 43173. [Google Scholar]
Lwin Z, Macfadden D, Al-Zahrani A, Atenafu E, Miller BA et al. (2013). Glioblastoma management in the temozolomide era: Have we improved outcome? Journal of Neurooncology 115: 303–310. [Google Scholar]
Ma J, Qiu Y, Yang L, Peng L, Xia Z et al. (2011). Desipramine induces apoptosis in rat glioma cells via endoplasmic reticulum stress-dependent CHOP pathway. Journal of Neurooncology 101: 41–48. [Google Scholar]
Ma L, Lee BH, Mao R, Cai A, Jia Y et al. (2014). Nicotinic acid activates the capsaicin receptor TRPV1: Potential mechanism for cutaneous flushing. Arteriosclerosis, Thrombosis, and Vascular Biology 34: 1272–1280. [Google Scholar]
Maciejewski-Lenoir D, Richman JG, Hakak Y, Gaidarov I, Behan DP, Connolly DT (2006). Langerhans cells release prostaglandin D2 in response to nicotinic acid. Journal of Investigative Dermatology 126: 2637–2646. [Google Scholar]
Masud A, Mohapatra A, Lakhani SA, Ferrandino A, Hakem R, Flavell RA (2007). Endoplasmic reticulum stress-induced death of mouse embryonic fibroblasts requires the intrinsic pathway of apoptosis. Journal of Biological Chemistry 282: 14132–14139. [Google Scholar]
Nakagawa T, Yuan J (2000). Cross-talk between two cysteine protease families. Journal of Cell Biology 150: 887–894. [Google Scholar]
Obacz J, Avril T, Le Reste PJ, Urra H, Quillien V et al. (2017). Endoplasmic reticulum proteostasis in glioblastoma—From molecular mechanisms to therapeutic perspectives. Science Signaling 10: eaal2323. [Google Scholar]
Quick QA, Faison MO (2012). CHOP and caspase 3 induction underlie glioblastoma cell death in response to endoplasmic reticulum stress. Experimental and Therapeutic Medicine 3: 487–492. [Google Scholar]
Rasmussen BK, Hansen S, Laursen RJ, Kosteljanetz M, Schultz H et al. (2017). Epidemiology of glioma: Clinical characteristics, symptoms, and predictors of glioma patients grade I–IV in the the Danish Neuro-Oncology Registry. Journal of Neurooncology 135: 571–579. [Google Scholar]
Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE et al. (1998). Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 280: 1763–1766. [Google Scholar]
Saito T, Sugiyama K, Takeshima Y, Amatya VJ, Yamasaki F et al. (2018). Prognostic implications of the subcellular localization of survivin in glioblastomas treated with radiotherapy plus concomitant and adjuvant temozolomide. Journal of Neurosurgery 128: 679–684. [Google Scholar]
Salazar M, Carracedo A, Salanueva IJ, Hernandez-Tiedra S, Lorente M et al. (2009). Cannabinoid action induces autophagy-mediated cell death through stimulation of ER stress in human glioma cells. Journal of Clinical Investigation 119: 1359–1372. [Google Scholar]
Samanta K, Kar P, Chakraborti T, Chakraborti S (2013). An overview of endoplasmic reticulum calpain system. In: Chakraborti S, Dhalla N (eds.Proteases in Health and Disease. Advances in Biochemistry in Health and Disease. New York, NY: Springer. [Google Scholar]
Sano R, Reed JC (2013). ER stress-induced cell death mechanisms. Biochimica et Biophysica Acta 1833: 3460–3470. [Google Scholar]
Spinsanti G, Zannolli R, Panti C, Ceccarelli I, Marsili L et al. (2008). Quantitative real-time PCR detection of TRPV1-4 gene expression in human leukocytes from healthy and hyposensitive subjects. Molecular Pain 4: 51. [Google Scholar]
Stevens HP, Ostlere LS, Begent RHJ, Dooley JS, Rustin MHA (1993). Pellagra secondary to 5-fluorouracil. British Journal of Dermatology 128: 578–580. [Google Scholar]
Szegezdi E, Fitzgerald U, Samali A (2003). Caspase-12 and ER-stress-mediated apoptosis: The story so far. Annals of the New York Academy of Sciences 1010: 186–194. [Google Scholar]
Tan Y, Dourdin N, Wu C, de Veyra T, Elce JS, Greer PA (2006). Ubiquitous calpains promote caspase-12 and JNK activation during endoplasmic reticulum stress-induced apoptosis. Journal of Biological Chemistry 281: 16016–16024. [Google Scholar]
Tunaru S, Kero J, Schaub A, Wufka C, Blaukat A et al. (2003). PUMA-G and HM74 are receptors for nicotinic acid and mediate its anti-lipolytic effect. Nature Medicine 9: 352–355. [Google Scholar]
Urano F, Wang X, Bertolotti A, Zhang Y, Chung P et al. (2000). Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287: 664–666. [Google Scholar]
Valdés-Rives SA, Casique-Aguirre D, Germán-Castelán L, Velasco-Velázquez MA, González-Arenas A (2017). Apoptotic signaling pathways in glioblastoma and therapeutic implications. BioMed Research International 2017: 7403747. [Google Scholar]
Weller M, Wick W, Aldape K, Brada M, Berger M et al. (2015). Glioma. Nature Reviews Disease Primers 1: 15017. [Google Scholar]
Wong CO, Chen K, Lin Yong Q, Chao Y, Duraine L et al. (2014). A TRPV channel in Drosophila motor neurons regulates presynaptic resting Ca2+ levels, synapse growth, and synaptic transmission. Neuron 84: 764–777. [Google Scholar]
Xu Y, Yang X, Mei S, Sun Y, Li J (2019). Acquisition of temozolomide resistance by the rat C6 glioma cell line increases cell migration and side population phenotype. Oncology Reports 42: 2355–2362. [Google Scholar]
Yang X, Mei S, Niu H, Li J (2017). Nicotinic acid impairs assembly of leading edge in glioma cells. Oncology Reports 38: 829–836. [Google Scholar]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |