DOI:10.32604/biocell.2022.019223
| BIOCELL DOI:10.32604/biocell.2022.019223 | |
| Article |
Genome-wide analysis of microRNAs induced and regulated by colchicine in mulberry
1Key Laboratory of Silkworm and Mulberry Genetic Improvement, Ministry of Agriculture, School of Biology and Technology, Jiangsu University of Science and Technology, Zhenjiang, 212100, China
2Sericultural Research Institute, Nanning, 530007, China
*Address correspondence to: Rongjun Fang, frj1971@163.com; Weiguo Zhao, wgzsri@126.com
#These authors contributed equally to this work
Received: 10 September 2021; Accepted: 25 November 2021
Abstract: A variety of plants were colchicine treated to double their chromosome number. Chromosomes are genetic carriers that determine the individual traits of organisms. The doubling of chromosomes will lead to modifications in plant morphology, physiology and genetics. To determine the response of mulberry trees induced by colchicine, using mulberry variety Yu-711 leaves as research materials, two small RNA libraries (control and experimental groups) were constructed. It was found that 45 known miRNA genes and 78 predicted novel miRNA genes in the sequence results. A comparison of data between the control group and the experimental group revealed 37 differentially expressed miRNA genes, 19 genes of which were up-regulated and 18 genes of which were down-regulated. Eight miRNAs were selected from 37 differentially expressed miRNA genes. These miRNAs were verified by quantitative real-time PCR, and the expression levels of these miRNAs were found to be consistent with those obtained by sequencing, which proved the accuracy of sequencing results. The PsRNATarget software was used to predict the target genes of 8 miRNAs. The Gene Ontology and Kyoto Encycopedia of Genes and Genomes analysis were used to collect the functions of target genes and confirm that target genes are mainly involved in biological processes, cell components and molecular functions. novel-77 miRNA was selected from 37 differentially expressed miRNAs. What will serve as a fundamental data in understanding mulberry miRNAs function in molecular biology research and further provides the molecular mechanism of mulberry gene regulation as induced by colchicine is that the prediction of novel_77 precursor sequence and the target gene (XM_010104258.2), as well as the secondary structure analysis.
Keywords: Chromosomes; Novel_77; qRT-PCR; Target genes; Colchicine
Mulberry is a perennial deciduous woody plant with high economic value, ecological value and medicinal value. As the only natural feed for silkworm, improving the yield and quality of mulberry leaves will directly affect the economic benefits of sericulture and silk industry. The traditional mulberry cross breeding cycle is long and targeting is low, so the breeding efficiency of new varieties is low. However, mulberry polyploid breeding has the advantages of short life, obvious effect and clear aim, and has good traits and economic yield, which has aroused great attention of mulberry breeders.
Colchicine is frequently used to generate polyploid plants and acts as a mitotic toxin, causing numerous mutagenic effects in plants (El-Nashar and Ammar, 2016). Colchicine function as microtubules in chromosome segregation and induces polyploidy by preventing chromosomes’ segregation during meiosis, resulting in half of the gametes (sex cells) containing double the chromosome number than usual (Manzoor et al., 2019). However, the other half of the gametes do not contain any chromosomes and yields embryos with doubled chromosome numbers (Manzoor et al., 2019). Colchicine induces mutation in plants and also double plant chromosomes. Plants mutant derived from colchicine are known as colchi-mutants (Ari et al., 2015). Varying concentrations of colchicine used to induce polyploidy in many plant species have been reported by several studies. In the case of campion (Lychnic senno), a lower concentration of 0.00001% has been used in inducing polyploidy (Castro et al., 2018). On the other hand, in ‘Maule’s quince (Chaenomeles japonica), a higher concentration of 1.5% has been successfully used to induce polyploidy (Castro et al., 2018). Since colchicine has a low affinity for tubulin in plant cells, increasing colchicine concentration can improve the binding force between colchicine and tubulin (Eng and Ho, 2019).
MicroRNAs (miRNAs) are a class of small, noncoding, single-stranded RNAs (Hu et al., 2021), which play important roles in regulating gene expression during plant development, signal transduction, and stress responses. The miRNA biogenesis is a multi-step process including transcription, processing, modification, and RISC loading. RNA polymerase II (Pol II) is responsible for transcribing microRNA (MIR) genes to produce miRNA precursors in plants and animals. MIR genes are transcribed into a pri-miRNA with a cap and a poly (A) tail in plants, which is processed into the stem-loop precursor, pre-miRNA, by a DCL protein. The pre-miRNA is further processed by DCL1 into a duplex of miRNA and miRNA*. The miRNA-miRNA* duplex is methylated on the 2’OH of the 3’ terminal nucleotides by HEN1 and then the plant miRNA is loaded into the ARGONAUTE1 (AGO1) complex leading to the cleavage of the target mRNA. It is well known that miRNAs play essential roles in plant growth and development and also control plant responses to biotic and abiotic stresses by regulating targeted gene expression (Zhou et al., 2019). It has been reported that the over-expression of miR156 in maize and Arabidopsis thaliana resulted in a longer juvenile period for these plants (Zeng et al., 2019). And miR477 may involve grape fruitripening by regulating its target genes (Jin et al., 2022). In addition, miR172 negatively regulates the cell fate specification in flower development of A. thaliana (Jia et al., 2014). Interestingly, in arabidopsis, miR160-targeted ARF10 expression is a positive factor for initiation and formation of callus and together with the miR160-targeted ARF16 expression controls the differentiation of distal columella stem cells and the formation of root cap in the primary root tip (Uwe et al., 2019). Plant microRNAs also play important roles in plant growth and development including leaf morphology and polarity, organ development, cell differentiation and proliferation, programmed cell death (Krishnatreya et al., 2020).
MicroRNAs negatively regulate target genes and inhibit their protein translation to participate in the regulation of the post-transcriptional expression of genes (Liu et al., 2021). As a negative regulator of gene expression, miRNAs in plants play an important role in response to biological and abiotic stresses. MicroRNAs mediate gene silencing by guiding argonaute (Ago) protein to the target site of 3’UTR of mRNA, which plays an important role in gene regulation at the post-transcriptional level (Chen et al., 2020). Since miRNA is usually located in upstream of regulation, its expression level affects the corresponding target genes, which is an important factor of plant polyploid phenotype changes (Ghani et al., 2014). The miRNAs regulate gene expression and induce the phenotype variation, which may play an important role in the occurrence of heterosis in the allotetraploid (Ghani et al., 2014).
At present, most of the studies on the artificial induction of mulberry chromosome doubling take the seedling germination of hybrid mulberry seeds or the organ parts of cultivated mulberry trees as materials, but this method is complicated, time-consuming and the success rate is not high. Mulberry polyploid studies mainly focus on the polyploid effect, but few studies on molecular aspects. In view of miRNAs play an important role in the regulation of abiotic stress in mulberry, it has not been characterized clearly how colchicine treatment regulates the expression of miRNA in plants and how to regulate the expression of downstream genes under abiotic stress.
The aim of this study was to identify and characterize microRNAs (miRNAs) in mulberry leaves induced by colchicine through small RNA sequencing. Expression levels of the selected miRNAs and targets were validated by quantitative real-time PCR (qRT-PCR) analysis. The experiment can not only provide theoretical basis and experimental materials for exploring excellent gene resources of mulberry and polyploid research of mulberry, but also enrich mulberry germplasm resources and further promote the development of sericulture industry. The completion of high-throughput and deep sequencing of the mulberry genome and the construction of a small RNA library laid a database foundation for identifying mulberry miRNA functions in the study of mulberry molecular biology. From total, 45 conserved miRNAs and 78 putative novel miRNAs were differentially expressed in leaves, respectively under colchicine stress. A total of 37 differentially expressed miRNAs identified with 19 up-regulated and 18 down-regulated. Subsequently, eight differentially expressed miRNAs were selected and verified by PCR and quantitative real-time PCR (qRT-PCR). Understanding a new miRNA precursor sequence and its secondary structure together with the target gene will provide a necessary foundation for identifying mulberry miRNA function in molecular biology research and further provided the molecular mechanism of mulberry induced regulation by colchicine.
Plant materials and colchicine induction treatment
Grafted Morus alba variety Yu-711 was used in this study, which was provided by the Institute of Sericulture, Chinese Academy of Agricultural Sciences, Zhenjiang, Jiangsu Province, China. Since high yield, quality and hereditary stability hybrid of the mulberry variety Yu-711, it was usually used on what studies on colchicine induced chromosome doubling in plants. We took mulberry leaves as sequencing material due to the root variety of mulberry variety Yu-711 is uncertain. The growing point of the mulberry seedlings was treated with 0.2% colchicine solution. The experiment was arranged with colchicine treatment as the main plot with 3 replicates. The experimental group was treated with colchicine solution while the control group was treated with water respectively. The leaf samples were harvested after 5, 10 and 15 day, and immediately frozen in liquid nitrogen, then stored at −80°C for following experiments. The reason why we choosed three time points (5, 10, 15 d) as the sampling was that three time points of the experiment were determined by the symptoms of mulberry stress. Mulberry trees showed significant changes in symptoms at these three time points. Flow cytometry (FCM) was used to estimate ploidy level of the mulberry after colchicine treatments (Dolezel and Bartos, 2005). The result of FCM was provided by Kunming Institute of Botany, Chinese Academy of Sciences.
Small RNA library construction and sequencing
Total RNA was isolated from colchicine-free and colchicine-treated leaves of mulberry using RNAiso Plus reagent (Takara, China) according to the manufacturer’s protocols. The mulberry samples were sent to Beijing Nuovo Zhiyuan Technology Co., Ltd. (China) for the small RNA sequencing. The Small RNA library were prepared directly from RNA samples which passed the quality and integrity testing using Small RNA Library Pre Kit. Since the 5’-small RNA has a complete phosphate group and the 3’-small RNA has a hydroxyl group, the total RNA is used as the starting sample and the linker is directly added to both ends of the small RNA. The cDNA was obtained by reverse transcription. The target cDNA fragments which were amplified by the polymerase chain reaction were separated by PAGE gel to eliminate the primer-dimer and other byproducts (Shangguan et al., 2020). Cutting the gel and recovering the target cDNA fragment to build the small RNA libraries which were used directly for high-throughput sequencing. Finally, the sequencing process was performed by using Illumina HiSeq 2000 technology following the manufacturer’s instructions.
Analysis of small RNA sequencing data
The clean small RNA reads were obtained after the removal of adaptor impurities and low-quality reads. The sRNA which the length from 18 nt to 30 nt was screened and then was further compared with sequences that the range was specified in the miRbase (http://www.mirbase.org/) to identify conserved miRNAs by the mirdeep2 software (Friedlnder et al., 2012). The characteristics of hairpin structure of miRNA precursor can be used to predict novel miRNA. The available software miREvo (Wen et al., 2012) and mirdeep2 (Friedlnder et al., 2012) were integrated to predict novel miRNA through exploring the secondary structure, the Dicer cleavage site and the minimum free energy of the small RNA tags unannotated in the former steps. During the development of miRNAs from precursors to mature bodies, the process is completed by Dicer digestion. Those with typical stem-loop structure were regarded as novel miRNAs.
Differential expression analysis of miRNAs
The input data of miRNA differential expression is readcount data obtained from miRNA expression level analysis. The expression level of the known and new miRNA in each sample was measured, and the expression level was normalized with TPM (Qu et al., 2021). To compare the miRNA abundance among the three sets of libraries and identify the differentially expressed miRNAs based on the number of genomic tags in each sample, we used TPM to normalize the expression level of small RNAs, since this kind of treatment can avoid the effect of quantitative accuracy of different sequencing amounts. Deseq2 (Love et al., 2014) based on negative binomial distribution was used for analysis. The overall distribution of differential miRNAs can be inferred from the volcano map. The differential miRNAs are screened by evaluating the difference multiple (foldchange) and the corrected significant level (Padj/Qvalue). And the default screening conditions for differential miRNA are: padj < 0.05. The blue dot indicates the miRNA with no significant difference, the red dot indicates the significantly up-regulated miRNA, and the green dot indicates the significantly down-regulated miRNA in the volcano maps of the differentially expressed miRNAs.
Prediction of miRNA target genes
The putative target genes of conserved and novel miRNAs were predicted using online software psRNATarget (http://plantgrn.noble.org/psRNATarget/) with default parameters. Then all predicted target genes of differently expressed miRNAs were mapped to Gene Ontology (GO, http://www.geneontology) terms. Moreover, the Genomes (KEGG, http://www.genome.jp/kegg/database) pathway enrichment analysis was performed to identify the involving biochemical metabolic pathways and signal transduction pathways of differentially expressed miRNA target genes.
qRT-PCR analysis was performed to confirm the expression level of selected miRNAs. Total RNAs from colchicine-free and colchicine-treated leaves of mulberry were extracted using RNAiso Plus reagent (Takara, China) following to the manufacturer’s protocols. The 6–8 bases which at the 3’reverse complement of the target miRNA were added to the 3’ end of the general stem-loop structure sequence to design the specific stem-loop reverse transcription primer of miRNAs. The specific stem-loop reverse transcription primer of miRNA was used to generate cDNA by reverse transcription using M-MLV Reverse Transcriptase kit (Takara, China) according manufacturer’s instructions. The procedures of qRT-PCR were performed on ABI Quant Studio 6 Flex System (Applied Biosystems, USA) using the SYBR qPCR SuperMix Plus (Novoprotein, China) and β-actin (Li et al., 2012) was used as the internal reference. Each 20 μL PCR contained 2 μL cDNA, 10 μL qPCR mix, 1 μL forward primer, 1 μL universal reverse primer and 6 μL ddH2O. The thermal cycling parameters were as follows: 95°C for 1 min, 40 cycles of denaturing at 95°C for 20 s and annealing extension at 60°C for 1 min, followed by melting curve analysis. The relative expression level of the target gene was calculated using the quantitative method (2-ΔΔCt)(Schmittgen and Livak, 2008).
Ploidy analysis of the mulberry
In this study, colchicine was used to induce mulberry seedlings to produce tetraploid mulberry. Results of the ploidy of mulberry are provided by Kunming Institute of Botany, Chinese Academy of Sciences. The result of flow cytometry showed that diploid mulberry was used in the control group and tetraploid mulberry was used in the experimental group (Table 1).

Sequencing information of small RNA library
A total of 14215322 and 21156195 raw reads data was obtained from both the control and experimental groups, respectively. To ensure the quality data is used for further analysis, raw reads were processed to remove low-quality reads, N bases (N means that the base information that cannot be determined) greater than 10% of reads, reads contaminated with 5’ linker, reads without 3’ linker sequence and insert, trim off 3’ linker sequence, and removal of polyA/T/G/C reads. The final clean reads and its proportion to the total number of raw reads were shown (Table 2). The sequence length distribution of small RNAs was collected (Fig. 1) and small RNAs within a certain length range was selected for subsequent analysis. The type and quantity of small RNAs were shown in Table 3.

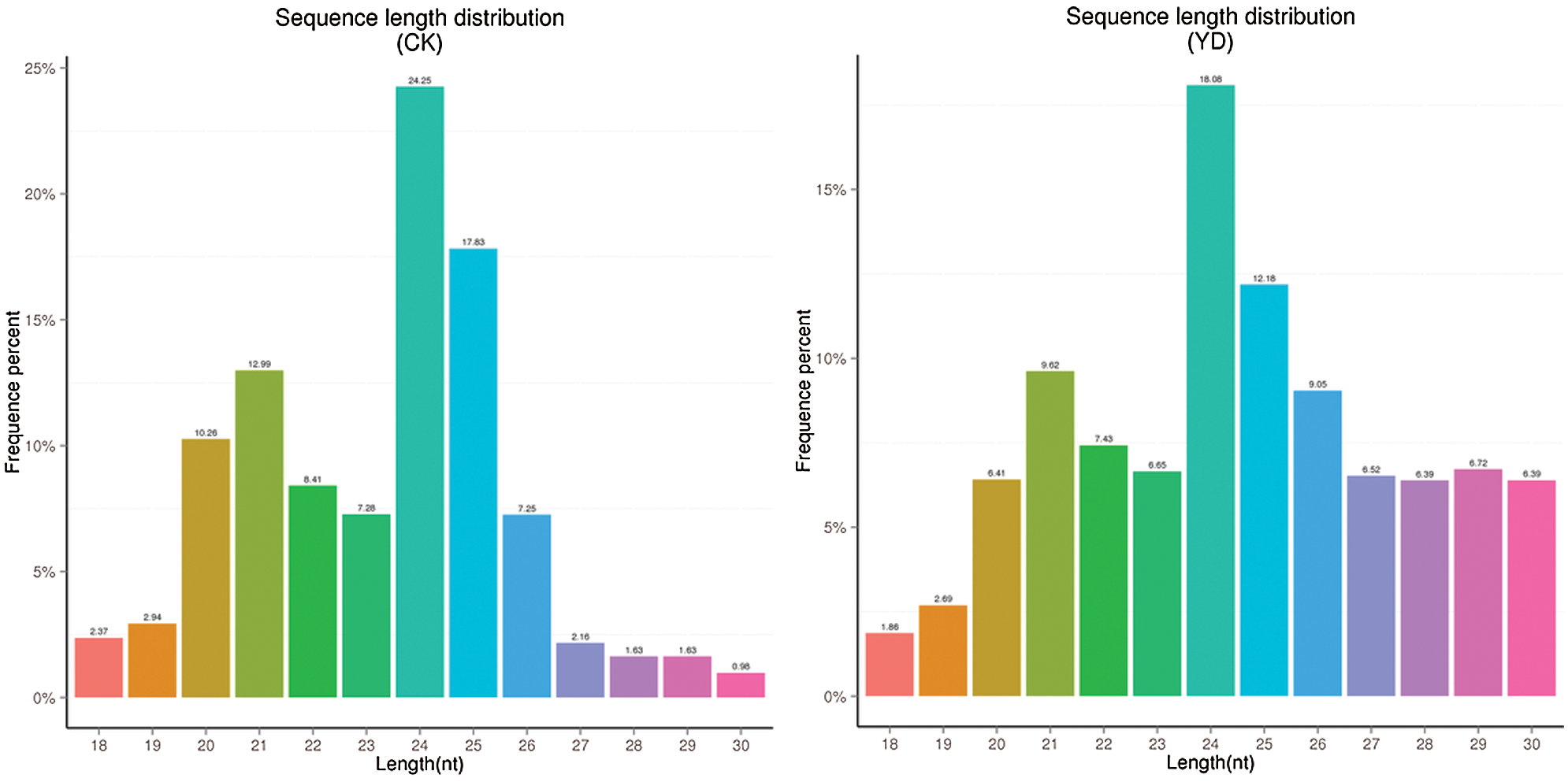
Figure 1: Size distribution of mulberry small RNA in the two small RNA libraries.

Expression profiles analysis of miRNAs
The development of miRNA from precursor to mature is completed by Dier digestion. The restriction site’s specificity makes the mature body sequence’s first base have a strong bias. To study the tetraploid of mulberry induced with colchicine, the length of the conserved miRNA was compared to the specified sRNA range sequence in miRbase. This was carried out to obtain the sequence length, the number of occurrences, and secondary structure of the known miRNA on each sample match. Further analysis on miRNA’s secondary structure, the restriction site, and energy characteristics was performed in predicting the novel miRNA in the sample. The two sRNA libraries, the comparison of known miRNA in each sample, and the predicted novel miRNA are shown in Table 4. A total of 45 known miRNAs and 45 precursor hairpin structures were identified in the sRNA library. Interesting, the library produced 78 novel miRNAs and 95 new hairpin structures (Table 4).

A total of 37 differential miRNAs were obtained in tetraploid mulberry trees induced by colchicine compared with the control group, among which 19 were up-regulated, and 18 were down-regulated. The expression level of each miRNA could be seen from the cluster analysis (Fig. 2). The red color indicated up-regulated expression, and the blue indicated down-regulated expression. It can be seen that the expression of tetraploid samples induced by colchicine exhibited both up-regulated down-regulation pattern when compared to the control group.

Figure 2: Cluster analysis of differentially expressed sRNA.
Enrichment analysis of miRNA target genes
The psRNATarget online software (http://plantgrn.noble.org/psRNATarget/) was used to predict miRNA target genes by using the default setting parameters). After obtaining the differentially expressed miRNAs between the groups, based on the corresponding relationship between the miRNAs and their targets, gene ontology (Young et al., 2010) and KEGG (Mao et al., 2005) enrichment analysis were performed on all target genes in each group of miRNAs. The target genes were selected according to the involvement in the pathway enrichment analysis. The involving pathway of the candidate target gene was identified through the gene ontology. These predicted target genes are mainly related to biological process involving oxidation-reduction process, cellular components involving nucleus and molecular function involving copper ions binding (Fig. 3).

Figure 3: Enriched GO Terms (YD vs. CK).
In living organisms, different genes perform biological functions together. The most important biochemical and signal transduction pathways involved in candidate target genes can be determined through significant enrichment of pathways. The KEGG candidate target gene concentration scatters plot is a graphical representation of the KEGG concentration analysis results. The enrichment in KEGG is measured by the enrichment factor, Q-value ranges (0~1), and the number of genes enriched by this pathway. The enrichment factor is the ratio of the number of genes located at the entry of the pathway to the total number of genes located at the pathway’s entry among all the annotated genes. There are the first top 20 enrichment pathways entries (Fig. 4). The analysis reveals that the genes were mainly enriched in ribosome, purine metabolism, biosynthesis of amino acid, homologous recombination, and plant hormone signal transduction (Fig. 5).
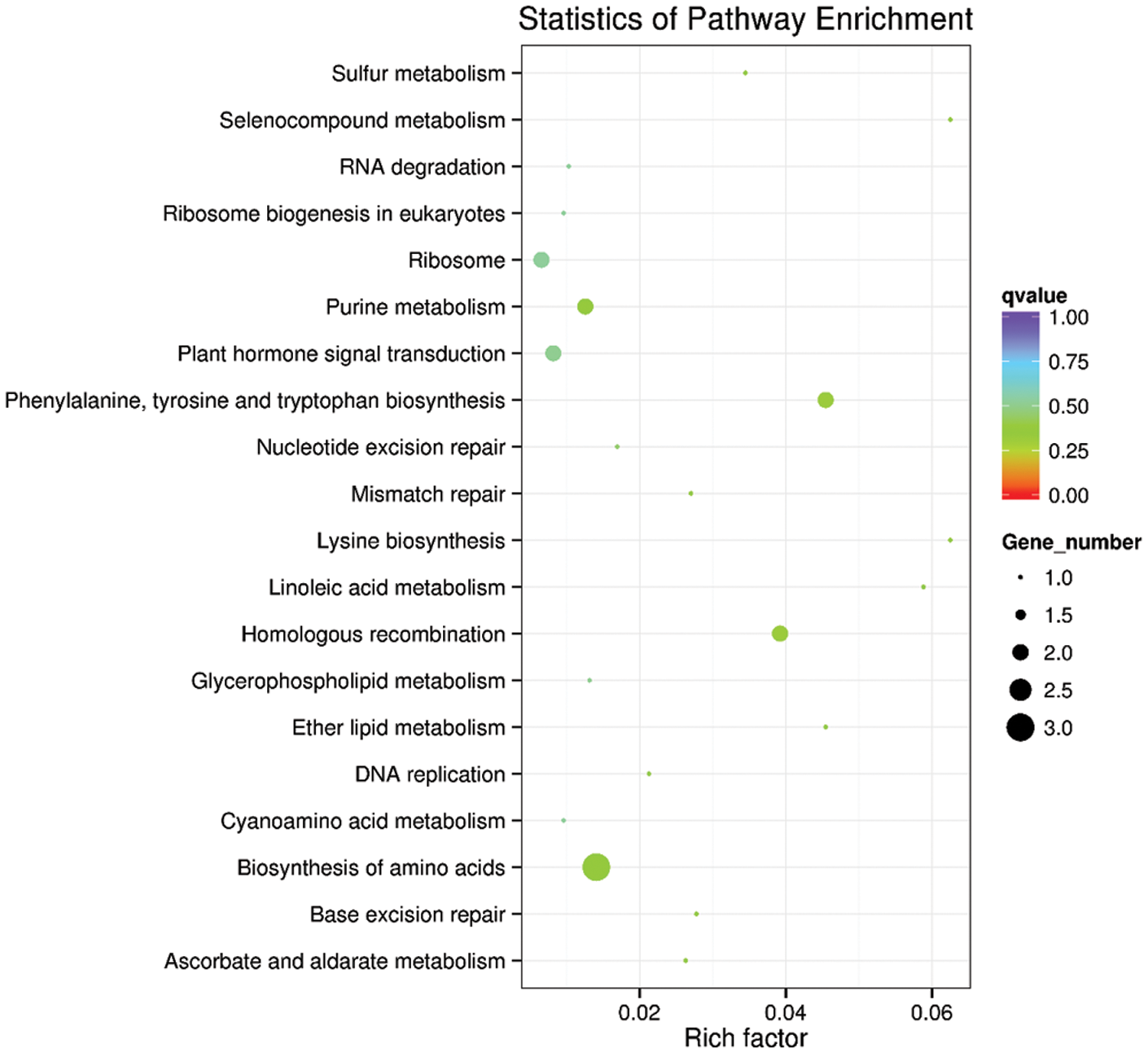
Figure 4: KEGG rich distribution map of the predicted target gene.

Figure 5: The relative expression level of miRNA after colchicine induction in mulberry.
The abscissa is the GO term at the next level of the three GO categories, and the ordinate is the number of candidate target genes annotated to the term (including the sub-terms of the term). The three different classifications represent the three basic classifications of Go terms (from left to right, biological processes, cellular components, and molecular functions).
The vertical axis represents the pathway, the horizontal axis represents the Rich factor, the size of the dots represents the number of candidate target genes in the pathway, and the color of the dots corresponds to different Q-value ranges.
qRT-PCR validation of miRNA expression
To identify miRNAs regulated by colchicine induction in mulberry leaves, the Dseq R software package (Likun et al., 2010; Love et al., 2014) was used to analyze the differential expression of miRNAs in the two groups of mulberry leaves. The p-values were adjusted using the Benjamini and Hochberg method. The corrected p-value of 0.05 was used as the default threshold for significant differential expression. A total of 37 differentially expressed miRNAs were obtained, of which 19 were up-regulated, and 18 were down-regulated. To verify the sequencing results, we designed special stem-loop qRT-PCR primers (Table 5) and selected eight (8) differentially expressed miRNAs for qRT-PCR (Fig. 5) using β-actin as the reference gene. The qRT-PCR results (Fig. 5) showed that the expression levels of all eight miRNAs selected could be detected in the mulberry leaf samples, and the up and down regulation trend of the verified miRNAs was consistent with the sequencing data, which proved the accuracy of the sequencing data.

Prediction of novel_77 miRNA and its target gene
According to the analysis of small RNA sequencing results, the novel-77 miRNA was selected for further study with significant differential expression in mulberry after colchicine induction. The mature sequence of the novel_77 (5’-UUGACAGAAGAGAGUGAGCAC-3’) was compared with the mulberry genome database (http://morus.swu.edu.cn/morusdb/) to find the location of novel_77 in the genome. Firstly, selecting the sequences that both before the 5’ end and after the 3’ end of the mature body sequence about 200 bp. Then, obtaining the potential precursor pre-novel_77 sequence of novel_77 according to the rule that the number of the base pair of the precursor was less than or equal to 4. Using the mfold software to predicte secondary structure of the potential precursor pre-novel_77 sequence based on the following rules are: (1) The folding free energy of miRNA precursor is less than –18 kcal/mol; (2) The asymmetric protrusion of miRNA/miRNA* double strands is less than or equal to 3; (3) miRNA/miRNA* mismatched base pairs are less than or equal to 4. Finally, the secondary structure of the novel_77 precursor sequence was determined. As shown in Fig. 6, the secondary structure has a typical stem-loop structure with a folding free energy of –39.80 kcal/mol, which meets the secondary structure standard. In addition, the online software psRNATarget was used to predicte the target gene of novel_77. The predicted target genes are shown in Table 6 (partial targets). The smaller the expected value, the more matching the miRNA and the predicted target gene sequence.

Figure 6: Secondary structure of novel_77 precursor sequence.

Polyploidization is a common phenomenon in the evolution of plants (Yang et al., 2021). Polyploid plants universally have the advantage over diploid plants in their responses to abiotic stresses (Fan et al., 2017). As the best agent to induce polyploids, colchicine acts on most plants and many parts of them, such as seeds, stem growth points, scion and callus (Sun et al., 2020). Colchicine has widely been used in the study of cytology, genetics and plant breeding, after the first success of doubling the chromosome number of plants such as in Datura in 1930s (Blakeslee and Avery, 1937).
Colchicine, as a mitotic inhibitor, causes the chromosome to stagnate in anaphase and cytokinesis of division mainly by inhibiting the formation of the spindle (Sun et al., 2020). Colchicine prevents spindle formation and cell division by interfering with the formation of microtubules. Although, colchicine usually prevents cell division by inhibiting microtubule formation, the molecular mechanism of plant response to colchicine treatment remains unclear. The study has shown that the high mortality rate of plants treated with colchicine is due to the large scale of down-regulation of phenylpropanoid synthesis genes (Zhou et al., 2017). The miRNA does play an important role in pathway of phenylpropanoid synthesis (Sun et al., 2020).
Early studies on Arabidopsis indicated that the potential targets for most miRNAs were transcription factors (Chen et al., 2012), and participated in complex gene regulatory networks. In Arabidopsis, miR397a probably targeted the laccase family members, whereas another family member, miR397b, displayed better complementarity with another casein kinase gene. The fact that miR397 can target laccase provides a unique tool to investigate laccase’s function in higher plants (Sunkar and Zhu, 2004). We identified the target of pau-miR398 in the present study. Another study suggests that the function of pau-miR398 in other plants are probably the same as those of miR397. The Pau-miR394 targeted gene of an F-box protein. F-box family proteins are critical determinants for controlling Skp1-Cullin-F box complex substrate selection and are a key regulator in many cell-signaling, transcription, and cell-cycle pathways (Kipreos and Pagano, 2000).
Wheat tetraploid has high salt tolerance. According to previous studies, miR397, miR398, miR399, and miR408 are expressed during drought, cold, abscisic acid, oxidative, and salt stress conditions (Munns and James, 2003; Sunkar et al., 2006; Jia et al., 2009). A similar situation happened with Populus tomentosa. Also, pau-miR858 was significantly highly expressed in the PT4 library targeting MYB transcription factors. pau-miR858 was widely involved in gene information processing and transcriptional responses to biological stress pathways. The target of Pau-miR408b is the pentapeptide peptide repeat protein, whose genes have specific characteristic resistance genes to the disease and their “nomad” character. The evolutionary expansion of plants may involve new molecular processes and selective pressures (Geddy and Brown, 2007). This finding indicates that the tetraploid of P. tomentosa may have better diploid resistance.
Colchicine has certain toxicity in the induction process, too high or too low concentration will affect the induction of polyploid. Tetraploid mulberry trees have high branches and leaves, high sugar content and good leaf quality. However, the yield of tetraploid mulberry trees is lower than that of diploid mulberry trees because of fewer springs and shorter rod height. In recent years, with the development of polyploid breeding research, it was found that some artificial tetraploids of Mulberry, especially those induced by seedling, not only have hypertrophy of branches and leaves, but also the number of tetraploids is close to diploid, so they have higher leaf yield, which may be directly planted and used in production. The multiples of polyploid breeding should be in a suitable range. Once beyond the suitable range, the increase of chromosome multiple would inhibit the growth of mulberry, and some economic traits of mulberry would become worse. It is generally believed that the target multiple of mulberry polyploid breeding should be in the range of triploid (3x) to hexaploid (6x). According to the current practice of mulberry polyploid breeding, triploid and tetraploid mulberry trees are the best.
After colchicine induction, malondialdehyde (MDA), superoxide dismutase (SOD), proline (PRO) and other physiological and biochemical indexes of mulberry showed that the content of MDA after induction was lower than that of the control group. The content of SOD and PRO were increased. The above results indicated that the antioxidant activity of mulberry after induction was stronger than that of the control group. We will show the relevant physiological results in another paper.
At present, many studies on polyploid mulberry have focused on its polyploid effects, but there are not many studies at the molecular level. miRNAs are small non-coding RNA molecules (18–24 nt) that play roles in the regulation of target-gene expression post-transcriptionally by binding to complementary target mRNAs which results in mRNA translational inhibition or degradation (Ysrafil et al., 2020). Many studies have shown that miRNA expression is closely related to plant growth and development. The loss of miRNA function can affect plant growth and development, such as plant size, flowering time and plant fertility. In addition, miRNAs play a vital role in the regulation of abiotic stress in plants. However, few reports have focused on abiotic stress response miRNAs at the mulberry genome level. The completion of high-throughput and deep sequencing of the mulberry genome and the construction of sRNA libraries are for identifying mulberry miRNA functions in mulberry polyploidy molecular biology. In our study, colchicine-induced mulberry samples and control samples were subjected to sRNA sequencing. A total of 45 known miRNAs and 62 predicted novel matured miRNAs were identified. The number of differentially expressed miRNAs was 37, of which 19 up-regulated and 18 down-regulated. Target genes of differently expressed miRNAs were mapped to Gene Ontology (GO, http://www.geneontology) terms and the Genomes (KEGG, http://www.genome.jp/kegg/database) pathway to determine the function of the differentially expressed genes. According to the GO terms, these predicted target genes are mainly involved in the oxidation-reduction process, nucleus, and copper ion binding. In the KEGG pathway, more target genes are gathered on the biosynthesis of amino acids pathway.
Artificially synthesized polyploids using colchicine could cause DNA mutation, but relevant studies have rejected this hypothesis and proven that polyploid genomic structural variation has nothing to do with colchicine (Parisod et al., 2010). DNA-SRAP analysis of the tetraploid Chrysanthe-mum induced by colchicine showed that 1.6% of new fragments and 1.1% of missing fragments appeared in newly synthesized tetraploid (Ri et al., 2016). A study on Elymus elongatus showed that 10% DNA deletion occurred in both artificial and natural formatting autopolyploid (Eilam et al., 2009). The target genes of some miRNAs might involve in the process of plant cell DNA deletion and genomic structural variation under colchicine treatment.
Colchicine is a toxic alkaloid and secondary metabolite. It prevents spindle formation and cell division by interfering with the formation of microtubules. miRNAs have played critical role in regulating cell response to various biotic and abiotic stress. We speculate that the miRNA plays an important role in colchicine induction of polyploid plants. To study the tetraploid of mulberry induced with colchicine, the length of the conserved miRNA was compared to the specified sRNA range sequence in miRbase to predicte the novel miRNA in the samples. According to the results of this study, we selected a new miRNA novel-77 and predicted its target gene (XM_010104258.2). There are currently no studies on the downstream target gene XM_010104258.2. However, the expression of this gene changed significantly in colchicine-induced tetraploid mulberry trees. As miRNAs participate in regulating gene expression via degradation of its target genes or translational inhibition, we speculated that miRNA novel-77 was negatively regulated with the expression level of target genes, and we will verify this prediction in subsequent experiments. Further studies are also needed to demonstrate the regulatory mechanisms between miRNAs and its target genes to develop polyploid mulberry trees which will help to increase mulberry leaf yield, enrich mulberry resources, and promote the further development of sericulture industry. Our results presented here will be also useful for further functional analysis of these novel miRNAs and their target genes.
In summary, studying the expression analysis of miRNA in mulberry leaves induced by colchicine revealed some information on both known and novel miRNA, which is vital to determine the miRNA functions in mulberry plants under different environmental conditions. These results will serve as a fundamental data for further understanding the various miRNA functions in the mulberry genome under environmental stresses. The miRNA novel-77 is the first to find in the experiment of colchicine inducing tetraploid plants. Based on the results of this study, we predicted the target genes of miRNA novel-77. We speculated that miRNA novel-77 was negatively regulated with the expression level of target genes and the target gene might involve in the process of plant cell DNA deletion and genomic structural variation under colchicine treatment. The result preliminarily revealed the miRNA regulation mechanism in mulberry under colchicine stress, and also provided valuable candidate miRNAs and target genes for further investigation to molecular mechanismtree about the mulberry induced by colchicine.
Acknowledgement: This work was supported by China Agriculture Research System of MOF and MARA, National Key R&D Program of China, Key Projects of International Scientific and Technological Innovation Cooperation (2021YFE0111100), Guangxi Innovation-Driven Development Project (AA19182012-2), Zhenjiang Science and Technology Support Project (GJ2021015).
Availability of Data and Materials: All data generated or analyzed during this study are included in this published article. Thus, the readers can freely access all the published data with proper acknowledgment and citation.
Author Contribution: The authors confirm contribution to the paper as follows: Mengmeng Wu and Peng Guo conceived the work and wrote the first draft. Yisui Shi contributed in the drawing of the tables. Danyan Zheng, Qiaonan Zhang, Xin Jin, Yuhua Wang and Qiang Lin critically reviewed the draft. Rongjun Fang and Weiguo Zhao reviewed the results and approved the final version of the manuscript.
Funding Statement: The authors received no specific funding for this study.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
Ari E, Djapo H, Mutlu N, Gurbuz E, Karaguzel O (2015). Creation of variation through gamma irradiation and polyploidization in Vitex agnus-castus L. Scientia Horticulturae 195: 74–81. DOI 10.1016/j.scienta.2015.08.039. [Google Scholar] [CrossRef]
Blakeslee AF, Avery AG (1937). Methods of inducing doubling of chromosomes in plants by treatment with colchicine. Journal of Heredity 12: 393–411. DOI 10.1093/oxfordjournals.jhered.a104294. [Google Scholar] [CrossRef]
Castro M, Castro S, Loureiro J (2018). Production of synthetic tetraploids as a tool for polyploid research. Web Ecology 18: 129–141. DOI 10.5194/we-18-129-2018. [Google Scholar] [CrossRef]
Chen L, Wang T, Zhao M, Tian Q, Zhang WH (2012). Identification of aluminum-responsive microRNAs in Medicago truncatula by genome-wide high-throughput sequencing. Planta 235: 375–386. DOI 10.1007/s00425-011-1514-9. [Google Scholar] [CrossRef]
Chen X, Raza SHA, Cheng G, Ma X, Zan L (2020). Bta-miR-376a targeting KLF15 interferes with adipogenesis signaling pathway to promote differentiation of qinchuan beef cattle preadipocytes. Animals 10: 2362. DOI 10.3390/ani10122362. [Google Scholar] [CrossRef]
Dolezel J, Bartos J (2005). Plant DNA flow cytometry and estimation of nuclear genome size. Annals of Botany 95: 99–110. DOI 10.1093/aob/mci005. [Google Scholar] [CrossRef]
Eilam T, Anikster Y, Millet E, Manisterski J, Feldman M (2009). Genome size in natural and synthetic autopolyploids and in a natural segmental allopolyploid of several Triticeae species. School of Plant Sciences and Food Security 52: 275–285. DOI 10.1139/G09-004. [Google Scholar] [CrossRef]
El-Nashar YI, Ammar MH (2016). Mutagenic influences of colchicine on phenological and molecular diversity of Calendula officinalis L. Genetics and Molecular Research 15: 7745. DOI 10.4238/gmr.15027745. [Google Scholar] [CrossRef]
Eng WH, Ho WS (2019). Polyploidization using colchicine in horticultural plants: A review. Scientia Horticulturae 246: 604–617. DOI 10.1016/j.scienta.2018.11.010. [Google Scholar] [CrossRef]
Fan G, Wang L, Dong Y, Zhao Z, Deng M et al. (2017). Genome of paulownia (Paulownia fortunei) illuminates the related transcripts, miRNA and proteins for salt resistance. Scientific Reports 7: 1285. DOI 10.1038/s41598-017-01360-9. [Google Scholar] [CrossRef]
Friedlnder MR, Mackowiak SD, Li N, Chen W, Nikolaus R (2012). miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Research 40: 37–52. DOI 10.1093/nar/gkr688. [Google Scholar] [CrossRef]
Geddy R, Brown GG (2007). Genes encoding pentatricopeptide repeat (PPR) proteins are not conserved in location in plant genomes and may be subject to diversifying selection. BMC Genomics 8: 1–13. DOI 10.1186/1471-2164-8-130. [Google Scholar] [CrossRef]
Ghani MA, Li J, Rao L, Raza MA, Cao L et al. (2014). The role of small RNAs in wide hybridisation and allopolyploidisation between Brassica rapa and Brassica nigra. BMC Plant Biology 14: 272. DOI 10.1186/s12870-014-0272-9. [Google Scholar] [CrossRef]
Hu H, Guo P, Zhao Q, Li H, Liu H, Ma C (2021). Upregulation of microRNA-451a improves endothelial cell function in atherosclerosis by direct targeting of macrophage migration inhibitory factor. BIOCELL 45: 995–1004. DOI 10.32604/biocell.2021.012851. [Google Scholar] [CrossRef]
Jia L, Zhang D, Qi X, Ma B, Xiang Z et al. (2014). Identification of the conserved and novel miRNAs in mulberry by high-throughput sequencing. PLoS One 9: e104409. DOI 10.1371/journal.pone.0104409. [Google Scholar] [CrossRef]
Jia X, Wang WX, Ren L, Chen QJ, Mendu V et al. (2009). Differential and dynamic regulation of miR398 in response to ABA and salt stress in Populus tremula and Arabidopsis thaliana. Plant Molecular Biology 71: 51–59. DOI 10.1007/s11103-009-9508-8. [Google Scholar] [CrossRef]
Jin H, Pei M, Guo D (2022). Phylogenetic analysis and target gene prediction of miR477 gene family in grape. BIOCELL 46: 941–949. DOI 10.32604/biocell.2022.016718. [Google Scholar] [CrossRef]
Kipreos ET, Pagano M (2000). The F-box protein family. Genome Biology 1: 1–7. DOI 10.1186/gb-2000-1-5-reviews3002. [Google Scholar] [CrossRef]
Krishnatreya DB, Moni BP, Bhaskar D, Sarma BK, Heena A et al. (2020). Mining of miRNAs from EST data in dendrobium nobile. Bioinformation 16: 245–255. DOI 10.6026/97320630016245. [Google Scholar] [CrossRef]
Li Q, Zhang Q, Han L, Yuan Z, Tan J et al. (2012). Molecular characterization and expression of As-nurp1 gene from Artemia sinica during development and in response to salinity and temperature stress. Biological Bulletin 222: 182–191. DOI 10.1086/BBLv222n3p182. [Google Scholar] [CrossRef]
Likun W, Zhixing F, Xi W, Xiaowo W, Xuegong Z (2010). DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 26: 136–138. DOI 10.1093/bioinformatics/btp612. [Google Scholar] [CrossRef]
Liu P, Zou A, Chen Q, Cheng B, Li Q (2021). Basing on microRNA-mRNA analysis identifies microRNA in exosomes associated with wound repair of diabetic ulcers. BIOCELL 45: 27–39. DOI 10.32604/biocell.2021.012601. [Google Scholar] [CrossRef]
Love MI, Huber W, Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology 15: 550. DOI 10.1186/s13059-014-0550-8. [Google Scholar] [CrossRef]
Manzoor A, Ahmad T, Bashir MA, Hafiz IA, Silvestri C (2019). Studies on colchicine induced chromosome doubling for enhancement of quality traits in ornamental plants. Plants 8: 194. DOI 10.3390/plants8070194. [Google Scholar] [CrossRef]
Mao X, Tao C, Olyarchuk JG, Wei L (2005). Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 21: 3787–3793. DOI 10.1093/bioinformatics/bti430. [Google Scholar] [CrossRef]
Munns R, James RA (2003). Screening methods for salinity tolerance: A case study with tetraploid wheat. Plant and Soil 253: 201–218. DOI 10.1023/A:1024553303144. [Google Scholar] [CrossRef]
Parisod C, Holderegger R, Brochmann C (2010). Evolutionary consequences of autopolyploidy. New Phytologist 186: 5–17. DOI 10.1111/j.1469-8137.2009.03142.x. [Google Scholar] [CrossRef]
Qu H, Liu Y, Jiang H, Liu Y, Chen L (2021). Identification and characterization of miRNAs associated with sterile flower buds in the tea plant based on small RNA sequencing. Hereditas 158: 26. DOI 10.1186/s41065-021-00188-8. [Google Scholar] [CrossRef]
Ri G, Haibin W, Bin D, Xiaodong Y, Sumei C et al. (2016). Morphological, genome and gene expression changes in newly induced autopolyploid chrysanthemum lavandulifolium (Fisch. ex Trautv.) Makino. International Journal of Molecular Sciences 17: 1690. DOI 10.3390/ijms17101690. [Google Scholar] [CrossRef]
Schmittgen TD, Livak KJ (2008). Analyzing real-time PCR data by the comparative CT method. Nature protocols 3: 1101–1108. DOI 10.1038/nprot.2008.73. [Google Scholar] [CrossRef]
Shangguan A, Zhou H, Sun W, Ding R, Li X et al. (2020). Cryopreservation induces alterations of miRNA and mRNA fragment profiles of bull sperm. Frontiers in Genetics 11: 419. DOI 10.3389/fgene.2020.00419. [Google Scholar] [CrossRef]
Sun FY, Liu L, Yu Y, Ruan XM, Sun G (2020). MicroRNA-mediated responses to colchicine treatment in barley. Planta 251: 1. DOI 10.1007/s00425-019-03326-9. [Google Scholar] [CrossRef]
Sunkar R, Kapoor A, Zhu JK (2006). Posttranscriptional induction of two Cu/Zn superoxide dismutase genes in arabidopsis is mediated by downregulation of miR398 and important for oxidative stress tolerance. Plant Cell 18: 2051–2065. DOI 10.1105/tpc.106.041673. [Google Scholar] [CrossRef]
Sunkar R, Zhu JK (2004). Novel and stress-regulated microRNAs and other small RNAs from Arabidopsis. Plant Cell 16: 2001–2019. DOI 10.1105/tpc.104.022830. [Google Scholar] [CrossRef]
Uwe D, Hilo A, Pérez-Pérez JM, Klopotek Y, Acosta M et al. (2019). Hajirezaei molecular and physiological control of adventitious rooting in cuttings: Phytohormone action meets resource allocation. Annals of Botany 123: 929–949. DOI 10.1093/aob/mcy234. [Google Scholar] [CrossRef]
Wen M, Shen Y, Shi S, Tang T (2012). miREvo: An integrative microRNA evolutionary analysis platform for next-generation sequencing experiments. BMC Bioinformatics 13: 140. DOI 10.1186/1471-2105-13-140. [Google Scholar] [CrossRef]
Yang F, Fernández Jiménez N, Majka J, Pradillo M, Pecinka A (2021). Structural maintenance of chromosomes 5/6 complex is necessary for tetraploid genome stability in Arabidopsis thaliana. Frontiers in Plant Science 12: 748252. DOI 10.3389/fpls.2021.748252. [Google Scholar] [CrossRef]
Young MD, Wakefield MJ, Smyth GK, Oshlack A (2010). Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biology 11: R14. DOI 10.1186/gb-2010-11-2-r14. [Google Scholar] [CrossRef]
Ysrafil Y, Astuti I, Anwar SL, Martien R, Sumadi FAN et al. (2020). MicroRNA-155-5p diminishes in vitro ovarian cancer cell viability by targeting HIF1α expression. Advanced Pharmaceutical Bulletin 10: 630–637. DOI 10.34172/apb.2020.076. [Google Scholar] [CrossRef]
Zeng W, Sun Z, Lai Z, Yang S, Chen H et al. (2019). Determination of the MiRNAs related to bean pyralid larvae resistance in soybean using small RNA and transcriptome sequencing. International Journal of Molecular Sciences 20: 2966. DOI 10.3390/ijms20122966. [Google Scholar] [CrossRef]
Zhou K, Fleet P, Nevo E, Zhang X, Sun G (2017). Transcriptome analysis reveals plant response to colchicine treatment during on chromosome doubling. Scientific Reports 7: 8503. DOI 10.1038/s41598-017-08391-2. [Google Scholar] [CrossRef]
Zhou M, Zheng S, Liu R, Lu L, Zhang C et al. (2019). The genome-wide impact of cadmium on microRNA and mRNA expression in contrasting Cd responsive wheat genotypes. BMC Genomics 20: 615. DOI 10.1186/s12864-019-5939-z. [Google Scholar] [CrossRef]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |