DOI:10.32604/biocell.2022.018166
| BIOCELL DOI:10.32604/biocell.2022.018166 | |
| Article |
Identification of differential mRNA and lncRNA expression in AcMNPV-infected Sf9 cells
School of Life Science, Sun Yat-sen University, Guangzhou, 510275, China
*Address correspondence to: Riqiang Deng, lssdrq@mail.sysu.edu.cn
Received: 04 July 2021; Accepted: 19 November 2021
Abstract: Sf9Sf9 are the ovarian cells of Spodoptera frugiperda that is the host of Autographa californica multiple nucleopolyhedrovirus (AcMNPV), and hence can serve as an effective test vehicle to understand the AcMNPV infection mechanism. In this study, through high-throughput sequencing technology using samples collected from Sf9 cells at different time points after AcMNPV infection, 3463 pieces of time-series differentially expressed RNA (1,200 mRNA and 2,263 lncRNA) are identified and justified by experimental verification of randomly selected samples from them, proving the validity of the bioinformatical analysis on this topic. Functional enrichment analysis and target prediction are performed on those differentially expressed RNA, from which the major functional enrichment distribution of those differentially expressed mRNA is derived. It has been found that the differential genes are mainly in the cellular anatomical entity and intracellular in terms of the cellular component, and in the binding and catalytic activity in terms of the molecular function. Also, the differential mRNA are mainly concentrated in global and overview maps, signal transduction, infectious diseases, and viral, etc. Moreover, those mRNA targeted by lncRNA are predicted. The correlation between those differentially expressed lncRNA and mRNA indicates that lncRNA is very likely playing an important role in the interaction between virus and host. Aided by an advanced co-expression analysis approach, the “hub” RNA is also identified. The study in this work pave the way for further analyzing and understanding how AcMNPV escapes from the host’s immunity, manipulates the host to realize the self-multiplication, and realizes the timely conversion between its two particle forms, laying the foundation for uncovering the host’s immune response process.
Keywords: AcMNPV; Sf9; lncRNA; mRNA; RNA-Seq
Abbreviations
| AcMNPV: | Autographa californica multiple nucleopolyhedrovirus |
| Sf9: | Spodoptera frugiperda cell |
| lncRNA: | Long non-coding RNA |
| mRNA: | Messenger RNA |
| EBSeqHMM: | A Bayesian approach for identifying gene-expression changes in ordered RNA-seq experiments |
| RNA-Seq: | Transcriptome sequencing technology |
| BV: | Budded virus |
| ODV: | Occlusion-derived virus |
| MOI: | Multiplicity of infection |
| hpi: | Hour post infection |
| qRT-PCR: | Real-Time Quantitative Reverse Transcription PCR |
| CABI: | Centre for Agriculture and Bioscience International |
| EFSA: | European Food Safety Authority |
| FAO: | Food and Agriculture Organization |
| SFMNPV: | Spodoptera frugiperda multiple nucleopolyhedrovirus |
Baculoviruses are pathogenic microorganisms that infect insects and invertebrates, making them their host. The particle form of the virus is Baculoviridae (Vago et al., 1974). Its main hosts are Lepidoptera (corn borer, etc.), Hymenoptera (bees, etc.), and Diptera (mosquitoes, etc.) (Slack and Arif, 2007). They are divided into four genera, namely, Alphabaculovirus, Betabaculovirus, Gammabaculovirus, and Deltabaculovirus, according to the insect hosts from which they were isolated and their biological characteristics (Jehle et al., 2006). Autographa californica multiple nucleopolyhedrovirus (AcMNPV) is the archetypal species of the alphabaculovirus. During the infection cycle, it produces two enveloped virion phenotypes with different structures and functions, which play different roles in virus pathogenesis: budded virus (BV) and occlusion-derived virus (ODV) (Rohrmann, 2013). The nucleocapsid structures of these viruses are similar, but the origin and composition of their envelope and their roles in the virus life cycle are different.
Spodoptera frugiperda (Lepidoptera: Noctuidae) is a major migratory insect from tropical and subtropical areas of America and one of the major hosts for AcMNPV. As the ovarian cells of Spodoptera frugiperda, Sf9 is also the target to infect by AcMNPV. Many researches (Sparks, 1979; Johnson, 1987; Barr and Manning, 1997; Matindoost et al., 2015; Yin et al., 2008) on AcMNPV and its host has been started by infecting these Sf9 cells with AcMNPV.
The baculovirus establishes a strict parasitic and adaptive relationship with its insect host during the long evolutionary process, and the information of the interaction is recorded and solidified in the viral genome (Horton and Burand, 1993). For example, polyhedrins need to be alkali-interpreted by releasing virions to initiate infection in the pH-alkaline larval midgut, the alkaline environment of the insect host’s midgut and alkaline solubility of the viral polyhedrin protein crystal is the result of the long-term evolution of both.
Baculoviruses interact with their hosts at multiple levels, but the details remain unclear. The expression of baculovirus genes has a time sequence (Friesen and Miller, 1985, 1986, 1987; Nissen and Friesen, 1989). It has been reported that the host plays an important role in the expression of baculovirus genes. For example, the expression of early baculovirus genes depends on the host’s transcription system and factors (Ooi and Miller, 1988). After the baculovirus unshells and enters the nucleus, DNAs that have not yet been replicated are transcribed in the first place. The early transcription of the virus is inhibited by muscarine carnitine, so host RNA polymerase II mediates most early gene transcription, leading to the activation and expression of the late gene, thus making the expression of viral genes orderly (Tjia et al., 1979). Host RNA polymerase is the most important host component in the early gene expression of the virus and plays a dominant role in the life cycle of the virus. In addition, a host phosphoprotein with a relative molecular weight of 30 ku has been identified, and found to have high affinity with AcMNPV polyhedroid protein promoter (Burma et al., 1994). After extensive analysis, Williams and Faulkner (1997). found that the host nuclear membrane was related to the formation of viral envelope (Williams and Faulkner, 1997).
The baculovirus also affects its host. For example, one of the characteristics of baculovirus overexpression in early gene expression to late gene expression is significant inhibition of host transcription. Some studies (Knudson and Tinsley, 1974; Bilimoria et al., 1986) have shown that in Sf9 cells infected with AcMNPV, host protein synthesis begins to decline between 6 and 10 h after infection and seems to stop completely after 24 h. In addition, baculovirus can also inhibit the apoptosis of host cells and affect the cycle process of host cells. Bertin et al. (1996) demonstrated that the P35 protein of baculovirus inhibits apoptosis induced by baculovirus infection. One of the functions of AcMNPV’s very early gene, IE2, has been shown to encode proteins that block cell cycles in various cell lines.
The interaction between baculovirus and host is reflected at multiple levels, from simple physical adsorption (Granados, 1978; Summers, 1971), invasion to complex manipulation of host metabolic system (Funk and Consigli, 1993), and induction of host cell apoptosis (Zhang et al., 2002, 2019), etc. Many interesting and important host-virus interaction mechanisms have been identified from fundamental studies of the biochemistry and molecular biology of baculoviruses (Manji, 2000; Miller and Lu, 1997; Cory et al., 2005; Joshi et al., 2010; Miller, 1997). These studies, to some extent, provide new strategies for biological pest control and eukaryotic expression vector systems.
A large part of functional genes of baculoviruses have been identified thus far, but the research on the relationship between the two main focuses on viruses, and there is less research on the function of host factors, so the work in this field is worthy of further development (Rohrmann, 2011; Friesen and Miller, 1986). In addition, many previous studies (Mccarthy and Theilmann, 2008; Cochran, 1983; Fang et al., 2009; Wu et al., 2018) have carried out relevant findings from the perspective of host genes or viral gene functions, while from the perspective of bioinformatics, there is no relevant report on the changes in the expression level of host genes after baculovirus infection. In this study, after the baculovirus infecting Sf9 cells, infected cells were collected at different time points for sequencing, and the sequencing results were analyzed to identify those messenger RNA (mRNA) and long non-coding RNA (lncRNA) that show expression features of sequential difference. Moreover, functional enrichment, target and co-expression analysis were performed to shed light on the correlation of mRNA and lncRNA. The molecular interaction between viral genes (and their products) and host cytokines are so exquisitely coordinated that even subtle changes can inhibit viral replication. This study provides some candidate genes that are closely related to the molecular regulation mechanism of Sf9 cells infected with AcMNPV. Our results will provide a useful resource for further analysis of baculovirus-host interactions, and understand the immune escape mechanism of the virus.
Sf9 (Spodoptera frugiperda IPLB-Sf21-AE clonal isolate 9) insect cells were cultured in Grace’s medium supplemented with 10% fetal bovine serum at 27°C. Groups of Sf9 cells were infected with AcMNPV at a multiplicity of infection (MOI) of 10, and subsequently, RNA samples were collected at 6, 12, 24, 36, 48, 60 and 72 h post-infection (hpi).
Total RNA was isolated from approximately 107 infected cells harvested using TRIzol reagent (Invitrogen, USA) according to the manufacturer’s protocol, and purified by using RNeasy mini kit (Qiagen, GmBH, Germany). Total RNA was dissolved in 50 μL RNase-free H2O. The RNA solution was digested for 30 min at 37°C with RNase-free DNase I (TaKaRa, Japan) to remove any contaminating genomic DNA. RNA integrity was analyzed by gel electrophoresis using a 1% agarose gel alongside an RNA marker. The samples were sent to Beijing Genomics Institute Shenzhen Co. (BGI-Shenzhen) (China) for sequencing using Solexa technology (Cuddapah et al., 2009; Nagalakshmi et al., 2008; Rougemont et al., 2008). In the subsequent sequencing test, RNA quality was checked at first. Only when the RNA integrity value (RIN) is larger than 8, indicating RNA quality and integrity meet the sequencing requirement, subsequent test could be continued. In this study, Illumina Novaseq 6000 was used for sequencing test, with a depth of 6G (20 M reads).
Raw data processing and transcriptome
Fastp (version 0.19.4) (Chen et al., 2018) was used to filter low-quality reads, cut adapters and quality control of raw FASTQ files to obtain clean reads. The clean reads of each sample were aligned against SLIVA (Quast et al., 2013) ribosomal RNA and AcMNPV genome (accession number NC_001623) with Bowtie2 (Langmead and Salzberg, 2012) to filter out ribosomal RNA and AcMNPV RNA.
Then, the unmapped reads of each sample were aligned against the Spodoptera frugiperda genome (NCBI, GCA_002213285.1) with HISAT2 (version 2.1.0) (Kim et al., 2015) and then subsequently assembled by StringTie (version 1.3.4d) (Pertea et al., 2015) separately. All the assemblies were merged into one transcriptome by TACO (Niknafs et al., 2017). Then, Salmon (version 0.11.2) (Patro et al., 2017) was applied to quantify the transcript expression of the transcriptome. To reduce noise, we include only transcripts with TPM > 0.1 and read count > 10.
BLASTP was used to compare the obtained sequences with the NR database, and the unmatched sequences were selected for subsequent analysis. PLEK (Li et al., 2014) was used to calculate the coding potential of the transcripts. Transcripts that had no coding potential, and harboured at least two exons, and had a length larger than 200 nt, were classified as lncRNAs. Transcripts that showed coding potential and/or had a similar sequence with proteins were classified as mRNAs. R package EBSeqHMM (Leng et al., 2015) was used to identify transcripts changing over time; FDR < 0.01 was considered as signature. The detailed flow is illustrated in Fig. 1.
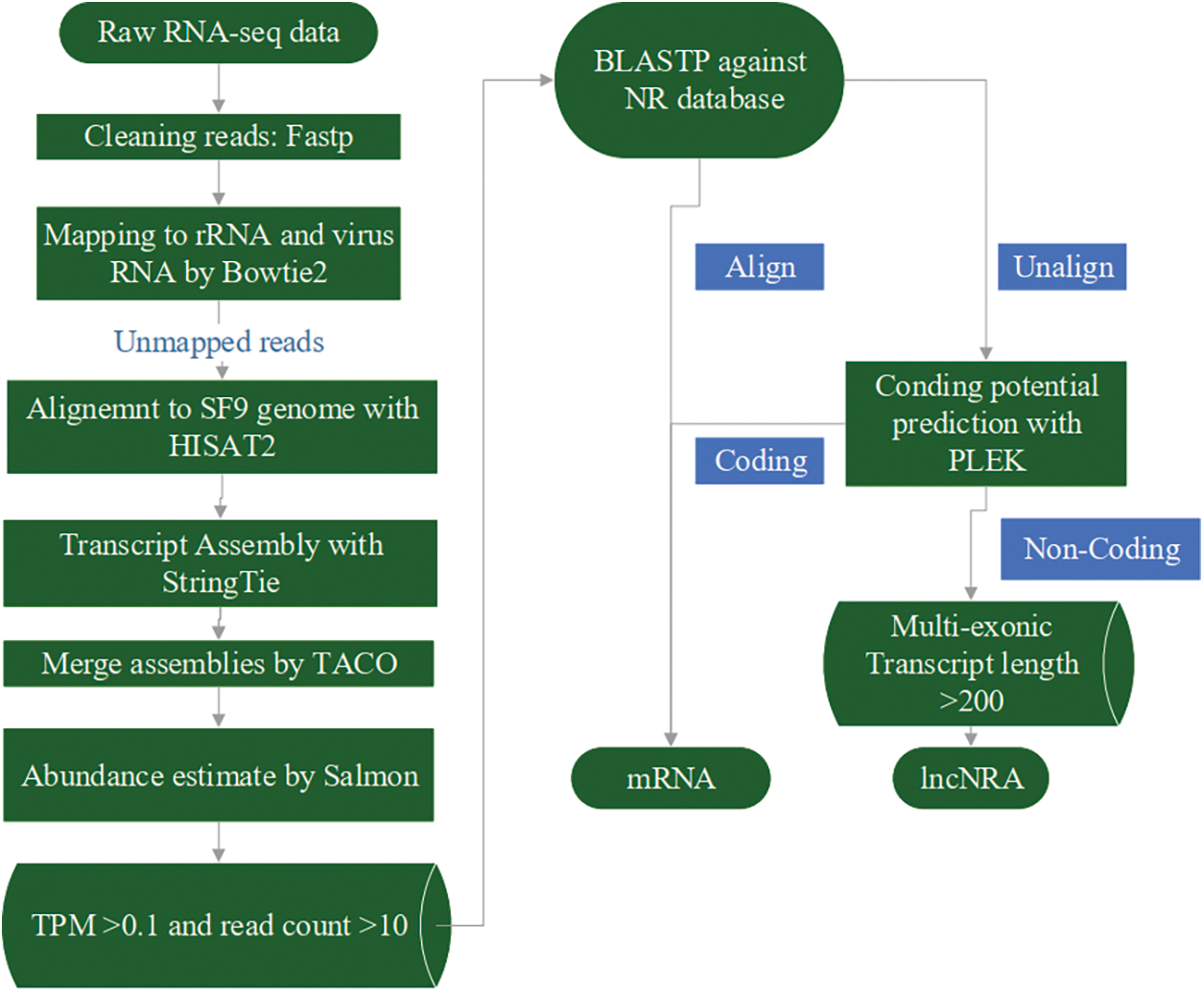
Figure 1: LncRNA and mRNA sequences were separated and assembled by processing RNA-seq data through a series of analytical processes. First, the raw data were filtered to remove rRNA and viral RNA. The obtained clean data were then compared with the Sf9 genome. Sequences with TPM expression levels greater than 0.1 and read count greater than 10 were compared with the NR database. The unmatched sequences were further analyzed using PLEK to differentiate lncRNA and mRNA.
Real-time quantitative Reverse Transcription-PCR (qRT-PCR) was performed to define the expression profile of mRNA and lncRNA. We compared 57,104 RNAs with eggNOG and other databases to find the sequence of selected genes and designed relevant primers based on the sequence information of selected genes. The mRNA and 5S rRNA were reversely transcribed using a random primer (Takara) and included in the respective sample. The qRT-PCR results were analyzed by utilizing the 2–ΔΔCT method. The experiment was repeated three times and averaged to show changes in the expression of host genes at different time points after infection.
GO and KEGG functional enrichment analysis methods
We performed a Gene Ontology (GO) analysis for biological processes, cellular components and molecular function and a KEGG pathway analysis (Kyoto Encyclopedia of Genes and Genomes http://www.genome.ad.jp/kegg) via enrich R package.
RNAPlex software is used in this work to predict the interaction between lncRNA and mRNA. It mainly calculates the minimum free energy according to the thermodynamic structure to predict the optimal base-pairing relationship (Tafer and Hofacker, 2008).
The weighted gene co-expression network analysis (WGCNA) technique is designed to search gene modules that are co-expressed and explore the correlation between gene network and targeted expression types and the core genes in the network (Langfelder and Horvath, 2008). This analysis applies to complex data models. For example, the response at different time points after pathogen infection can be well analyzed. In this study, with the help of this analysis, the lncRNA with time-series difference and the “hub” RNA will be identified.
Overview of RNA sequencing (RNA-seq)
After standardizing the raw data, a total of 24,230,852.75 clean reads that match the Spodoptera frugiperda genome were obtained through sequencing and filtering out ribosomal RNA and AcMNPV RNA of samples of the 8-time points.
Identification of lncRNA and mRNA
The obtained clean reads were assembled into 57,104 non-redundant RNA transcripts, including 19,982 lncRNA and 37,122 mRNA, as shown in Figs. 2A–2C. In general, GC content and length can be well differentiated.
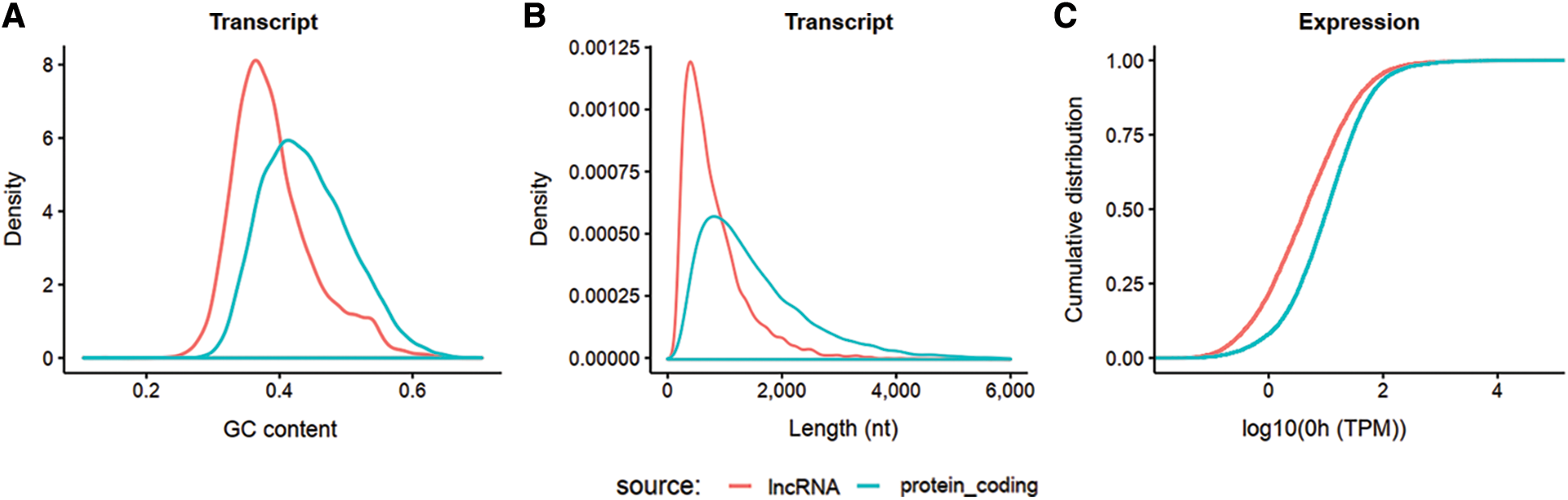
Figure 2: Isograph of GC content, length, and gene expression of lncRNA and mRNA. RNA meeting the requirement that its transcripts present in at least 2 samples, counts > 10, TPM > 0.1 for each sample, was used. lncRNA and mRNA can be distinguished from each other in all three aspects. A: GC percentage distribution comparison between lncRNA and protein coding RNA transcripts. B: Length distribution comparison between lncRNA and protein coding RNA transcripts. C: Cumulative gene expression (TPM) distribution of lncRNA and protein coding RNA at 0 h. The horizontal axis is log10 (TPM) at 0 h, and the vertical axis is the accumulation of its probability density.
Sequence expression differences of infected Sf9 cells
Aided by further EBSeqHMM analysis, it was found that 1,200 mRNA and 2,263 lncRNA showed differences in temporal expression after AcMNPV infection. Furthermore, we randomly selected 4 RNA, including 1 mRNA and 3 lncRNA, for further experimental validation.
As shown in Fig. 3, the overall trend of the selected lncRNA and mRNA was the same whether the results were analyzed by EBSeqHMM or by qRT-PCR. After virus infection, XP_022825542.1, XP_022823371, XP_022827228.1, and XP_022814930.1 exhibit an overall downward trend in both the bioinformatics and qRT-PCR predictions. In contrast, XP_022827228.1 exhibit an overall upward trend in both the bioinformatics and qRT-PCR predictions.
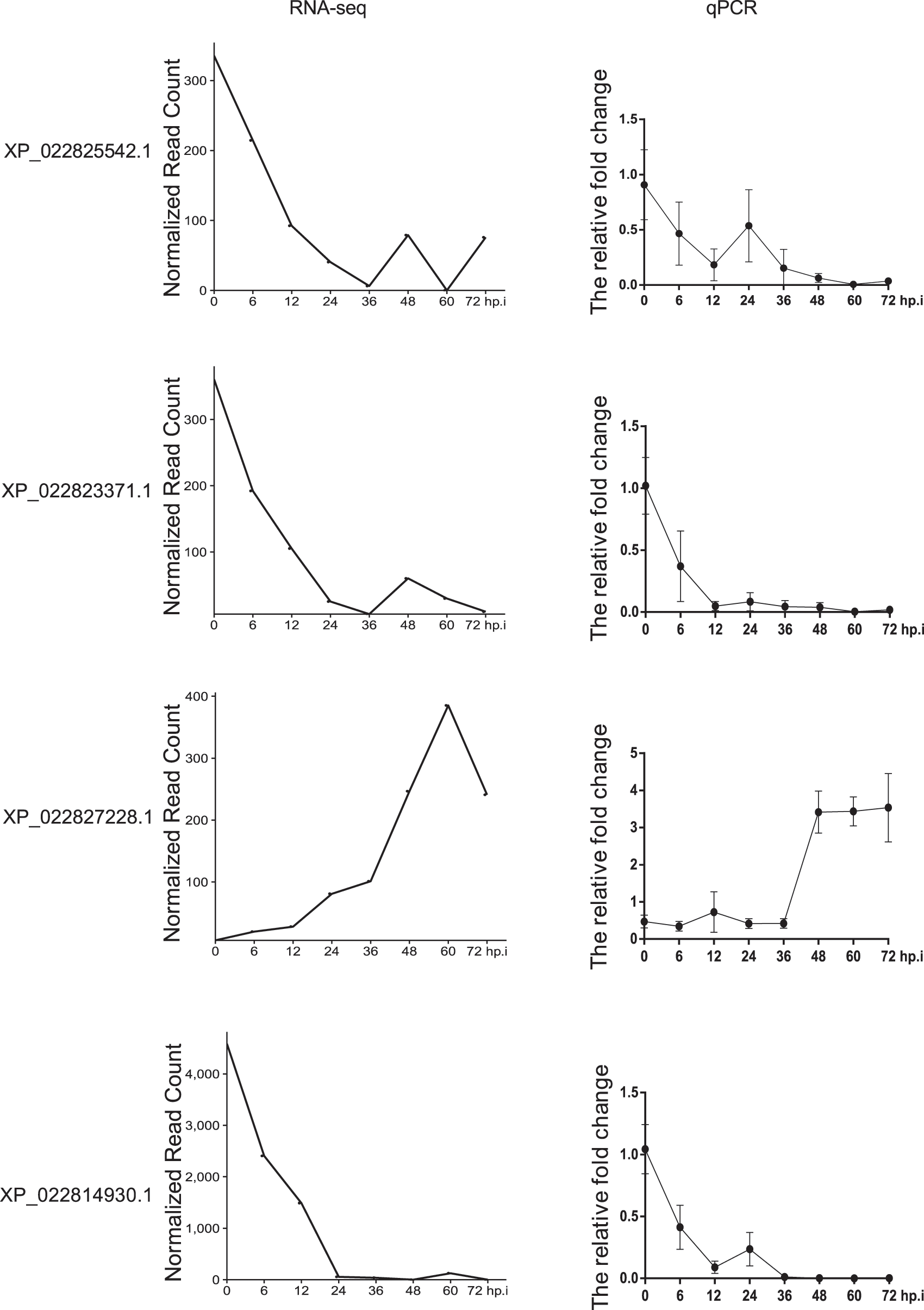
Figure 3: Comparison between bioinformatics and qPCR results of lncRNA XP_022825542.1, XP_022823371, and XP_022827228.1, and mRNA XP_022814930.1, showing an overall downward trend for XP_022825542.1, XP_022823371, and XP_022814930.1, and an overall upward trend for XP_022827228.1, in both the bioinformatics and qRT-PCR predictions. The bioinformatics analysis was carried out by EBSeqHMM, and qPCR is the experimental methodology. After the cells were infected with the virus, cells at the time points involved in the bioinformatics analysis were collected to extract RNA for reverse transcription, and then qPCR was performed.
The result indicates that among the obtained 19,982 lncRNA, 2,263 showed differences in temporal expression, accounting for 11.33%, while only 1,200 of the obtained 37,122 mRNA showed differences in temporal expression, accounting for 3.23%, implying that many non-coding RNA played a role in the interaction between viruses and hosts.
GO and KEGG functional enrichment outcome
We analyzed GO and KEGG for the differential mRNAs, as shown in Fig. 4. In the GO analysis, the biological process difference of these differential mRNAs is mainly reflected on the biological regulation, and cellular and metabolic processes. There are 188, 177, and 64 genes exhibiting sequential differential expressions in the metabolic process, cellular process, and biological regulation, respectively. We found that the differential gene is mainly in the cellular anatomical entity and intracellular in terms of the cellular component, and in the binding and catalytic activity in terms of the molecular function. There are 159, 94, 209, and 186 genes exhibiting sequential differential expressions in the cellular anatomical entity, intracellular, binding, and catalytic activity, respectively.
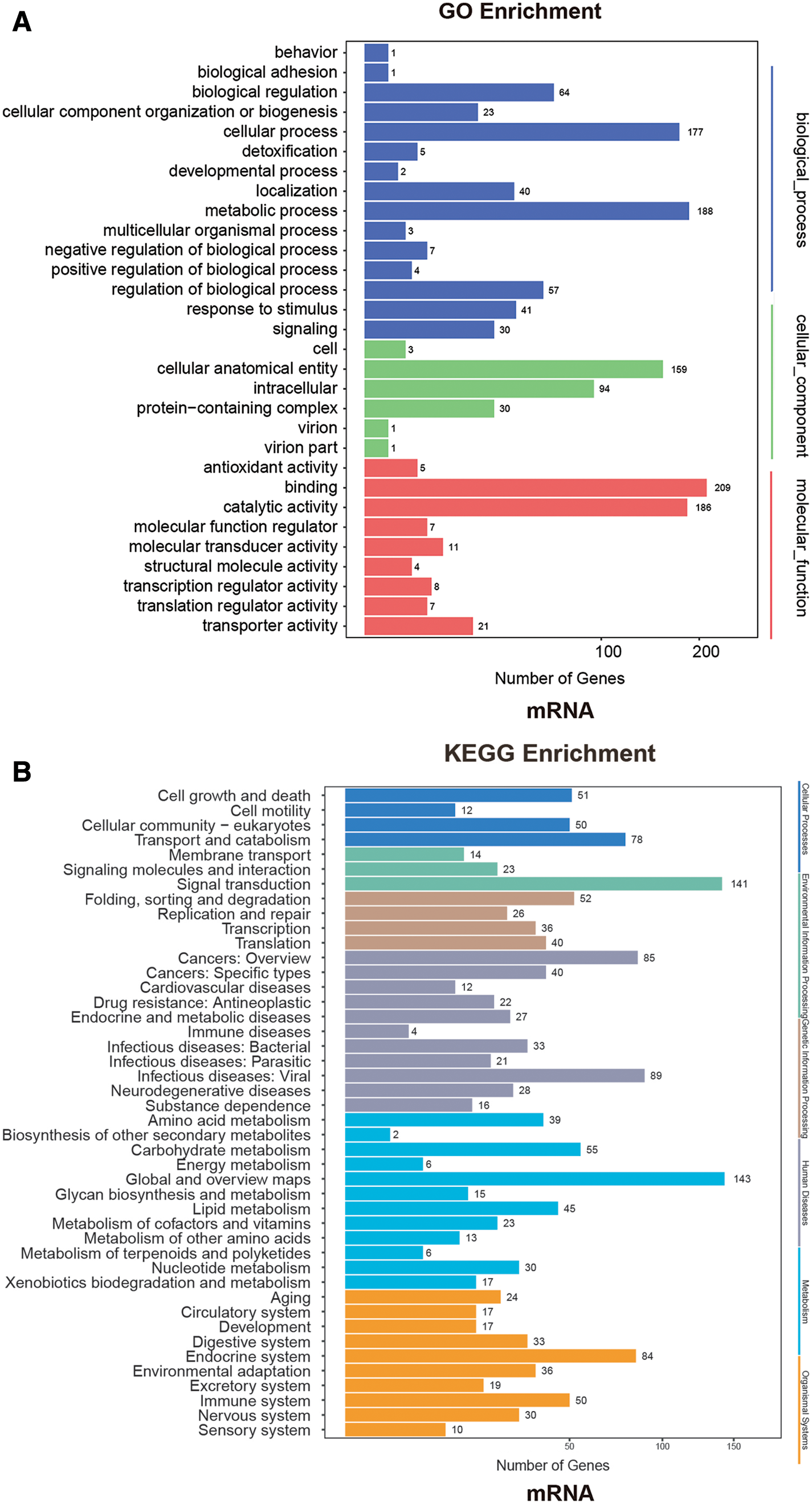
Figure 4: A: GO enrichment analysis of differentially expressed mRNA. B: KEGG enrichment analysis of differentially expressed mRNA.
In the KEGG enrichment analysis, we found that the differential mRNAs were mainly concentrated in global and overview maps, signal transduction, and infectious diseases (viral), etc. There are 143, 141, and 89 genes exhibiting sequential differential expressions in the global and overview maps, signal transduction, and infectious diseases (viral), respectively.
In this study, differentially expressed lncRNAs and mRNAs were obtained through analysis, and these differentially expressed mRNAs may be directly regulated by lncRNAs. Thus, this study analyzed the targets of differential lncRNAs in these differential mRNAs with the help of RNAPlex. As shown in Fig. 5A, the target gene of the lncRNA TU187 (XP_022825542.1) is TU42049. However, due to incomplete annotation of SF9 gene, the function of this gene is not clear. Figs. 5B–5D shows that there are three target genes of the lncRNA TU23998 (XP_022823371.1), one is TU20268, which is a UDP-glycosyltransferase, the second is TU42142, which is an embryonic development factor, and the third is TU50365, which is a predicted homologue with ATPase. Therefore, its target is related to the energy metabolism and growth of the host. However, the target of XP_022827228.1 was not found in the mRNAs with time-series variation characteristics obtained in our analysis this time. This may be because we involved more time points and narrow conditions, and it was only found in the genes with differentially changed genes to find its target.
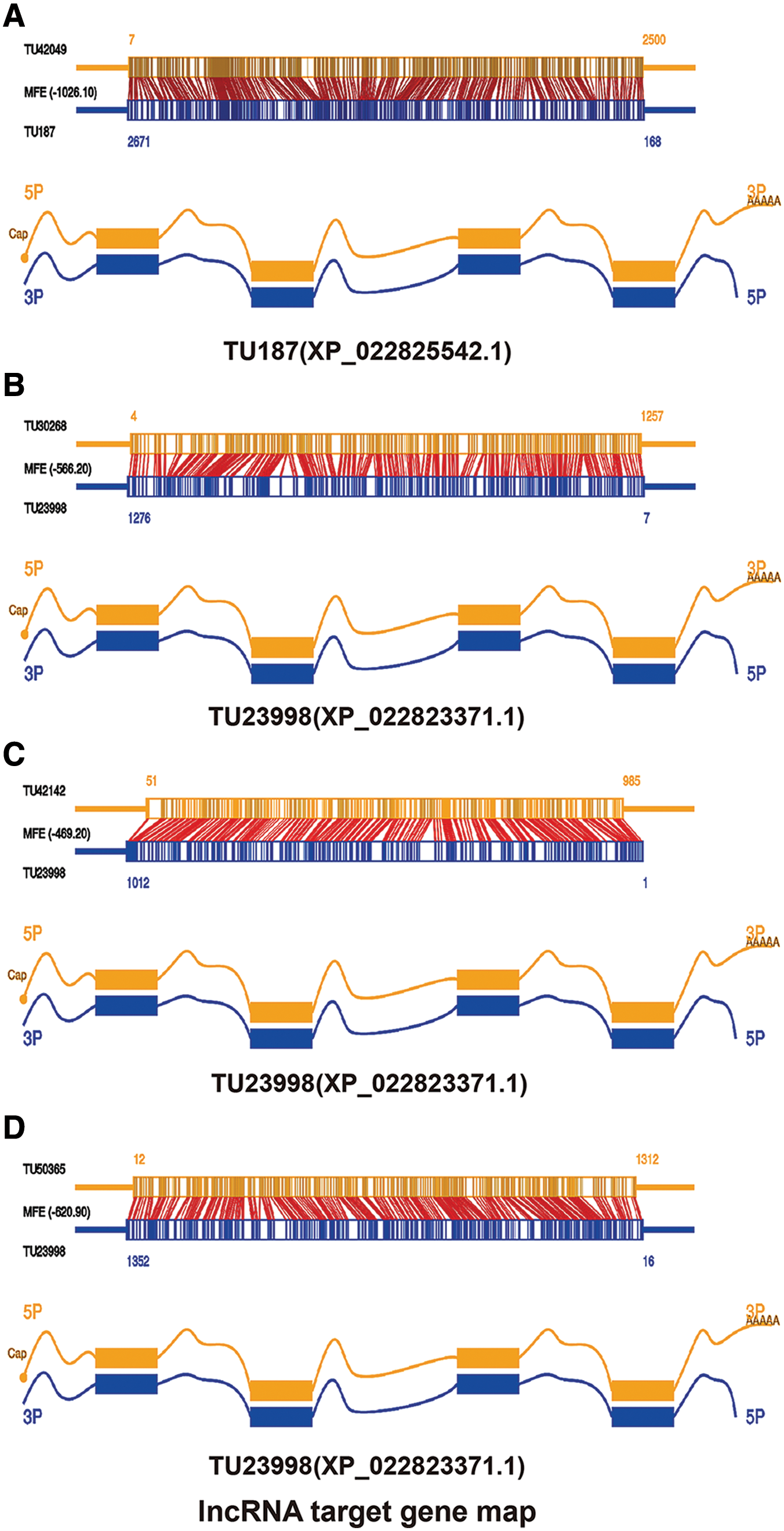
Figure 5: A: TU187 lncRNA and its target gene TU42049. B–D: TU23998 lncRNA and its 3 target genes TU20268, TU42142, and TU50365.
With the help of WGCNA, we analyzed the co-expression of these different lncRNAs and mRNAs. First, the results of WGCNA were analyzed, and the genes of each cluster were enriched by GO and Pathway analysis. Weight > 0.6 is used to filter the interaction relationship pairs, and then the interaction network graph is built up with Cytoscape software, as shown in Fig. 6. We can further see that some important RNAs are differentially expressed, such as TU32028 and TU33799 in mRNA. The former is a ribonucleoside diphosphate reductase subunit M2, which is an important gene associated with DNA synthesis, and the latter is a phosphodiesterase, which plays an important role in signal transduction and other processes. Among lncRNAs, TU29511 and TU42509 were co-expressed by GTP-binding proteins and ethanolamine kinase in the former, and casein kinase ATP-binding proteins in the latter.
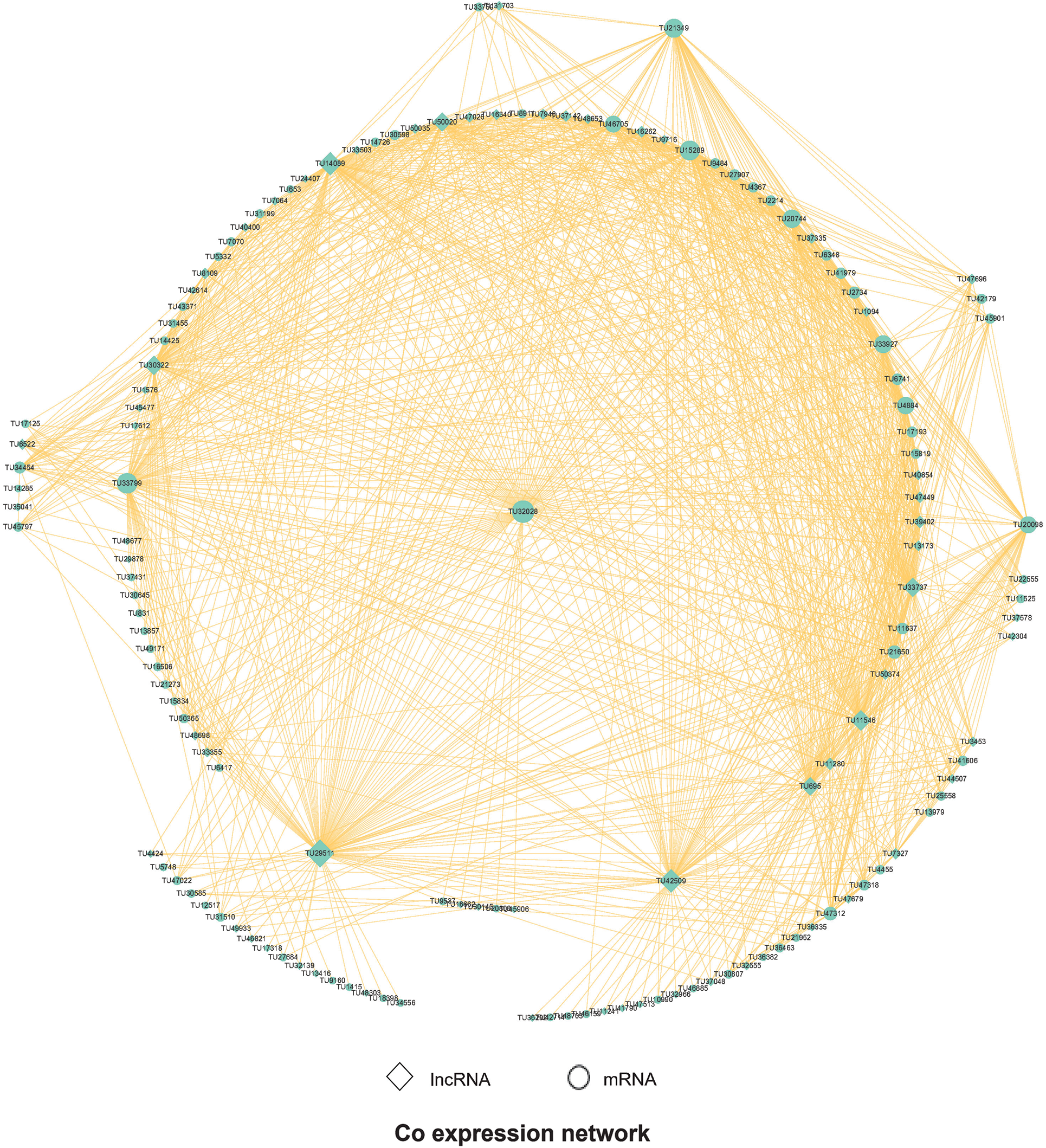
Figure 6: WGCNA Analysis shows the Co expression network. Note that original vector file has been uploaded as a supplementary material.
Spodoptera frugiperda is extremely harmful to crops. In 2016, for the first time, Spodoptera frugiperda was found in Africa and rapidly swept across 44 countries in the south of the Sahara within two years (Goergen et al., 2016; Chapman et al., 2017). According to the Centre for Agriculture and Bioscience International (CABI), a survey of 12 African maize-producing countries shows that Spodoptera frugiperda is responsible for losses of between 8.3 million and 20.6 million tons, or $2.5 billion to $6.2 billion USD, of maize production each year. In addition, more than $13 billion USD of crops are still at risk, causing major damage to agricultural production in Africa (Stokstad, 2017; Early et al., 2018). In 2017, the European Food Safety Authority (EFSA) listed Spodoptera frugiperda as a quarantine pest (Jeger et al., 2017). The Food and Agriculture Organization (FAO) of the United Nations has also issued a global warning, putting it on the world’s top 10 blacklisted plant pests (Food and Agriculture Organization of the United Nations, 2018). The work on prevention and control has not stopped, and biological control, as an important method, has also attracted researchers’ attention. Great progress in preventing and controlling Spodoptera frugiperda has been achieved by utilizing viruses, bacteria, nematode and other pathogenic microorganisms, such as Spodoptera frugiperda multiple nucleopolyhedrovirus (SFMNPV) and Beauveria bassiana et. (Roger et al., 2017; Casmuz et al., 2010). When the pathogenic microorganisms, such as B. bassiana, Metarhizium rileyi and Caenorhabditis elegans, were mixed with insecticides, the prevention and control effect of Spodoptera frugiperda could be significantly improved.
Researchers have successively found the encoded mRNA and function of the AcMNPV gene. AcMNPV-miR-1 is considered to target and down-regulate the expression of viral gene odv-e25, accelerate polyhedra formation, and promote viral infection efficiency in Trichoplusia ni larvae (Zhu et al., 2013, 2016). AcMNPV-miR-3 is located on the opposite strand of the viral gene ac101 coding sequence in the AcMNPV genome, and it can be detected at 6 h post-infection and accumulated to a peak around 12 h post-infection in AcMNPV-infected Sf9 cells. Five viral genes (ac101, ac23, ac25, ac86, and ac98) were verified to be regulated by AcMNPV-miR-3 (Jiao et al., 2019). As a parasite of Spodoptera frugiperda, AcMNPV is essential in the biological control of Spodoptera frugiperda in a safe and efficient way. Thus, in-depth study from gene level is an important step to understand their interaction.
In this study, by sequencing samples collected at different time points after infecting the host cells with AcMNPV, those time-series differentially expressed mRNA and lncRNA are identified. Four types of RNA are randomly selected and verified against experiment, proving that the experiment results is consistent with bioinformatics analysis, and hence that the bioinformatics analysis/methodology employed in this work is valid and reliable. Through GO enrichment analysis, we found that in the aspect of biological process, the differentially expressed genes are concentrated in the biological process, cellular process, metabolic process and regulation of biological process. This finding indicates that after the virus infecting hosts, the cell growth will be impacted. Knudson and Tinsley (1974) observed in their study that after the host is infected with baculovirus, DNA synthesis of the host is quickly inhibited. In the aspect of cellular component, they are mainly in the cellular anatomical entity, indicating that after infection with baculovirus, the host body composition is greatly affected. Volkman et al. (1986) and Monsma et al. (1996) pointed out that the transmission of BV, a particle form of baculovirus, from cell to cell in the host depends on endocytosis, during which the cell membrane, one of the physical components of the host cell, changes. Binding and catalytic activity are found to be the two major aspects of molecular function, indicating that after infection, the activities of many enzymes of the host are affected, thus affecting the normal life process of the host. For instance, it has been reported that the specific structure of AcMNPV’s homologous region 1 (hr1) enables it to bind to a 38 ku (kilo units) host nuclear protein, which may play an important role in enhancer function of hr1 (Kim et al., 2015).
In the KEGG enrichment analysis, it can be found that the time-series differentially expressed mRNA is enriched in global and overview maps, and signal transduction, etc., indicating that by regulating the signal transduction, the virus regulates the host so as to evade immunity and realize its own proliferation. The transformation of the two morphologic particles of the virus may also be closely related to signal transduction. As pointed out by many researchers, signal transduction is closely related to cell proliferation (Wiernas et al., 2010; Seuwen and Pouysségur, 1992). It has also been found in the study to some virus that signal transduction is essential in the growth and activity of the virus (Kieser, 2010; Vainionpää et al., 1991; Gupta and Vayuvegula, 1987; Hijikata et al., 1999). There has been limited research on the signal transduction of virus that infect insets. The results in this study indicates that signal transduction plays an important role in the self- proliferation after the host being infected by AcMNPV. The high enrichment result in this study indicates that the virus regulates the signal transduction of the host after it infects the host, thereby changing the cell function and directing it to the direction that is conducive to its own proliferation.
Based on GO and KEGG analysis, we obtained the differential expression of the host after virus infection at the macroscopic level. Instead of the conventional comparison of the difference before and after infection, we monitored and recorded the dynamically varying difference post infection. Those filtered differential genes could play important roles in the procedure of virus infecting hosts and realizing self-proliferation. For example, the 64 differential genes, enriched in the biological regulation, have high chance to impact the host life activity and hence the virus proliferation.
It has been reported that after AcMNPV infects the host, host DNA synthesis is quickly inhibited (Knudson and Tinsley, 1974), host transcription will be significantly suppressed (Ooi and Miller, 1988), and host protein synthesis begins to decline at 6 h–10 h after infection, and seems to stop completely after 24 h (Carstens et al., 1979; Wood, 1980). At later stages the cell ruptures, releasing polyhedra (Blissard, 1996). However, further studies and understanding are needed on what the status of the regulation network is, which mRNA and lncRNA are involved. For example, researchers already know that BV enters cells through endocytosis (Volkman and Goldsmith, 1985; Habib et al., 1996), but the details and molecular mechanisms of this process need to be further studied. In this study, added by RNAplex analysis, we obtained correlation between differentially expressed lncRNA and mRNA, laying the foundation for future exploration of the regulation network.
Aided by WGCNA, the co-expression of those differential lncRNA and mRNA was analyzed further, and hub RNA are identified, such as TU32028 in mRNA, which is essential in the DNA synthesis, and is part of the p53 signaling pathway, and TU29511 and TU42509 in lncRNA, which are related to GTP-binding protein and ethanolamine kinase etc., and casein kinase ATP binding protein, etc., respectively. Apparently, those differentially expressed RNA occupy an important position in the signal transduction. For the host, it also needs to rely on a variety of signal transduction pathways to meet its own needs.
How the virus uses these transduction pathways, so that the host evolves toward the direction that benefits the virus to achieve its proliferation is worth discussing. Bioinformatics analysis in this study provides a prerequisite for such a discussion.
The molecular interaction between virus and host is very delicate and coordinated. The smooth expression of virus genes is not only related to the virus itself, but also closely related to host factors. The understanding on how this regulation is conducted, and which gene plays the key role become the pre-requisite of uncovering the mechanism of this phenomena.
Although researchers have conducted many studies of AcMNPV and its host, at present the understanding of the regulating mechanism of a series of virus life process in the host cells is relatively limited, including the temporal transcriptional regulation mechanism of host genes during infection. The sequencing and annotation of host’s gene group were started relatively late. In 2014, Kakumani et al. (2014) sequenced the genome of Sf21, which is the separated ovarian cell line of Spodoptera frugiperda, by using the second-generation sequencing technique, which was the first genome data of Sf2 (Kakumani et al., 2014). In 2017, scientists resequenced maize and rice strains of the same ovarian cell line (Sf21), yielding a genome of about 396 Mb (Gouin et al., 2017). In the same year, Nandakumar et al. (2017) also sequenced the genome of another cell line (Sf9) of Spodoptera frugiperda, which obtained a total of 451 Mb of genome data. The assembly result and sequencing quality were better than the former two. In 2019, Liu et al. (2019) sequenced the entire genome of Spodoptera frugiperda that had invaded China and assembled the sequencing results to the chromosome level. The samples were collected from Yunnan and Guangdong provinces in China. This study obtained Spodoptera frugiperda genome size of about 536 Mb, with N50 reaching more than 14 Mb, and more than 80% of the genome data were assembled on the 31 chromosomes. In other words, the gene sequencing on hosts has not been paid sufficient attention until recent years, and annotation work lacks detailed report, which also prevents further in-depth analysis in this study. As the annotation work continues to improve, the data and analysis in this study serve as the first step toward a thorough understanding of the interaction between virus and hosts.
By high-throughput sequencing of RNA from Sf9 cells infected by AcMNPV, we found 1,200 mRNA and 2,263 lncRNA showing different temporal expression. Results from the experimental group are basically identical with those from the bioinformatic analysis. The overall trend is consistent, though there are differences at individual points in time, which may be associated with the variation in the repetition count of the experiment and sequencing analysis, and can be improved by more sequencing in further studies. Through GO and KEGG analysis, we obtained the enrichment information of differential genes, which enabled us to have a certain understanding of the classification of differential genes and the complex interrelationship between genes and metabolites from a macro perspective. This provides the opportunity to further study the genes that play a key role in host-virus interaction and contribute to a comprehensive understanding of the interaction between virus and host. We also analyzed the target genes of the obtained differential lncRNAs in the differential mRNAs by RNAplex. The last but not the least, we analyzed the co-expression of the differential lncRNAs and mRNAs by WGCNA, and identified those “hub” RNAs. This study paves the way for further in-depth analysis and understanding of the interaction between virus and host post AcMNPV infection, and the regulation mechanism of virus involved life processes.
Availability of Data and Materials: The datasets generated during and/or analysed during the current study are available in the NCBI database (PRJNA736178).
Authors’ Contribution: Tiejun Zhao and Mengqiu Chen performed the experiment and analysis. Tiejun Zhao, Riqiang Deng and Xunzhang Wang wrote the manuscript.
Funding Statement: The authors received no specific funding for this study.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
Barr AJ, Manning DR (1997). Agonist-independent activation of Gz by the 5-hydroxytryptamine1A receptor co-expressed in Spodoptera frugiperda cells: Distinguishing inverse agonists from neutral antagonists. Journal of Biological Chemistry 272: 32979–32987. [Google Scholar]
Bertin J, Mendrysa SM, Lacount DJ, Gaur S, Friesen PD (1996). Apoptotic suppression by baculovirus p35 involves cleavage by and inhibition of a virus-induced ced-3/ice-like protease. Journal of Virology 70: 6251–6259. [Google Scholar]
Bilimoria SL, Liu HS, Reinish AJ (1986). Gene expression of baculovirus in semiperssive insect cell lines. In: Fundamental and Applied Aspects of Invertebrate Pathology, pp. 51–54. Wageningen: Foundation of the Fourth International Colloquium of Envertebrate Pathlogy. [Google Scholar]
Blissard GW (1996). Baculovirus-insect cell interactions. Cytotechnology 20: 73–93. [Google Scholar]
Burma S, Mukherjee B, Jain A, Habib S, Hasnain SE (1994). An unusual 30-kDa protein binding to the polyhedrin gene promoter of Autographa californica nuclear polyhedrosis virus. Journal of Biological Chemistry 269: 2750–2757. [Google Scholar]
Carstens EB, Tjia ST, Doerfler W (1979). Infection of Spodoptera frugiperda cells with Autographa californica nuclear polyhedrosis virus I. Synthesis of intracellular proteins after virus infection. Virology 99: 386–398. [Google Scholar]
Casmuz A, Juárez ML, Socías MG, Murúa MG, Prieto S, Medina S, Willink E, Gastaminza G (2010). de los hospederos del gusano cogollero del maíz, Spodoptera frugiperda (Lepidoptera: Noctuidae). Revista De La Sociedad Entomológica Argentina 69: 209–231. [Google Scholar]
Chapman D, Purse BV, Roy HE, Bullock JM (2017). Global trade networks determine the distribution of invasive non-native species. Global Ecology and Biogeography 26: 907–917. [Google Scholar]
Chen S, Zhou Y, Chen Y, Gu J (2018). Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34: 884–890. [Google Scholar]
Cochran MA (1983). Faulkner P. location of homologous DNA sequences interspersed at five regions in the baculovirus AcMNPV genome. Journal of Virology 45: 961–970. [Google Scholar]
Cory JS, Green BM, Paul RK, Hunter-Fujita F (2005). Genotypic and phenotypic diversity of a baculovirus population within an individual insect host. Journal of Invertebrate Pathology 89: 101–111. [Google Scholar]
Cuddapah S, Barski A, Cui K, Schones DE, Wang Z et al. (2009). Native chromatin preparation and Illumina/Solexa library construction. Cold Spring Harb Protocols 2009: 1–7. [Google Scholar]
Early R, Gonzalez-Moreno P, Murphy ST, Day R (2018). Forecasting the global extent of invasion of the cereal pest Spodoptera frugiperda, the fall armyworm. NeoBiota 40: 25–50. [Google Scholar]
Food and Agriculture Organization of the United Nations (2018). Fall Armyworm Likely to Spread from India to Other Parts of Asia with South East Asia and South China Most at Riskhttps://www.fao.org/news/story/en/item/1148819/icode/ [Google Scholar]
Fang M, Nie Y, Theilmann DA (2009). AcMNPV EXON0 (AC141) which is required for the efficient egress of budded virus nucleocapsids interacts with beta-tubulin. Virology 385: 496–504. DOI 10.1016/j.virol.2008.12.023. [Google Scholar] [CrossRef]
Friesen PD, Miller LK (1985). Temporal regulation of baculovirus RNA: Overlapping early and late transcripts. Journal of Virology 54: 392–400. DOI 10.1128/jvi.54.2.392-400.1985. [Google Scholar] [CrossRef]
Friesen PD, Miller LK (1986). The regulation of baculovirus gene expression. Current Topics in Microbiology and Immunology 131: 31–49. DOI 10.1007/978-3-642-71589-1. [Google Scholar] [CrossRef]
Friesen PD, Miller LK (1987). Divergent transcription of early 35- and 94-kilodalton protein genes encoded by the HindIII K genome fragment of the baculovirus Autographa californica nuclear polyhedrosis virus. Journal of Virology 61: 2264–2272. DOI 10.1128/jvi.61.7.2264-2272.1987. [Google Scholar] [CrossRef]
Funk CJ, Consigli RA (1993). Phosphate cycling on the basic protein of Plodia interpunctella granulosis virus. Virology 193: 396–402. [Google Scholar]
Goergen G, Kumar PL, Sankung SB, Togola A, Tamò M et al. (2016). First report of outbreaks of the fall armyworm Spodoptera frugiperda (J E Smith) (Lepidoptera, Noctuidaea new alien invasive pest in West and Central Africa. PLoS One 11: e0165632. [Google Scholar]
Gouin A, Bretaudeau A, Nam K, Gimenez S, Aury JM et al. (2017). Two genomes of highly polyphagous lepidopteran pests (Spodoptera frugiperda, Noctuidae) with different host-plant ranges. Scientific Reports 7: 11816. [Google Scholar]
Granados RR (1978). Early events in the infection of Hiliothis zea midgut cells by a baculovirus. Virology 90: 170–174. [Google Scholar]
Gupta S, Vayuvegula B (1987). Human immunodeficiency virus-associated changes in signal transduction. Journal of Clinical Immunology 7: 486–489. DOI 10.1007/BF00915060. [Google Scholar] [CrossRef]
Habib S, Pandey S, Chatterji U, Burma S, Hasnain SE (1996). Bifunctionality of the AcMNPV Homologous Region Sequence (hr 1Enhancer and ori functions have different sequence requirements. DNA and Cell Biology 15: 737–747. DOI 10.1089/dna.1996.15.737. [Google Scholar] [CrossRef]
Hijikata M, Tsuchihara K, Marusawa H, Shimotohno K (1999). Regulation of cell proliferation by hepatitis C virus. Tanpakushitsu Kakusan Koso Protein Nucleic Acid Enzyme 44: 1543–1551. [Google Scholar]
Horton HM, Burand JP (1993). Saturable attachment sites for polyhedron-derived baculovirus on insect cells and evidence for entry via direct membrane fusion. Journal of Virology 67: 1860–1868. [Google Scholar]
Jeger M, Bragard C, Caffier D, Candresse T, MacLeod A (2017). Pest categorisation of Spodoptera frugiperda. EFSA Journal 15: e04927. [Google Scholar]
Jehle JA, Lange M, Wang H, Hu Z, Wang Y, Hauschild R (2006). Molecular identification and phylogenetic analysis of baculoviruses from Lepidoptera. Virology 346: 180–193. [Google Scholar]
Jiao Y, Wang J, Deng R, Yu X, Wang X (2019). AcMNPV-miR-3 is a miRNA encoded by Autographa californica nucleopolyhedrovirus and regulates the viral infection by targeting ac101. Virus Research, 267: 49–58. [Google Scholar]
Johnson SJ (1987). Migration and the life history strategy of the fall armyworm, Spodoptera frugiperda in the western hemisphere. International Journal of Tropical Insect Science 8: 543–549. [Google Scholar]
Joshi L, Davis TR, Mattu TS, Rudd PM, Dwek RA, Shuler ML, Wood HA (2010). Influence of Baculovirus-host cell interactions on complex N-linked glycosylation of a recombinant human protein. Biotechnology Progress 16: 650–656. [Google Scholar]
Kakumani PK, Malhotra P, Mukherjee SK, Bhatnagar RK (2014). A draft genome assembly of the army worm, Spodoptera frugiperda. Genomics 104: 134–143. [Google Scholar]
Kieser A (2010). Signal transduction by the Epstein-Barr virus oncogene latent membrane protein 1 (LMP1). Signal Transduction 7: 20–33. [Google Scholar]
Kim D, Langmead B, Salzberg SL (2015). HISAT: A fast spliced aligner with low memory requirements. Nature Methods 12: 357–360. [Google Scholar]
Knudson DL, Tinsley TW (1974). Replication of a nuclear polyhedrosis virus in a continuous cell culture of Spodoptera frugiperda: Purification, assay of infectivity, and growth characteristics of the virus. Journal of Virology 14: 934–944. [Google Scholar]
Langfelder P, Horvath S (2008). WGCNA: An R package for weighted correlation network analysis. BMC Bioinformatics 9: 559. [Google Scholar]
Langmead B, Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nature Methods 9: 357–359. DOI 10.1038/nmeth.1923. [Google Scholar] [CrossRef]
Leng N, Li Y, McIntosh BE, Nguyen BK, Duffin B, Tian S, Thomson JA, Dewey CN, Stewart R, Kendziorski C (2015). EBSeq-HMM: A Bayesian approach for identifying gene-expression changes in ordered RNA-seq experiments. Bioinformatics 31: 2614–2622. DOI 10.1093/bioinformatics/btv193. [Google Scholar] [CrossRef]
Li A, Zhang J, Zhou Z (2014). PLEK: A tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinformatics 15: 311. DOI 10.1186/1471-2105-15-311. [Google Scholar] [CrossRef]
Liu H, Lan T, Fang D, Gui F, Liu X (2019). Chromosome level draft genomes of the fall armyworm, Spodoptera frugiperda (Lepidoptera: Noctuidaean alien invasive pest in China. bioRxiv. DOI 10.1101/671560. [Google Scholar] [CrossRef]
Manji GA (2000). Baculovirus and Host Factors Regulating Apoptosis: Mechanistic Clues on Viral Op-IAP Anti-Apoptotic Function. Madison: The University of Wisconsin. [Google Scholar]
Matindoost L, Nielsen L, Reid S (2015). Intracellular trafficking of baculovirus particles: A quantitative study of the HearNPV/HzAM1 cell and AcMNPV/Sf9 cell systems. Viruses 7: 2288–2307. DOI 10.3390/v7052288. [Google Scholar] [CrossRef]
Mccarthy CB, Theilmann DA (2008). AcMNPV ac143 (odv-e18) is essential for mediating budded virus production and is the 30th baculovirus core gene. Virology 375: 277–291. DOI 10.1016/j.virol.2008.01.039. [Google Scholar] [CrossRef]
Miller LK, Lu A (1997). The Molecular Basis of Baculovirus Host Range. Springer US. https://link.springer.com/chapter/10.1007%2F978-1-4899-1834-5_9. [Google Scholar]
Miller LK (1997). Baculovirus interaction with host apoptotic pathways. Journal of Cellular Physiology 173: 178–182. [Google Scholar]
Monsma SA, Oomens AG, Blissard GW (1996). The GP64 envelope fusion protein is an essential baculovirus protein required for cell-to-cell transmission of infection. Journal of Virology 70: 4607–4616. [Google Scholar]
Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, Gerstein M, Snyder M (2008). The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 320: 1344–1349. [Google Scholar]
Nandakumar S, Ma H, Khan AS (2017). Whole-genome sequence of the Spodoptera frugiperda Sf9 insect cell line. Genome Announcements 5: e00829-17. [Google Scholar]
Niknafs YS, Pandian B, Iyer HK, Chinnaiyan AM, Iyer MK (2017). TACO produces robust multisample transcriptome assemblies from RNA-seq. Nature Methods 14: 68–70. [Google Scholar]
Nissen MS, Friesen PD (1989). Molecular analysis of the transcriptional regulatory region of an early baculovirus gene. Journal of Virology 63: 493–503. DOI 10.1128/jvi.63.2.493-503.1989. [Google Scholar] [CrossRef]
Ooi BG, Miller LK (1988). Regulation of Host RNA levels during baculovirus infection. Virology 166: 515–523. DOI 10.1016/0042-6822(88)90522-3. [Google Scholar] [CrossRef]
Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nature Methods 14: 417–419. DOI 10.1038/nmeth.4197. [Google Scholar] [CrossRef]
Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nature Biotechnology 33: 290–295. DOI 10.1038/nbt.3122. [Google Scholar] [CrossRef]
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glockner FO (2013). The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Research 41: 590–596. DOI 10.1093/nar/gks1219. [Google Scholar] [CrossRef]
Roger D, Phil A, Melanie B, Tim B, Victor C et al. (2017). Fall armyworm: Impacts and implications for Africa. Outlooks on Pest Management 28: 196–201. DOI 10.1564/v28_oct_02. [Google Scholar] [CrossRef]
Rohrmann GF (2011). Baculovirus Molecular Biology. Bethesda, MD: National Center for Biotechnology Information. [Google Scholar]
Rohrmann GF (2013). Baculovirus Molecular Biology. Bethesda, MD: National Center for Biotechnology Information. [Google Scholar]
Rougemont J, Amzallag A, Iseli C, Farinelli L, Xenarios I, Naef F (2008). Probabilistic base calling of Solexa sequencing data. BMC Bioinformatics 9: 1–12. DOI 10.1186/1471-2105-9-431. [Google Scholar] [CrossRef]
Seuwen K, Pouysségur J (1992). Protein-controlled signal transduction pathways and the regulation of cell proliferation. Advances in Cancer Research 58: 75–94. DOI 10.1016/S0065-230X(08)60291-2. [Google Scholar] [CrossRef]
Slack J, Arif BM (2007). The baculoviruses occlusion-derived virus: Virion structure and function. Advances in Virus Research 69: 99–165. DOI 10.1016/S0065-3527(06)69003-9. [Google Scholar] [CrossRef]
Sparks AN (1979). A review of the biology of the fall Armyworm. The Florida Entomologist 62: 82–87. DOI 10.2307/3494083. [Google Scholar] [CrossRef]
Stokstad E (2017). New crop pest takes Africa at lightning speed. Science 356: 473–474. [Google Scholar]
Summers MC (1971). Electron microscopic observations on granulosis virus entry, uncoating and replication processes during infection of the midgut cells of Trichoplusia ni. Journal of Ultrastructure Research 35: 606–625. [Google Scholar]
Tafer H, Hofacker IL (2008). RNAplex: A fast tool for RNA-RNA interaction search. Bioinformatics 24: 2657–2663. [Google Scholar]
Tjia ST, Carstens EB, Doerfler W (1979). Infection of Spodoptera frugiperda cells with Autographa californica nuclear polyhedrosis virus: II. The viral DNA and the kinetics of its replication. Virology 99: 399–409. [Google Scholar]
Vago C, Aizawa K, Ignoffo C, Martignoni ME, Tinsley TW (1974). Editorial: Present status of the nomenclature and classification of invertebrate viruses. Journal of Invertebrate Pathology 23: 133–134. [Google Scholar]
Vainionpää R, Hyypiä T, Akerman KE (1991). Early signal transduction in measles virus-infected lymphocytes is unaltered, but second messengers activate virus replication. Journal of Virology 65: 6743–6748. [Google Scholar]
Volkman LE, Goldsmith PA, Hess RT (1986). Alternate pathway of entry of budded Autographa californica nuclear polyhedrosis virus: Fusion at the plasma membrane. Virology 148: 288–297. [Google Scholar]
Volkman LE, Goldsmith PA (1985). Mechanism of neutralization of budded Autographa californica nuclear polyhedrosis virus by a monoclonal antibody: Inhibition of entry by adsorptive endocytosis. Virology 143: 185–195. [Google Scholar]
Wiernas TK, Davis TL, Griffin BW, Sharif NA (2010). Effects of bradykinin on signal transduction, cell proliferation, and cytokine, prostaglandin E2 and collagenase-1 release from human corneal epithelial cells. British Journal of Pharmacology 123: 1127–1137. [Google Scholar]
Williams GV, Faulkner P (1997). Cytological changes and viral morphogenesis during baculovirus infection. Springer US. https://link.springer.com/chapter/10.1007%2F978-1-4899-1834-5_4. [Google Scholar]
Wood HA (1980). Autographa californica nuclear polyhedrosis virus-induced proteins in tissue culture. Virology 102: 21–27. DOI 10.1016/0042-6822(80)90066-5. [Google Scholar] [CrossRef]
Wu K, Shirk PD, Taylor CE, Furlong RB, Shirk BD, Pinheiro DH, Siegfried BD, He ZY (2018). CRISPR/Cas9 mediated knockout of the abdominal–A homeotic gene in fall armyworm moth (Spodoptera frugiperda). PLoS One 13: 1–16. [Google Scholar]
Yin J, Hu ZP, Song DX, Zhong J (2008). Insertional disruption of AcMNPV p95 gene did not affect virus replication in Sf9 cells. Journal of Fudan University (Natural Science) 47: 302–305. [Google Scholar]
Zhang B, Liang A, Fu Y (2019). Ac34 protein of AcMNPV promoted progeny virus production and induced the apoptosis in host Sf9 cells. Biotechnology Letters 41: 147–158. DOI 10.1007/s10529-018-2623-3. [Google Scholar] [CrossRef]
Zhang P, Yang B, Dai XJ, Pang Y, Zhong J, Su DM (2002). Apoptosis of Spodoptera litura cells induced by AcMNPVie-1 gene. Acta Biochimica Et Biophysica Sinica 34: 707–711. [Google Scholar]
Zhu M, Wang J, Deng R, Wang X (2016). Functional regulation of an Autographa californica nucleopolyhedrovirus-encoded MicroRNA, AcMNPV-miR-1, in baculovirus replication. Journal of Virology 90: 6526–6537. DOI 10.1128/JVI.00165-16. [Google Scholar] [CrossRef]
Zhu M, Wang J, Deng R, Xiong P, Liang H, Wang X (2013). A microRNA encoded by Autographa californica nucleopolyhedrovirus regulates expression of viral gene ODV-E25. Journal of Virology 87: 13029–13034. DOI 10.1128/JVI.02112-13. [Google Scholar] [CrossRef]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |