| Congenital Heart Disease |
DOI: 10.32604/CHD.2020.011500
ARTICLE
Long Term Follow-Up of Ebstein’s Anomaly—What to Expect in Adult Life?
1Department of Cardiology, Hospital de Santa Marta, Centro Hospitalar Universitário de Lisboa Central, Lisbon, 1169-024, Portugal
2Department of Cardiothoracic Surgery, Hospital de Santa Marta, Centro Hospitalar Universitário de Lisboa Central, Lisbon, 1169-024, Portugal
3Department of Paediatric Cardiology, Hospital de Santa Marta, Centro Hospitalar Universitário de Lisboa Central, Lisbon, 1169-024, Portugal
*Corresponding Author: Tânia Branco Mano. Email: taniabmano@gmail.com
Received: 14 May 2020; Accepted: 28 May 2020
Abstract: Introduction: Due to the low prevalence and wide variation of severity of Ebstein’s Anomaly (EA), long-term follow-up data are scarce. The aim was to evaluate the long-term outcome of an adult population with EA. Methods: Retrospective analysis of EA adults followed in the past 42 years in a tertiary congenital heart disease outpatient clinic. Predictors of complications and mortality were assessed. Results: We studied 53 patients: 53% females, mean age 46 ± 19 years, 36% cyanotic, 55% diagnosed at adult age. Tricuspid regurgitation was moderate or severe in 33% and 46%, respectively, and during follow-up (mean 12 ± 10 years) 11 patients had right ventricular dysfunction. We found an association between New York Heart Association class and cyanosis (p = 0.041) and severity of tricuspid regurgitation (p = 0.02). The most frequent symptom was palpitations (57%), with 29 patients exhibiting rhythm disturbances (62% supraventricular tachycardia). Thromboembolic events were found in 23% and were associated with atrial septal defect or patent foramen ovale (p = 0.017) and arrhythmia diagnosis (p = 0.011). Nine patients required tricuspid valve surgery and two underwent cardiac transplantation. In 25 pregnancies, 48% developed fetal complications. Total of 14 deaths (2.4 deaths per 10 patients-years) occurring at a mean age of 49 ± 18 years, of cardiac cause in more than half of the cases and 29% of sudden death. No significant differences were found in the mortality rate of patients presenting with severe tricuspid regurgitation, with or without surgical management. Conclusion: Ebstein’s Anomaly is often diagnosed in adulthood. It is accompanied by high morbidity, especially arrhythmias, and non-negligible mortality with relevance in assessing the risk of sudden death.
Keywords: Ebstein’; s anomaly; sudden cardiac death; pregnancy; congenital heart disease
Ebstein’s Anomaly (EA), first described by Wilhelm Ebstein in 1866, is a rare form (<1%) of congenital heart disease (CHD), estimated at 1 per 20 000 live births [1,2].
It is a right ventricular myopathy with failure of tricuspid valve delamination [1] characterized by: 1) adherence of the septal and posterior leaflets to the underlying myocardium; 2) apical displacement of the functional annulus (towards the right ventricle apex and/or outflow tract); 3) dilatation of the “atrialized” portion of the right ventricle; 4) redundancy, fenestrations and tethering of the anterior leaflet and dilatation of the right atrioventricular junction [3].
It is commonly associated with other cardiac anomalies such as atrial septal defect (ASD) or patent foramen ovale (PFO) (80–94%), right ventricular outflow tract obstruction, left-sided heart disease (39%) and ventricular pre-excitation (10–38%) [1,2,4].
The presentation varies from the symptomatic neonate to the asymptomatic adult, depending on the severity of tricuspid valve dysfunction and associated heart abnormalities. The most significant clinical presentations in adulthood include arrhythmia and heart failure (right or left-sided) [2,5].
To improve outcome, structural repair and/or ablation therapy are often indicated, but optimal timing and inconsistent results are still challenging [6].
Due to the low prevalence and wide variation of anatomic and haemodynamic severity of EA, data of patients with long-term follow-up are scarce. Therefore, we aimed to characterize and evaluate the outcome of an adult population with EA and assess predictors of long-term complications and mortality.
A retrospective analysis was performed, including all adult patients with EA followed in the adult CHD outpatient clinic of our tertiary centre, between January of 1977 and December of 2019.
We evaluated demographic, clinical, imaging and electrocardiographic characteristics from clinical files and mortality data from a national database.
Diagnosis was considered in the paediatric age if made until 18 years of age. The clinical presentation that led to the diagnosis was divided into echocardiographic screening (asymptomatic), heart failure symptoms (exercise intolerance, dyspnea or peripheral oedema), palpitations, arrhythmia or syncope.
New York Heart Association (NYHA) Functional Classification was used to evaluate the severity of heart failure symptoms at follow-up.
The chest radiograph presented at the end of follow-up were analysed in relation to cardiothoracic index: <0.50, between 0.50–0–60 and >0.60.
When available, we analysed two distinct echocardiographic data: the first transthoracic echocardiogram (TTE) that led to the diagnosis and the last TTE, completed before tricuspid valve surgery or at the end of follow-up period (if conservative management).
The diagnosis of EA was previously made by the presence of apical distal displacement of the septal or posterior leaflet (in adults ≥0.8 cm/m2 body surface area) measured in the apical four-chamber view [6]. Tricuspid valve regurgitation was graded as mild, moderate or severe based on the recommendations of the European Association of Cardiovascular Imaging [7]. For longitudinal function evaluation, tricuspid annular plane systolic excursion (TAPSE) was measured by 2-dimensional echocardiography-guided M-mode recordings from the apical four-chamber view, with the cursor placed at the junction of the right atrial and right ventricular myocardium [8]. Qualitative assessment of the functional (non-atrialized distal part) right ventricle function were also address by expert echocardiographers. In patients who underwent cardiac magnetic resonance (CMR), the functional right ventricle was used for right ventricle measurements and contours were drawn to the insertion of the displaced tricuspid valve leaflets. Ejection fraction was calculated from end-diastolic and end-systolic volumes indexed to body surface area.
Electrocardiogram available at the end of follow-up was analysed for rhythm, conduction system abnormalities and pre-excitation pattern. An arrhythmic event was defined as presence of documented supraventricular tachycardia (atrioventricular reentrant tachycardia-ARVT, atrioventricular nodal reentrant tachycardia-AVNRT, atrial fibrillation-AF, atrial flutter-AFL or atrial tachycardia), ventricular arrhythmia (ventricular fibrillation, sustained ventricular tachycardia and/or episodes of non-sustained ventricular tachycardia) or frequent symptomatic ventricular premature beats (≥10% in 24-hour Holter monitoring).
Regarding thromboembolic events, cardioembolic stroke was defined in the presence of a potential intracardiac source of embolism, in the absence of cerebrovascular disease in a patient with non-lacunar stroke. Cases of pulmonary embolism and deep vein thrombosis were confirmed by computed tomography pulmonary angiogram and by doppler ultrasound, respectively.
Additionally, pregnancy outcomes were assessed from clinical file data, namely preeclampsia, eclampsia, acute heart failure, arrhythmia or fetal complications (spontaneous abortion, premature birth, stillbirth, congenital heart defect).
The statistical analysis was performed using SPSS Statistics version 22 (IBM SPSS, Chicago, IL). Data for categorical variables are reported as frequency and percentage (%) and continuous variables are expressed as mean ± standard deviation. Continuous variables that were not normally distributed are reported as median and range (minimum and maximum). Pearson’s chi-square or Fisher’s exact test were applied for categorical variables. The Student’s t test or the Wilcoxon-Mann-Whitney test was used for continuous variables. The predictors of mortality and complications were assessed with univariate Cox proportional hazards analysis (forward stepwise). Kaplan-Meier curves were used to estimate survival.
3.1 Patients Characteristics and Follow-Up
Baseline characteristics of the 53 adults enrolled, with a mean follow-up of 12 ± 10 years, are presented in Tab. 1. The mean age at last follow-up was 47 ± 19 years, with a slightly higher prevalence of female gender (53%).

The diagnosis was made at the pediatric age in 45% of patients. However, the presentation in adulthood did not differ from paediatrics: echocardiographic screening (44% vs. 40%, p = 0.817), palpitations, arrhythmia or syncope (36% vs. 35%, p = 0.919) and heart failure symptoms (20% vs. 25%, p = 0.774).
We were able to obtain echocardiography data at diagnosis in 46 patients (87%). The mean apical displacement of the tricuspid valve was 34.5 ± 14.4 mm, and tricuspid regurgitation was mild, moderate or severe in 22%, 33% and 46% of patients, respectively. In 80% of the cases there was an associated congenital heart defect, more frequently (51%) an ASD or PFO.
At the last follow-up, a cardiothoracic ratio ≥0.50 or ≥0.65 was present in 30 (57%) and 11 (21%) patients, respectively. At last follow-up or before valve surgery, 11 (21%) patients showed right ventricle dysfunction (mean TAPSE 12 ± 3 mm). Twelve patients underwent CMR (mean right ventricle ejection fraction 49 ± 5.9%). In two cases, there were discrepancies between the right ventricle function evaluated by echocardiography and by CMR, with lower values assessed by the latter (right ventricle ejection fraction of 38 and 40%), although not reaching statistical significance (p = 0.582). The remaining ten patients (83%) had preserved right ventricle function evaluated by CMR.
Three patients had moderate left ventricle systolic function impairment (mean left ventricle ejection fraction of 42%) and one patient (2%) had left ventricular non-compaction. Right atrial volumes were significantly enlarged (mean 62.8 ± 50.0 ml/m2).
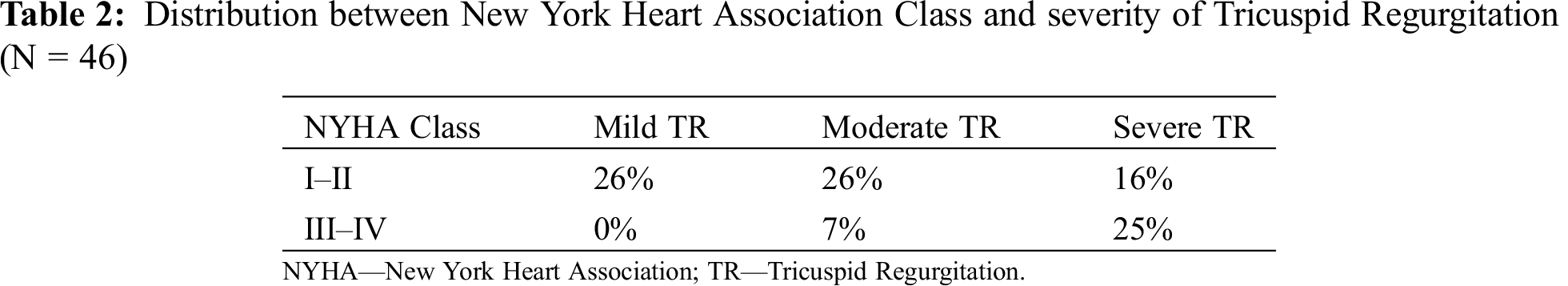
Regarding clinical data, most patients were in NYHA class I or II, and cyanosis was present in 36% of the population. The most frequent symptom was palpitations (57%), followed by exercise intolerance (37%) or peripheral oedema (17%). We found a relation between NYHA class and cyanosis (p = 0.041) and severity of tricuspid regurgitation (p = 0.020) – (Tab. 2).
Most of the patients (91%) received cardiovascular medication during follow-up: anticoagulation (38%), antiplatelet therapy (30%), beta-blockers (36%), diuretics (28%), amiodarone (22%) or digoxin (2%). No cases of infective endocarditis were documented, although most patients received antibiotic prophylaxis for dental procedures.
During follow-up, thromboembolic phenomena occurred at a rate of 1.8% events/year: eight patients with cardioembolic stroke, two cases of pulmonary embolism, two lower extremity deep vein thrombosis and one patient with subclavian thrombosis as consequence of an intravenous catheter.
We found an association of thromboembolic episodes with ASD or PFO (OR 7.31; 95% Confidence Interval [CI] 1.370–38.979; p = 0.017) and arrhythmia diagnosis (OR 13.08; 95% CI: 1.480–115.541; p = 0.011).
Thirty-one (58%) patients had right bundle branch block and 10 (19%) pre-excitation pattern in the 12-lead electrocardiogram. During follow-up, 5% of arrhythmic events/year were noticed, corresponding to a total of twenty-nine (55%) patients with arrhythmia diagnosis, 62% of which were supraventricular tachycardia: AVRT (N = 9), AF (N = 7) and AFL (N = 6). In the remaining patients, episodes of ventricular tachycardia (N = 2) or symptomatic ventricular premature beats (N = 8) were documented. One patient with ventricular tachycardia (50%), 4 patients with AFL (66%), and 3 (43%) of those with AF had previous cardiac intervention (tricuspid valve repair/replacement or atrial septal defect closure).
The majority of patients were treated with antiarrhythmics drugs (66%): beta-blockers (N = 10), amiodarone (N = 8), propafenone (N = 3), digoxin (N = 1) and/or calcium channel blockers (N = 1). Five patients (2 with AF and 3 with AFL) required an electrical cardioversion during follow-up.
In the subgroup with pre-excitation, 7 patients (70%) had evidence of an arrhythmia: AVRT (N = 6), AFL (N = 1) and ventricular tachycardia (N = 1). Electrophysiological studies were conducted in 5 patients with AVRT, one of them with concomitant documented AFL. All had percutaneous ablation of multiple accessory pathways (all right accessory pathways: 4 posterolateral, 1 posterior, 1 mesoseptal and 1 anterior) with an acute success rate of 60%. The patient with ventricular tachycardia had prosthetic tricuspid valve and right ventricle disfunction and implanted a cardioverter-defibrillator (ICD) for secondary prevention while waiting for cardiac transplant.
Furthermore, three patients were submitted to radiofrequency ablation of cavotricuspid isthmus-dependent AFL (success rate 66%) and one patient had surgical ablation of AF (with concomitant valvular tricuspid intervention). The remaining patient with palpitations and syncope, with concomitant coronary artery disease and biventricular dysfunction, underwent an electrophysiological study with ventricular tachycardia induction followed by ICD implantation.
Additionally, one patient needed implantation of a dual-chamber pacemaker due to brady-tachycardia syndrome.
Nine patients (17%) required tricuspid valvular surgery, most of them (89%) for symptomatic severe tricuspid regurgitation. All had right ventricle dysfunction before surgery, except one patient with left atrial myxoma, moderate tricuspid regurgitation and preserved right ventricle function. Two cases underwent bioprosthetic valve replacements and seven a tricuspid valve repair (40% with cone procedure). The mean age at the time of surgery was 47.9 ± 13.0 years (26–67 years). There was no early surgical mortality.
Regarding long-term follow-up after surgery, one patient was submitted to cardiac transplantation 22 years after tricuspid valve replacement and three patients died (mean 10 ± 3 years after surgery), one of cardiac cause (heart failure).
Another patient had cardiac transplantation at the age of 47 years due to biventricular dysfunction, without previous cardiac surgery.
Atrial septal defects were surgically corrected in 6 patients, in 3 cases simultaneously with valve surgery. Percutaneous closure of atrial septal defect was performed in two other patients. The main reason for closure was paradoxical embolism and/or cyanosis.
The survival rate was 90% and 63% at 10- and 20- years follow-up, respectively. The mean survival in patients with right ventricle dysfunction was not different (17.7 ± 4 years) to patients with preserved right ventricle function (26.8 ± 2 years, p = 0.116). The survival curves during follow-up related to the presence of arrhythmia, severe tricuspid regurgitation, and surgical management are illustrated in Fig. 1. In this cohort, there were a total of 14 deaths (mean age 49 ± 18 years), at a rate of 2.4 deaths per 10 patients-years. The mortality causes were: sudden cardiac death (n = 4), heart failure (n = 2), non-cardiac death (n = 2), after heart transplant (n = 2) and of unknown cause (n = 4).
In the sudden death subgroup, all had cardiothoracic index >0.50 and mild to moderate tricuspid regurgitation and three (75%) were cyanotic, with a mean haemoglobin of 16.6 g/dL. None of these patients had previous documented arrhythmia nor ventricle dysfunction.
We did not find significant differences in the mortality rate of patients with severe tricuspid regurgitation who were managed conservatively vs. surgically (p = 0.267). Also, we did not find a significant association between mortality or sudden death and the previous diagnosis of an arrhythmia (p = 0.316).

Figure 1: Survival rates of adult patients with Ebstein’s Anomaly during follow-up time
In total population, the mean cumulative survival was 25.1 ± 2.2 years. Regarding tricuspid regurgitation severity, the mean follow-up survival was 24.6 ± 3 years in non-severe vs. 25.1 ± 3 years in severe tricuspid regurgitation (p = 0.468). The presence of an arrhythmia did not also confer a statistically significant change in mean survival rate (26.3 ± 3 vs. 20.1 ± 2 years in patients without arrhythmic events, p = 0.327). In patients with severe tricuspid regurgitation, conservative management confer a non-statistically significant higher survival (mean 26.5 ± 2 years) comparing to surgical intervention (mean 17.5 ± 3 years, p = 0.185).
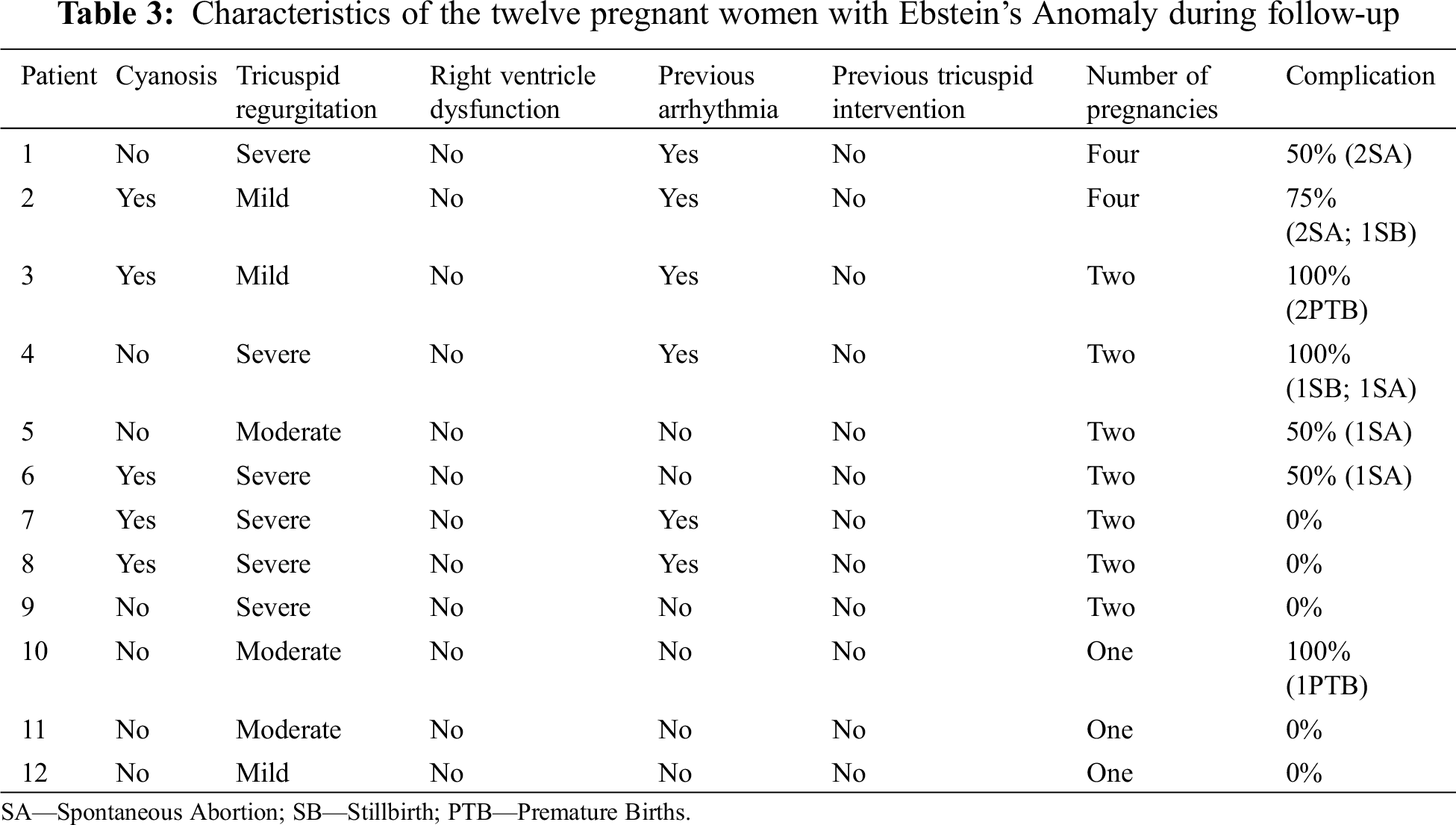
Twelve women became pregnant (43% of female population), with a total of 25 pregnancies. Fetal complications were documented in 12 pregnancies (48%): 7 spontaneous abortions, 3 premature births and 2 stillbirths. One child was born with mitral valve prolapse and another with EA. The latter had a genetic study that confirmed a family mutation of the β-myosin heavy chain 7 (MHC 7) gene (Tab. 3).
There were no maternal deaths or complications, such as preeclampsia, eclampsia or acute heart failure. We did not find any association or predictor of fetal complications, namely presence of cyanosis, arrhythmia, cardiothoracic index, tricuspid valve regurgitation or ventricle dysfunction.
Our study characterizes the long-term outcomes of adults with EA followed in a tertiary centre. To the best of our knowledge, this is the largest Portuguese report on this subject [9].
EA is a rare CHD, with significant very long-term overall mortality in our study. The risk factor more consistently associated with poor outcome, being an independent predictor of early and late mortality and for reoperation, is severe right ventricle dysfunction previous to surgery [1]. In our population, we observed a non-statistical significant lower survival during follow-up in patients with right ventricle dysfunction. Several authors suggested early surgical intervention, before the development of significant right heart enlargement, hoping to prevent progressive right ventricular dysfunction [1,2,5,10–12]. Serial assessments of right ventricle volume and function by echocardiography and, more recently, by cardiac magnetic resonance, can be helpful in determining deterioration over a period of time [13].
Regarding severe tricuspid regurgitation, we did not find statistically significant differences in the mortality rate of patients managed conservatively or surgically. Our survival rate of 90% and 63% at 10- and 20- years of follow-up is also in agreement with a cohort of 81 adult patients who underwent surgical intervention [5], but significantly lower than other recent report (91% survival at 20 years) [11].
On the other hand, 17% patients had tricuspid valvular surgery, similar to the cohort of Luu et al. [4] In our tertiary centre, the first two tricuspid valvular surgeries were bioprothesis replacements before 1988. Since then, seven other patients had tricuspid valve repair with annuloplasty and/or cone reconstruction. Surgical repair techniques have evolved markedly and thus comparison between different series is challenging, together with the fact that outcomes after surgical repair procedures are dependent on the experience of the surgical centre. Our peri-operative mortality was 0% and published data suggest that survival is increasing as advances in diagnosis, surgical techniques and postoperative care are made [12,13].
Nowadays, tricuspid valve replacement remains a good option when repair cannot be accomplished, particularly in older patients (>60 years) with massive right ventricle or annular dilatation. Bioprosthetic tricuspid valves are preferred and have a good durability, with one patient in our cohort needing a redo surgery after twenty-three years. Cardiac transplantation is reserved for patients with biventricular dysfunction who are considered at too high risk for conventional surgery, as was the case of two of our patients [1].
Careful assessment should be taken in deciding to close atrial septal defects (simultaneous at the time of tricuspid surgery or with percutaneous closure alone). Some patients require the defect as a “pop-off” valve to decompress the right ventricle volume and closing it may worsen right ventricle function [1,2].
In addition to hemodynamic burden of the valve defect, Ebstein’s patients had an extraordinarily high incidence of tachyarrhythmias, often with a clinical presentation with an arrhythmic event [14–18]. As expected, the most noteworthy clinical event in our adult population was arrhythmia (55%), followed by thromboembolic events (23%).
In concordance with our cohort, the most common arrhythmias are supraventricular. Wolff-Parkinson-White syndrome is more frequently associated with EA than with any other CHD. Structural and histologic abnormalities around the right atrioventricular junction potentiate a higher prevalence of accessory pathways, that are mostly right-sided (posterolateral, posterior and posteroseptal along the abnormal tricuspid valve ring) and multiple. Moreover, Ebstein’s physiology can make these patients more vulnerable to hypotension and worsened cyanosis during any type of tachycardia. Historically, success of accessory pathways ablation is typically lower in EA than in normal structural hearts, because of the anatomical pathway locations, cardiac enlargement causing catheter instability and signal complexity. Nowadays, even with progresses in three-dimensional mapping and ablative technology or intracardiac echocardiography, the acute success rate has only slightly improved, and recurrence rate after a first procedure remains high (20–40%, 40% in our cohort). The dilated right atrium and, in some patients, the left atrium (with long standing right-to-left shunting) also contribute to the development of other atrial tachycardias, such as AFL and AF. Also, in these cases, the ablation procedure is challenging, with the highest recurrence in AFL (40–60%, 33% in our cohort) [14–18].
Intrinsic monomorphic ventricular tachycardia from the “atrialized” portion of the right ventricle muscle is relatively rare, but Ebstein’s patients with advanced degrees of ventricular dysfunction, or after tricuspid valve surgery, can develop more rapid and disorganized ventricular tachycardia or ventricular fibrillation, culminating in sudden death [19]. Statistics are limited by the rarity of this malformation and the fact that sudden death in this population can be hemodynamic, arrhythmic, thromboembolic, or any combination of the above [19,20]. In large collaborative studies focused on sudden arrhythmic death in CHD, Ebstein’s subjects account for <5% of all events [19]. A report describing the Mayo Clinic experience over the past four decades measured a 10-, 50-, and 70-year cumulative incidences of sudden death from birth of 0.8%, 8.3%, and 14.6%, respectively [20]. The approximate incidence of 0.2%/year is comparable to that seen in Tetralogy of Fallot and much higher than expected in a non-EA population of a similar age and gender. That study also identified a prior history of ventricular tachycardia or syncope, heart failure, pulmonary stenosis, tricuspid valve surgery and haemoglobin >15 g/dL to be strong predictors of sudden death in a multivariable analysis [20]. In our cohort, accounting that not all autopsy reports were available for consultation, at least 4 patients died of sudden death during follow-up. Although we did not find any statistically significant association with sudden death, most of these patients were cyanotic (75%) and had haemoglobin levels >15 g/dL. It is postulated that the element of cyanosis, with correspondingly poorer right ventricle function, higher right atrium pressures and worse pulmonary perfusion can be a possible mechanism for the worse prognosis [20]. Also, in concordance with the findings of Attenhofer Jost et al. [3], the extent of tricuspid regurgitation and right ventricle enlargement were not predictive of future sudden death events. In fact, all our patients with sudden death had mild to moderate tricuspid regurgitation. As already mentioned, more advanced imaging techniques such as three-dimensional echocardiography and cardiac magnetic resonance may allow for a more reliable determination of right ventricle size and function [20].
Two patients in our study had ICD for secondary prevention. In the study mentioned above, none of the patients in the primary prevention subgroup received appropriate shocks [20]. There are no specific guidelines for ICD implantation in EA, combined with the technical difficulties of the procedure (risk of paradoxical embolus or of worsening valve function because of leads across an abnormal or reconstructed tricuspid valve) make the decision of implantation in primary prevention a case-by-case one. There is a need for future studies and creation and validation of a sudden death risk calculator specific to EA patients [14,15,20].
In a cohort of young adults, it is also imperative to analyse maternal and fetal complications. Women with EA should undergo pre-pregnancy counselling with a multidisciplinary team with expertise in CHD, because of their increased risk of right heart failure, arrhythmias and sudden death, and also of low birth weight and fetal loss, as demonstrated in our population. NYHA class > II, cyanosis, prior arrhythmia events, higher cardiothoracic index and tricuspid regurgitation severity were associated with adverse pregnancy outcomes in previous reports [5,21].
The risk of CHD in offspring is 6–8%, similar to our study, but familiar cases of EA are rare. Case-control studies suggest genetic, reproductive and environmental risk factors in the basis of this anomaly that are not yet fully understood. There are heterogeneous genetic factors, but the mutation in the gene encoding β-MYH7 (which commonly causes cardiomyopathy) found in the offspring of one of the patients, has already been described in several families with EA and left ventricular noncompaction [3,5,22].
The approach to the management of patients with EA is influenced by the patient’s age and clinical presentation. As we notice in our study, EA is still a high-risk population demanding close clinical follow-up and multimodality imaging techniques. Most of our cohort (72%) was in NYHA class I or II, nevertheless requiring cardiovascular medication (91%).
This was a retrospective study from a single centre with limited sample size that makes the analysis less robust. There was a significant proportion of missing data, mostly in the first years of follow-up, that did not have computerized data and, despite our attempts, the cause of death of 4 patients remained undetermined. Also, we did not analyse data from cardiopulmonary exercise test and most of the patients did not performed cardiac magnetic resonance that would further characterize this population.
During our very long follow-up time, there were substantial refinement in management, evaluation, and surgical techniques that were not addressed. However, we are a centre of reference for CHD and this study reflects daily clinical practice in our country.
EA is a rare CHD, often diagnosed in adulthood. Many patients have no symptoms and require only monitoring while others are symptomatic, requiring supportive medical therapy and surgical intervention. EA is accompanied by high morbidity, especially arrhythmias, and non-negligible cardiac mortality. The structural alterations related with EA are frequently associated both with supraventricular and ventricular arrhythmias, with lower success rates in percutaneous ablation procedures and with relevance in assessing the risk of sudden death.
Acknowledgement: We thank the Centro Hospitalar Universitário Lisboa Central for the opportunity to perform this study.
Data Sharing: No more data will be shared.
Funding Statement: The authors received no specific funding for this study.
Conflict of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
1. Dearani, J. A., Mora, B. N., Nelson, T. J., Haile, D. T., O’Leary, P. W. (2015). Ebstein anomaly review: what’s now, what’s next? Expert Review of Cardiovascular Therapy, 13(10), 1101–1109. [Google Scholar]
2. Safi, L. M., Liberthson, R. R., Bhatt, A. (2016). Current management of Ebstein’s anomaly in the adult. Current Treatment Options in Cardiovascular Medicine, 18(9), 56. [Google Scholar]
3. Attenhofer Jost, C. H., Connolly, H. M., Dearani, J. A., Edwards, W. D., Danielson, G. K. (2007). Ebstein’s Anomaly. Circulation, 115(2), 277–285. [Google Scholar]
4. Luu, Q., Choudhary, P., Jackson, D., Canniffe, C., McGuire, M. et al. (2015). Ebstein’s anomaly in those surviving to adult life-A single centre experience. Heart, Lung and Circulation, 24(10), 996–1001. [Google Scholar]
5. Arya, P., Beroukhim, R. (2014). Ebstein anomaly: assessment, management, and timing of Intervention. Current Treatment Options in Cardiovascular Medicine, 16(10), 338. [Google Scholar]
6. Baumgartner, H., Bonhoeffer, P., De Groot, N. M., de Haan, F.,Deanfield, J. E. et al. (2010). ESC Guidelines for the management of grown-up congenital heart disease (New Version 2010). European Heart Journal, 31(23), 2915–2957. [Google Scholar]
7. Lancellotti, P., Tribouilloy, C., Hagendorff, A., Popescu, B. A., Edvardsen, T. et al. (2013). Recommendations for the echocardiographic assessment of native valvular regurgitation: an executive summary from the European Association of Cardiovascular Imaging. European Heart Journal of Cardiovascular Imaging, 14(7), 611–644. [Google Scholar]
8. Therrien, J., Henein, M. Y., Li, W., Somerville, J., Rigby, M. (2000). Right ventricular long axis function in adults and children with Ebstein’s malformation. International Journal of Cardiology, 73(3), 243–249. [Google Scholar]
9. Patrício, L., Branco, L. M., Ferreira, M. L., Agapito, A. F., Catarino, C. et al. (1991). Ebstein’s anomaly. Clinical Aspects and Surgical Therapy. Portuguese Journal of Cardiology, 10(4), 325–330. [Google Scholar]
10. Kim, H. Y., Jang, S. Y., Moon, J. R., Kim, E. K., Chang, S. A. et al. (2015). Natural course of adult Ebstein anomaly when treated according to current recommendation. Journal of Korean Medical Science, 31(11), 1749–1754. [Google Scholar]
11. Hetzer, R., Hacke, P., Javier, M., Miera, O., Schmitt, K. et al. (2015). Long-term impact of various techniques for tricuspid repair in Ebstein’s anomaly. Journal of Thoracic and Cardiovascular Surgery, 150(5), 1212–1219. [Google Scholar]
12. Belli, E., Rabot, M., Petit, J., Gouton, M. (2017). Ebstein’s anomaly in adults: modified cone reconstruction of the tricuspid valve is associated with promising outcomes. Archives of Cardiovascular Disease, 110(5), 325–333. [Google Scholar]
13. Qureshi, M. Y., O’Leary, P. W., Connolly, H. M. (2018). Cardiac imaging in Ebstein anomaly. Trends in Cardiovascular Medicine, 28(6), 403–409. [Google Scholar]
14. Walsh, E. P. (2018). Ebstein’s anomaly of the tricuspid valve—A natural laboratory for reentrant tachycardias. JACC: Clinical Electrophysiology, 4(10), 1271–1288. [Google Scholar]
15. Sherwin, E. D., Abrams, D. J. (2017). Ebstein anomaly. Cardiac Electrophysiology Clinics, 9(2), 245–254. [Google Scholar]
16. Hassan, A., Tan, N. Y., Aung, H., Connolly, H. M., Hodge, D. O. et al. (2017). Outcomes of atrial arrhythmia radiofrequency catheter ablation in patients with Ebstein’s anomaly. Europace, 20(3), 535–540. [Google Scholar]
17. Roten, L., Lukac, P., de Groot, N.,Nielsen, J. C., Szili-Torok, T. et al. (2011). Catheter ablation of arrhythmias in Ebstein’s anomaly: a multicenter study. Journal of Cardiovascular Electrophysiology, 22(12), 1391–1396. [Google Scholar]
18. Orczykowski, M., Derejko, P., Bodalski, R., Urbanek, P., Zakrzewska-Koperska, J. et al. (2017). Radiofrequency catheter ablation of accessory pathways in patients with Ebstein’s anomaly: at 8 years of follow-up. Cardiology Journal, 24(1), 1–8. [Google Scholar]
19. Koyak, Z., Harris, L., de Groot, J. R.,Silversides, C. K., Oechslin, E. N. et al. (2012). Sudden cardiac death in adult congenital heart disease. Circulation, 126(16), 1944–1954. [Google Scholar]
20. Attenhofer Jost, C. H., Tan, N. Y., Hassan, A., Vargas, E. R., Hodge, D. O. et al. (2018). Sudden death in patients with Ebstein anomaly. European Heart Journal, 39(21), 1970–1977. [Google Scholar]
21. Kanoh, M., Inai, K., Shinohara, T., Shimada, E., Shimizu, M. et al. (2018). Influence of pregnancy on cardiac function and hemodynamics in women with Ebstein’s anomaly. Acta Obstetricia et Gynecologica Scandinavica, 97, 1025–1031. [Google Scholar]
22. Yuan, S. M. (2017). Ebstein’ anomaly: genetics, clinical manifestations, and management. Pediatrics and Neonatology, 58(3), 211–215. [Google Scholar]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |