DOI:10.32604/biocell.2022.018638
| BIOCELL DOI:10.32604/biocell.2022.018638 | |
| Review |
ER exit pathways and the control of proteostasis: Crucial role of the UPR, COPII, and ER-phagy in the secretory pathway
1Dipartimento di Medicina, Chirurgia e Odontoiatria “Scuola Medica Salernitana”, Università degli Studi di Salerno, Baronissi, 84034, Italy
2Dipartimento di Farmacia, Università degli Studi di Salerno, Fisciano, 84084, Italy
*Address correspondence to: Paolo Remondelli, premondelli@unisa.it
Received: 07 August 2021; Accepted: 13 October 2021
Abstract: The endoplasmic reticulum (ER) is the site of entry of all proteins that function in the secretory pathway including the extracellular environment. Because it controls the folding of newly synthesized secretory proteins, the ER is indispensable for the maintenance of proteostasis in the secretory pathway. Within the ER and, in part, in post-ER compartments, the quality control of protein folding is under the regulation of the unfolded protein response (UPR) pathways. The UPR strategy is to enhance protein folding, increase the ER degradation pathway of misfolded proteins, and allow the exit from the ER of only correctly folded proteins. The latter is controlled by the multimeric complex COPII, which also provides some of the components for ER-phagy the only route for the disposal of protein aggregates. In this overview, we wish to contribute to the introduction of new perspectives in the study of the mechanisms underlying the control of proteostasis within the secretory pathway.
Keywords: Proteostasis; ER stress; Unfolded protein response; COPII; ER-phagy
The endoplasmic reticulum (ER) is a cell compartment common to all eukaryotic cells. It is involved in multiple functions, including protein synthesis, folding, processing, and transport, lipid metabolism, calcium storage, and detoxification (Lemmer et al., 2021). The ER membranes are in continuity with the nuclear envelope (NE), from which they unravel in a peripheral ER (pER) made of flat sheets and widely distributed tubular networks (English et al., 2009; Wilkinson, 2019). ER tubules are shaped by specialized proteins (Shibata et al., 2010; Friedman et al., 2011; Voeltz and Barr, 2013; Westrate et al., 2015; Grimaldi et al., 2018), are highly dynamic (Lee and Chen, 1993), and establish connections with almost all the cell membranes throughout contact sites (CS), where signals and molecules are exchanged to support metabolism and control of subcellular compartments (Phillips and Voeltz, 2016; Moltedo et al., 2019; Amodio et al., 2021). The specialized ER domain, called rough ER (rER), accommodates ribosomes and translocation complexes, from where many of newly synthesized proteins enter the secretory pathway. While the entrance is allowed by sorting signals, ER exit requires that newly synthesized proteins have acquired their correct conformation and this ensures that post-ER compartments receive proteins in their folded and potentially active state. To make this result effective, the ER is supplied by a complex machinery—the quality control (QC) of protein folding—made of chaperones and enzymes. QC requires that ER chaperones, which make posttranslational modifications, recognize and retain folding intermediates within the ER. Retention lasts until newly synthesized proteins acquire their native conformation (Braakman and Hebert, 2013; Balchin et al., 2016; Cherepanova et al., 2016). The activity of QC ensures the proteostasis in the secretory pathway in physiological conditions and during ER stress, a condition characterized by the accumulation of unfolded or misfolded proteins within the ER. QC operates a very severe selection, so that about 12%–15% of newly synthesized proteins in eukaryotes are eliminated (Duttler et al., 2013; Wang et al., 2013) by the ER-associated degradation (ERAD) in physiological conditions (Brodsky, 2012; Forrester et al., 2019). In contrast, when folding is biased by mutations or by environmental insults that impair QC function, unfolded proteins form insoluble aggregates, which cannot be cleared by ERAD (Trombetta and Parodi, 2003; Chen et al., 2011; Guerriero and Brodsky, 2012; Shao and Hegde, 2016; Tao and Conn, 2018). In this case, to avoid toxic effects for the cell, the alternative route for degradation is a specific form of autophagy, called ER-phagy. The ER-phagy is the autophagic degradation of the ER, in which fragments of the ER are enclosed by autophagosomes and fused with lysosomes (macro-ER-phagy) or are directly engulfed by lysosomal membrane invaginations (micro-ER-phagy). Alternatively, lysosomes can directly fuse with ER-derived vesicles for degradation (‘vesicular delivery’) (Chino and Mizushima, 2020). Both ERAD and autophagy are directly dependent on the activity of the signaling pathway known as “unfolded protein response” (UPR). However, emerging evidence shows that components of the multiprotein complex COPII, which forms vesicles packaged with correctly folded proteins released from the QC, are also required for the ER-phagy, suggesting that cell survival could also depend on this crucial factor of the secretory pathway.
The UPR and the Multitasking Control of Protein Folding
The UPR is activated by three distinct transducers, namely the protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK), the inositol-requiring enzyme 1 (IRE1), and the activating transcription factor 6 (ATF6) (Hetz et al., 2020). These three pathways work in concert to potentiate QC when the ER fills up with malformed proteins during ER stress. In this state, the 78-kDa glucose-regulated protein (GRP78), a chaperone that normally keeps locked each transducer, releases these proteins, thereby favoring the activation of the UPR. The UPR affect proteostasis within the secretory pathway by different mechanisms as follows: i) the regulation of ER protein folding load by decreasing protein translation; ii) the improvement of protein folding within the ER through the transcriptional activation of QC factors; iii) the degradation of terminally misfolded proteins through the induction of ERAD or ER-phagy; iv) the regulation of COPII-mediated export of cargo proteins from the ER. In this way, the UPR ensures the folding and trafficking of secretory proteins in properly folded, functional conformations and prevents the aberrant secretion of non-native protein conformations (Plate and Wiseman, 2017; Wu and Rapoport, 2018). In particular, the UPR potentiates the QC machinery by activating the expression of ER enzymes and chaperones, such as lectins calreticulin (CLR) and calnexin (CLNX), oxidoreductases including protein disulfide isomerase (PDI) and ER-resident protein 57 (ERp57), and chaperone GRP78 (Sun and Brodsky, 2019). At the same time, the UPR works in concert with the ERAD machinery whose components are upregulated by IRE1 and ATF6 pathways, as occurs for suppressor/enhancer of Lin-12-like (SEL1L), E3 ligase hydroxymethylglutaryl reductase degradation 1 (HRD1), mannosidase ER degradation enhancing alpha-mannosidase-like (EDEM), and transcription factor erythroid 2-like 1 (Nfe2l1) (Bartelt et al., 2018; Hwang and Qi, 2018; Wu and Rapoport, 2018). The upregulation of these factors not only potentiates the QC system but can also be implicated in the attenuation of UPR signaling. For example, PDIA6 displays oxidoreductase activity but is also involved in the attenuation of IRE1 signaling (Eletto et al., 2014). The kinase/endoribonuclease IRE1 induces the stable expression of XBP1, which exerts its transcriptional activity on a subset of genes linked to protein folding, secretion, ERAD, and lipid synthesis (Yoshida et al., 2001; Calfon et al., 2002; Acosta-Alvear et al., 2007). In addition, IRE1α could reduce expression of secretory proteins by specifically degrading ER-localized mRNAs, ribosomal RNA, and miRNAs throughout the regulated IRE1-dependent decay (RIDD) (Hollien et al., 2009; Maurel et al., 2014). In addition, the kinase PERK unburdens the QC machinery by greatly reducing the load of potentially unfolded proteins through decrease of protein translation by the eukaryotic translation initiation factor 2α (eIF2α) phosphorylation. Finally, the transcription factor ATF6 activates the expression of several chaperones and enzymes, including GRP78, as well as the transcription of XBP1 mRNA (Yoshida et al., 2001). Still under the UPR control, QC also is effective in the post-ER compartments. Indeed, unfolded proteins escaping the COPII check-point or exiting the ER by bulk-flow are retrieved from the Golgi QC (GQC) and plasma membrane QC (PMQC) to the ER, where they are finally addressed to ERAD for degradation (Travers et al., 2000; Karagoz et al., 2019; Preissler and Ron, 2019). Thus, the goal of all the events endorsed by the UPR is to return to normality. Alternatively, if QC is overtaken by unfolding and cellular homeostasis is compromised, the UPR promotes cell death (Chen et al., 2011; Wolff et al., 2014; Schneider and Bertolotti, 2015). Thus, the health of the entire secretory pathway and, therefore, of the entire cell is at stake within the ER, and in this context, a crucial role is played by the UPR, which carefully modulates folding assistance and degradation. Recently, COPII components have been shown to play an important role in the QC scenario. On one hand, crucial components of this multiprotein complex can favor the ER exit of folded proteins and, on the other hand, they can recognize protein aggregates accumulating within the ER, thereby mediating ER-phagy.
The Conventional Role of COPII Carriers in Exiting the ER
COPII carriers bud from restricted domains of the ER membrane, known as transitional ER (tER) or ER exit sites (ERESs) (Budnik and Stephens, 2009). COPII assembly initiates with the GDP/GTP exchanges by Sec12 on the Sar1 protein, which in its GTP-bound form inserts its amphiphilic α-helix into the ER membrane (Barlowe and Schekman, 1993; Weissman et al., 2001). Sar1 together with Sec23/Sec24 dimers forms the pre-budding complex, which starts membrane curvature and engages cargo proteins interacting with Sec24 isotypes (Hicke et al., 1992; Barlowe et al., 1994; Matsuoka et al., 1998; Miller et al., 2002; Miller et al., 2003; Lee et al., 2005). Finally, Sec13/Sec31 heterodimers complete membrane curvature and COPII vesicle formation (Fath et al., 2007; Stagg et al., 2008). Since their discovery in yeast cells, it has become clear through subsequent work that COPII components play important roles in the development of human diseases (Lu and Kim, 2020), so much that the work of their discoverer Randy Shekman has been acknowledged with the 2013 Nobel Prize in Physiology and Medicine. Indeed, mutations in genes encoding Sec23A lead to the accumulation of unsecreted procollagen within the ER and, consequently, to collagen deposition defects followed by skeletal and developmental alterations, as revealed in a rare genetic disease known as cranio-lenticulo-sutural dysplasia (LSD) (Boyadjiev et al., 2006). Similarly, pre-chylomicrons, large lipid particles, accumulate in the ER of enterocytes when the COPII component SAR1B is mutated, causing the chilomicron retention disease (CRD) (Jones et al., 2003). A recent work has shown that an SEC31A mutation causes a neurological syndrome characterized by developmental delay, microcephaly, and epilepsy (Halperin et al., 2019). It has been revealed that the vesicular coat COPII plays a central role in the control of proteostasis within the secretory pathway. Normally, COPII mediates budding of carrier vesicles to ensure that only those proteins that passed the QC are packaged into vesicles. This step is fundamental for the structural and functional maintenance not only of post-ER compartments but also of the extracellular environment (Amodio et al., 2009; Gomez-Navarro and Miller, 2016). Indeed, in driving vesicle formation, COPII ensures by Sec24 isotypes that only folded transmembrane proteins or soluble cargo molecules are sorted and packaged by specific cargo receptors, respectively. The flux of COPII vesicle carriers exiting the ER is regulated by different factors according to secretory demand (Subramanian et al., 2019) or to the ER stress conditions, in which the abundance of COPII vesicles is reduced, giving rise to alterations in cargo secretion and structural defects of post-ER compartments (Amodio et al., 2009; Amodio et al., 2013; Amodio et al., 2017).
The Uncanonical Role of COPII in ER-Phagy
Recent evidence shows that COPII subunits can initiate ER-phagy at specific sites of the ER tubular network, the ER-phagy sites (ERPHSs), which are distinct from ERESs (Cui et al., 2019; Subramanian et al., 2019). Characteristically, ERPHSs contain only one of the Sec24 paralogs (Sfb3/Lst1 in yeast or SEC24C in mammals), which binds to ER-phagy receptors such as ATG40, which in turn interact with Atg8/LC3, ubiquitin-like protein essential for autophagosome biogenesis. The formation of ERPHS also depends on Lnp1, an ER shaping protein that resides in ER tubules and stabilizes the three-way junctions that arise upon tubular fusion (Cui et al., 2019). Therefore, Lst1/SEC24C-Sec23 and Sec24-Sec23 complexes sort cargoes into two distinct routes, the secretory route from ERESs or the ER-phagy from ERPHSs. Thus, the two pathways are independent; in concert with QC and ERAD, which are UPR-controlled, they contribute to the maintenance of proteostasis in the secretory pathway (Fig. 1).
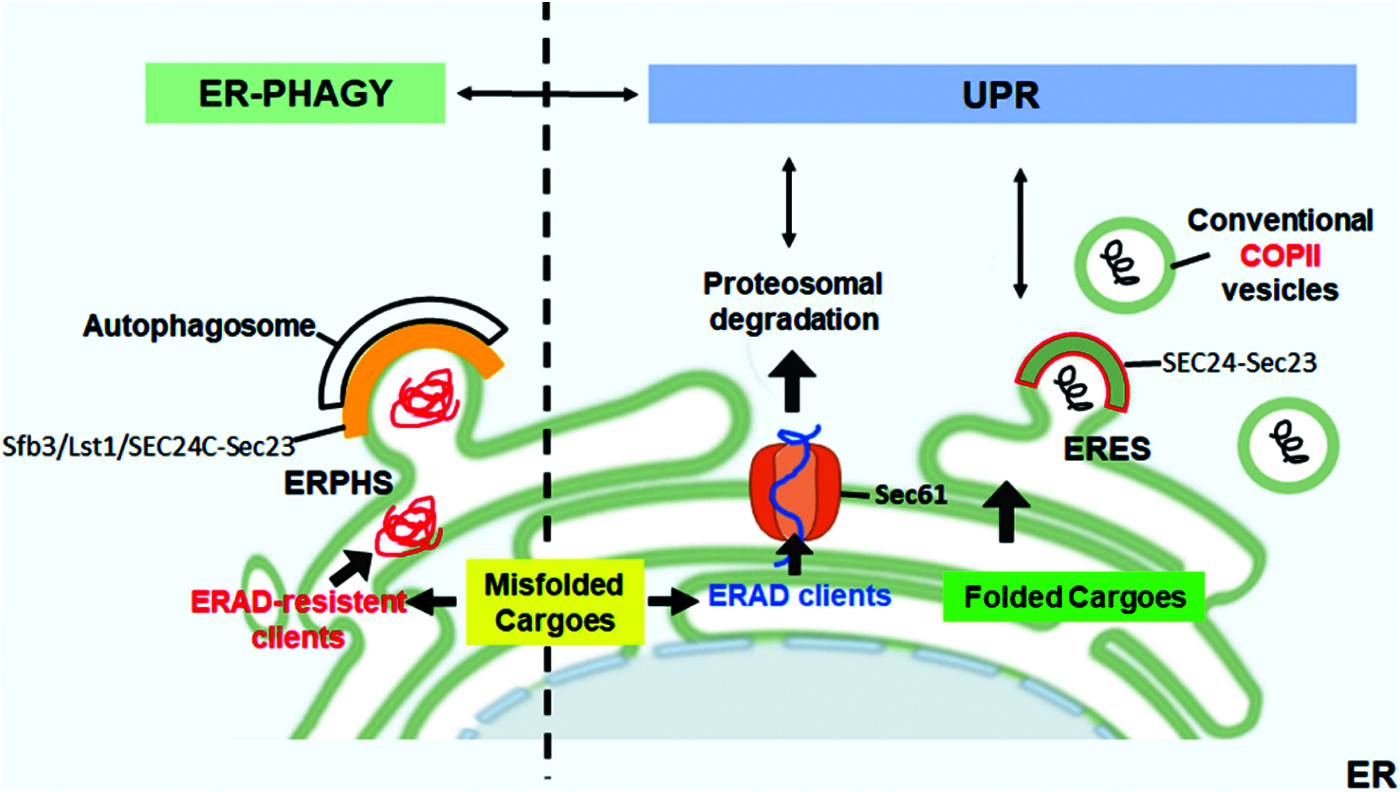
Figure 1: ER-phagy and UPR in the control of ER proteostasis. Correctly folded proteins are packaged at ER exit sites (ERESs) into conventional COPII coat containing SEC24-Sec23. The accumulation of misfolded proteins within the ER activates the UPR that upregulates ERAD to allow the retrotranslocation and proteasomal degradation of the misfolded ERAD clients. Aggregation-prone proteins that are resistant to ERAD are segregated into dedicated ER subdomains (ER-phagy sites, ERPHS) and are cleared by ER-phagy. At ERPHS, the COPII coat subunits Sec23-SEC24C (in mammals)/Sfb3/Lst1 (in yeast) interact with an ER-phagy receptor to initiate autophagosome biogenesis.
The Cross Talk between the UPR and the ER-Phagy in the Control of the Fate of Cargo Proteins
There is strong evidence for the existence of a cross talk between the UPR and the ER-phagy in the control of the fate of cargo proteins (Smith et al., 2018; Ji et al., 2019; Loi et al., 2019; Zielke et al., 2020). However, besides the recognized role of the UPR in protein folding and ERAD control, the mechanisms underlying the involvement of the UPR in the modulation of COPII-mediated QC remain unclear. Different experimental works have identified a direct correlation between the UPR signaling and the regulation of COPII proteins expression and assembly (Higashio and Kohno, 2002; Sato et al., 2002; Farhan et al., 2008; Teske et al., 2011). Other works have suggested that the UPR indirectly affects COPII-mediated export from the ER by impacting the amount of transport-competent proteins inside the ER (Amodio et al., 2009; Amodio et al., 2013; Shaheen, 2018; Subramanian et al., 2019). There is growing evidence for high specificity of ER-phagy in the disposal of protein aggregates; however, the role of the Lst1/SEC24C-Sec23 complex at the ERPHSs in the recruitment of such misfolded aggregates has not yet been clarified. Indeed, different misfolded cargo proteins are degraded by ER-phagy mechanisms. In particular, it is known that the calnexin chaperone recognizes misfolded procollagen I (PCI) and interacts with the ER-phagy receptor FAM134B to deliver PCI to degradation through ER-phagy (Omari et al., 2018; Forrester et al., 2019). Another ER-phagy receptor, RTN3L, is involved in the degradation of proinsulin protein aggregates (Cunningham et al., 2019). ER-phagy seems to be also involved in the degradation of 1-antitrypsin Z (ATZ) polymers (Itakura et al., 2012) through the fusion of ER-derived vesicles with endolysosomes. Importantly, recent works have supported the interplay between the ER stress and the ER-phagy in the disposal of protein aggregates (Smith et al., 2018; Loi et al., 2019; Zielke et al., 2020). Smith et al. (2018) demonstrated that the ER-phagy receptor cell cycle progression protein 1 (CCPG1) is upregulated during ER stress to remove protein aggregates in the pER. In the same way, ER-phagy is directly implicated in the recovery from cyclopiazonic acid-induced ER stress, in which ER-derived vesicles are directly engulfed by endolysosomes (Loi et al., 2019). However, it is still unknown how the UPR and ER-phagy are coordinated and how the outgoing proteins are sorted to the appropriate ER subdomain. In our opinion, it is important to consider that the two heterodimers Lst1/SEC24C-Sec23 and Sec24-Sec23 have Sec23 protein as the common partner, suggesting that the availability of this protein could be important in modulating the two alternative pathways. Curiously, according to our previous work, the amount of COPII vesicles is drastically reduced under ER stress (Fig. 2), and such a reduction impairs ER-to-Golgi trafficking, thereby leading to structural alterations of post-ER compartments (Amodio et al., 2009; Amodio et al., 2013). Moreover, under such conditions, Sec23a cycling at ERES is highly accelerated, presumably to compensate for the amount of Sec23 required for the ER-phagy. Interestingly, under the ER stress, the number of ERESs revealed by the marker Sec16 (Fig. 2) is un-altered (Amodio et al., 2013), which is in agreement with the fact that ERPHSs rather than ERESs initiate the ER-phagy response. On these grounds, the relationship between ER exit at ERESs and ER-phagy should be investigated in more detail. Such studies could open paths toward novel therapeutic approaches for intervention in diseases related to defective ER protein folding, such as Parkinson disease, Alzheimer dementia, Fabry’s disease, cystic fibrosis, and many others (Wang and Kaufman, 2016; Needham et al., 2019).
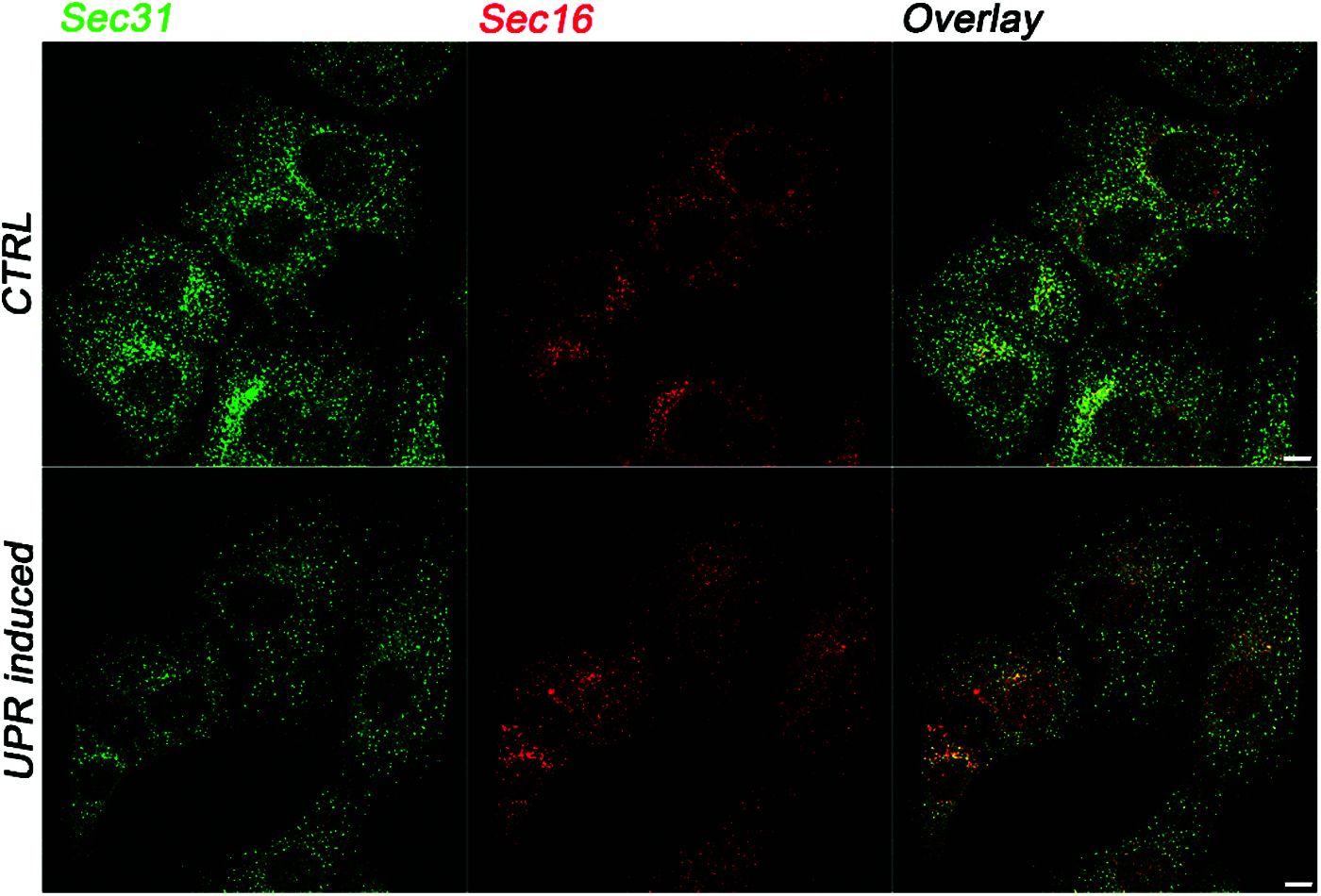
Figure 2: The UPR-dependent regulation of COPII vesicles formation at ERES. Representative immunofluorescence images showing the intracellular distribution of the COPII coat marker Sec31 and the ERES marker Sec16 in control (CTRL) conditions or in UPR-induced cells. In UPR-induced cells, the number of Sec31-labeled spots is drastically reduced compared to control cells (Amodio et al., 2013). In contrast, UPR does not induce changes in the intracellular pattern of Sec16. Immunofluorescence was conducted on Huh7 cells as previously described (Amodio et al., 2017; Pagliara et al., 2020; Rapa et al., 2021). Briefly, Huh7 cells seeded on glass coverslips were either not treated (CTRL) or treated with 300 nM thapsigargin for 2 h and processed for immunofluorescence with anti-Sec31 (BD Transduction Laboratories) and anti-Sec16 (Bethyl Laboratories) specific antibodies. Scale bar: 10 µm.
Overall, the QC within the ER responsible for ensuring that only adequately folded cargoes enter the secretory pathway relies on the concerted activity of the ER-phagy and UPRs, wherein COPII components are involved. However, the precise mechanisms need to be further investigated.
Authors’ Contribution: The authors confirm contribution to the paper as follows: GA and VP conceived the work and wrote the first draft. PR and OM critically reviewed the draft. All authors contributed to drafting the work, revised the final manuscript, and approved submission.
Funding Statement: This work was supported by POR FERS Regione Campania 2014–2020 ASSE 1 O.S 1.2; grant System Innovation for Cancer Early Diagnosis (SICED).
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
Acosta-Alvear D, Zhou Y, Blais A, Tsikitis M, Lents NH, Arias C, Lennon CJ, Kluger Y, Dynlacht BD (2007). XBP1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Molecular Cell 27: 53–66. DOI 10.1016/j.molcel.2007.06.011. [Google Scholar] [CrossRef]
Amodio G, Margarucci L, Moltedo O, Casapullo A, Remondelli P (2017). Identification of cysteine ubiquitylation sites on the Sec23A protein of the COPII complex required for vesicle formation from the ER. Open Biochemistry Journal 11: 36–46. DOI 10.2174/1874091X01711010036. [Google Scholar] [CrossRef]
Amodio G, Pagliara V, Moltedo O, Remondelli P (2021). Structural and functional significance of the endoplasmic reticulum unfolded protein response transducers and chaperones at the mitochondria-ER contacts: A cancer perspective. Frontiers in Cell and Developmental Biology 9: 641194. DOI 10.3389/fcell.2021.641194. [Google Scholar] [CrossRef]
Amodio G, Renna M, Paladino S, Venturi C, Tacchetti C, Moltedo O, Franceschelli S, Mallardo M, Bonatti S, Remondelli P (2009). Endoplasmic reticulum stress reduces the export from the ER and alters the architecture of post-ER compartments. International Journal of Biochemistry & Cell Biology 41: 2511–2521. DOI 10.1016/j.biocel.2009.08.006. [Google Scholar] [CrossRef]
Amodio G, Venditti R, De Matteis MA, Moltedo O, Pignataro P, Remondelli P (2013). Endoplasmic reticulum stress reduces COPII vesicle formation and modifies Sec23a cycling at ERESs. FEBS Letters 587: 3261–3266. DOI 10.1016/j.febslet.2013.08.021. [Google Scholar] [CrossRef]
Balchin D, Hayer-Hartl M, Hartl FU(2016). In vivo aspects of protein folding and quality control. Science 353: 43–54. DOI 10.1126/science.aac4354. [Google Scholar] [CrossRef]
Barlowe C, Orci L, Yeung T, Hosobuchi M, Hamamoto S, Salama N, Rexach MF, Ravazzola M, Amherdt M, Schekman R (1994). COPII: A membrane coat formed by Sec proteins that drive vesicle budding from the endoplasmic reticulum. Cell 77: 895–907. DOI 10.1016/0092-8674(94)90138-4. [Google Scholar] [CrossRef]
Barlowe C, Schekman R (1993). SEC12 encodes a guanine-nucleotide-exchange factor essential for transport vesicle budding from the ER. Nature 365: 347–349. DOI 10.1038/365347a0. [Google Scholar] [CrossRef]
Bartelt A, Widenmaier SB, Schlein C, Johann K, Goncalves RLS et al. (2018). Brown adipose tissue thermogenic adaptation requires Nrf1-mediated proteasomal activity. Nature Medicine 24: 292–303. DOI 10.1038/nm.4481. [Google Scholar] [CrossRef]
Boyadjiev SA, Fromme JC, Ben J, Chong SS, Nauta C et al. (2006). Cranio-lenticulo-sutural dysplasia is caused by a SEC23A mutation leading to abnormal endoplasmic-reticulum-to-Golgi trafficking. Nature Genetics 38: 1192–1197. DOI 10.1038/ng1876. [Google Scholar] [CrossRef]
Braakman I, Hebert DN (2013). Protein folding in the endoplasmic reticulum. Cold Spring Harbor Perspectives in Biology 5: a013201. DOI 10.1101/cshperspect.a013201. [Google Scholar] [CrossRef]
Brodsky JL (2012). Cleaning up: ER-associated degradation to the rescue. Cell 15: 1163–1167. DOI 10.1016/j.cell.2012.11.012. [Google Scholar] [CrossRef]
Budnik A, Stephens DJ (2009). ER exit sites—Localization and control of COPII vesicle formation. FEBS Letters 583: 3796–3803. DOI 10.1016/j.febslet.2009.10.038. [Google Scholar] [CrossRef]
Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D (2002). IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415: 92–96. DOI 10.1038/415092a. [Google Scholar] [CrossRef]
Chen B, Retzlaff M, Roos T, Frydman J (2011). Cellular strategies of protein quality control. Cold Spring Harbor Perspectives in Biology 3: a004374. DOI 10.1101/cshperspect.a004374. [Google Scholar] [CrossRef]
Cherepanova N, Shrimal S, Gilmore R (2016). N-linked glycosylation and homeostasis of the endoplasmic reticulum. Current Opinion in Cell Biology 41: 57–65. DOI 10.1016/j.ceb.2016.03.021. [Google Scholar] [CrossRef]
Chino H, Mizushima N (2020). ER-Phagy: Quality control and turnover of endoplasmic reticulum. Trends in Cell Biology 30: 384–398. DOI 10.1016/j.tcb.2020.02.001. [Google Scholar] [CrossRef]
Cui Y, Parashar S, Zahoor M, Needham PG, Mari M et al. (2019). A COPII subunit acts with an autophagy receptor to target endoplasmic reticulum for degradation. Science 365: 53–60. DOI 10.1126/science.aau9263. [Google Scholar] [CrossRef]
Cunningham CN, Williams JM, Knupp J, Arunagiri A, Arvan P, Tsai B (2019). Cells deploy a two-pronged strategy to rectify misfolded proinsulin aggregates. Molecular Cell 75: 442–456. DOI 10.1016/j.molcel.2019.05.011. [Google Scholar] [CrossRef]
Duttler S, Pechmann S, Frydman J (2013). Principles of cotranslational ubiquitination and quality control at the ribosome. Molecular Cell 50: 379–393. DOI 10.1016/j.molcel.2013.03.010. [Google Scholar] [CrossRef]
Eletto D, Dersh D, Gidalevitz T, Argon Y (2014). Protein disulfide isomerase A6 controls the decay of IRE1alpha signaling via disulfide-dependent association. Molecular Cell 53: 562–576. DOI 10.1016/j.molcel.2014.01.004. [Google Scholar] [CrossRef]
English AR, Zurek N, Voeltz GK (2009). Peripheral ER structure and function. Current Opinion in Cell Biology 21: 596–602. DOI 10.1016/j.ceb.2009.04.004. [Google Scholar] [CrossRef]
Farhan H, Weiss M, Tani K, Kaufman RJ, Hauri HP (2008). Adaptation of endoplasmic reticulum exit sites to acute and chronic increases in cargo load. EMBO Journal 27: 2043–2054. DOI 10.1038/emboj.2008.136. [Google Scholar] [CrossRef]
Fath S, Mancias JD, Bi X, Goldberg J (2007). Structure and organization of coat proteins in the COPII cage. Cell 129: 1325–1336. DOI 10.1016/j.cell.2007.05.036. [Google Scholar] [CrossRef]
Forrester A, De Leonibus C, Grumati P, Fasana E, Piemontese M et al. (2019). A selective ER-phagy exerts procollagen quality control via a Calnexin-FAM134B complex. EMBO Journal 38: e99847. [Google Scholar]
Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK (2011). ER tubules mark sites of mitochondrial division. Science 334: 358–362. DOI 10.1126/science.1207385. [Google Scholar] [CrossRef]
Gomez-Navarro N, Miller E (2016). Protein sorting at the ER-Golgi interface. Journal of Cell Biology 215: 769–778. DOI 10.1083/jcb.201610031. [Google Scholar] [CrossRef]
Grimaldi M, Stillitano I, Amodio G, Santoro A, Buonocore M, Moltedo O, Remondelli P, D’Ursi AM (2018). Structural basis of antiviral activity of peptides from MPER of FIV gp36. PLoS One 13: e0204042. DOI 10.1371/journal.pone.0204042. [Google Scholar] [CrossRef]
Guerriero CJ, Brodsky JL (2012). The delicate balance between secreted protein folding and endoplasmic reticulum-associated degradation in human physiology. Physiological Reviews 92: 537–576. DOI 10.1152/physrev.00027.2011. [Google Scholar] [CrossRef]
Halperin D, Kadir R, Perez Y, Drabkin M, Yogev Y et al. (2019). SEC31A mutation affects ER homeostasis, causing a neurological syndrome. Journal of Medical Genetics 56: 139–148. DOI 10.1136/jmedgenet-2018-105503. [Google Scholar] [CrossRef]
Hetz C, Zhang K, Kaufman RJ (2020). Mechanisms, regulation and functions of the unfolded protein response. Nature Reviews Molecular Cell Biology 21: 421–438. DOI 10.1038/s41580-020-0250-z. [Google Scholar] [CrossRef]
Hicke L, Yoshihisa T, Schekman R (1992). Sec23p and a novel 105-kDa protein function as a multimeric complex to promote vesicle budding and protein transport from the endoplasmic reticulum. Molecular Biology of the Cell 3: 667–676. DOI 10.1091/mbc.3.6.667. [Google Scholar] [CrossRef]
Higashio H, Kohno K (2002). A genetic link between the unfolded protein response and vesicle formation from the endoplasmic reticulum. Biochemical and Biophysical Research Communications 296: 568–574. DOI 10.1016/S0006-291X(02)00923-3. [Google Scholar] [CrossRef]
Hollien J, Lin JH, Li H, Stevens N, Walter P, Weissman JS (2009). Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. Journal of Cell Biology 186: 323–331. DOI 10.1083/jcb.200903014. [Google Scholar] [CrossRef]
Hwang J, Qi L (2018). Quality control in the endoplasmic reticulum: Crosstalk between ERAD and UPR pathways. Trends in Biochemical Sciences 43: 593–605. DOI 10.1016/j.tibs.2018.06.005. [Google Scholar] [CrossRef]
Itakura E, Kishi-Itakura C, Mizushima N (2012). The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 151: 1256–1269. DOI 10.1016/j.cell.2012.11.001. [Google Scholar] [CrossRef]
Ji CH, Kim HY, Heo AJ, Lee SH, Lee MJ et al. (2019). The N-Degron pathway mediates ER-phagy. Molecular Cell 75: 1058–1072. DOI 10.1016/j.molcel.2019.06.028. [Google Scholar] [CrossRef]
Jones B, Jones EL, Bonney SA, Patel HN, Mensenkamp AR et al. (2003). Mutations in a Sar1 GTPase of COPII vesicles are associated with lipid absorption disorders. Nature Genetics 34: 29–31. DOI 10.1038/ng1145. [Google Scholar] [CrossRef]
Karagoz GE, Acosta-Alvear D, Walter P (2019). The unfolded protein response: Detecting and responding to fluctuations in the protein-folding capacity of the endoplasmic reticulum. Cold Spring Harbor Perspectives in Biology 11: a033886. DOI 10.1101/cshperspect.a033886. [Google Scholar] [CrossRef]
Lee C, Chen LB (1993). Motility and construction of the endoplasmic reticulum in living cells. Subcellular Biochemistry 21: 343–352. DOI 10.1007/978-1-4615-2912-5. [Google Scholar] [CrossRef]
Lee MC, Orci L, Hamamoto S, Futai E, Ravazzola M, Schekman R (2005). Sar1p N-terminal helix initiates membrane curvature and completes the fission of a COPII vesicle. Cell 122: 605–617. DOI 10.1016/j.cell.2005.07.025. [Google Scholar] [CrossRef]
Lemmer IL, Willemsen N, Hilal N, Bartelt A (2021). A guide to understanding endoplasmic reticulum stress in metabolic disorders. Molecular Metabolism 47: 101169. DOI 10.1016/j.molmet.2021.101169. [Google Scholar] [CrossRef]
Loi M, Raimondi A, Morone D, Molinari M (2019). ESCRT-III-driven piecemeal micro-ER-phagy remodels the ER during recovery from ER stress. Nature Communications 10: 50–58. DOI 10.1038/s41467-019-12991-z. [Google Scholar] [CrossRef]
Lu CL, Kim J (2020). Consequences of mutations in the genes of the ER export machinery COPII in vertebrates. Cell Stress and Chaperones 25: 199–209. DOI 10.1007/s12192-019-01062-3. [Google Scholar] [CrossRef]
Matsuoka K, Orci L, Amherdt M, Bednarek SY, Hamamoto S, Schekman R, Yeung T (1998). COPII-coated vesicle formation reconstituted with purified coat proteins and chemically defined liposomes. Cell 93: 263–275. DOI 10.1016/S0092-8674(00)81577-9. [Google Scholar] [CrossRef]
Maurel M, Chevet E, Tavernier J, Gerlo S (2014). Getting RIDD of RNA: IRE1 in cell fate regulation. Trends in Biochemical Sciences 39: 245–254. DOI 10.1016/j.tibs.2014.02.008. [Google Scholar] [CrossRef]
Miller EA, Beilharz TH, Malkus PN, Lee MC, Hamamoto S, Orci L, Schekman R (2003). Multiple cargo binding sites on the COPII subunit Sec24p ensure capture of diverse membrane proteins into transport vesicles. Cell 114: 497–509. DOI 10.1016/S0092-8674(03)00609-3. [Google Scholar] [CrossRef]
Miller E, Antonny B, Hamamoto S, Schekman R (2002). Cargo selection into COPII vesicles is driven by the Sec24p subunit. EMBO Journal 21: 6105–6113. DOI 10.1093/emboj/cdf605. [Google Scholar] [CrossRef]
Moltedo O, Remondelli P, Amodio G (2019). The mitochondria-endoplasmic reticulum contacts and their critical role in aging and age-associated diseases. Frontiers in Cell and Developmental Biology 7: 172. DOI 10.3389/fcell.2019.00172. [Google Scholar] [CrossRef]
Needham PG, Guerriero CJ, Brodsky JL (2019). Chaperoning endoplasmic reticulum-associated degradation (ERAD) and protein conformational diseases. Cold Spring Harbor Perspectives in Biology 11: a033928. DOI 10.1101/cshperspect.a033928. [Google Scholar] [CrossRef]
Omari S, Makareeva E, Roberts-Pilgrim A, Mirigian L, Jarnik M, Ott C, Lippincott-Schwartz J, Leikin S (2018). Noncanonical autophagy at ER exit sites regulates procollagen turnover. Proceedings of the National Academy of Science USA 115: 10099–10108. DOI 10.1073/pnas.1814552115. [Google Scholar] [CrossRef]
Pagliara V, Donadio G, de Tommasi N, Amodio G, Remondelli P, Moltedo O, Dal Piaz F (2020). Bioactive Ent-Kaurane diterpenes oridonin and irudonin prevent cancer cells migration by interacting with the actin cytoskeleton controller Ezrin. International Journal of Molecular Sciences 21: 7186. DOI 10.3390/ijms21197186. [Google Scholar] [CrossRef]
Phillips MJ, Voeltz GK (2016). Structure and function of ER membrane contact sites with other organelles. Nature Reviews Molecular Cell Biology 17: 69–82. DOI 10.1038/nrm.2015.8. [Google Scholar] [CrossRef]
Plate L, Wiseman RL (2017). Regulating secretory proteostasis through the unfolded protein response: From function to therapy. Trends in Cell Biology 27: 722–737. DOI 10.1016/j.tcb.2017.05.006. [Google Scholar] [CrossRef]
Preissler S, Ron D (2019). Early events in the endoplasmic reticulum unfolded protein response. Cold Spring Harbor Perspectives in Biology 11: a033894. DOI 10.1101/cshperspect.a033894. [Google Scholar] [CrossRef]
Rapa SF, Magliocca G, Pepe G, Amodio G, Autore G, Campiglia P, Marzocco S (2021). Protective effect of pomegranate on oxidative stress and inflammatory response induced by 5-fluorouracil in human keratinocytes. Antioxidants (Basel) 10: 203. DOI 10.3390/antiox10020203. [Google Scholar] [CrossRef]
Sato M, Sato K, Nakano A (2002). Evidence for the intimate relationship between vesicle budding from the ER and the unfolded protein response. Biochemical and Biophysical Research Communications 296: 560–567. DOI 10.1016/S0006-291X(02)00922-1. [Google Scholar] [CrossRef]
Schneider K, Bertolotti A (2015). Surviving protein quality control catastrophes—From cells to organisms. Journal of Cell Science 128: 3861–3869. DOI 10.1242/jcs.173047. [Google Scholar] [CrossRef]
Shaheen A (2018). Effect of the unfolded protein response on ER protein export: A potential new mechanism to relieve ER stress. Cell Stress and Chaperones 23: 797–806. DOI 10.1007/s12192-018-0905-2. [Google Scholar] [CrossRef]
Shao S, Hegde RS (2016). Target selection during protein quality control. Trends in Biochemical Science 41: 124–137. DOI 10.1016/j.tibs.2015.10.007. [Google Scholar] [CrossRef]
Shibata Y, Shemesh T, Prinz WA, Palazzo AF, Kozlov MM, Rapoport TA (2010). Mechanisms determining the morphology of the peripheral ER. Cell 143: 774–788. DOI 10.1016/j.cell.2010.11.007. [Google Scholar] [CrossRef]
Smith MD, Harley ME, Kemp AJ, Wills J, Lee M, Arends M, von Kriegsheim A, Behrends C, Wilkinson S (2018). CCPG1 Is a non-canonical autophagy cargo receptor essential for ER-phagy and pancreatic ER proteostasis. Developmental Cell 44: 217–232. DOI 10.1016/j.devcel.2017.11.024. [Google Scholar] [CrossRef]
Stagg SM, LaPointe P, Razvi A, Gurkan C, Potter CS, Carragher B, Balch WE (2008). Structural basis for cargo regulation of COPII coat assembly. Cell 134: 474–484. DOI 10.1016/j.cell.2008.06.024. [Google Scholar] [CrossRef]
Subramanian A, Capalbo A, Iyengar NR, Rizzo R, di Campli A et al. (2019). Auto-regulation of secretory flux by sensing and responding to the folded cargo protein load in the endoplasmic reticulum. Cell 176: 1461–1476. DOI 10.1016/j.cell.2019.01.035. [Google Scholar] [CrossRef]
Sun Z, Brodsky JL (2019). Protein quality control in the secretory pathway. Journal of Cell Biology 218: 3171–3187. DOI 10.1083/jcb.201906047. [Google Scholar] [CrossRef]
Tao YX, Conn PM (2018). Pharmacoperones as novel therapeutics for diverse protein conformational diseases. Physiological Reviews 98: 697–725. DOI 10.1152/physrev.00029.2016. [Google Scholar] [CrossRef]
Teske BF, Wek SA, Bunpo P, Cundiff JK, McClintick JN, Anthony TG, Wek RC (2011). The eIF2 kinase PERK and the integrated stress response facilitate activation of ATF6 during endoplasmic reticulum stress. Molecular Biology of the Cell 22: 4390–4405. DOI 10.1091/mbc.e11-06-0510. [Google Scholar] [CrossRef]
Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P (2000). Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell 101: 249–258. DOI 10.1016/S0092-8674(00)80835-1. [Google Scholar] [CrossRef]
Trombetta ES, Parodi AJ (2003). Quality control and protein folding in the secretory pathway. Annual Review of Cell and Developmental Biology 19: 649–676. DOI 10.1146/annurev.cellbio.19.110701.153949. [Google Scholar] [CrossRef]
Voeltz GK, Barr FA (2013). Cell organelles. Current Opinion in Cell Biology 25: 403–405. DOI 10.1016/j.ceb.2013.06.001. [Google Scholar] [CrossRef]
Wang F, Durfee LA, Huibregtse JM (2013). A cotranslational ubiquitination pathway for quality control of misfolded proteins. Molecular Cell 50: 368–378. DOI 10.1016/j.molcel.2013.03.009. [Google Scholar] [CrossRef]
Wang M, Kaufman RJ (2016). Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 529: 326–335. DOI 10.1038/nature17041. [Google Scholar] [CrossRef]
Weissman JT, Plutner H, Balch WE (2001). The mammalian guanine nucleotide exchange factor mSec12 is essential for activation of the Sar1 GTPase directing endoplasmic reticulum export. Traffic 2: 465–475. DOI 10.1034/j.1600-0854.2001.20704.x. [Google Scholar] [CrossRef]
Westrate LM, Lee JE, Prinz WA, Voeltz GK (2015). Form follows function: The importance of endoplasmic reticulum shape. Annual Reviews of Biochemistry 84: 791–811. DOI 10.1146/annurev-biochem-072711-163501. [Google Scholar] [CrossRef]
Wilkinson S (2019). ER-phagy: Shaping up, destressing the endoplasmic reticulum. FEBS Journal 286: 2645–2663. DOI 10.1111/febs.14932. [Google Scholar] [CrossRef]
Wolff S, Weissman JS, Dillin A (2014). Differential scales of protein quality control. Cell 157: 52–64. DOI 10.1016/j.cell.2014.03.007. [Google Scholar] [CrossRef]
Wu X, Rapoport TA (2018). Mechanistic insights into ER-associated protein degradation. Current Opinion in Cell Biology 53: 22–28. DOI 10.1016/j.ceb.2018.04.004. [Google Scholar] [CrossRef]
Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K (2001). XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107: 881–891. DOI 10.1016/S0092-8674(01)00611-0. [Google Scholar] [CrossRef]
Zielke S, Kardo S, Zein L, Mari M, Covarrubias-Pinto A et al. (2020). ATF4 links ER stress with reticulophagy in glioblastoma cells. Autophagy 17: 2432–2448. [Google Scholar]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |