DOI:10.32604/cmc.2021.016081
| Computers, Materials & Continua DOI:10.32604/cmc.2021.016081 | |
| Article |
BitmapAligner: Bit-Parallelism String Matching with MapReduce and Hadoop
1Department of Computer Science, COMSATS University Islamabad, Wah, 47040, Pakistan
2Department of Computer Science, AIR University Islamabad, Aerospace and Aviation Campus Kamra, 43570, Pakistan
3Department of Computer Science, National Textile University, Faisalabad, 37610, Pakistan
4Department of Industrial Engineering and Research Center for Electrical and Information Technology, Seoul National University of Science and Technology, Nowon-Gu, Seoul, 01811, Korea
5Data Science and Cyber Analytics Research Group, Edinburgh Napier University, Edinburgh, EH11 4DY, UK
*Corresponding Author: Junaid Rashid. Email: junaidrashid062@gmail.com
Received: 22 December 2020; Accepted: 19 March 2021
Abstract: Advancements in next-generation sequencer (NGS) platforms have improved NGS sequence data production and reduced the cost involved, which has resulted in the production of a large amount of genome data. The downstream analysis of multiple associated sequences has become a bottleneck for the growing genomic data due to storage and space utilization issues in the domain of bioinformatics. The traditional string-matching algorithms are efficient for small sized data sequences and cannot process large amounts of data for downstream analysis. This study proposes a novel bit-parallelism algorithm called BitmapAligner to overcome the issues faced due to a large number of sequences and to improve the speed and quality of multiple sequence alignment (MSA). The input files (sequences) tested over BitmapAligner can be easily managed and organized using the Hadoop distributed file system. The proposed aligner converts the test file (the whole genome sequence) into binaries of an equal length of the sequence, line by line, before the sequence alignment processing. The Hadoop distributed file system splits the larger files into blocks, based on a defined block size, which is 128 MB by default. BitmapAligner can accurately process the sequence alignment using the bitmask approach on large-scale sequences after sorting the data. The experimental results indicate that BitmapAligner operates in real time, with a large number of sequences. Moreover, BitmapAligner achieves the exact start and end positions of the pattern sequence to test the MSA application in the whole genome query sequence. The MSA’s accuracy is verified by the bitmask indexing property of the bit-parallelism extended shifts (BXS) algorithm. The dynamic and exact approach of the BXS algorithm is implemented through the MapReduce function of Apache Hadoop. Conversely, the traditional seeds-and-extend approach faces the risk of errors while identifying the pattern sequences’ positions. Moreover, the proposed model resolves the large-scale data challenges that are covered through MapReduce in the Hadoop framework. Hive, Yarn, HBase, Cassandra, and many other pertinent flavors are to be used in the future for data structuring and annotations on the top layer of Hadoop since Hadoop is primarily used for data organization and handles text documents.
Keywords: Next-generation sequencing; multiple sequence alignment; MapReduce; Hadoop; whole-genome; big data; bit-parallelism
Multiple sequence alignment (MSA) involves identifying and comparing the homologous regions among the various sequences in deoxyribonucleic acid (DNA), ribonucleic acid (RNA), and amino acid (AA). MSA perceives the genome mutations that are beneficial for the design of drugs in the pharmaceutical industry, with rare side effects. Therefore, MSA plays a crucial role in the life sciences, health, and society. The recent development of automated next-generation sequencers (NGS) has transformed the biological domain, where data repositories containing a large extent of raw sequences and a low expenditure of DNA profiling have facilitated research and improvement in the field of bioinformatics. The associated data repositories contain big data, such as gene sequences on a large scale. The MSA algorithms must be executed in parallel and distributed over the Hadoop cluster to handle big data efficiently [1].
Moreover, MSA accuracy is also essential as many bioinformatics systems and procedures are dependent on the MSA results. Subsequently, the NGS machines now provide the genome profiles to be analyzed for biological purposes. MSA is a universal example of any mutation in the genome sequence and is a major issue in the field of biology. Therefore, finding precise solutions is essential, along with designing an efficient and accurate approach to achieve the required research objectives. The large amount of data produced by the NGS platforms and the number of repeated reads introduce new challenges in developing novel solutions considering the sequence alignment algorithm’s time and space complexity.
The recent advances in high-throughput sequencing (HTS) utilized a different process to analyze the multi-variant publicly accessible databases such as the Human Genome Project [2], the one thousand Genomes Project, and the Genome 10K Project. The downstream analysis of multiple associated sequences has become a bottleneck for the growing genomic data due to the issues with storage and space utilization in the domain of bioinformatics. The measures employed to analyze the performance of the MSA methods are biological accuracy and computational complexity. Biological accuracy refers to the correspondence between multiple sequences and the actual outcomes, presenting the insertions-deletions (indels) and the gaps found in the exact positions. However, the computational analysis quantifies the execution in terms of the time and utilization of the CPU in memory in the local drive of the Hadoop distributed file system (HDFS). The pipeline of MSA approaches and hybrid techniques with distributed and parallel computing can improve the MSA applications’ running time quality and competency [3].
Furthermore, large data sequences must be processed with high biological accuracy [4]. There are no significant issues encountered if the number of pattern sequences, n, is smaller than the long pattern sequences, L, as most of MSA approaches deal with longer pattern readsr than n. However, the algorithms being used in the current techniques handle a large number of pattern reads as well as longer sequence reads [5].
In cloud computing and big data analytics technology, HDFS overcomes the memory complexity and provides easy access for large data sequences. This study employs the bit-parallelism extended shifts (BXS) in MapReduce, which reduces the amount of time consumed and executes many data sequences [6,7]. The study analyzes the sequence alignment applications using two input pattern sequences and whole-genome sequences to find similar regions. Any insertions or deletions that occur at any position of a specific gene sequence in the whole genome result in the pertinent disease. The insertion or omission of a single base pair has a significant biological impact, due to which biologists require mapping algorithms to analyze the sequence reads. Therefore, it is essential to analyze the vast datasets using exact string-matching algorithms based on either a dynamic or exact approach. They must be capable of solving biological problems without being overwhelmed by large data sequences. An MSA algorithm has been designed and implemented with minor modifications as a hybrid approach since the conventional methods do not work well with large amounts of data. The BXS algorithm is implemented on the parallel and distributed system of Apache’s Hadoop and MapReduce technologies to form a generalized framework of MSA. This generalized framework is flexible and efficient in analyzing large sequences by considering the time and space complexity as well as the biological accuracy. The main objective of the proposed approach is to provide an efficient and exact solution to handle a large amount of sequence data using the MapReduce-based MSA method. The working of the proposed method is as follows. The proposed technique considers a reference file as an input and then considers another test file. Both the files are controlled and managed by the HDFS in the Hadoop framework. Before processing the sequence alignment, the proposed technique converts the test file (the whole-genome sequence) into binaries and organizes the messy sequence according to the length of the pattern reads. If the test file is too large (e.g., greater than 5 GB), it can be divided into small chunk files, e.g., 5 GB into 1 GB files, and further divided into 128 MB files (default block size on HDFS) to process the data efficiently on a computer having a processor with fewer cores. The data is then ready to be executed in order to map (perform sequence alignment on) the pattern and the reference sequence to obtain the result of a similar region. The experimental results indicate that the proposed BitmapAligner operates in real-time with many sequences and produces the exact position of a similar region in the test file (whole-genome sequence).
This study proposes BitmapAligner, using the BXS algorithm and MapReduce, to solve the sequence alignment problem. The limitations of the existing MSA algorithms are addressed and the MapReduce-based bitmask approach is used to solve the genomic problem. The contributions of the paper can be summarized as follows:
1. BitmapAligner converts the input sequence base pair (bp) data into binaries and builds/stores it into the Hadoop framework’s memory cluster to overcome the computational complexity.
2. BitmapAligner performs mapping and shuffles the genome binaries to align each pattern read in a whole-genome sequence.
3. BitmapAligner identifies the longest pattern that allows for the following MapReduce actions to be performed on the candidates: map, shuffle, and reduce. It thus selects the best results and focuses on the biological accuracy, using a bitmask and the extended shift approach to find the corresponding optimal results.
The remainder of this paper is organized as follows. Section 2 describes the related work in this field. Section 3 presents the proposed methodology and the design of BitmapAligner. Section 4 provides the details of the experimental results. Lastly, the conclusion and the future scope are presented in Section 5.
Big data analytic technologies can potentially resolve many bioinformatics issues involving time complexity, memory spacing, and data storage by using large sets of genomes. Performance is a significant issue for billions of base-pairs in the sequences, which can be resolved with the combination of MapReduce and HDFS [8]. The use of MapReduce has become increasingly widespread as the most preferable solution for big data problems. It exploits the parallelism among the computing nodes to process large amounts of data sets. MapReduce consists of two main phases: Map and Reduce.
For the past few decades, researchers have developed the string-matching algorithms of four major techniques: 1) character comparison, 2) bit-parallelism, 3) automata, and 4) packed string matching. The MSA algorithms are classified into five different groups depending on the index property: 1) hash tables, 2) suffix trees, 3) bitmask indexing, 4) packed string matching, and 5) merge sorting [9,10].
Recently, various tools have been developed for MSA applications using the hash-based indexing and the seeds-and-extend approaches commonly used in contemporary techniques [11,12], as shown in Fig. 1. In contrast to these methods, BXS strongly authenticates the usage of bitmask indexing as an exact solution for MSA without any risk of error.
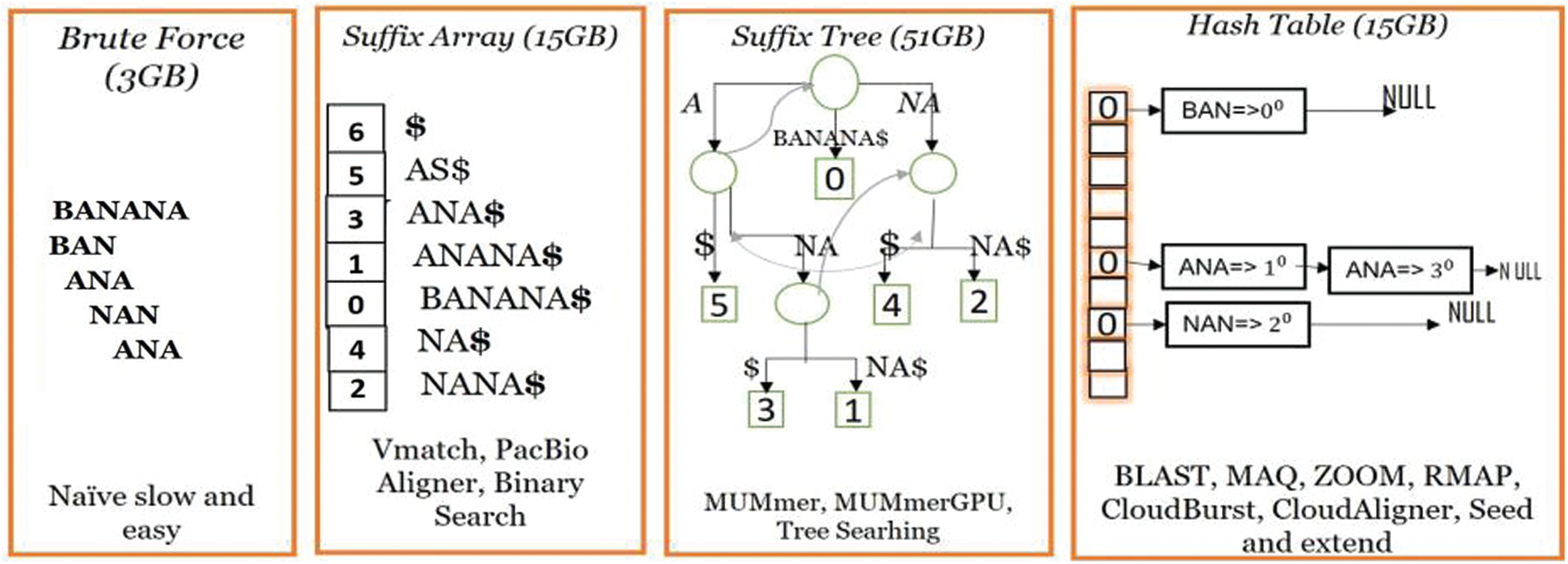
Figure 1: Paradigm of the existing MSA approaches
The short read sequences [13] and the paired alignment approaches [14] provide high accuracy and speed; however, they are limited when applied to large sequences. Similarly, techniques like SOAP (short oligonucleotide alignment program), MrsFAST (A cache-oblivious algorithm for short-read mapping) [15], and BioPig face the same challenges while processing large scale data. Additionally, CloudAligner [16] is proposed to find mismatches and bisulfite mapping, and the seed-and-extend algorithm is employed to MapReduce to increase the processing speed of MSA, resulting in improved performance when compared to CloudBurst [17] and RMAP [18]. Comparatively, BulkAligner [19] performed better than CloudAligner and CloudBurst when implemented in the MapReduce function due to its De-Bruijn graph strategy, which works for long sequences. It features a unique ability to search for long pattern sequences in the whole-genome reference sequence while calculating the similar regions of the pattern sequence.
Approaches such as [20–22], which work for single nucleotide polymorphisms (SNPs), genotyping, and personal genomics, are developed using the MapReduce implementations. Most short-read MSA packages are open-source. However, packages such as NovoAlign and Eland are available for commercial usage. Due to the production rate of large-scale data, traditional bioinformatics tools are unsuitable for handling large amounts of data. Therefore, research has focused on the development of both new and existing tools to obtain improved results. In this context, the Hadoop-based MapReduce functions are utilized in the MSA algorithm to resolve big data-related issues.
Unfortunately, the approaches that reduce the risk of errors in the sequence alignment process cannot provide dynamic and exact solutions. The strategies such as Halign [23] and Halvade [24] address the sequence alignment issue but do not focus on the error-free alignment. Similarly, the consistency-based MSA methods [25] provided pairwise alignment and introduced big data technologies. The algorithm is limited when applied for large sequences and cannot entirely reduce the error risk resulting from the alignment. BLAST is widely used for sequence alignment. In [26], the existing algorithms such as Needleman–Wunsch, Smith–Waterman, and BLAST are employed along with Hadoop or other big data technologies to scale down the time, memory memory memory and the CPU consumption. Spark MSNA (Multiple Sequence Nucleotide Alignment) services are used to compare the suffix tree approach [27]. FASTdoop [28] is able to load the FASTA and FASTQ input files for bioinformatics applications on the MapReduce framework. BitmapAligner considers the FASTA file as input, converts it into binaries, and sequences the whole data suitably for the suggested algorithm. Additionally, the biological accuracy along with the computational accuracy and efficiency considered as the primary objectives.
MSA is employed in the Spark framework to obtain optimized and efficient results based on the suffix tree and the center-star strategy. This technique is used by SparkBWA, which is another tool that has high speed and addresses the MSA on a large scale by using Spark [29].
Conversely, most of the MSA applications presented in the previous studies are based on the seeds-and-extend and other conventional error-prone methods. Such methods are efficient and maintain a good balance between performance and accuracy. However, the accuracy of these methods is reduced when they are applied for big data. More importantly, from the literature review, it is observed that employing the BXS in the MapReduce framework can optimize the biological accuracy and the computing infrastructure of MSAs and increase the efficiency. Therefore, the MapReduce function in the Hadoop framework is employed in this study to obtain the objectives of BitmapAligner.
This section provides the details of the core problem to explain the MSA process. The MSA package is designed with relevant features to help biologists analyze the DNA sequences for usage. Consequently, the BXS package is implemented with MapReduce while the HDFS file system processes large-scale data sequences in a Linux operating system environment. The proposed approach, BitmapAligner, identifies the homology regions with their possible counts in the genome sequence. The computational complexity of the proposed approach is also considered, and the analysis is presented to further understand the comparative effects of the BNDM (backward non-deterministic directed acyclic word graph matching) algorithm.
Fig. 2 shows the complete paradigm and flow of the proposed approach. The step-by-step process of BitmapAligner is outlined as follows.
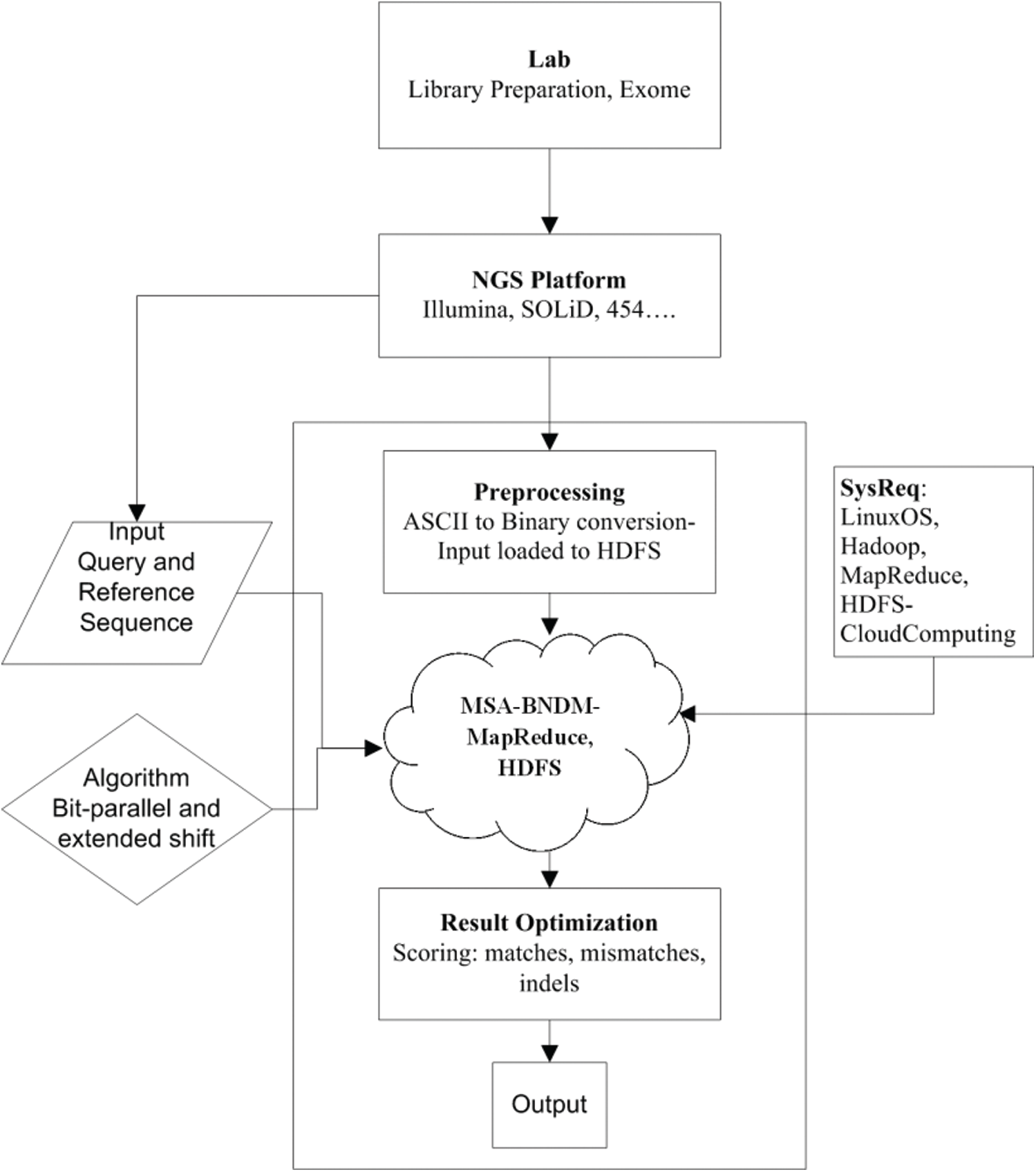
Figure 2: Paradigm of the proposed method
Step 1: BitmapAligner uses nucleotide sequences as pattern sequences to find the start and end positions in the whole-genome sequence. The pattern sequence is defined as a concerned gene sequence, while the whole-genome sequence is considered as a complete DNA profile. The genome data libraries are initially prepared in the biological incubation labs through the given specimen i.e., blood, skin, or hair. The NGS then processes the sequence data into a readable format. The genome sequences profiling as ATGC is generated into a readable format through the NGS platforms, such as Illumina, ABiSOLID, Live Sciences, and Roche 454. In this study, the sequence data of the Illumina sequencer are obtained from the publicly accessible NCBI (National Center for Biotechnology Information) database. The produced sequences are then normalized to reduce the irrelevant data sequences. There are two input files used in the MSA analysis: the first one is used as a pattern (query) sequence and the other is used as a test file (whole-genome). BitmapAligner searches for several pattern sequences in the whole-genome sequence (test file).
Step 2: The next step involves pre-processing the data sequences, including those encoded into binaries (ASCII conversion). BitmapAligner takes a reference file and a test file from the user, and then manages and organizes them in the HDFS. Before processing the sequence alignment, the proposed algorithm converts the test file (whole-genome sequence) into binaries of equal length, line by line. If the test file is relatively large (greater than 5 GB), it divides the input file into smaller files (5 GB into 1 GB), and each file is used to process the data efficiently on a computer with a few computing cores. The small-sized sequence data are then processed more efficiently by BitmapAligner with fewer cores in its processor. Otherwise, it would generate a storage error in the HDFS local memory because large-scale data requires more processing memory. Therefore, the large files are split into smaller files to test BitmapAligner, and the application is implemented on the Linux operating system. The Hadoop framework is essentially used to process large-scale data and the BXS is a sequence alignment algorithm that identifies similar patterns within the repeated number of sequences with complete accuracy; this is validated during the test experiment.
Step 3: The BXS is implemented through a MapReduce-based distributed environment. Numerous pattern sequences, used as the concerned gene sequences to find the start and end positions in the target sequence, are taken as the inputs. These start and end positions located in the target genome show the similar region, which is the primary focus of biologists to analyze any mutations that may be present. BitmapAligner accurately detects similar regions in the target sequence. The biologists considered these detected regions to determine whether the genes are mutated or normal.
Step 4: In this step, the BXS, which is an extended form of BNDM, is employed in the MapReduce functionality. The BXS is a bitmask in the binary block property, which enables the identification of similar locations without generating any errors, while the MapReduce implementations handle a large amount of data. The BXS is a dynamic and exact approach used for biological problems. Therefore, this method is implemented with MapReduce in a Java environment and is executed over the Hadoop framework. The optimized MSA results are obtained for large-scale datasets. Fig. 3 depicts the detailed specifications and the architecture of the designed BitmapAligner, which deals with large amounts data and utilizes the memory space of the HDFS. These features enhanced the performance of the tool in solving the MSA-related problems.
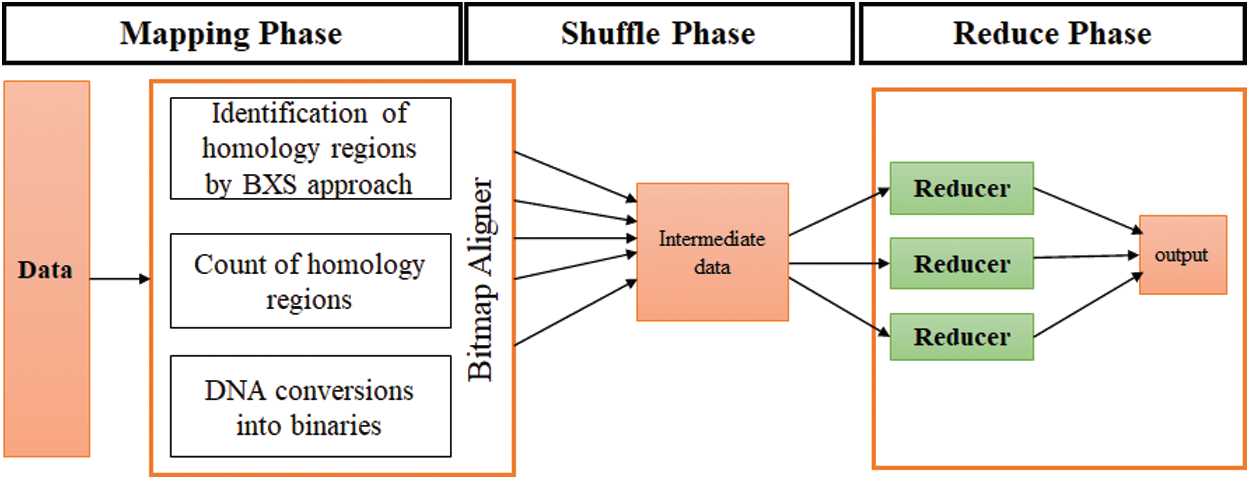
Figure 3: Architecture of Bitmapaligner
Step 5: After designing the basic structure of BitmapAligner, the MSA application worked through MapReduce in three phases: a) mapping phase, b) shuffle phase, and c) reduce phase. BitmapAligner gathers all the similar regions of the sequences being identified through the BXS approach in the mapping phase. The output of the mapping phase is in the form of key-value pairs. In the shuffle phase, the intermediate results are shuffled and sorted. The in-node combiner function is then used to gather the results of the map phase. The combiner aggregates the same keys and sends the results to the reducer. Lastly, the reduce phase discards the irrelevant sequences and produces the desired results for the MSA application.
Step 6: The final step includes the output, which contains the matches and the mismatches observed in the genome sequence that indicate the pattern sequence’s correct location within the whole-genome sequence. The results provide information on whether there is any mutation in the genome sequence. The pattern sequence’s correct location is viewed as both the start and end positions in the whole-genome sequence (test file).
The experimental setup and the discussion on the user inputs and the proposed algorithm results are presented in the following sub-sections.
Apache Hadoop 2.7.3 version is installed in the Linux operating system. Hadoop is a large cluster of commodity hardware which incorporates and analyzes large-scale data for MSA. It also supports distributed file systems and the MapReduce paradigm. HDFS features high fault tolerance and is capable of large-scale data storage. These services facilitate high throughput access to process the genome’s large-scale data, e.g., billions of base pairs. The data node configured with Hadoop is created on a disk with a capacity of 18.58 GB, in which the DFS utilizes 46.01 MB and the remaining space of the DFS is 11.31 GB.
The BXS algorithm is implemented in Java (.jar file) to be employed in the MapReduce phases (mapping, shuffling, and reduction). The input files are placed and managed in the HDFS before the execution of the MSA application. BitmapAligner fetches the inputs for the sequence alignment after executing the command.
4.2 Computational Complexity of BXS Algorithm
The adopted MapReduce and HDFS implementations achieved efficiency in space and time utilization while processing big data. To create bitmask indexing, the BXS encodes the genome sequences into binary sequences. The sequence alignment is then applied to the binary sequences, removing the error risk. It also helps in handling lengthy pattern sequences and producing accurate matching without any error risk. Therefore, the bit-masking approach is employed for multiple sequence alignment. A set of pattern sequences of equal length, R, which should be aligned in a whole-genome sequence, is shown in Eq. (1):
Eq. (2) shows the class distribution set, R′, which is used to construct the class distribution for each pattern sequence [30].
where
where n is a constant and
The complexity given above is valid for random pattern sequences and whole-genome sequences. The pattern sequence, R′, is structurally organized and is not random. The algorithm handles the repetitive short reads (patterns) better than the random short reads. The probability of a random text substring corresponds to the pattern for any position that is lower than the iterative short reads. The optimal time complexity of this string matching algorithm is defined in Eq. (3); the BXS is thus worse than the optimal by a factor of
The short read patterns are selected as a query sequence and a genome sample is selected as a reference sequence dataset1 from the publicly available Gene Expression Omnibus (GEO) database. The real high-throughput genomic data of Homo sapiens, platform ID GPL10295, which is produced by the Illumina sequencer at the Dana-Farber Cancer Institute, Boston USA, is retrieved from this dataset. The series GSE21257, containing genome-wide gene expression profiling on the pre-chemotherapy biopsies of osteosarcoma patients who developed metastases within 5 years for n = 34, where n is the number of patients, is specifically analyzed in this study. The patients developed metastases within 5 years (n = 19). The growth of the bone can be interpreted from the several genes that are discovered, and these gene sequences are used as a biomarker signature consisting of various similar functional genes in the FASTA2 format. During the experiment, it is observed that searching the similar regions is helpful in promoting further studies into the treatment of any genetic disease if the mutation occurs in any genes, as the genes follow a similar functional behavior.
The short-read MSA provides guidelines for further medical treatments to address the genetic problems that may cause cancer to increase cancer patients’ survival rate. The retrieved results are also evaluated using different datasets gathered from online databases for analysis and annotations. Various performance parameters are used to determine the next-generation MSAs’ performance next-generation MSAs’ performance, integrated with cloud virtual machines, to handle large amounts of data and simplify the accessibility in the distributed environment. The sequence file format is FastQ or FASTA, which is produced by the NGS platform as the input. The computed results include the score of the matches, the mismatches, or the indels, where the output is generated in the .txt format by BitmapAligner.
The biological accuracy and the computational time is also analyzed to evaluate the proposed BitmapAligner. A generalized framework model is developed to include a modified dynamic-based BXS and is employed in the MapReduce implementations. The HDFS, which works with parallel execution using multiple computing nodes to process the large amount of genomic data, is also included in the proposed technique. BitmapAligner is a combination of the BXS, MapReduce, and the HDFS. The BXS provides accuracy in the MSA calculations of the large-scale data sequences. MapReduce and the HDFS are used to store and manage data on a single machine. The raw data sequences must also be normalized to maintain the quality of the data. It is observed from the experiments that BitmapAligner can increase the flexibility of the sequence alignment as per the user requirements.
The MSA application’s primary objectives in the bioinformatics field, which include the biological and the computational accuracy, achieved by employing the BXS in MapReduce. The biological accuracy refers to the accurate identification of the start and end positions of the pattern sequences. The computational accuracy involves handling the large amount of data sequences for the MSA problem. MSA plays a vital role in the medical field in providing guidelines at the molecular level. The biological accuracy enables identifying the index value of a similar region within the large amount of sequence data. Therefore, BitmapAligner adapts the BXS approach, which provides the exact location of the test file pattern.
The BitmapAligner pipeline is performed on Hadoop 2.0 installed in a Linux operating system and is coded in Java. While testing BitmapAligner, it is observed that the locations of the pattern sequences found in the whole-genome sequence are relatively accurate. The exact locations are identified by the property of bit-mask indexing blocks of the BXS algorithm applied in BitmapAligner. This algorithm presents good performance even with longer gene sequences when compared to the various string-matching algorithms, such as naive binary search, tree searching, and the seeds-and-extend approach. The major limitation of a multiple sequence alignment is processing the 100K pattern gene sequences in the whole-genome sequence. The MapReduce function of Hadoop is able to handle this large-scale task efficiently.
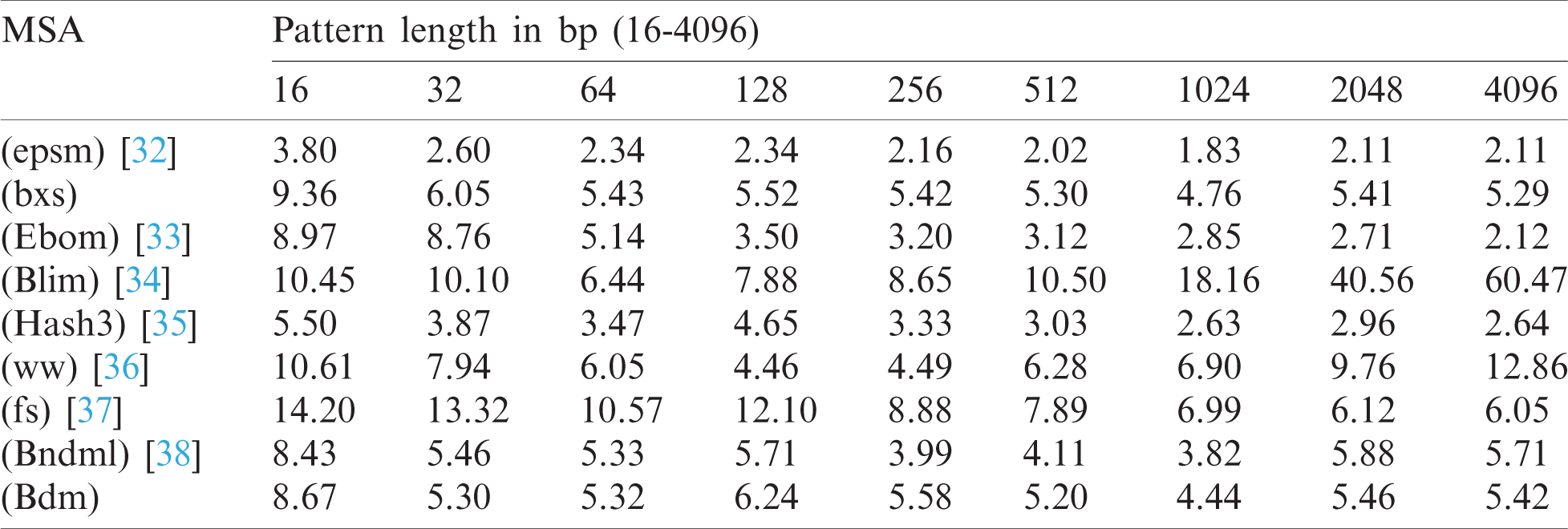
The test cases accredited to BitmapAligner is denoted by either capital letters (ATCG) or small letters (ATCG) when providing the inputs of the nucleotides. In addition, it constitutes the feature to convert both the files in the same level of characters to execute the MSA command successfully. Moreover, BitmapAligner splits the large-sized input file into smaller equal-sized files while storing them in the HDFS to process a large number of data sequences. The large-sized file is split to avoid the java heap space error due to the low storage capacity of the RAM, which does not read larger files. It is also observed that an efficient hardware specifications system is required to reduce the time consumption and improve the memory optimization. The proposed BitmapAligner can also efficiently process multiple files simultaneously. The test data of the E. coli from the Large Canterbury Corpus of 4 MB with different pattern lengths from 16 to 4096 bp (base pairs) are used to compare BitmapAligner with other various string-matching algorithms, as shown in Tab. 1, to obtain empirical insights. This test validation is performed by using the String Matching Algorithms Research Tool (SMART). SMART contains comprehensive online string-matching algorithms to test and validate the performance of the concerned algorithm. Consequently, all the string matching algorithms are implemented in the C language and are divided into four groups: 1) bit-parallelism, 2) packed string matching, 3) character comparisons, and 4) deterministic automata (further classification is possible) [31].
SMART is used in the experiment to compare the various string-matching algorithms. It is observed from Tab. 1, that the algorithms consumed a larger amount of time for smaller pattern lengths than the longer patterns. Furthermore, the traditional string-matching algorithms are efficient due to the limited size of the data sequences. The BXS algorithm, which is tested over Hadoop clusters, shows good performance for the large-sized data. The selection of the algorithm is based on providing an error-free and exact solution.
Table 1: Comparison of various MSA approaches (running time in milliseconds)
It is observed from Fig. 4, that the BXS improves the accuracy and efficiency of the running time when compared to Window Wide (WW) and Fastest Search (FS).
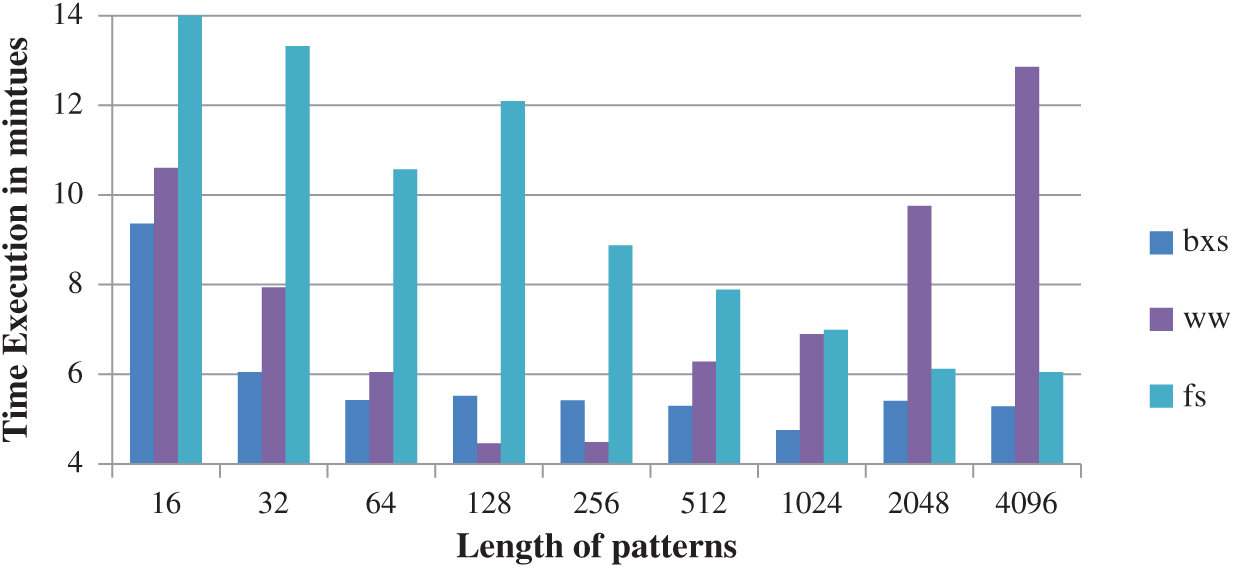
Figure 4: Comparison of bxs with WW and FS
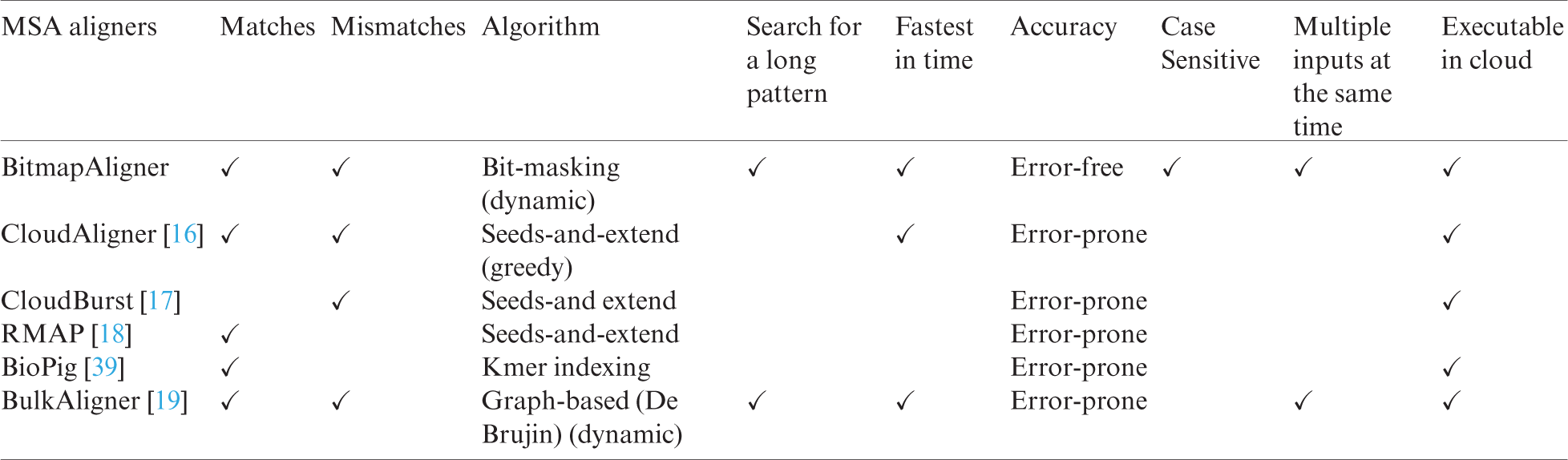
The performance of the proposed BitmapAligner is compared with its counterparts, CloudAligner, CloudBurst, RMAP, BioPig, and BulkAligner, to validate MSA for large data files. BitmapAligner presents greater biological accuracy, identifies the pattern sequences character by character, fetches the position, and counts the homologous gene sequence in the whole-genome sequence. BitmapAligner and its counterpart aligners’ parameters are shown in Tab. 2, indicating that BitmapAligner is error-free due to its bit-masking approach in the MSA application.
Table 2: Comparison of BitmapAligner with its counterparts
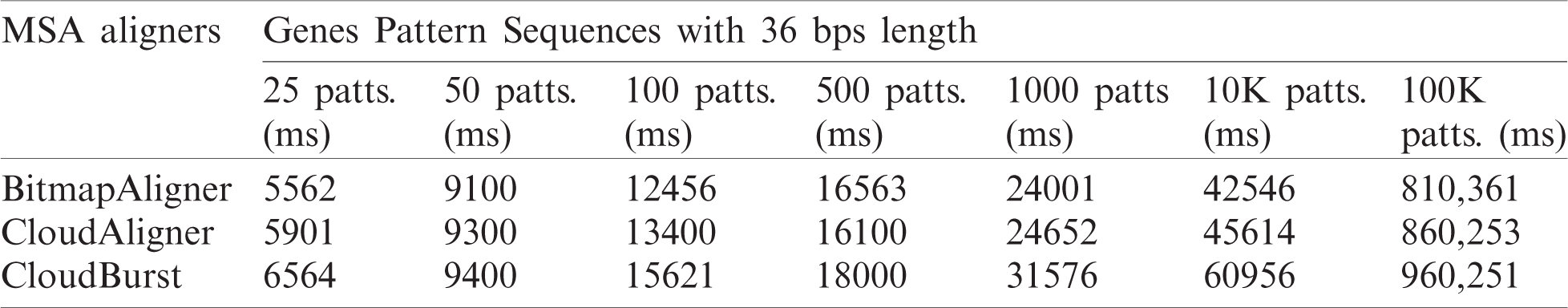
Tab. 3 provides the experimental results of the comparison between BitmapAligner and its counterpart aligners with the variation in the number of pattern sequences. There are 100K pattern sequences used to find such sequences in the whole-genome sequence file. The 100K pattern is divided into 25, 50, 100, 500, 1000, 10, and 100K pattern sequences to test and evaluate milliseconds’ running time. Each pattern sequence consists of 36 bps in length. During the validation of this experiment, the biological accuracy of BitmapAligner is established, where the accurate position is fetched from the target sequence; the computational accuracy is also established, which refers to the in-memory and the time efficiency.
Table 3: Running time of BitmapAligner and its counterparts (in milliseconds)
As the number of computing cores is increased, the computation time of processing the MSA in BitmapAligner is improved significantly for a large amount of data, which is the desired property of the proposed BitmapAligner. Tab. 4 shows that an increased number of computing cores is inversely proportional to the execution time of BitmapAligner. Increasing the RAM capacity also improves the processing time of BitmapAligner.
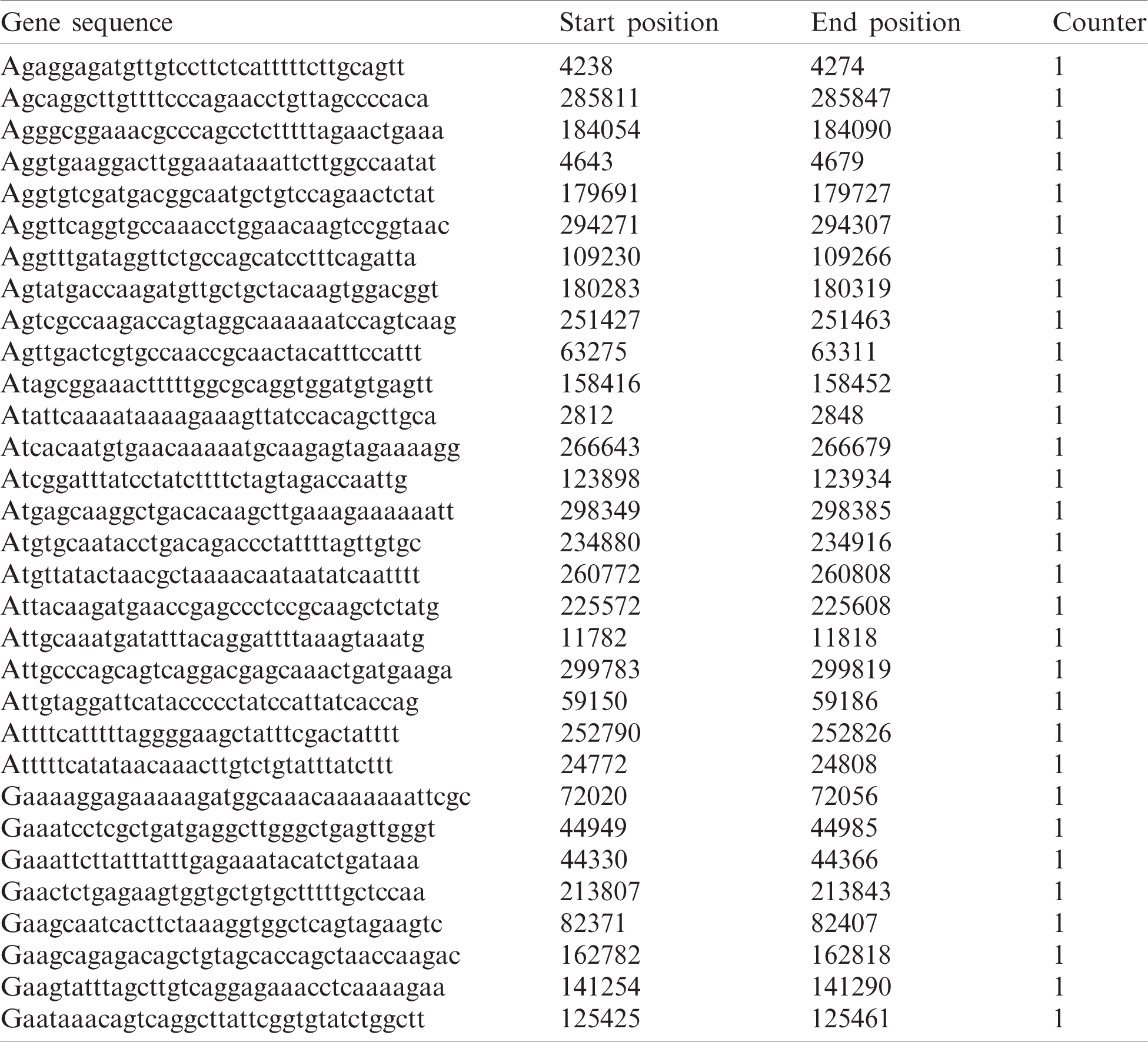
The proposed method is tested on a heterogeneous dataset containing 100K short reads aligned with Streptococcus. Notably, a molecular biomarker signature comprises 45 specific genes that support bone growth and development. If there is a mutation in any of these genes, it causes bone cancer. For medical treatment, the normal signature genes are mapped with the mutated genes to identify the mutated occurrence. Tab. 5 presents the output generation of BitmapAligner, including the gene sequence, start position, end position, and counts of the homology region. The data sequences of these gene sequences are tested and taken from the Gene Expression Omnibus database. Several string matching algorithms are tested through variant patterns aligned with the E. coli genome of 4 MB available in the NCBI database. The performance of the proposed method is measured by the accuracy and the time and space utilization.
Table 4: Execution time of BitmapAligner as the number of computing cores is varied

Table 5: Result generation by BitmapAligner
The NGS sequence data production has been improved and is less expensive due to the NGS platforms’ advancements. Therefore, a large amount of genome data is progressively produced, creating issues in processing large amounts of data for downstream analysis, particularly for MSA. This study proposed BitmapAligner, which employs the BXS algorithm with bitmask indexing and MapReduce for MSA in the Hadoop framework. BitmapAligner successfully achieves the objectives of space and time complexity using arithmetic and logical operations with bit-mask indexing instead of the traditional seeds-and-extend approach. BitmapAligner is tested on 100K patterns that are used as query sequences and a Streptococcus genome sample is used as a reference sequence, from www.sanger.ac.uk. The validation test of the real data on BitmapAligner presented improved performance in identifying the homology regions (the start and end positions) of the specific genes in the whole-genome sequence, thus achieving this study’s objectives. The 4.18 MB data size of pattern reads and 32.4 MB size of the genome sample is tested. BitmapAligner is compared with various existing algorithms, exhibiting the bit-masking technique’s improved performance compared to the seeds-and-extend, FS, WW, and Blim approaches. BitmapAligner also showed better performance and greater accuracy for longer patterns.
Researchers are constantly challenged by the new and increasing types of big data (whole-genome sequences) obtained from the advanced NGS platforms. A majority of the MSA applications are still dependent on the Hadoop-based MapReduce to process large amounts of data. However, Hadoop is primarily used for data organization and handles text documents; therefore, Hive, Yarn, HBase, Cassandra, and many other pertinent flavors are used for data structuring and annotations the top layer of Hadoop.
Funding Statement: This work was supported in part by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. 2018R1C1B5084424), and in part by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (No. 2019R1A6A1A03032119).
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
1In the field of https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc = GSE21257
2In the field of bioinformatics, FASTA format is used as a text-based format to represent either nucleotide sequences or peptide sequences.
1. J. Daugelaite, A. O. Driscoll and R. D. Sleator, “An overview of multiple sequence alignments and cloud computing in bioinformatics,” ISRN Biomathematics, vol. 2013, no. 6, pp. 1–14, 2013. [Google Scholar]
2. A. Bogojeska, S. Kalajdziski and L. Kocarev, “Next-generation DNA sequencing technology, challenges and bioinformatics approaches for sequence alignment 2 next generation sequencing technologies,” ICT Innovations, vol. 2010, pp. 271–280, 2010. [Google Scholar]
3. J. Luo, M. Wu, D. Gopukumar and Y. Zhao, “Big bata application in biomedical research and health care: A literature review,” Biomedical Informatics Insights Biomedical Informatics Insights, vol. 8, pp. 1–10, 2016. [Google Scholar]
4. G. Cattaneo, U. Ferraro, R. Giancarlo and G. Roscigno, “An effective extension of the applicability of alignment-free biological sequence comparison algorithms with Hadoop,” Journal of Supercomputing, vol. 73, no. 4, pp. 1467–1483, 2017. [Google Scholar]
5. A. Cinti, F. M. Bianchi, A. Martino and A. Rizzi, “A novel algorithm for online inexact string matching and its FPGA implementation,” Cognitive Computation, vol. 12, no. 2, pp. 369–387, 2020. [Google Scholar]
6. L. Santana-Quintero, H. Dingerdissen, J. Thierry-Mieg, R. Mazumder and V. Simonyan, “HIVE-hexagon: High-performance, parallelized sequence alignment for next-generation sequencing data analysis,” PLoS One, vol. 9, no. 6, pp. 1–11, 2014. [Google Scholar]
7. S. Pabinger, A. Dandar, M. Fisher, R. Snajder, M. Sperk et al., “A survey of tools for variant analysis of next-generation genome sequencing data,” Briefing in Bioinformatics, vol. 15, no. 2, pp. 256–278, 2014. [Google Scholar]
8. M. A. Sarwar, A. Rehman and J. Ferzund, “Database search, alignment viewer and genomics analysis tools: Big data for bioinformatics,” International Journal of Computer Science and Information Security, vol. 14, no. 12, pp. 317–328, 2016. [Google Scholar]
9. N. Malhis, Y. S. N. Butterfield, M. Ester and S. J. M. Jones, “Slider-maximum use of probability information for alignment of short sequence reads and SNP detection,” Bioinformatics, vol. 25, no. 1, pp. 6–13, 2009. [Google Scholar]
10. N. Malhis and S. J. M. Jones, “High quality SNP calling using Illumina data at shallow coverage,” Bioinformatics, vol. 26, no. 8, pp. 1029–1035, 2010. [Google Scholar]
11. I. Khurram, M. M. Fraz, M. Shahzad and N. M. Rajpoot, “Dense-captionnet: A sentence generation architecture for fine-grained description of image semantics,” Cognitive Computation, vol. 2020, pp. 1–17, 2020. [Google Scholar]
12. S. Michael and S. Alignment, “Sequence alignment,” Bioinformatics, vol. 16, pp. 425–438, 2010. [Google Scholar]
13. B. Langmead, C. Trapnell, M. Pop and S. L. Salzberg, “Ultrafast and memory-efficient alignment of short DNA sequences to the human genome,” Genome Biology, vol. 10, no. 3, pp. R25, 2009. [Google Scholar]
14. B. Langmead and S. L. Salzberg, “Fast gapped-read alignment with bowtie 2,” Nature Methods, vol. 9, no. 4, pp. 357–359, 2012. [Google Scholar]
15. R. Li, Y. Li, K. Kristiansen and J. Wang, “SOAP: Short oligonucleotide alignment program,” Bioinformatics, vol. 24, no. 5, pp. 713–714, 2008. [Google Scholar]
16. T. Nguyen, W. Shi and D. Ruden, “CloudAligner: A fast and full-featured mapreduce based tool for sequence mapping,” BMC Research. Notes, vol. 4, no. 1, pp. 1–7, 2011. [Google Scholar]
17. M. C. Schatz, “CloudBurst: Highly sensitive read mapping with mapreduce,” Bioinformatics, vol. 25, no. 11, pp. 1363–1369, 2009. [Google Scholar]
18. A. D. Smith, W. Y. Chung, E. Hodges, J. Kendall, G. Hannon et al., “Updates to the RMAP short-read mapping software,” Bioinformatics, vol. 25, no. 21, pp. 2841–2842, 2009. [Google Scholar]
19. J. Lee, Y. Yeu, H. Roh, Y. Yoon and S. Park, “BulkAligner: A novel sequence alignment algorithm based on graph theory and trinity,” Information Sciences, vol. 303, no. S1, pp. 120–133, 2015. [Google Scholar]
20. M. Ruffalo, T. Laframboise and M. Koyutürk, “Comparative analysis of algorithms for next-generation sequencing read alignment,” Bioinformatics, vol. 27, no. 20, pp. 2790–2796, 2011. [Google Scholar]
21. G. Lunter and M. Goodson, “Stampy: A statistical algorithm for sensitive and fast mapping of illumina sequence reads,” Genome Research, vol. 21, no. 6, pp. 936–939, 2011. [Google Scholar]
22. R. Zhou and L. Niu, “Feature selection of network data VIA l 2, p regularization,” Cognitive Computation, vol. 12, no. 6, pp. 1217–1232, 2020. [Google Scholar]
23. Q. Zou, Q. Hu, M. Guo and G. Wang, “Sequence analysis halign: Fast multiple similar DNA/RNA sequence alignment based on the centre star strategy,” Bioinformatics, vol. 31, no. 15, pp. 2475–2481, 2018. [Google Scholar]
24. D. Decap, J. Reumers, C. Herzeel, P. Costanza and J. Fostier, “Sequence analysis halvade: Scalable sequence analysis with mapreduce,” Bioinformatics, vol. 31, no. 15, pp. 2482–2488, 2018. [Google Scholar]
25. S. He, J. Huang and X. He, “Collective neurodynamic optimization for image segmentation by binary model with constraints,” Cognitive Computation, vol. 12, no. 6, pp. 1265–1275, 2020. [Google Scholar]
26. M. Gaikwad and S. Ahirrao, “BLAST using big data technologies: A survey,” in Fourth Int. Conf. on Computing Communication Control and Automation, Pune, India, pp. 1–5, 2018. [Google Scholar]
27. V. Vineetha, C. L. Biji and A. S. Nair, “SPARK-MSNA: Efficient algorithm on apache spark for aligning multiple similar DNA/RNA sequences with supervised learning,” Scientific Report, vol. 9, no. 1, pp. 1–11, 2019. [Google Scholar]
28. U. F. Petrillo, G. Roscigno, G. Cattaneo and R. Giancarlo, “FASTdoop: A versatile and efficient library for the input of FASTA and FASTQ files for mapreduce hadoop bioinformatics applications,” Bioinformatics, vol. 33, no. 10, pp. 1575–1577, 2017. [Google Scholar]
29. J. M. Abuín, J. C. Pichel, T. F. Pena and J. Amigo, “SparkBWA : Speeding up the alignment of high-throughput DNA sequencing data,” PloS one, vol. 11, no. 5, pp. 1–21, 2016. [Google Scholar]
30. B. Durian, H. Peltola, L. Salmela and J. Tarhio, “Bit-parallel search algorithms for long patterns,” Lecture Notes Computer Science, vol. 6049, pp. 129–140, 2010. [Google Scholar]
31. N. Zemmal, N. Azizi, M. Sellami, S. Cheriguene, A. Ziani et al., “Particle swarm optimization based swarm intelligence for active learning improvement: Application on medical data classification,” Cognitive Computation, vol. 12, no. 5, pp. 991–1010, 2020. [Google Scholar]
32. Z. Yao, F. Hongbo, H. Qingsong and L. Lijun, “Fast string matching algorithms for very short patterns,” in IET, Shenzhen, pp. 1–64, 2012. [Google Scholar]
33. S. Faro and T. Lecroq, “Efficient variants of the backward-oracle-matching algorithm,” International Journal of Foundation of Computer Science, vol. 20, no. 6, pp. 967–984, 2009. [Google Scholar]
34. K. Simov, P. Koprinkova and A. Popov, “A reservoir computing approach to word sense disambiguation,” Cognitive Computation, vol. 12, pp. 1–10, 2020. [Google Scholar]
35. T. Lecroq, “Fast exact string matching algorithms,” Information Processing Letter, vol. 102, no. 6, pp. 229–235, 2007. [Google Scholar]
36. L. He, B. Fang and J. Sui, “The wide window string matching algorithm,” Theoretical Computer Science, vol. 332, no. 1, pp. 391–404, 2005. [Google Scholar]
37. D. Cantone and S. Faro, “Fast-Search: A new efficient variant of the boyer-moore string matching algorithm,” in Int. Workshop on Experimental and Efficient Algorithms, Rome, Italy, pp. 47–58, 2007. [Google Scholar]
38. G. Navarro and M. Raffinot, “A bit-parallel approach to suffix automata: Fast extended string matching,” in Annual Symp. on Combinatorial Pattern Matching, Piscataway, NJ, USA, pp. 14–33, 1998. [Google Scholar]
39. H. Nordberg, K. Bhatia, K. Wang and Z. Wang, “BioPig: A hadoop-based analytic toolkit for large-scale sequence data,” Bioinformatics, vol. 29, no. 23, pp. 3014–3019, 2013. [Google Scholar]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |