DOI:10.32604/cmc.2022.018860
| Computers, Materials & Continua DOI:10.32604/cmc.2022.018860 | |
| Article |
An Improved Convolutional Neural Network Model for DNA Classification
1Department of Information Technology, College of Computer and Information Sciences, Princess Nourah Bint Abdulrahman University, Riyadh, Saudi Arabia
2Department of Electronics and Electrical Communications Engineering, Faculty of Electronic Engineering, Menoufia University, Menoufia, 32952, Egypt
3Department of Computer Science and Engineering, Faculty of Electronic Engineering, Menoufia University, Menoufia, 32952, Egypt
*Corresponding Author: Naglaa. F. Soliman. Email: nfsoliman@pnu.edu.sa
Received: 24 March 2021; Accepted: 11 June 2021
Abstract: Recently, deep learning (DL) became one of the essential tools in bioinformatics. A modified convolutional neural network (CNN) is employed in this paper for building an integrated model for deoxyribonucleic acid (DNA) classification. In any CNN model, convolutional layers are used to extract features followed by max-pooling layers to reduce the dimensionality of features. A novel method based on downsampling and CNNs is introduced for feature reduction. The downsampling is an improved form of the existing pooling layer to obtain better classification accuracy. The two-dimensional discrete transform (2D DT) and two-dimensional random projection (2D RP) methods are applied for downsampling. They convert the high-dimensional data to low-dimensional data and transform the data to the most significant feature vectors. However, there are parameters which directly affect how a CNN model is trained. In this paper, some issues concerned with the training of CNNs have been handled. The CNNs are examined by changing some hyperparameters such as the learning rate, size of minibatch, and the number of epochs. Training and assessment of the performance of CNNs are carried out on 16S rRNA bacterial sequences. Simulation results indicate that the utilization of a CNN based on wavelet subsampling yields the best trade-off between processing time and accuracy with a learning rate equal to 0.0001, a size of minibatch equal to 64, and a number of epochs equal to 20.
Keywords: DNA classification; CNN; downsampling; hyperparameters; DL; 2D DT; 2D RP
Technological advances in DNA sequencing allowed sequencing of the genome at a low cost within a reasonable period. These advances induced a huge increase in the available genomic data. Bioinformatics addresses the need to manage and interpret the data that is massively generated by genomic research. Computational DNA classification is among the main challenges, which play a vital role in the early diagnosis of serious diseases. Advances in machine learning techniques are expected to improve the classification of DNA sequences [1]. Recently, survey studies have been presented by Leung et al. [2], Mamoshina et al. [3], and Greenspan et al. [4]. These studies discussed bioinformatic applications based on DL. The first two are limited to applications in genomic medicine and the latter to medical imaging. The DL is a relatively new field of artificial intelligence, which achieves good results in the areas of big data processing such as speech recognition, image recognition, text comprehension, translation, and genomics.
There are several contributions based on DL in the fields of medical imaging and genomic medicine. However, the DNA sequence classification issue has received little attention. For an in-depth study of DL in bioinformatics, we can consider the review study conducted by Seonwoo et al. [5]. In addition, several studies have been devoted to the utilization of CNNs and recurrent neural networks (RNNs) in the field of bioinformatics and DNA classification [6,7].
The classification task based on CNNs depends on several layers. Tab. 1 provides a list of the basic functions of a variety of CNN layers [5].

Rizzo et al. [8] presented a DNA classification approach that depends on a CNN, and the spectral representation of DNA sequences. From the results, they found that their approach provided similar and good results between 95% and 99% at each taxonomic level. Moreover, Rizzo et al. [1] suggested a novel algorithm that depends on CNNs with frequency chaos game representation (FCGR). The FCGR was utilized to convert the original DNA sequence to an image before feeding it to the CNN model. This method is considered as an expansion of the spectral representation that was reported to be efficient. This work is a continuation of the work of Rizzo et al. [1] for the classification of DNA sequences using a deep neural network, and chaos game representation, except for the addition of downsampling layers that can achieve the best trade-off between performance and time of processing, which is the main contribution of this work. The proposed approach is an improved form of the CNN to obtain better classification accuracy.
A weakness of the convolutional layer performance is that it reports the exact position of features in the input. Slight shifts in the features located in the input image contribute to different feature maps. The pooling layer is used to resize the feature maps to overcome this problem. A simplified representation of the features observed in the input is the outcome of using a pooling layer. In practice, max-pooling works better than average pooling for computer vision fields such as image recognition [9]. We can handle this issue in signal processing by using downsampling methods such as 2D RP, two-Dimensional two-Directional Random Projection (
Dimensionality reduction methods can be briefly categorized into two classes, namely subspace and feature selection. Subspace methods include Principal Component Analysis (PCA), Linear Discriminant Analysis (LDA), Random Projection (RP), etc. The RP can be free from training and much faster. Some extensions of one-dimensional RP (1D RP), including two-dimensional RP (2DRP) [10], two-directional two-dimensional RP
The authors of [13] used 2D schemes instead of 1D ones to reduce computational complexity and storage costs. In addition, in [10], the authors proposed
Feature selection methods depend on different spectral transformations such as two-dimensional Discrete Cosine Transform (2D DCT), and two-dimensional Discrete Wavelet Transform (2D DWT) to extract the features to reduce the amount of data, thereby simplifying the subsequent classification problem, and hence decision-making. Adaptive selection/weighting of features/coefficients is typically used for dimensionality reduction and performance improvement. The features that achieve high discrimination [14], high accuracy [15], and low correlation [12] should be selected and provided with high weights. The number of selected features is less than that of the original features. Feature selection methods have several advantages compared with subspace methods, such as PCA. Sometimes, feature selection methods can be fast and training-free, while it is comparable to the subspace methods in terms of accuracy. Furthermore, the selected features maintain their original forms. So, it is easy to observe the true values of the features. The authors in [16] proposed a novel approach for face and palmprint recognition in the DCT domain. In addition, the utilization of fusion rules is also an important tool to reduce computational complexity and storage costs [17].
The rest of this paper is organized as follows. Section 2 presents the proposed CNN models based on different downsampling layers. The max-pooling, DT, and RP are explained in Sections 3–5, respectively. Section 6 introduces the dataset. The results and discussions are given in Section 7. Finally, Section 8 gives the concluding remarks.
2 The Proposed CNNs Based on Different Downsampling Layers
We designed the proposed architecture, inspired by Rizzo et al. [1] architecture that has been reported as an efficient architecture for bacteria classification. We have added one convolutional layer followed by DT or 2D RP or
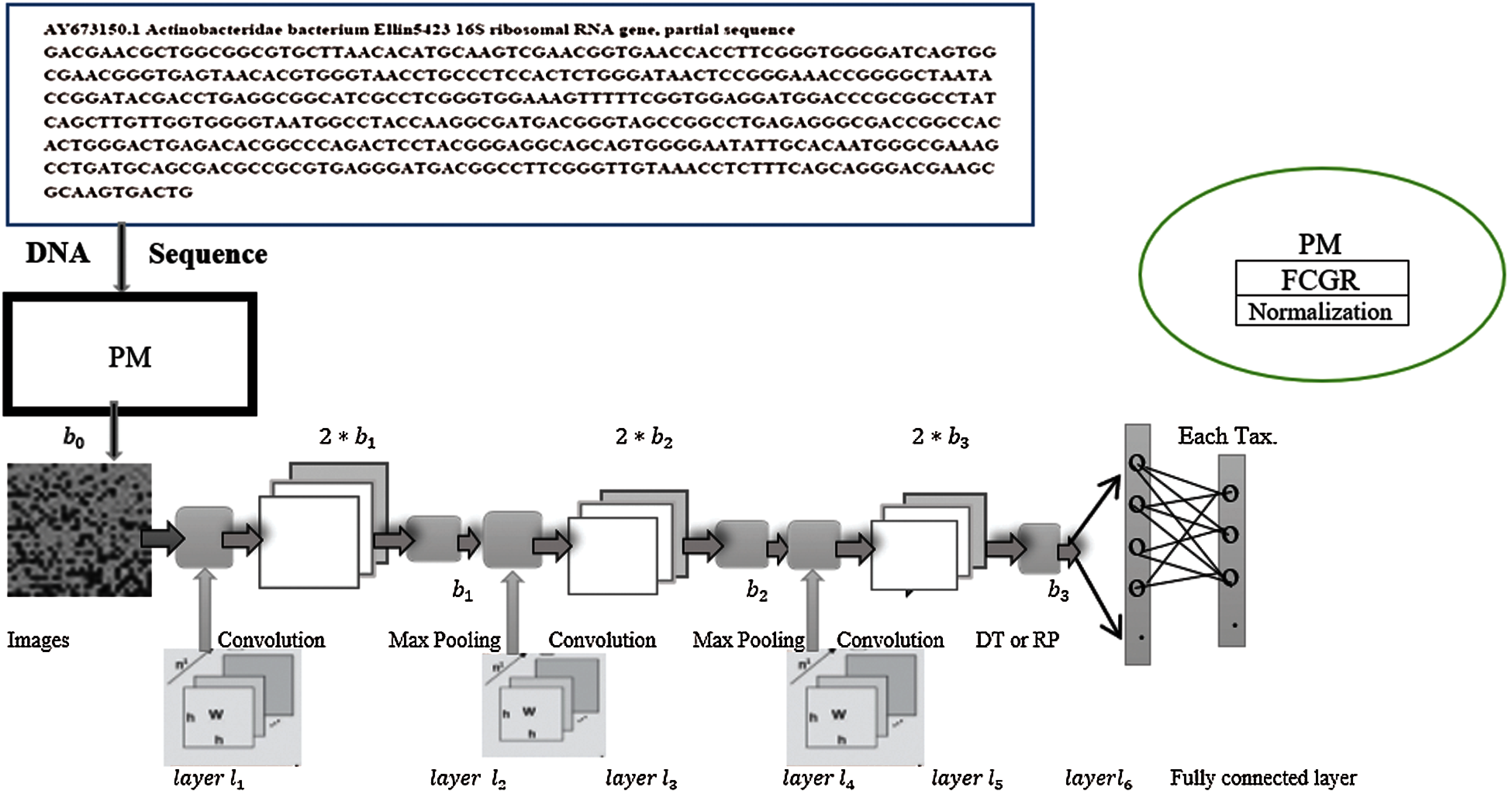
Figure 1: The architecture of the proposed model
After the convolutional layers, a set of the output images is generated; each of them of dimension
For example, let k = 6. Hence,
Figure 2: The architecture of the classifier
The downsampling layer is another name for the pooling layer. It reduces the dimensionality of data, by dividing the input into rectangular pooling regions. The max-pooling computes the maximum of each region
while the average pooling function can be expressed as:
where
Let us examine the effect of the max-pooling, when a 4 × 4 matrix input image is used, as shown in Fig. 3b.
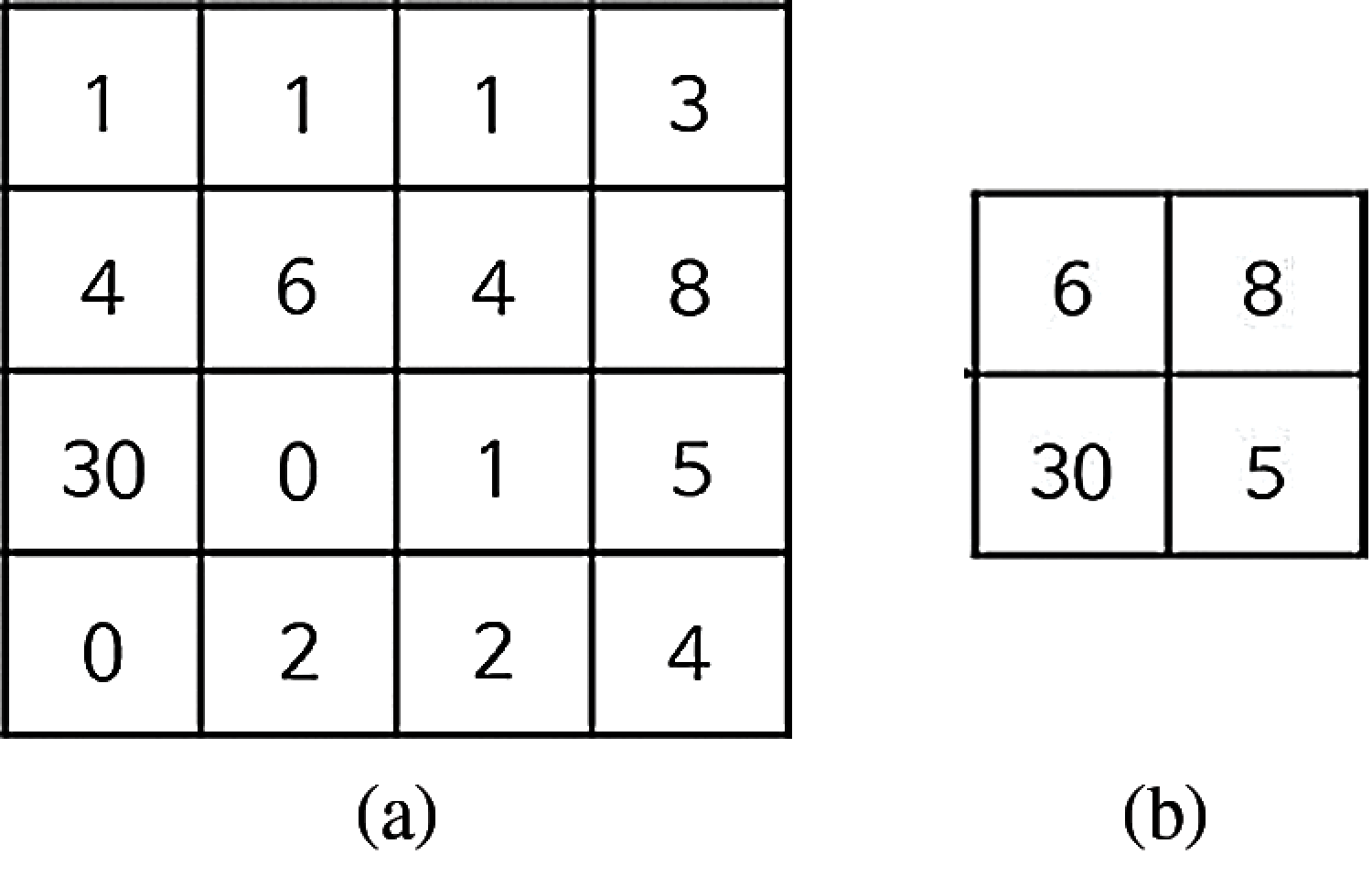
Figure 3: The pooling of a 4 × 4 matrix (a) The 4 × 4 matrix (b) The effect of max-pooling
In the case of an irregular nature of DNA sequences, k-mers recognition, the effective downsampling layer increases the ability of the CNN to achieve high performance. Anyway, the classification results do not critically depend on the feature extraction stage, but strongly depend on how these features are reduced.
Since the FCGR converts the DNA sequences into the form of images, we can apply the spectral transformations (Discrete Fourier Transform (DFT), Discrete Cosine Transform (DCT), and Discrete Wavelet Transform (DWT)) for downsampling, and the feature extraction stage for DNA images. The reason for applying these transformations emerges from their wide and effective use for extracting features, decorrelation, ordering, and dimensionality reduction purposes in the fields of speech, image, and bio-signal processing [19]. In signal processing, the DCT [20] can reveal the discriminative characteristics of the signal, namely, its frequency components. It is considered as a separable linear transformation. The basic idea of the DT is to select a certain sub-band after implementing the transformation. For example, the DCT can be implemented on the numerical sequence representing the DNA, and certain coefficients from the DCT can be selected to represent the whole sequence. The definition of the two-dimensional DCT for an input image A is given by:
where
and
while M and N are the row and column lengths of A, respectively.
The wavelet transform is faster and more efficient than the Fourier transform in capturing the essence of data [20]. Therefore, there is a growing interest in utilizing the wavelet transform to analyze biological sequences. The DWT is investigated to predict the similarity accurately and reduce computation complexity compared to the DCT and the DFT techniques.
The wavelet transform has been a very novel method for analyzing and processing of non-stationary signals such as bio-signals in which both time- and frequency-domain information are required. The wavelet analysis is often used for compression and de-noising of signals without appreciable degradations. The wavelet transform can be used to analyze the sequences at different frequency bands. In 2D DWT, the image is decomposed into four sub-bands. After filtering, the signal is downsampling by 2. In this work, the DWT is employed to reduce the dimensionality of features by performing the single-level 2D wavelet decomposition. The decomposition is conducted using a particular wavelet filter. Then, approximation coefficients (LL) can be selected. For example, let the first convolutional layer
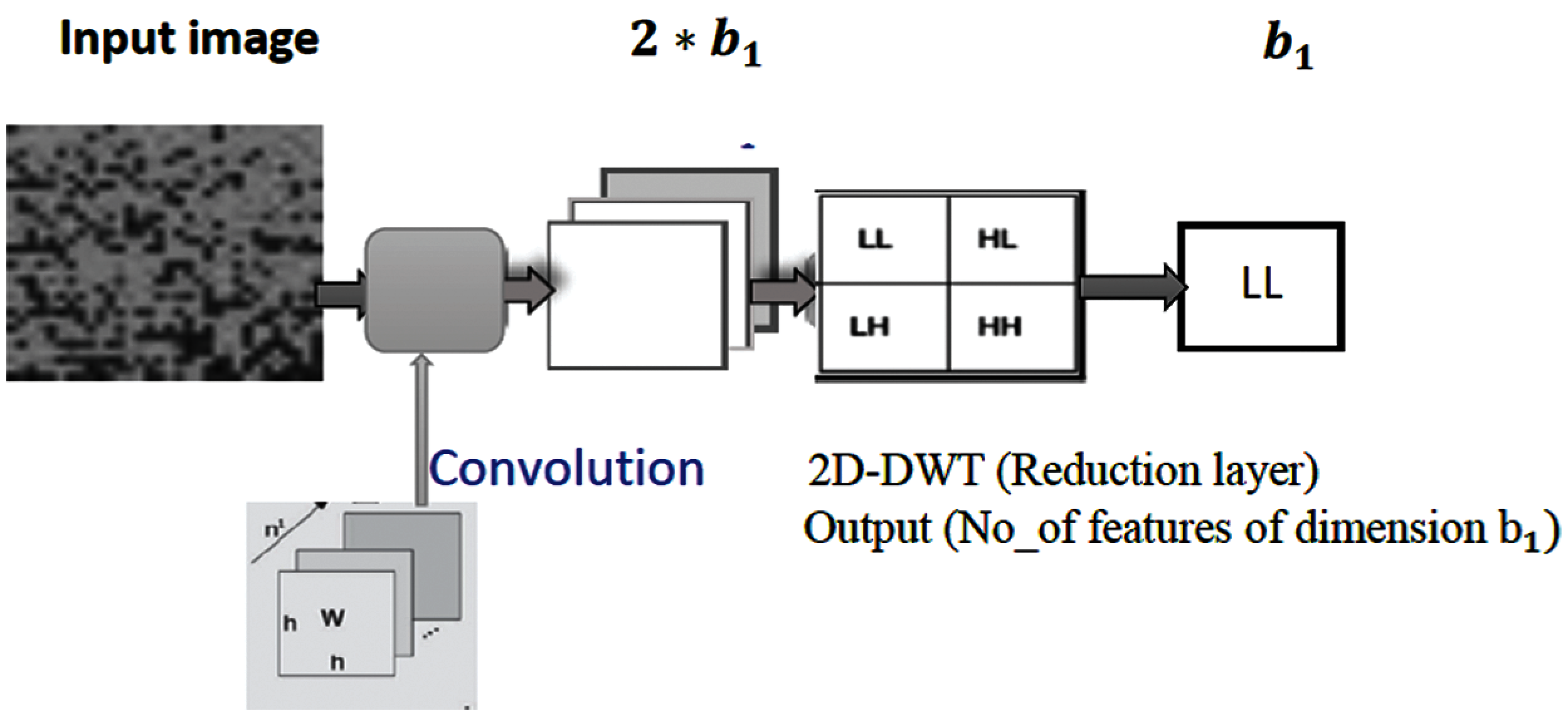
Figure 4: The proposed DWT pooling
5 Two-Dimensional Random Projection (2D-RP)
This method achieves the dimensionality reduction with low computational cost [21,22]. If the original dataset is represented by the matrix
where
The following stages of the RP are written using Matlab 2018a:
• Set the input as the features map
• Reshape the input to
• Create a
• for j = 1:n
• Y(:, j) = R×X(:, j);
• End for.
• Output=
5.2 Two Directional Two Dimensional Random Projection
The 2D RP can be implemented simultaneously in two directions, that is called
where R and C are the left mapping matrix for column-direction and right mapping matrix for row-direction, respectively and h
The dimensionality reduction is the main purpose of pooling layers as introduced in the previous sections. In this work, the DWT and DCT are proposed to make the pooling layer to satisfy this purpose and add more details to feature maps. Hybrid methods that combine

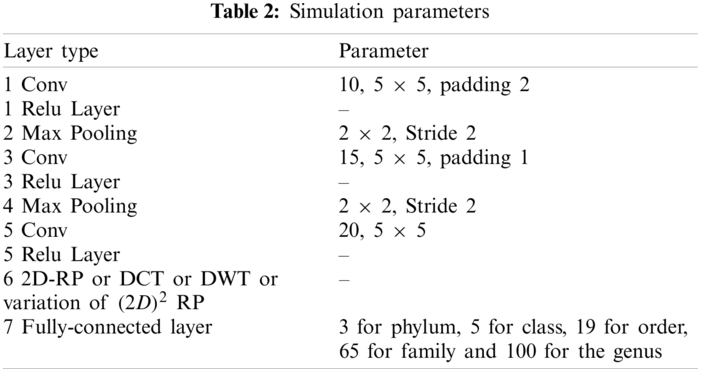
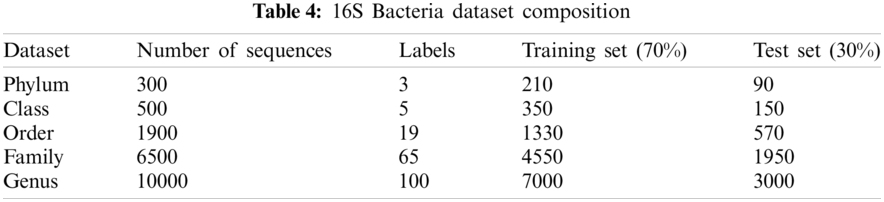
Data were obtained from the Ribosomal Database Project (RDP) [23], Release 11. A file in the FASTA record was obtained from the repository, which includes data on 1423984 outstanding bacterial gene sequences. For each bacterium, we have data on which taxonomic categories belong to certain genetic sequences. In addition, we have information on the phylum, class, order, family, and genus of a given 16S rRNA gene sequence. The bacterial genome contains the small-subunit ribosomal RNA transcript and is useful as a general genetic marker. It is often used to determine bacterial diversity, identification, and genetic similarity, and it is the basis for molecular taxonomy [24]. Two different sequences were used for comparison; (a) full-length sequences with a length of approximately 1200 – 1500 nucleotides and (b) 500 bp DNA sequence fragments. The total set of data includes sequences of the 16SrRNA gene of bacteria belonging to 3 different phylum, 5 different classes, 19 different orders, 65 different families, and 100 different genera, as shown in Tab. 4.
One of the key parameters that affect the DNA classification based on CNN is avoiding dimensionally problem and the sensitivity to the positions of the features. Even though the complex nature of DNA sequences is improved by convolutional layers, it is still necessary to ensure that the multi-layer CNN feature map has as suitable dimensions as possible. Therefore, there is a bad need to provide a downsampling layer that improves the generation ability of the original features. In this work, the CNN is utilized as a choice for deep learning, FCGR is applied for data preprocessing method, and different types of downsampling layers are introduced, such as DCT, DWT, 2D RP,
7.1 Comparison between Different Types of Downsampling Based on CNN
The effectiveness of different downsampling layers has been investigated to classify bacterial sequences to reach the highest possible accuracy. First, the given DNA sequences have been mapped using the FCGR algorithm with k = 6, 7, and 8. Then, the proposed CNN models based on different downsampling layers have been trained for each taxon. These models are.
• Model_1 (Max-CNN): Rizzo paper [1].
• Model_2 (RP-CNN): CNN classification followed by max-pooling or 2D RP.
• Model_3 (DWT-CNN): CNN classification followed by max-pooling or DWT.
• Model_4 (DCT-CNN): CNN classification followed by max-pooling or DCT.
•
• Model_6 (
•
To demonstrate the effectiveness of the proposed models, two simulation experiments are conducted. In the first case, the efficiency of the prediction for each taxonomic level is measured separately by taking into account the whole bacteria sequence. In the second case, instead of the whole sequence, we consider only the 500 bp long sequences. The simulation results are demonstrated in Tabs. 5–7, and Fig. 5 introduces the experimental results for the full-length DNA sequences, while Tabs. 8–10 and Fig. 6 present the results for 500 bp-length sequences. The classification is obtained for the same sequence with the representation of images at different values of k. From these tables and figures, it is clear that the proposed CNN model based on DWT and
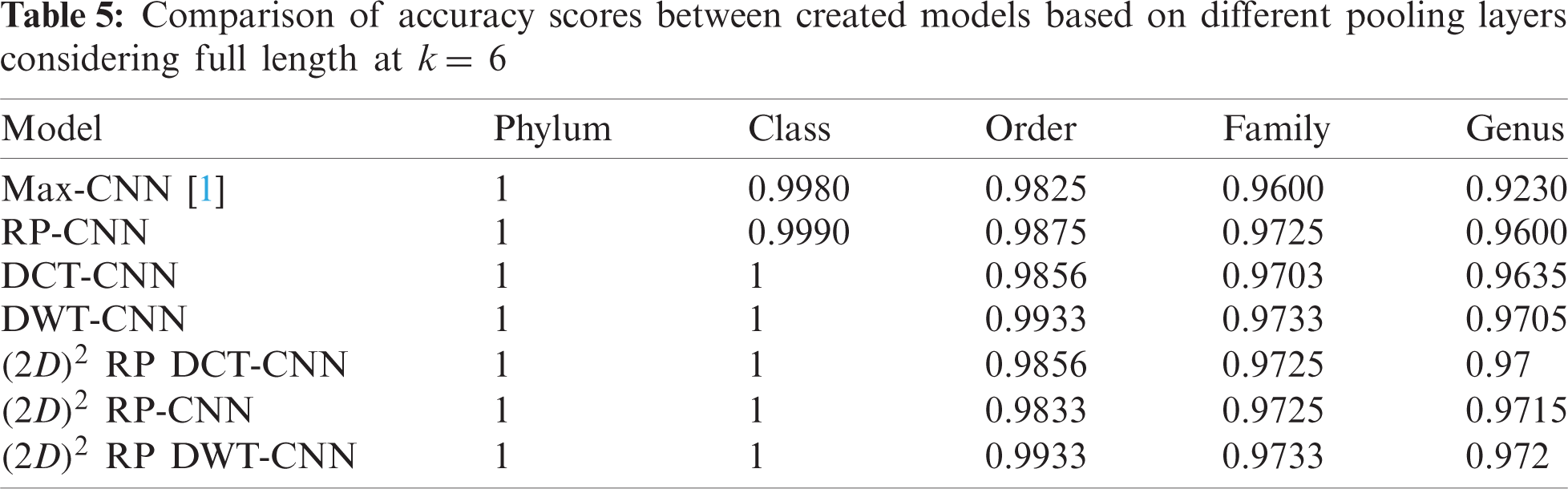
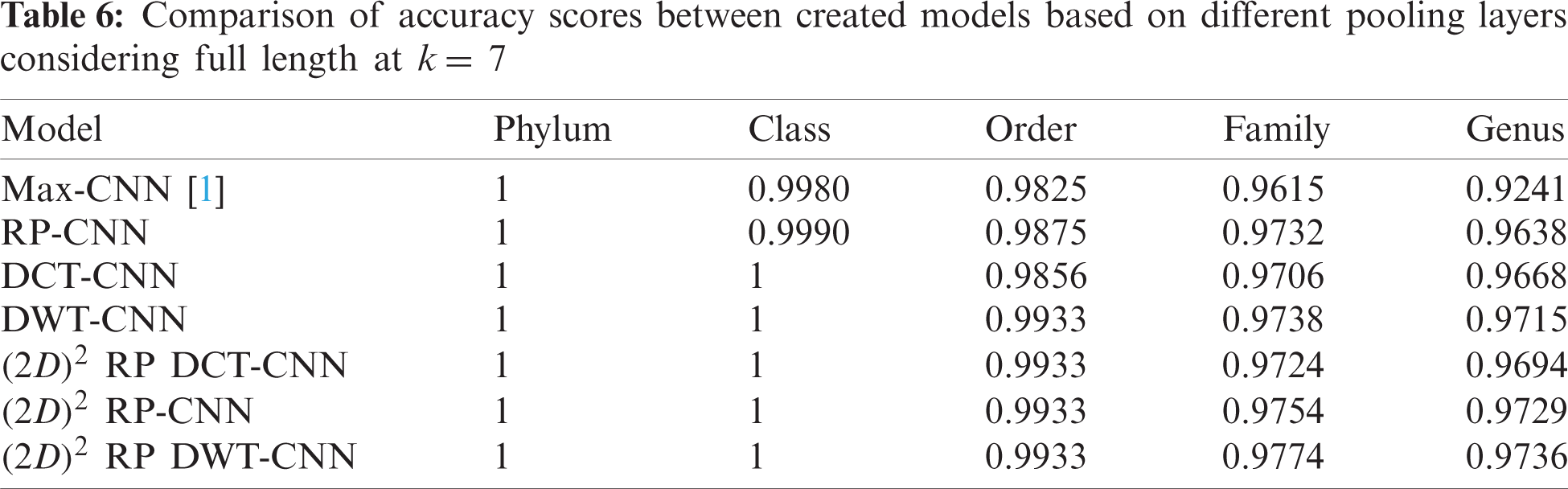
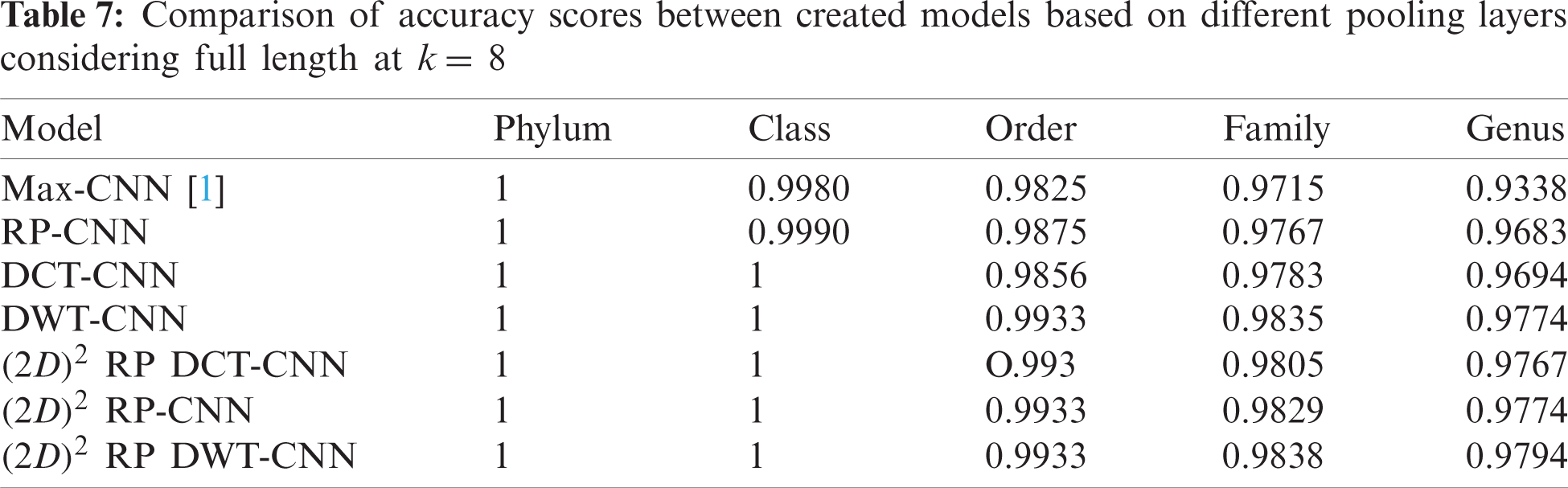
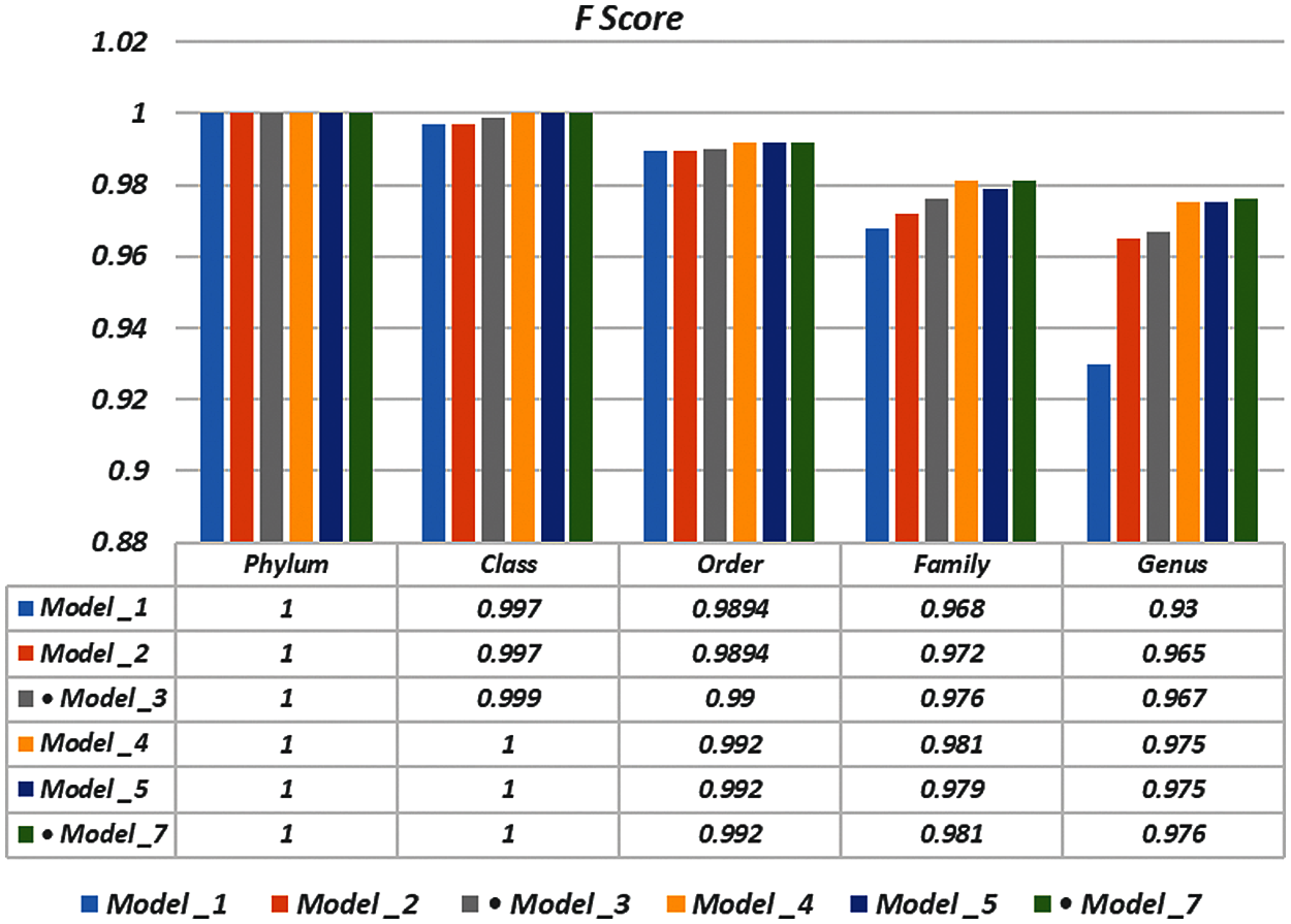
Figure 5: F-scores of the proposed model at k = 8, for the full length case
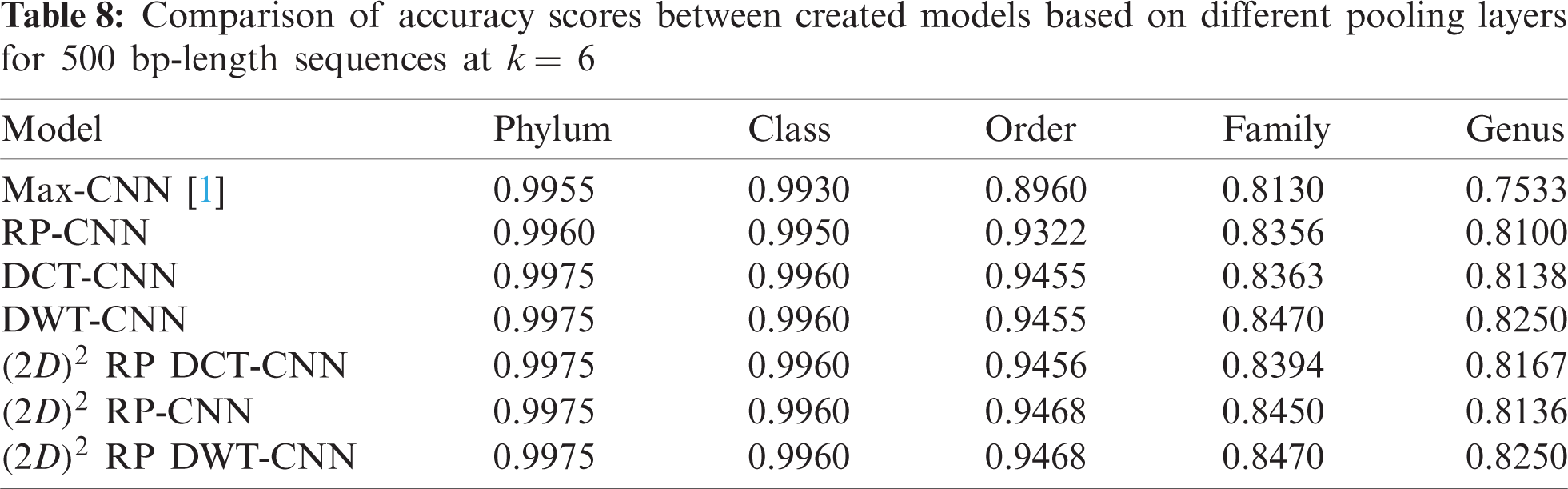
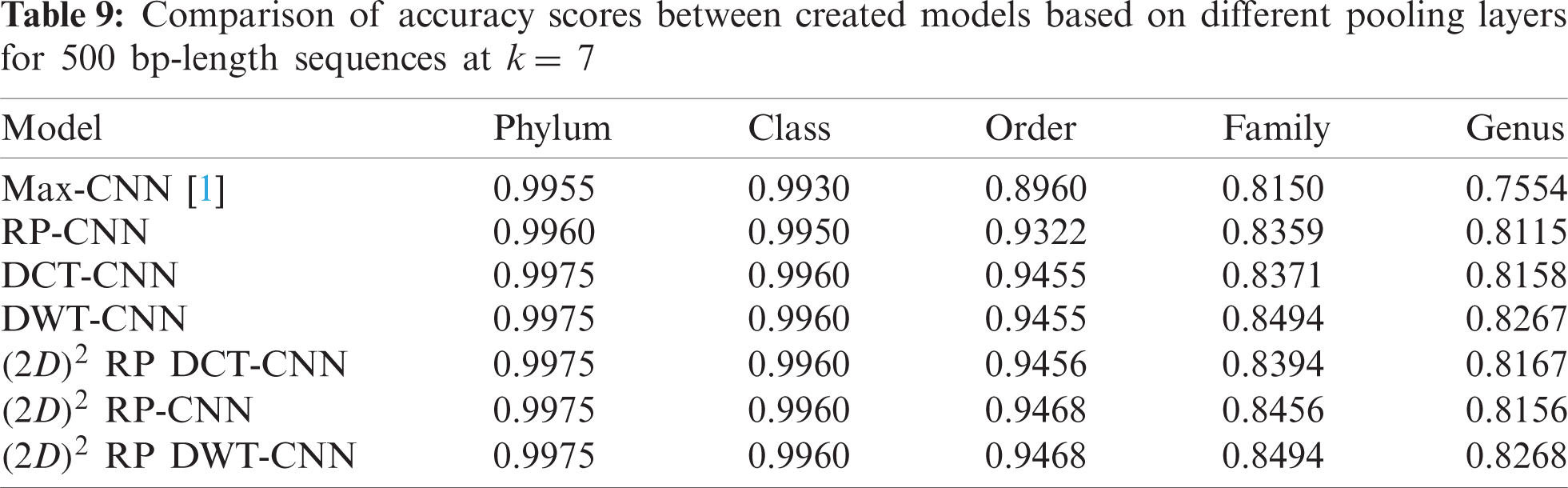
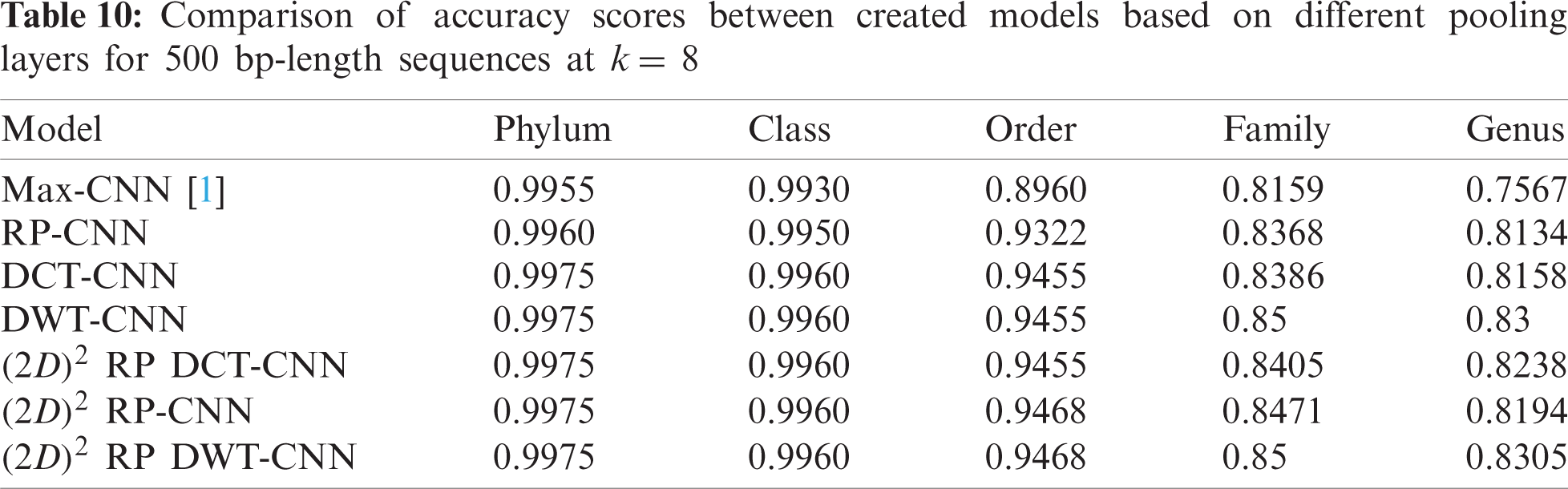
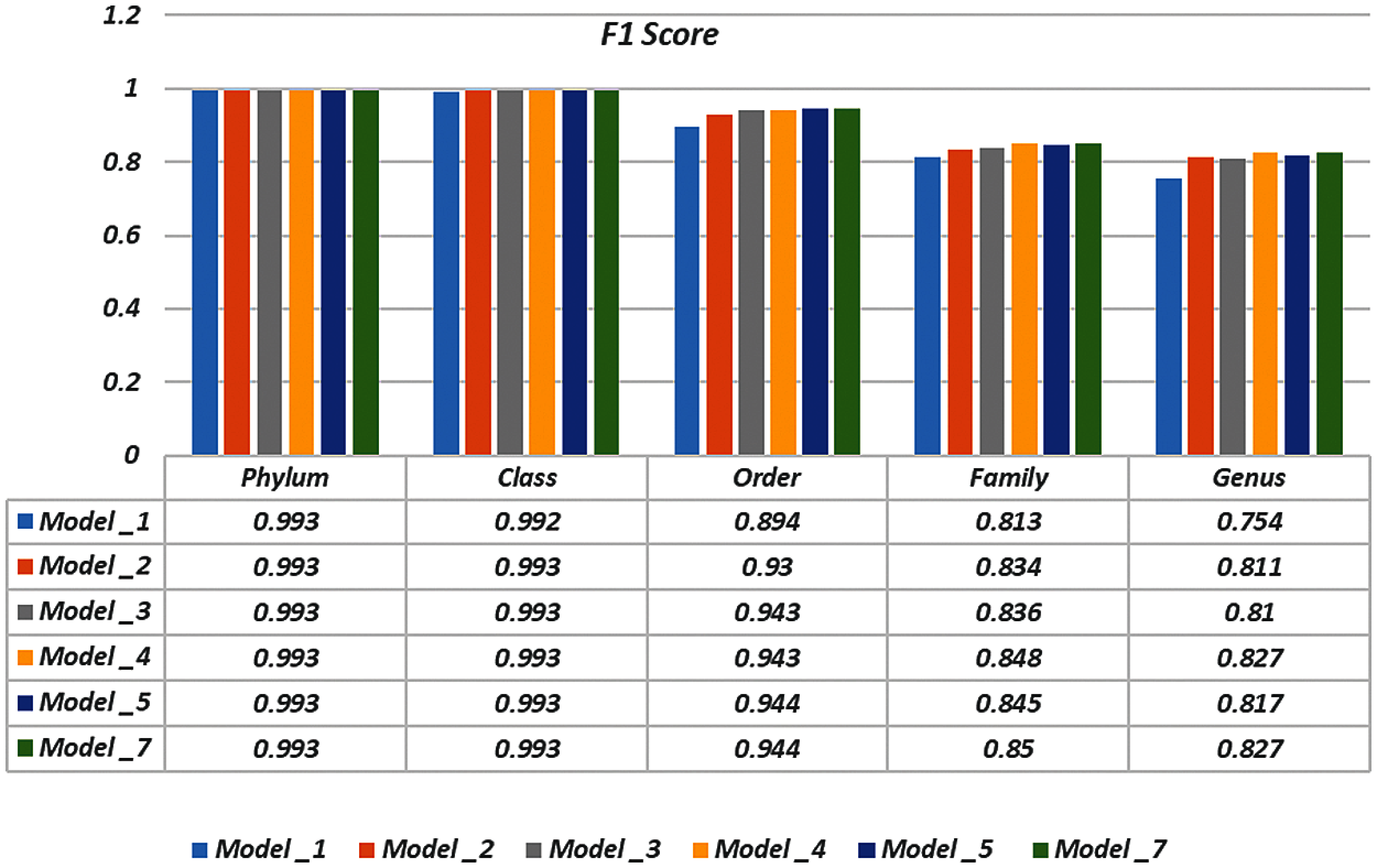
Figure 6: F-scores of the proposed model at k = 8, for 500 bp-length sequences
Tabs. 11 and 12 present comparisons between the performance of the proposed
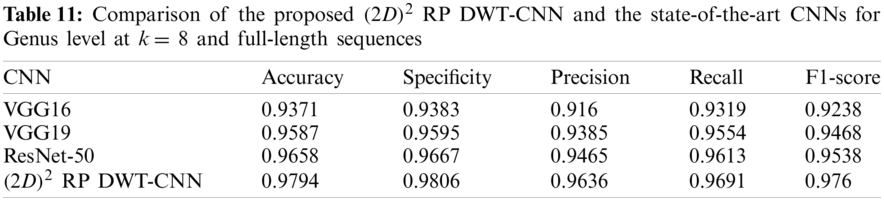
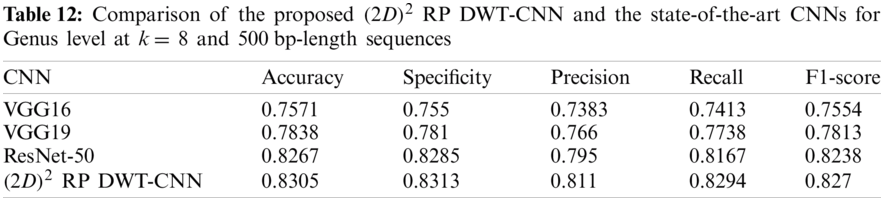
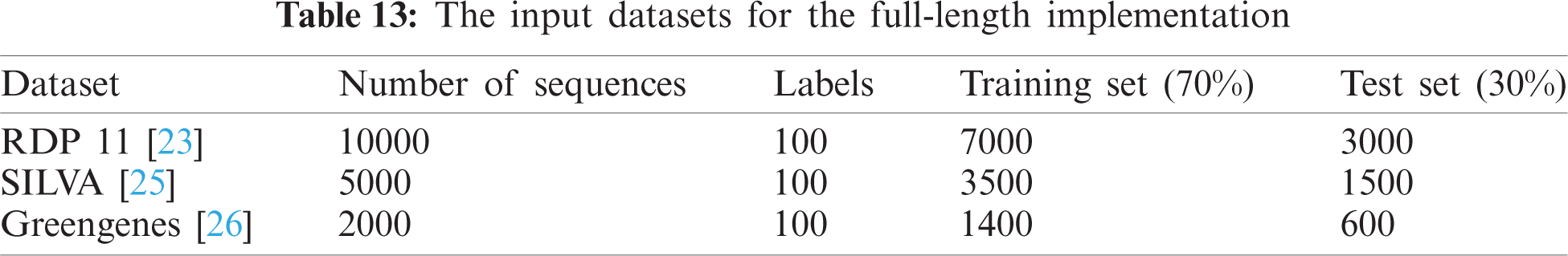
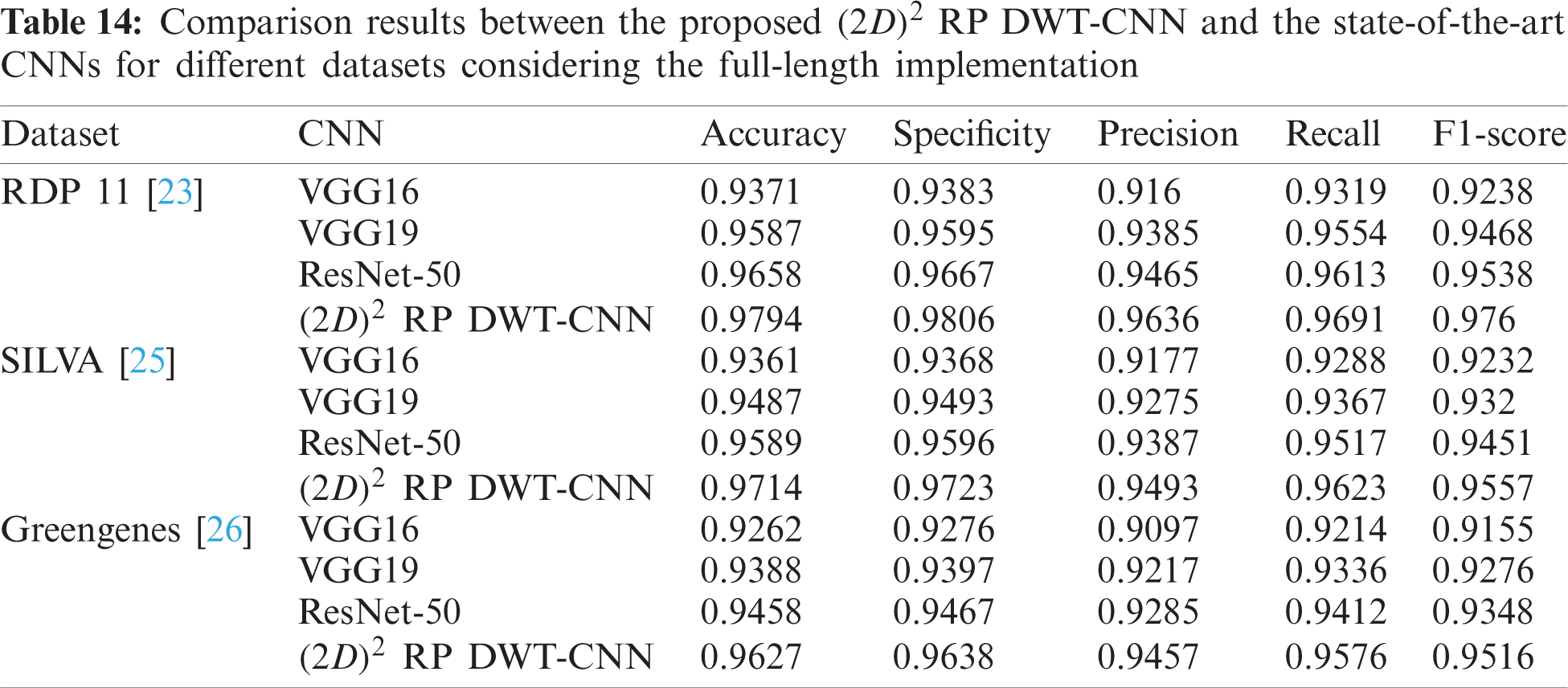
Tab. 13 indicates the different datasets used for the full-length implementation. Tab. 14 summarizes the experimental results for the proposed model and the state-of-the-art models. It is shown that the proposed model is superior, and it achieves a classification accuracy equal to 97.94% against 97.14%, 96.27%, and 96.27% for RDP 11, SILVA dataset [25], and greengenes dataset [26], respectively.
The training process may be quite difficult due to the enormous number of initial variables called hyperparameters. These values are defined before the start of the learning process. Some examples of hyperparameters include the learning rate, the minibatch size, and the number of epochs. In this paper, some changes in hyperparameters are applied to iteratively configure and train the proposed model. This section can be divided into subsections as follows:
In this subsection, the effect of the learning rate on the CNNs with different downsampling layers at the genus level is investigated in the case of full-length and 500 bp-length sequences. These downsampling layers include Max-CNN, RP-CNN, DCT-CNN, DWT-CNN,
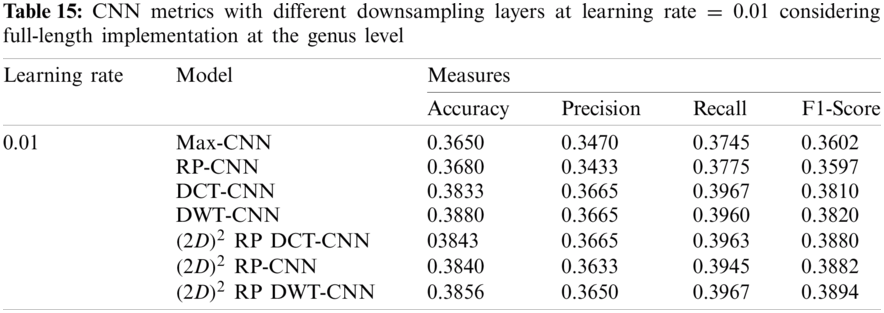
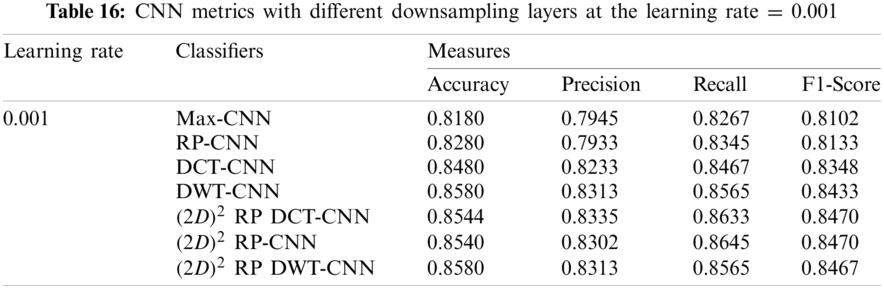
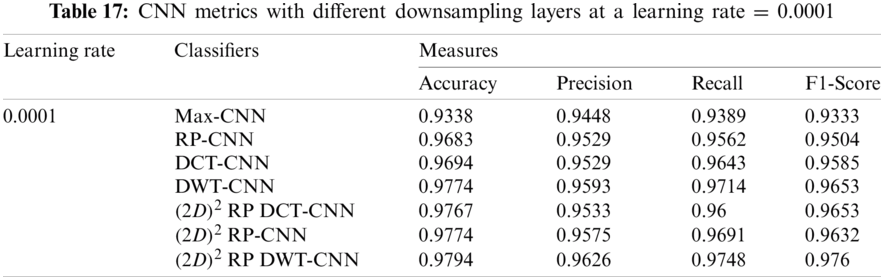
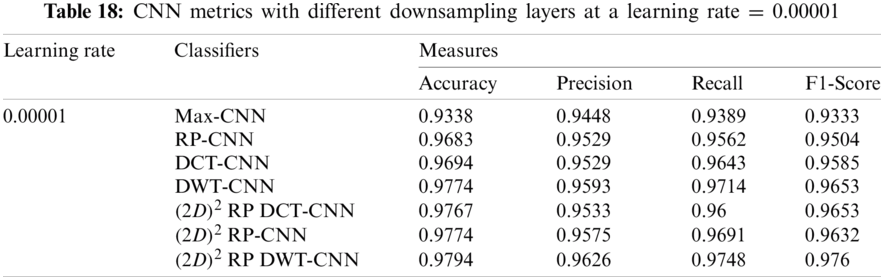
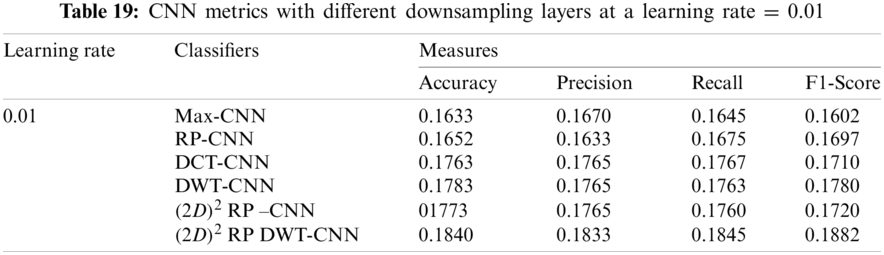
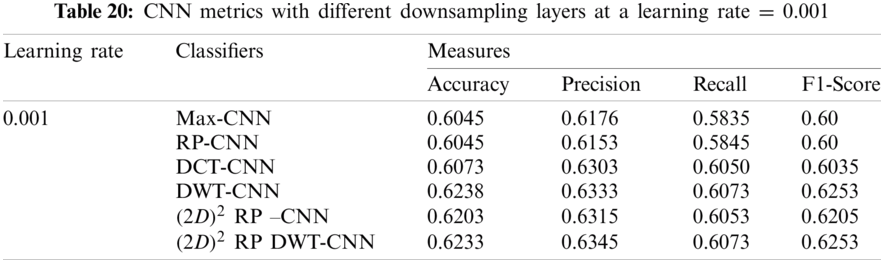
It can be noted that the highest accuracy is obtained at the learning rate equal to 0.0001 and 0.00001, but processing time increases, where 0.0001 learning rate has a processing time less than that of the 0.00001 learning rate. The same comparison is conducted for 500 bp-length sequences to trust the achieved results as demonstrated in Tabs. 19–21. Therefore, at a 0.0001 learning rate, superior accuracy for the training set can be attained for any length of the DNA sequences.
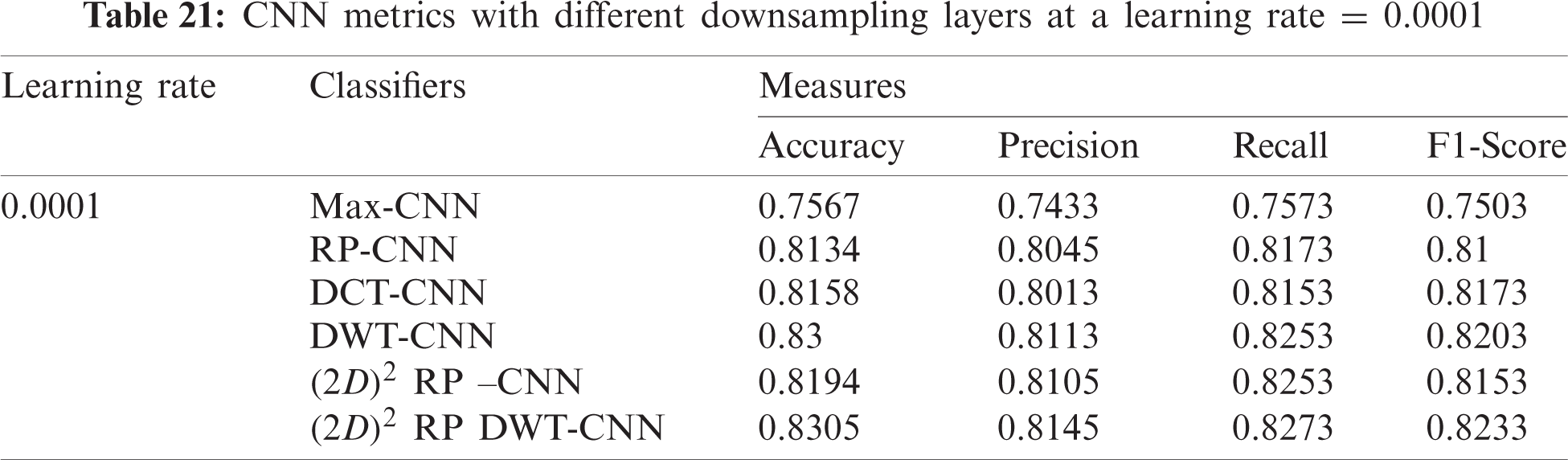
7.2.2 Mini-batch Size and Number of Epochs
In this subsection, the evaluation using different mini-batch sizes is investigated in the training process against different iterations for the proposed
From the mentioned results, we can conclude that the best performance of the proposed DWT-CNN model is achieved at the learning rate equal to 0.0001 and the mini-batch size equal to 64. We can select a suitable number of epochs considering these values. Fig. 8 reveals the training progress of
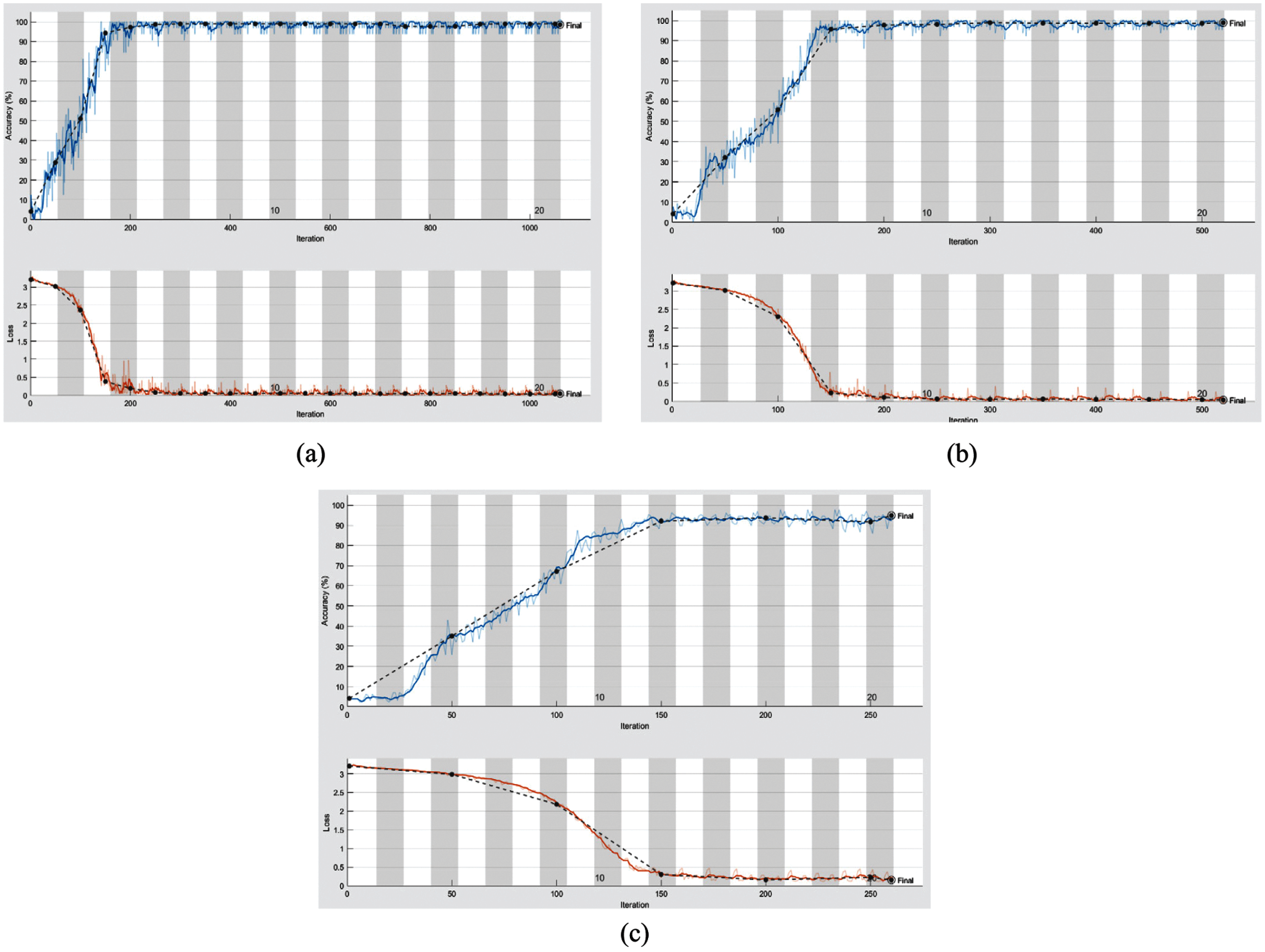
Figure 7: Training progress of
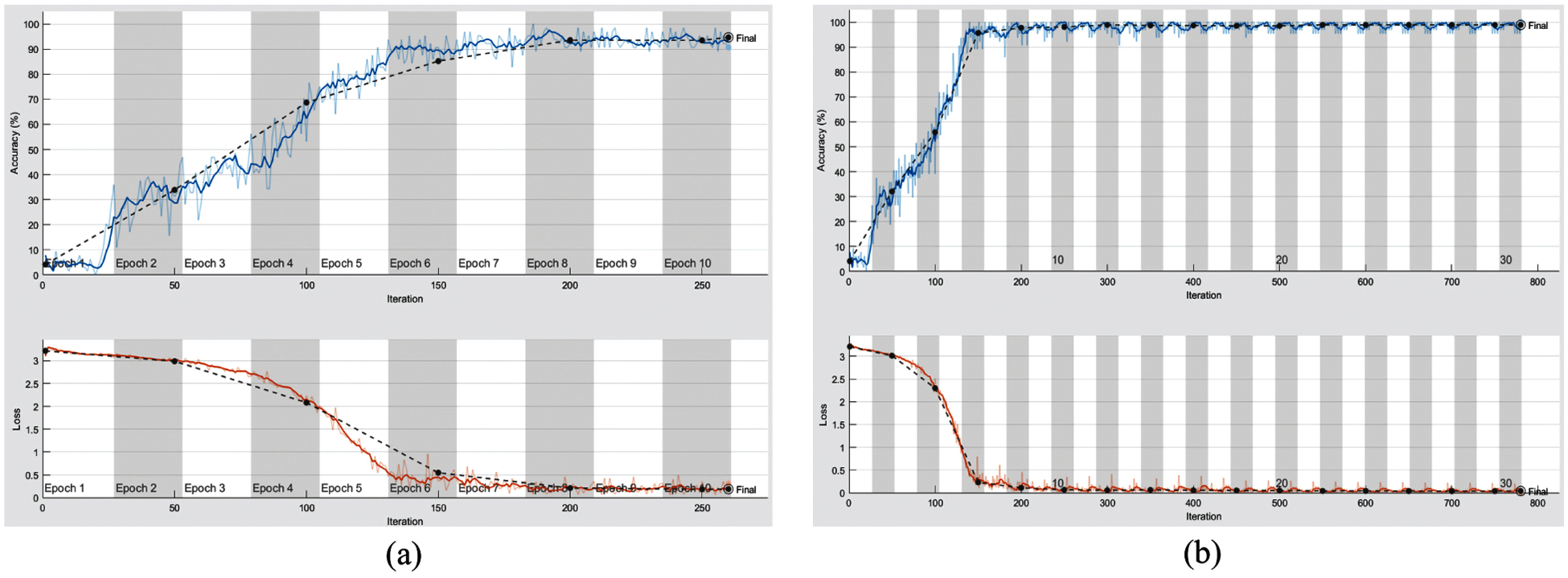
Figure 8: Training progress of

8 Conclusions and Future Research Directions
This paper presented two contributions to the bacterial classification of DNA sequences. The first one is represented in the proposed models for bacterial classification using an improved CNN. In these models, the 2D RP,
Acknowledgement: The authors would like to thank the support of the Deanship of Scientific Research at Princess Nourah Bint Abdulrahman University.
Funding Statement: This research was funded by the Deanship of Scientific Research at Princess Nourah Bint Abdulrahman University through the Fast-track Research Funding Program.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
1. R. Rizzo, A. Fiannaca, M. Rosa and A. Urso, “Classification experiments of DNA sequences by using a deep neural network and chaos game representation,” in Proc. Int. Conf. on Computer Systems and Technologies, Palermo, Italy, pp. 222–228, 2016.
2. M. Leung, A. Delong, B. Alipanahi and B. Frey, “Machine learning in genomic medicine: A review of computational problems and data sets,” Proceedings of the IEEE, vol. 104, no. 1, pp. 176–197, 2016.
3. P. Mamoshina, A. Vieira, E. Putin and A. Zhavoronkov, “Applications of deep learning in biomedicine,” Molecular Pharmaceutics, vol. 13, no. 5, pp. 1445–1454, 2016.
4. H. Greenspan, B. Ginneken and R. Summers, “Guest editorial deep learning in medical imaging: Overview and future promise of an exciting new technique,” IEEE Transactions on Medical Imaging, vol. 35, no. 5, pp. 1153–1159, 2016.
5. M. Seonwoo, L. Byunghan and Y. Sungroh, “Deep learning in bioinformatics,” Briefings in Bioinformatics, vol. 18, no. 5, pp. 851–869, 2017.
6. Y. LeCun, Y. Bengio and G. Hinton, “Deep learning,” Nature, vol. 521, no. 7553, pp. 436–444, 2015.
7. G. Bosco and M. Gangi, “Deep learning architectures for DNA sequence classification,” in Proc. 11th Int. Workshop of Fuzzy Logic and Soft Computing Applications, Naples, Italy, pp. 162–171, 2017.
8. R. Rizzo, A. Fiannaca, M. Rosa and A. Urso, “A deep learning approach to DNA sequence classification,” in Proc. Int. Meeting on Computational Intelligence Methods for Bioinformatics and Biostatistics, Springer, Cham, vol. 9874, pp. 129–140, 2016.
9. I. Goodfellow, Y. Bengio and A. Courville, Deep Learning (Adaptive Computation and Machine Learning Series). The MIT Press, 2016.
10. L. Leng, S. Zhang, X. Bi and M. Khan, “Two-dimensional cancelable biometric schemes,” in Proc. of Int. Conf. on Wavelet Analysis and Pattern Recognition, Xi'an, China, pp. 164–169, 2012.
11. A. Alarifi, M. Amoon, M. Aly and W. El-Shafai, “Optical PTFT asymmetric cryptosystem-based secure and efficient cancelable biometric recognition system,” IEEE Access, vol. 8, pp. 221246–221268, 2020.
12. L. Leng and J. Zhang, “Palmhash code vs. palmPhasor code,” Neurocomputing, vol. 108, pp. 1–12, 2013.
13. L. Leng and J. Zhang, “Palmhash code for palmprint verification and protection,” in Proc. of 25th IEEE Canadian Conf. on Electrical and Computer Engineering, Montreal, QC, Canada, pp. 1–4, 2012.
14. L. Leng, M. Li, C. Kim and X. Bi, “Dual-source discrimination power analysis for multi-instance contactless palmprint recognition,” Multimedia Tools and Applications, vol. 76, pp. 333–354, 2017.
15. S. Chakraborty and V. Gupta, “DWT based cancer identification using EIIP,” in Proc. of IEEE Second Int. Conf. on Computational Intelligence & Communication Technology, Ghaziabad, India, pp. 718–723, 2016.
16. L. Leng, J. Zhang, M. Khan, X. Chen and K. Alghathba, “Dynamic weighted discrimination power analysis: A novel approach for face and palmprint recognition in the DCT domain,” International Journal of Physical Sciences, vol. 5, no. 17, pp. 467–471, 2010.
17. L. Leng, M. Li and A. Teoh, “Conjugate 2DpalmHash code for secure palm-print-vein verification,” in Proc. of 6th IEEE Int. Congress on Image and Signal Processing, Hangzhou, China, pp. 1705–1710, 2013.
18. Y. Wang, K. Hill, S. Singh and L. Kari, “The spectrum of genomic signatures: From dinucleotides to chaos game representation,” Gene, vol. 346, pp. 173–185, 2005.
19. Y. Liu, “Wavelet feature selection for microarray data,” in Proc. IEEE/NIH Life Science Systems and Applications Workshop, Bethesda, MD, USA, pp. 205–208, 2007.
20. A. Tsonis and P. Kumar, “Wavelet analysis of DNA sequences,” Phys Rev E Stat Phys Plasmas Fluids Relat Interdiscip Topics, vol. 53, no. 2, pp. 1828–1834, 1996.
21. R. Wu, S. Yang, D. Leng and Z. Luo, “Random projected convolutional feature for scene text recognition,” in Proc. IEEE 15th Int. Conf. on Frontiers in Handwriting Recognition, Shenzhen, China, pp. 132–137, 2016.
22. P. Wojcik and M. Kurdziel, “Training neural networks on high-dimensional data using random projection,” Pattern Analysis and Applications, vol. 22, no. 3, pp. 1221–1231, 2019.
23. rRNA sequences, [Online]. Available: https://rdp.cme.msu.edu, (Access date 11 January 2021).
24. M. Balvočiūtė and D. Huson, “SILVA, RDP, greengenes, NCBI, and OTT—How do these taxonomies compare?,” BMC Genomics, vol. 18, no. 114, pp. 1–8, 2017.
25. P. Yilmaz, L. Parfrey, P. Yarza, J. Gerken, E. Pruesse et al., “The silva and all-species living tree project (ltp) taxonomic frameworks,” Nucleic Acids Research, vol. 42, no. D1, pp. D643–D648, 2014.
26. D. McDonald, M. Price, J. Goodrich, E. Nawrocki, T. DeSantis et al., “An improved green genes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea,” ISME Journal, vol. 6, no. 3, pp. 1–8, 2012.
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |