| Congenital Heart Disease |
DOI: 10.32604/CHD.2020.012910
ARTICLE
Severe Right Ventricular Dysplasia with Absent Pulmonary Valve Syndrome and Tricuspid Atresia: A Literature Review
1Nicklaus Children’s Hospital, Department of Cardiology 2nd Floor, Miami, FL 33155, USA
2Herbert Wertheim School of Medicine, Florida International University, Miami, FL 33130, USA
3Kidz Medical Services, Miami, FL 33176, USA
*Corresponding Author: Kinjal Parikh. Email: kinjal723@gmail.com
Received: 17 July 2020; Accepted: 20 August 2020
Abstract: This is a newborn male prenatally diagnosed with severe right ventricular (RV) hypertrophy and depressed function, aneurysmal dilation of the main pulmonary artery and tachyarrhythmia. Postnatally, he required immediate intubation and inotropic support. Echocardiogram revealed a large dysplastic RV, absent pulmonary valve syndrome (APVS), markedly dilated pulmonary arteries and tricuspid atresia (TA). The trabecular portion of the RV was excessively trabeculated and severely dilated. Inflow and infundibular walls were thin, with multiple infundibular aneurysms. There was APVS with free regurgitation and massively dilated pulmonary arteries. The RV bulged into the LV, though there was no outflow tract obstruction. The LV had mildly depressed systolic function. Computed tomography angiography showed marked dilation of the main and branch pulmonary arteries, with compression of the airway. This, along with profound anasarca, prohibited weaning of ventilatory support. Ventricular tachycardia contributed to low cardiac output. Genetic testing revealed a heterozygous variant in the desmoplakin (DSP) gene, which is associated with familial arrhythmogenic RV dysplasia and dilated cardiomyopathy. The parents opted to withdraw care. Severe RV dysplasia associated with APVS and TA has previously been reported, however the degree of RV dilation with primitive myocardium in this case is profound. Further, presence of both fetal and postnatal ventricular tachycardia contributing to low cardiac is a novel presentation. This demonstrates that the overall poor prognosis was multifactorial.
Keywords: Dysplastic right ventricle; absent pulmonary valve syndrome; tricuspid atresia; arrhythmogenic right ventricular dysplasia
Absent pulmonary valve syndrome (APVS) with tricuspid atresia (TA) is a rare cardiac anomaly occurring in less than 1% of infants with congenital heart disease [1]. This complex can be further complicated by the presence of a primitive and hypertrophied right ventricle (RV), which represents an embryologic failure of the development and refinement of the RV musculature as well as pulmonary valve [2]. This constellation of findings has previously been reported, with varying degrees of RV dysplasia and outcomes (Tab. 1). Despite the presence of severely dilated pulmonary arteries and bronchial compression, as well as a functional single ventricle due to tricuspid atresia, palliation has been successfully performed [3]. However, a comprehensive evaluation of such cases with co-existing conditions, such as RV dysplasia or a tachyarrhythmia, have not been previously reviewed. We are presenting a case of APVS in the setting of functional TA, intact ventricular septum (IVS) and an unusual degree of severe RV dysplasia with a ventricular dysrhythmia. Fetal echocardiography assisted in the evaluation by predicting the presence of hemodynamic compromise at birth. Postnatal echocardiography and computed tomography angiography (CTA) confirmed the complex and unusual anatomy.
Previously Reported Cases
Table 1: Detailed descriptions of previously reported cases with tricuspid atresia and absent pulmonary valve syndrome
At 24 weeks gestation, a newborn male was prenatally diagnosed by fetal echocardiography to have severe RV hypertrophy with depressed function, aneurysmal dilation of the main pulmonary artery and tachyarrhythmia. The RV cavity was moderately hypoplastic with decrease tricuspid valve inflow (Fig. 1A), despite the appearance of adequate antegrade flow across the pulmonary valve. The hypertrophied interventricular septum appeared to bulge into the left ventricular (LV) outflow tract, without significant obstruction. The LV cavity retained normal dimensions and function. A tortuous ductus had bidirectional flow (Figs. 1B and 1C). There was aneurysmal dilation of the ascending aorta and main pulmonary artery. Some degree of hemodynamic compromise was evidenced by a small pericardial effusion around RV free wall, as well as abnormal ductus venosus and umbilical venous Doppler patterns.
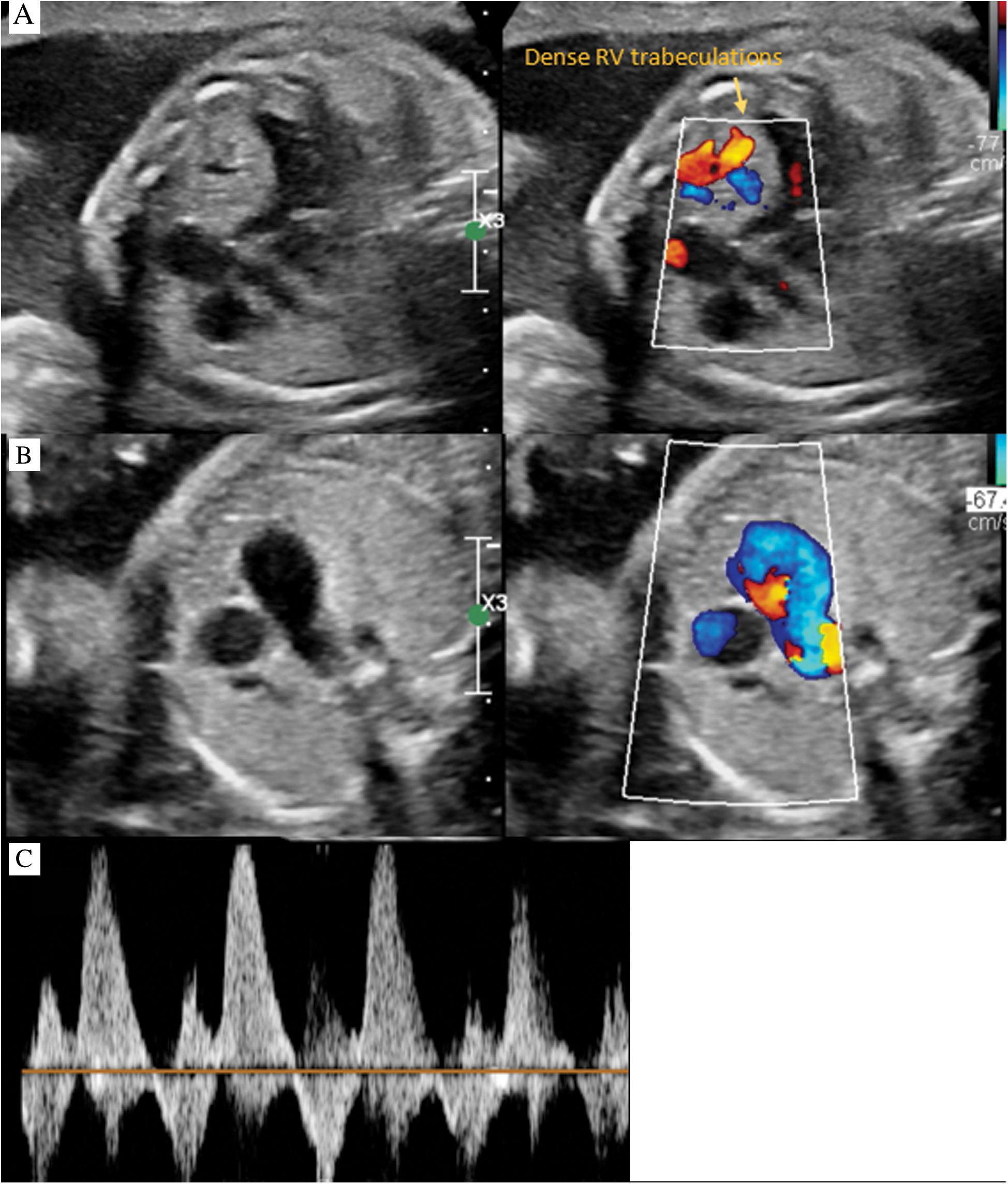
Figure 1: Fetal echocardiography. A: Four chamber color compare with cardiomegaly, RV dysplasia and dense trabeculations. B: 3 vessel view color compare with bidirectional flow in MPA. C: Doppler interrogation of MPA demonstrating bidirectional flow
Delivery was scheduled at the closest birthing center with the surgical team prepared to initiate extracorporeal membrane oxygenation. At 39 weeks gestation, he underwent elective cesarean section. Birth weight was 3 kilograms. The infant was born limp and cyanotic. APGAR scores were 2, 5 and 5, at 1, 5 and 10 minutes respectively. He ultimately required intubation, epinephrine and prostaglandin, and was briskly transferred to the cardiac intensive care unit. Postnatal transthoracic echocardiogram revealed APVS with free pulmonary regurgitation and markedly dilated pulmonary arteries, near membranous TA, an enlarged and dysplastic RV with compression of the LV, no LV outflow tract obstruction, mildly depressed LV systolic function and a large, tortuous patent ductus arteriosus (PDA) with right to left flow in systole and left to right in diastole (Figs. 2A–2D).
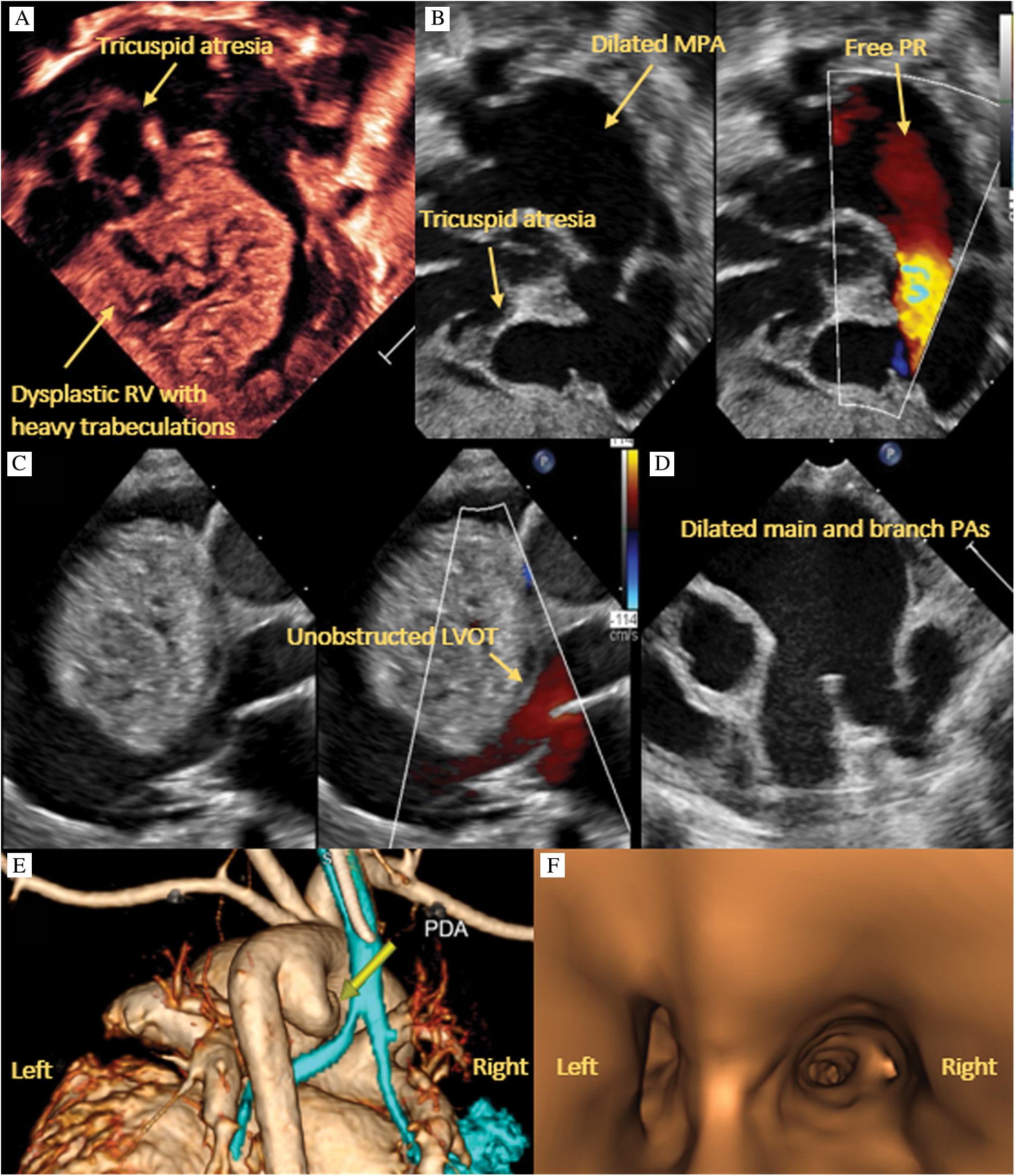
Figure 2: Transthoracic echocardiography. A: 4 chamber view of near membranous tricuspid atresia and a large, dysplastic RV with multiple trabeculations. B: Right anterior oblique view of absent pulmonary valve with free regurgitation as well as thin walled RV inflow and outflow tracts. C: Parasternal long axis view of large, dysplastic RV impinging on the LV but without obstruction. D: Parasternal short axis view of dilated main and branch pulmonary arteries. E: CTA 3D reconstruction showing compression of the left mainstem bronchus. F: CT airway reconstruction showing left bronchus narrowing due to external compression
After extensive discussion and review of published case reports, palliation with main pulmonary artery ligation and placement of a Blalock-Thomas-Taussig shunt seemed plausible. CTA confirmed profound dilatation of the main and branch pulmonary arteries. While the trachea was of normal caliber, there was marked compression of the left mainstem, bilateral upper lobe and right lower lobe bronchi (Figs. 2E–2F). The significant bronchial compression and anasarca prohibited weaning of ventilatory support. Further, idioventricular tachycardia, managed with lidocaine and amiodarone due to breakthrough episodes, contributed to a low cardiac output state. Genetic evaluation ultimately revealed a heterozygous variant in the desmoplakin (DSP) gene. This particular variant has not been previously reported. However, the pathogenic variant in DSP is associated with autosomal dominant familial arrhythmogenic RV dysplasia (ARVD) and dilated cardiomyopathy. After extensive discussion regarding the unknown prognosis and potential comorbidities, the parents opted to withdraw care.
APVS is an intricate and rare cardiac anomaly, consisting of dysplastic or complete absent pulmonary valve leaflets [13]. This leads to torrential pulmonary regurgitation, causing significant volume overload to the right heart structures, as well as variable degree of pulmonary stenosis. In the more common subcategory of Fallot type, there is a ventricular septal defect (VSD) with markedly dilated pulmonary arteries and no PDA. On the contrary, a smaller percentage of APVS are categorized as non-Fallot type, with no VSD and a milder degree of dilated pulmonary arteries due to pulmonary blood run off through a PDA. Typically, APVS can be surgically palliated with a biventricular repair. However, the addition of tricuspid valve stenosis or atresia with RV hypoplasia and an IVS mandates a single ventricle palliation [9]. This combination of anomalies was first reported in 1973, with the 12 day old infant undergoing a Waterston shunt but ultimately passing away [4]. Since then, additional case reports have been sporadically documented in literature (Tab. 1). An extensive and thorough review of the literature allowed for identification and comprehensive analysis of these clinical cases. The diagnosis, presentation, clinical course, outcomes as well as postmortem findings were evaluated to augment the understanding of this unique constellation of findings.
RV dysplasia was described in a few of the reported cases. While definitively unknown, it has been suggested that an arrest in embryologic development results in primitive RV myocardium with atretic or near atretic tricuspid and pulmonary valves. The dysplasia can be a spectrum. Postmortem examinations showed that a lack of myocardial cells in the RV, as well as hypertrophy, cavitations and sinusoids in the RV free wall and septum [8]. Further, the morphogenic mechanism of TA with IVS occurring with a dysplastic RV appears to be distinct from classic TA with a VSD and hypoplastic RV. Detailed autopsy analysis showed that in the prior defect, the atretic TV is positioned above the RV, while in classic TA, it is above the ventricular septum. Based on these findings, it has been proposed that the abnormal RV and ventricular myocardium plays a role in the development of tricuspid and pulmonary valve anomalies [8]. This concept was further validated in a subsequent case report and autopsy findings [9].
The clinical outcomes of the neonates with APVS, TA, IVS and a dysplastic RV are poor [7]. Per detailed literature review, fetal demise has been documented twice. In one, a stillborn infant was born at 32 weeks gestation. The diagnosis was made around 21 weeks gestation. Pregnancy course had been unremarkable until the mother noted decreased fetal movements. Postmortem microscopic findings showed ventricular myocardial necrosis, with sinusoids and calcifications in the RV and subendocardial fibrosis with autolysis in the LV [7]. A second case reported premature labor at 27 weeks due to polyhydramnios, with still birth. The cardiac anomaly also included a dilated LV with mitral regurgitation and a coronary arteriovenous fistula from the RCA to the RA [10].
Even in the absence of RV dysplasia, outcomes are poor. In a 2014 retrospective review by Szwast et al, the presence of TA with APVS/IVS was strongly correlated with heart transplantation (p = 0.003). Markedly abnormal coronary arteries caused ischemic changes and hence left ventricular dysfunction, while bowing of hypertrophied ventricular septum led to left ventricular outflow tract obstruction with poor cardiac output. In the setting of ventricular dysfunction or hydrops fetalis, families have opted for termination [16].
This particular case report highlights a rare constellation of cardiac defects consisting of APVS, TA, and a dysplastic RV with hypertrophy. While cases have been previously documented (Tab. 1), this report delineates the effects of prenatal diagnosis on delivery management as well as postnatal hemodynamics on clinical outcomes. Further, none of the previous documented cases reported ventricular arrhythmia as a finding. In the setting of primitive RV myocardium, it is not irrational to see rhythm disturbances. The dysrhythmia would exacerbate the already existing, hemodynamic instability in a patient with complex congenital heart disease, as is the scenario in our case report. Lastly, the genetic investigation revealed a gene mutation in the DSP gene, which is associated with ARVD. This rare inherited disorder of the myocardium is a common cause of life-threatening arrhythmias and sudden cardiac death in adolescents and athletes. Prevalence has been reported to be 1:1000 to 1:500, usually by autosomal dominant inheritance pattern with variable penetrance. Affecting primarily the right ventricular muscle, the condition entails progressive myocardium replacement by fatty or fibrofatty tissue, contributing to conduction abnormalities. It is now understood that ARVD is a cardiac desmosomal disorder, with effects on both ventricles. As protein complexes that link adjacent cells, desmosomes are a crucial element of the intercalated disc proteins. They maintain structural integrity, cell adhesion, calcium homeostasis and electrical conductivity [17]. A 2017 analysis of genome Aggregation Database identified DSP as one of the five most commonly mutated genes in ARVD. Interestingly, pathologic variants in DSP genes were the least represented in the database. The authors suggested that these variants may have more lethal effects compared to the other genes, and would explain the low prevalence in the database due to natural selection [18]. Though not much is known in the neonatal presentation, the findings of a dysplastic RV with ventricular arrhythmia in our patient may be a presentation of the heterogeneous phenotype of ARVD.
In conclusion, the presence of RV dysplasia with APVS and TA has been documented in previous case reports, typically with pre or postnatal demise. This particular report demonstrates a case with a quite unusual degree of severe RV hypertrophy and dilation with primitive myocardium, associated with a ventricular rhythm and genetic mutation in desmoplakin gene. The concurrent arrhythmia contributed to the low cardiac output state. Based on structural, hemodynamic and genetic findings, the overall poor prognosis was multifactorial.
Data Sharing: Individual deidentified participant data (including data dictionaries) will not be shared.
Funding Statement: The author(s) received no specific funding for this study.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
1. Grewal, D. S., Chamoli, S. C., Saxena, S. (2013). Absent pulmonary valve syndrome: antenatal diagnosis. Medical Journal Armed Forces India, 70(2), 198–200. DOI 10.1016/j.mjafi.2013.07.002.
2. Mori, K., Ando, M., Nakazawa, M., Momma, K., Takao, A. (1992). Imperforate tricuspid valve with dysplasia of the right ventricular myocardium, pulmonary valve and coronary artery: a clinocopathological study of nine cases. Pediatric Cardiology, 13(1), 24–29.
3. Futuki, A., Fijuwara, K., Yoshizawa, K., Sakazaki, H. (2018). Absent pulmonary valve syndrome with tricuspid atresia, ventricular septal defect, and aneurysmal dilated pulmonary artery: a case report of successful fontan completion. World Journal for Pediatric and Congenital Heart Surgery, 9(1), 101–104. DOI 10.1177/2150135116659651.
4. Marin-Garcia, J., Roca, J., Blieden, L. C., Lucas, R. V.Jr., Edwards, J. E. (1973). Congenital absence of pulmonary valve associated with tricuspid atresia and intact ventricular septum. Chest, 64(5), 658–661. DOI 10.1378/chest.64.5.658.
5. Quero-Jimenez, M., Maitre azcarate, M. J., Alvarez Bejarano, H., Vazquez Martul, E. (1975). An anatomical study of 17 cases. European Journal of Cardiology, 3(4), 337–348.
6. Cox, J. N., de Seigneux, R., Bolens, M., Haenni, B., Bopp, P. et al. (1976). Tricuspid atresia, hypoplastic right ventricle, intact ventricle septum and congenital absence of the pulmonary valve. Helvetica Paediatrica Acta, 30(4–5), 389–398.
7. Freedom, R. M., Patel, R. P., Bloom, K. R., Duckworth, J. W., Silver, M. M. et al. (1979). Congenital absence of the pulmonary valve associated with imperforate membrane type of tricuspid atresia, rv tensor apparatus and intact ventricular septum: a curious developmental complex. European Journal of Cardiology, 10(3), 171–196.
8. Litovsky, S., Choy, M., Park, J., Parrish, M., Waters, B. et al. (2000). Absent pulmonary valve with tricuspid atresia or severe tricuspid strenosis: report of three cases and review of the literature. Pediatric and Developmental Pathology, 3(4), 353–366. DOI 10.1007/s100249910050.
9. Lopes, K. R. M., Mennes, F., Delazoide, A. L., Iserin, F., Azancot, A. (2007). Prenatal diagnosis of absent pulmonary valve with membranous tricuspid atresia and intact ventricular septum: report of one case and review of literature. Prenatal Diagnosis, 27(10), 973–975. DOI 10.1002/pd.1799.
10. Joshi, A. N., Rane, H. S., Kamble, R. C., Mestry, P. J., Maniar, H. et al. (2010). Prenatal diagnosis of absent pulmonary valve syndrome. Journal of Ultrasound Medicine, 29(5), 823–829. DOI 10.7863/jum.2010.29.5.823.
11. Lato, K., Gembrunch, U., Geipel, A., Lachmann, R., Schneider, M. et al. (2010). Tricuspid atresia with absent pulmonary valve and intact ventricular septum: intrauterine course and outcome of an unusual congenital heart defect. Ultrasound Obstetrical Gynecology, 35(2), 243–245.
12. Best, T. H., Vyas, H. V., Jaquiss, R. D., Sachdeva, R. (2011). Tricuspid atresia with absent pulmonary valve syndrome and intact ventricular septum: a rare association. Congenital Heart Disease, 35(4), 393–396.
13. Wertascchnigg, D., Jaeggi, M., Chitayat, D., Shannon, P., Ryan, G. et al. (2013). Prenatal diagnosis and outcome of absent pulmonary valve syndrome: contemporary single-center experience and review of the literature. Ultrasound Obstetric Gynecology, 41(2), 162–167. DOI 10.1002/uog.11193.
14. Miki, Y., Tanakam, T., Oshima, Y. (2017). Functional near-tricuspid atresia in a patient with absent pulmonary valve and intact ventricular septum. Cardiology in the Young, 27(2), 391–393.
15. Monacci, F., Bondi, T., Canessa, C., Chiappa, E. (2019). Absent pulmonary valve with intact ventricular septum mimicking tricuspid valve atresia: Prenatal diagnosis and postnatal course. The Journal of Obstetrics and Gynaecology Research, 45(3), 714–718.
16. Szwast, A., Tian, Z. T., McCann, M., Soffer, D., Combs, J. et al. (2014). Anatomic variability and outcome in prenatally diagnosed absent pulmonary valve syndrome. Annals of Thoracic Surgery, 98(1), 152–158. DOI 10.1016/j.athoracsur.2014.03.002.
17. Blusztein, D. I., Zenter, D., Thompson, T., Jayadeva, P., Liang, D. et al. (2018). Arrhythmogenic right ventricular cardiomyopathy: a review of living and deceased probands. Heart Lung Circulation, 28(7), 1034–1041. DOI 10.1016/j.hlc.2018.07.017.
18. Hall, C., Sutanto, H., Dalageorgou, C., McKenna, W. J., Syrris, P. et al. (2018). Frequency of genetic variants associated with arrhythmogenic right ventricular cardiomyopathy in the genome aggregation database. European Journal of Human Genetics, 26(9), 1312–1318. DOI 10.1038/s41431-018-0169-4.
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |