DOI:10.32604/cmc.2022.023974
| Computers, Materials & Continua DOI:10.32604/cmc.2022.023974 | |
| Article |
Intelligent Approach for Clustering Mutations’ Nature of COVID-19 Genome
1Computer Science and Engineering, Women Institute of Technology, Uttarakhand, 248007, India
2Chitkara University Institute of Engineering and Technology, Chitkara University Punjab, Punjab, 140401, India
3Division of Innovation & Entrepreneurship, Lovely Professional University, Punjab, 144411, India
4Computer Science and Engineering, Chandigarh University, Punjab, 140413, India
5Faculty of Computing and Information Technology, King Abdulaziz University, Jeddah, 37848, Saudi Arabia
6Computer Science and Engineering, Lovely Professional University, Punjab, 144411, India
7Higher Polytechnic School, Universidad Europea del Atlántico, Santander, 39011, Spain
8Universidad Internacional Iberoamericana, Campeche, 24560, Mexico, C.P
9Universidade Internacional do Cuanza Bairro Kaluanda, Bié, Angola
*Corresponding Author: Divya Anand. Email: divyaanand.y@gmail.com
Received: 28 September 2021; Accepted: 10 January 2022
Abstract: In December 2019, a group of people in Wuhan city of Hubei province of China were found to be affected by an infection called dark etiology pneumonia. The outbreak of this pneumonia infection was declared a deadly disease by the China Center for Disease Control and Prevention on January 9, 2020, named Novel Coronavirus 2019 (nCoV-2019). This nCoV-2019 is now known as COVID-19. There is a big list of infections of this coronavirus which is present in the form of a big family. This virus can cause several diseases that usually develop with a serious problem. According to the World Health Organization (WHO), 2019-nCoV has been placed as the modern generation of Severe Acute Respiratory Syndrome (SARS) and Middle East Respiratory Syndrome (MERS) coronaviruses, so COVID-19 can repeatedly change its internal genome structure to extend its existence. Understanding and accurately predicting the mutational properties of the genome structure of COVID-19 can form a good leadership role in preventing and fighting against coronavirus. In this research paper, an analytical approach has been presented which is based on the k-means cluster technique of machine learning to find the clusters over the mutational properties of the COVID-19 viruses’ complete genome. This method would be able to act as a promising tool to monitor and track pathogenic infections in their stable and local genetics/hereditary varieties. This paper identifies five main clusters of mutations with
Keywords: nCoV-2019; SARS-CoV-2; COVID-19; genome structure; etiology; COVID-19 mutations; COVID-19 genomes
At the end of the year 2019, around 31 December 2019, a group of people in Wuhan province of China began to develop diseases resulting from acute respiratory disease, whose root cause was believed to be pneumonia [1–4]. But given the rapid increase of this infection in the people, the Health Department of China called this infection fatal to the lives of the people and named it nCoV-2019. Acute respiratory diseases associated with coronavirus infection came to be known as SARS-CoV-2 by the WHO [5–7]. Because this species of infection is related to SARS, which was known as SARS-CoV in the year 2002, which is related to the genome structure of the SARS-CoV virus [8,9]. This genome structure is a fundamental factor in continuous change like the coronavirus. According to the most recent WHO report the total cumulative numbers of confirmed cases of COVID-19 are over 196,553,009 and a cumulative total of confirmed death 4,200,412.
Coronaviruses have to develop genetic proofreading mechanisms to maintain the Ribonucleic Acid (RNA) genome sequence for a long time. Despite this mechanism being lacking in the Sars-CoV-2 virus, this infection enters the logo’s cells with the help of mutant cellular receptors in the spike protein, which infects the internal cells as well as the tissue tropics and pathogen infections, also strongly affected. During the outbreak of SARS-CoV infection in the years 2002–2003, one such mutation-mediated adaptation to intermediate civet host infection as well as to inter human transmission was found.
In this way, by classifying the similarities between different mutations of the infection, the infection can be classified into different groups (represented in Fig. 1) with the help of their genome sequence. The main contributions of this research work are summarized in the following points:
• To create a detailed background about the structure and mutational nature of the coronavirus.
• The main objective of this research is to analyze the coronavirus genome structure and its sequence through machine learning techniques.
• Building a model using the k-mean clustering technique on complete genomic sequences of SARS-CoV-2 strains capable of clustering through the mutational nature of the COVID-19 virus.
This paper is organized in such a way; Section 2 covers a detailed background of the coronavirus variants and their entire family including their genome structure and sequence. In addition, Section 3 covers the machine learning techniques used under this research work to analyze the genome structure of the COVID-19 virus, including the datasets considered. The next Section 4 represents the calculated results, their discussion, and the result verification process. In the last Section 5, the research is concluded with the calculation of 5 clusters after applying the model to the datasets considered.
Coronaviruses (CoVs) cover a wide range of human and animal infections. It is a single standard positive-sense RNA virus with the largest viral genome ever recorded and belongs to the families Coronaviridae and Nidovirales. The coronavirus infection spreads rapidly across a wide range of populations, among various vertebrates, and as feathered animals come into contact with each other. The virus is capable of causing a variety of diseases, for example, most commonly infecting the respiratory, intestinal, liver, and sensory systems, as shown in Fig. 1. The first human coronavirus was identified in the mid-1960s, which was named human coronavirus (HCoV). The incidence of diseases related to CoV-HKU1 infection has been found in the youth of the United States, but the effect of this virus has been seen less in adults. The series of coronaviruses consists of three exceptionally pathogenic coronaviruses known as SARS-CoVs, MERS-CoVs, and SARS-CoV-2, which evolved individually in 2002, 2012, and 2019, respectively. These coronaviruses have caused severe respiratory diseases in people around the world, leading to many deaths [10]. In short, the coronavirus is divided into 4 genera named α-CoV, β-CoV, γ-CoV, and δ-CoV respectively.

Figure 1: Phylogeny of coronavirus (CoVs), phylogenetic tree of 50 coronaviruses developed by a neighbor binding technique using MEGA 5.0 of partial nucleotide sequences of RNA polymerase (RNA-pol) subordinates
Dividing this chain further, the β variant of coronavirus has been divided into four categories named A, B, C, and D habitats respectively, which are shown in detail in Fig. 2. There are seven types of coronaviruses that have the potential to spread more infection than the human coronavirus. Among them, the α-coronaviruses type HCoV-229E, and HCoV-NL63 are placed in this category, the β-type categories have HCoV-OC43, and HCoV-HKU1 as A-type, SARS-CoV and SARS-CoV2 as B-type, and MERS-CoV are placed in the C type categories respectively [5].

Figure 2: Development program for deep pathogen infection and proposed disease
In order to formulate the SARS-CoV-2 genomic classifier, a thorough consideration of the potential of the SARS-CoV-2 genome as opposed to SARS-CoV is needed. According to the genome structure of coronaviruses, its virions contain three important and basic encoder proteins, which have been named S-glycoprotein, M-glycoprotein, and E-protein, respectively. S-glycoproteins (for spikes) are extremely large (approximately 200 K) in size capable of forming the annoying peplomers (15–20 nm) found in the viral envelope. The same M-glycoprotein is an extracellularly translocated glycoprotein and consists of an inner layer of phosphorylated nucleocapsid protein (N). Additionally, the E protein is a minor transmembrane, and some coronaviruses also contain an additional envelope protein with clumping and esterase (HE) capability.
The 30 kb positive-strand single-stranded RNA genome is the largest known viral RNA genome. It terminates at the 5′-terminus and is polyadenylated and irradiated at the 3′-terminus. Because of their size, their individual properties are declared through a highly complex process, in which all 5′ sequences with identical endings are delivered to a set of established mRNAs. This can then be followed by a comprehensive review of the causes of the recombination of heterologous RNA. The 5′ end of the genome contains a sequence of untranslated (UTRs) of 65 to 98 nucleotides, called leading RNAs, which are also present in the 5′ portions of all genomic mRNAs. RNA is another 200 to 500 nucleotide untranslated sequence created by a move (A) tail at the 3′ end of the genome. Both untranslated regions are important in directing RNA replication and translation.
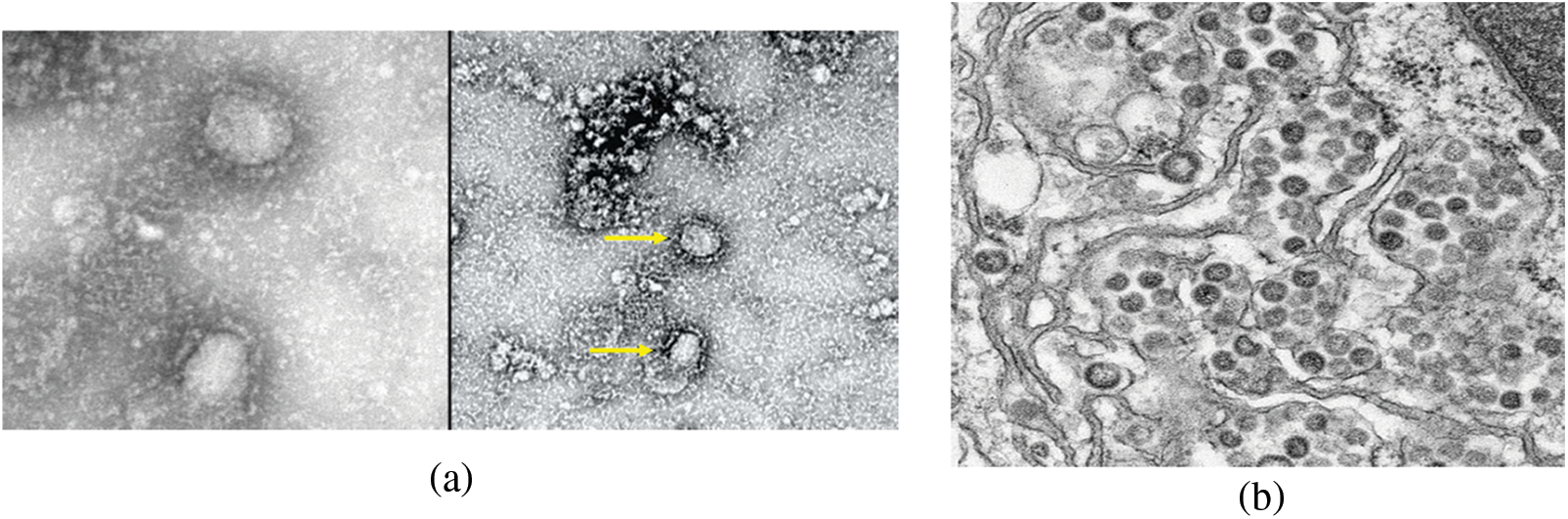

Figure 3: Morphological structure of the coronavirus. (a) Negative contrast electron microscopy of destructive SARS-CoV strain, (b) Middle East Respiratory Syndrome (MERS-CoV) strain, (c) Model structure of the coronavirus virus, which causes supercoiling of viral nucleocapids under the envelope is made [11]
By carefully studying the genome structure of the coronavirus, a total of 7 to 14 open reading frames (ORF) are found in it, starting from the 5′-end on the genome structure. In this structure, gene 1 comprises up to 66% of the genome which accounts for the largest number of genome structures and has a structural size of about 20–22 kb. This gene 1 is formed by combining two coverage ORFs (1A and 1B). These ORFs act together in the structure as a viral RNA-pol. The structure of the genome uses the following four properties of auxiliary proteins from the 5′-end to the 3′-end, known as S (spike), E (envelope), M (membrane), and N (nucleocapsid) as presented in Fig. 3. These structural properties of the gene are associated with certain ORFs that encode non-helper proteins such as HE glycoproteins. Each trait in the combined strategy in notably number, nucleotide sequence, quality request and differs among coronaviruses, but these are stored in the same serogroup [12]. Sars-CoV contributes to the presence of some short ORFs in the 3′ end of the genome’s structure, which is very difficult to contain in the structure of an individual coronavirus [13]. With the help of these partial ORFs, 8 new helper proteins are more likely to be transmitted. All reactive antibodies to the Sars-CoV protein have been detected in sera taken from patients infected with sera. Their presence suggests that these proteins are communicated by transfection of the genome, thereby facilitating the spread of the infection.
Coronavirus infection changes the genome structure of the virus first into two polyproteins called ORFs 1a and later 1b, which are indistinguishable at the N-terminus. One of these polyproteins aids in elongation at the C-terminal. These growths at the terminals of the genome are anticoagulants of proteins in the replication complex. All coronaviruses belonging to these terminals can encode a chymotrypsin-protease, also known as mPro (primary protein) or 3CLPro. This type of virus also shows some similarities to the picornavirus 3C protease. The proteins present in the terminals are also responsible for the management of the remaining polyproteins, with the help of which more than 16 different types of non-helper proteins are formed. The structure of Sars-CoV contains the largest amount of these non-native proteins, forming a multifunctional protein with the activities of the nsp3 protease and ADP-ribose 1′ phosphate at the action end. In addition to these terminal proteins, two proteins (NSP7 and NSP8) also form a similar cylindrical structure, which may play an important role in the assimilation of coronavirus RNA. These additional proteins are also capable of forming a single chain RNA restriction protein (NSP9). One of the polyproteins of the genome structure (ORF1b) can encode an RNA-pol and a multifunctional helicase protein subordinate to viral RNA. With their help, regardless of the activities of the helicase, it provides functions to the triphosphate 5′ NTPase and dNTPase of protein terminals.
SARS-CoV-2 coronavirus infections have been reported to affect a direct single-stranded positive RNA genome sequence. The main reason for this is that the SARS-CoV-2 coronavirus is composed of a major sequence of the genome. The proteins that encode ORF 1a and b from RNA replication have properties of all non-structural proteins (NSPs) and basic structure proteins (SPs). This property is the only major genomic sequence of coronavirus replication, which is approximately 265 bp in size, and heterogeneity plays a fundamental role in the expression of the quality of the coronavirus during its sub-genomic replication [12]. ORF 1a and b encode knockoffs of poly-proteins required for replication and transcription of viral RNA [13]. Expression of the C-proximal bit of ORF 1a and b requires the translocation of the (−1) ribosomal frame. The first non-structural protein (nsp) encoded by ORF 1a and b is a papain-like protein (PL proteinase, nb3). Nsp3 is a fundamental and most important part of the replication and interpretation complex. The protein Plk1 in Nsp3 cleaves NPs and sections in an insensitive intrinsic reaction, which further enhances cytokine expression [14,15]. Nsp4 encodes in ORF 1a and b which is responsible for shaping double-layered vesicles (DMVs). Other nsp are 3CLPro protease (3-chymotrypsin-like proteinase, 3CLPro) and nsp6. The 3CLPro proteins are responsible for the management of the C-terminus from nsp4 to nsp16 in all coronaviruses [16]. Therefore, the moderate structure and reactive location of the 3CLPro structure may be an attractive focus for antiviral drugs [1,3]. The unregulated function of nsp3, nsp4, and nsp6 may promote DMV [17].
The replication of the RNA structure of SARS coronavirus infection is unique, as the structure of this virus includes two subordinate RNA-pol. The primary RNA-pol is the early nonstructural protein 12 (nsp12), and its second RNA-pol is nsp8. In place of nsp12, nsp8 plays an important role in the replication and translation of SARS-CoV-2. In the same SARS coronavirus, nsp7 and nsp8 contain a polynomial RNA-pol for both initiation and initial expansion of a complex [18]. The nonstructural protein 8 is also bound to the ORF b motif protein. The SARS coronavirus nsp9 protein binds to replication RNA and associates with nsp8 for its actions [19].
The S-spike protein of the auxiliary proteins of the genome structure is classified as a glycoprotein, which consists of two distinct types of regions known as S1 and S2 [20]. The S1 spike protein binds to the ACE2 receptor to initiate and extend infection early in infection, as well as bind the virion to cell films. This is due to adaptive changes occurring in the glycoprotein S-spike protein, which expands after infection in the host cell endosomes [21]. The S-spike protein is once again cleaved with the help of the CTSL cathepsin, which can jointly cleave the S2 peptide, initiating the conjugation of the fibrils within the endosomes. On the other hand, the S2 spike protein helps isolate the viral assembly proteins, virions, and adjustments of cell films in a segment of the genome. In particular, these spike proteins are more effective for the Sars-CoV-2 coronavirus because the furin-like cleavage site is found in the spike glycoprotein of the Sars-CoV-2 virus. Detection of furin during infection and consideration of the extent of pyrolysis in the affected area is critical, which is why the state of zoonotic contamination has been linked to infection. The E-protein (envelope) interacts with the M-layer proteins in the mature compartment of the host cell, as the M-proteins primarily possess cellular immunogenicity [22]. This M-layer protein and nucleoprotein (ORF9A) during viral binding in association with the viral genome accommodates a positive chain viral RNA genome into a helical ribonucleocapsid (RNP) [23]. Thus, these subgenera play an important role in the translation of viral RNA as well as in improving the ability of viral replication.
The spread of the coronavirus pandemic and its clinical evidence shows that Sars-CoV-2 infection has lower ground transmission efficiency and pathogenicity than Sars-CoV infection transmission [24]. However, the high transmission equipment in the infection of Sars-CoV-2 is misleading. Deoxyribonucleic Acid (DNA) disposition testing of infection using single nucleotide polymorphisms (SNPs) is often used for evolutionary investigations and may be particularly helpful in considering genomic alterations of coronavirus infection. Because a major reason for more mutation of SARS-CoV-2 can also be RNA polymerized. RNA is also prone to errors in genomic replication.
Understanding the changes in the genomic structure of the infection is of utmost importance to understand the progression of Sars-Cov-2 infection expansion, through this research configuring SNP genotyping techniques to a machine learning model and analyzing the effects of Sars-CoV-2 infection. Examine the genotype changes of infection during expansion. The results of the investigation suggest that the genotype of the infection is not reasonably consistent among the total Sars-CoV-2 genome. This genotyping study reveals a pair of deep-travel mutations present in the Sars-CoV-2 genome. Variation in deep-visited SNPs may adjust to virus infection and correlate with loss of infection. Mutations are located in S-protein, RNA-pol, RNA base, and nucleoproteins, which are the main proteins for antibody viability. Thus, high recessive SNP variants are an important variable in creating a vaccine to prevent SARS-CoV-2 coronavirus disease.
The experimental work was performed by using the machine learning algorithm that provides better results in classifying the genome sequence of coronaviruses. This research work focused on the k-mean unsupervised machine learning clustering approach with major concerns as follows:
• Because of the intricacy of the data, including different variables which made it was hard to order that infection into particular marks.
• This unsupervised learning algorithm has a fast convergence rate.
Genome sequencing marks sense for requests to DNA nucleotides or targets in a genome: thus, requests for As, Cs, Gs, and Ts that makes up the DNA of a living creature. The genome of humans is prepared of over 3 billion hereditary i.e., genetic letters. This current research input sequence consists of Fast Adaptive Shrinkage Threshold Algorithm (FASTA) data format. FASTA is essentially a coded text language of nucleotide and amino acid array useful in bioinformatics. Since the spread of input information is FASTA (DNA) and coronavirus is an RNA type of infection, we need to convert DNA into RNA.
The curved double helix structure of DNA allows it to be released into a stepping stool molded structure, as presented in Fig. 4. This stepping stool structure is made of composite engineered letters called bases. Four of these are available in DNA alone: adenine, thymine, guanine, and cytosine. Adenine links thymine and guanine to cytosine. These bases are talked about independently with As, Ts, Gs, and Cs.
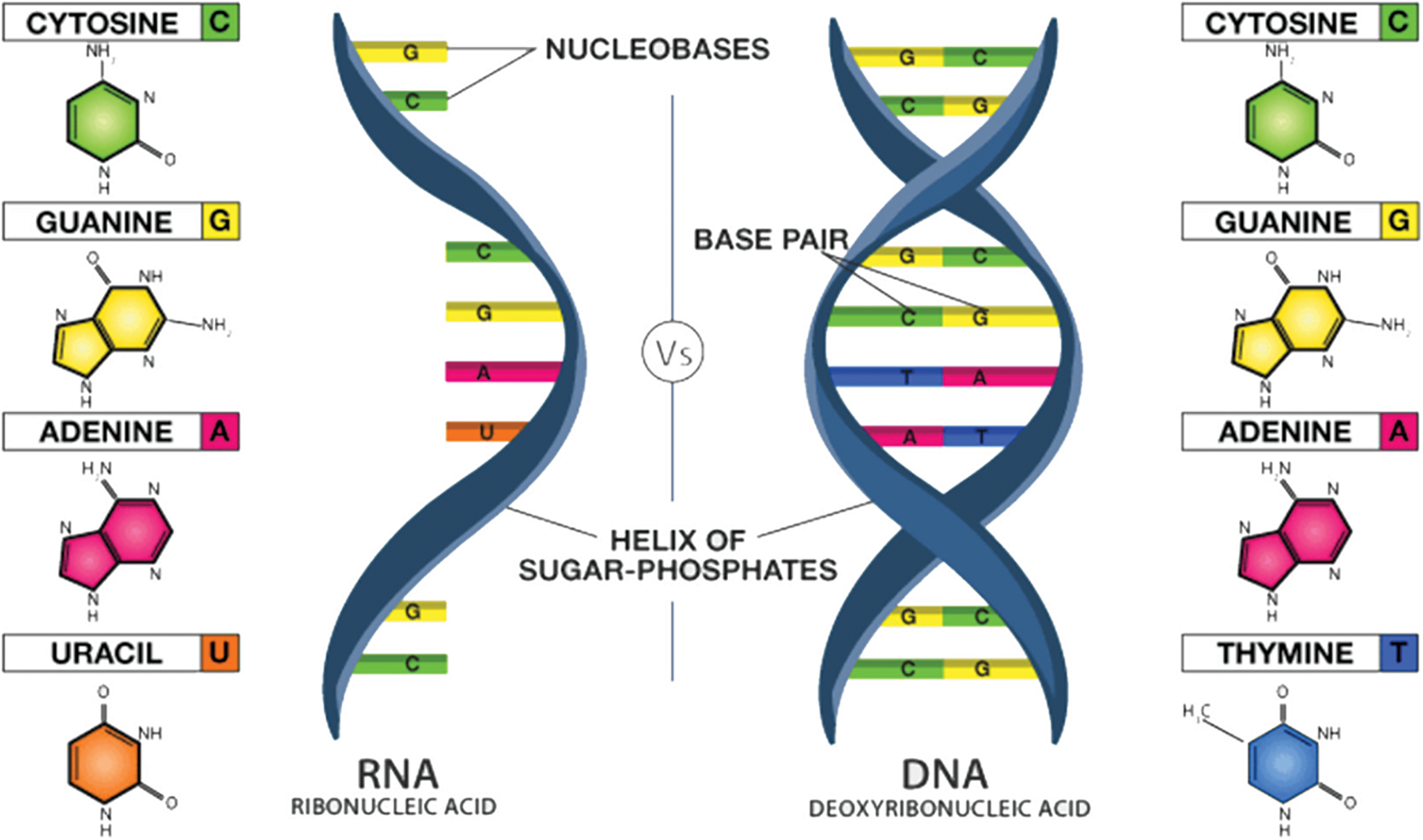
Figure 4: Polymer structure of RNA and DNA [10,12]
The following steps are used to convert DNA to RNA as follows:
• Transcribe DNA into mRNA (ATTAAAGGTT… => AUUAAAGGUU…), where T (thymine) is replaced by U (uracil) as shown in Fig. 5, so we start with AUUAGGGUU using translation function i.e., transcribe () from the bio-python library.
• Translate the mRNA sequence into an amino-acid sequence using a bio-python library function translate ()-called the STOP codon, a successful protein separator.
• DNA basically controls how the infection unfolds.
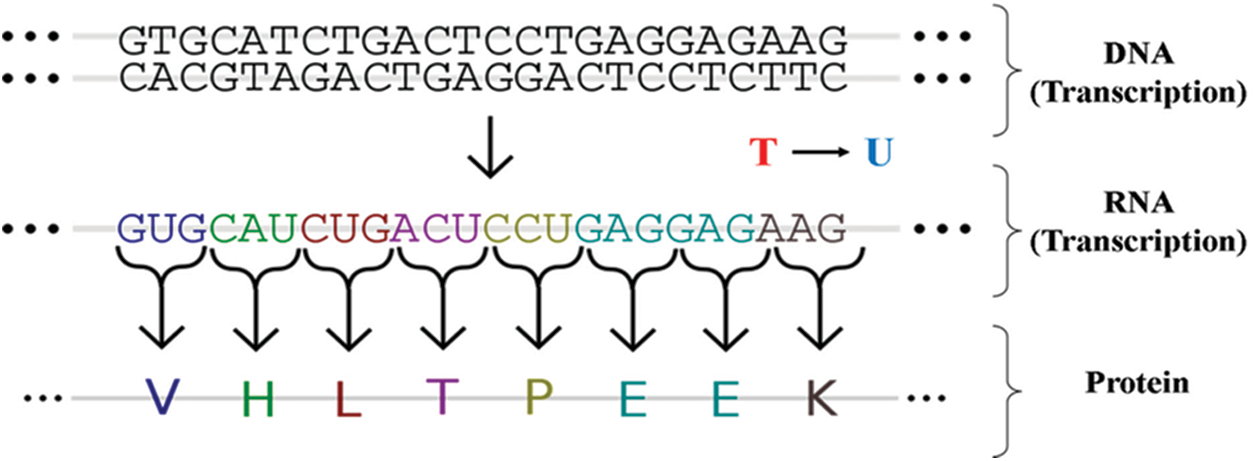
Figure 5: Transmission of information from FASTA (DNA) to coronavirus RNA and subsequent proteins
The k-Means Cluster Algorithm is a non-hierarchical clustering method of machine learning used to examine the characteristics of objects and divide them into groups. The basic objective of k-means clustering calculation is to determine the number of clusters that form most rapidly over time.
This clustering process starts with the specific information
In the second step of the algorithm, a data cluster will be a member of k if the value of that data’s distance from centroid k is the smallest compared to the distance to the second centroid. It can be determined through the formula Eq. (2).
In the third step of the algorithm, the data is classified which belonging to each cluster. The centroid value can be determined by finding the mean value of the data, which helps locate the members of the cluster using the formula described in Eq. (3).
where,
The sequences of all available severe acute respiratory syndrome coronavirus-2 isolate 2019-nCoV/USA-AZ1/2020 complete genomes, available as of 11th February, 2020, were downloaded from the GISAID database (genome data presented in Tabs. 1 and 2 with head sample variable descriptions) have been made on 31st March, 2020. The 3CLpro gene sequences were isolated from the whole genome sequence and converted into protein arrays using the ExPASy Server decryption device. The adjusted genes are localized by the SARS-CoV-2 reference genome (USA-AZ1; NCBI reference sequence/GSAID: MN997409). A total of 29,903 bp complete genomic sequences of SARS-CoV-2 strains are available in the referenced dataset. The dataset only includes complete genomes of high coverage.
The reference coronavirus genome sequence represents 263 mutations of its own to increase the survival rate within a few weeks. The data consists of rows and columns of size 263 × 12 respectively, the top data is described as follows:
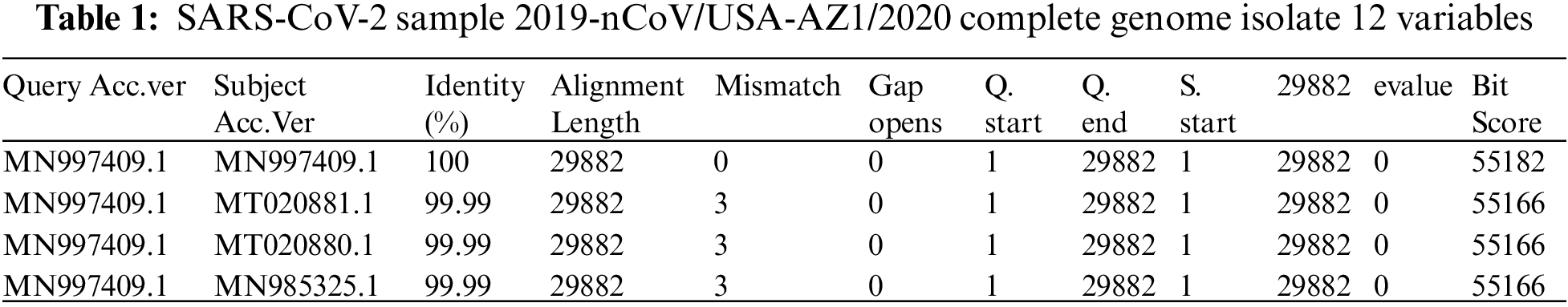
The details of some important columns of data are as follows:

4.1 k-Means Centroid Calculation
k-means clustering is implemented by exploiting numerical values from data points and then applying distance measurement formulas to calculate sequence length, rate of similarity, and the interval between data points. Since the data points are assumed to be a total of 10 columns, random initialization of centroids has been used through this experimental work. After 5 iterations of these algorithms converge, the data values are sequential and thus predict the best centers of classification. The performance metric of the algorithm is fixed at an estimated value of 0.65352 (as shown in Fig. 6) which claims that the algorithm reaches the best point of convergence. In another test of the experiment, we considered 100 sequences of the genome, which cover the maximum amount of information of the sequences, to perform an optimal clustering procedure. One experiment was performed with less than 100 sequences, but we observed that the sequences lost their genetic information, probably due to an inappropriate mutation or some environmental problem.
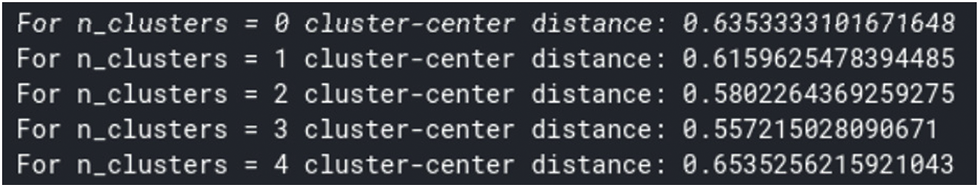
Figure 6: Silhouette method of computing centroid
The functionality of the k-means clustering algorithm can be accelerated to k-means ++ clustering with the help of Python programming. In the first iteration process, it can be seen that the clusters are randomly oriented according to the data as the centroids are randomly allocated in this process.
4.2 Calculation of Cluster Formation
The k-means algorithm follows 2 phases for the formation of clusters:
• Phase 1: A distance metric, here we used the silhouette distance metric to calculate the distance from each data point to the center point.
• Phase 2: Properly labeling each centromere based cluster so that the classification is easily visualized
Run 5 iterations through this research algorithm to form the optimal cluster at the last iteration (as shown in Fig. 7). The implemented k-means clustering algorithm can be accelerated usingk-means ++ metrics in a python environment.
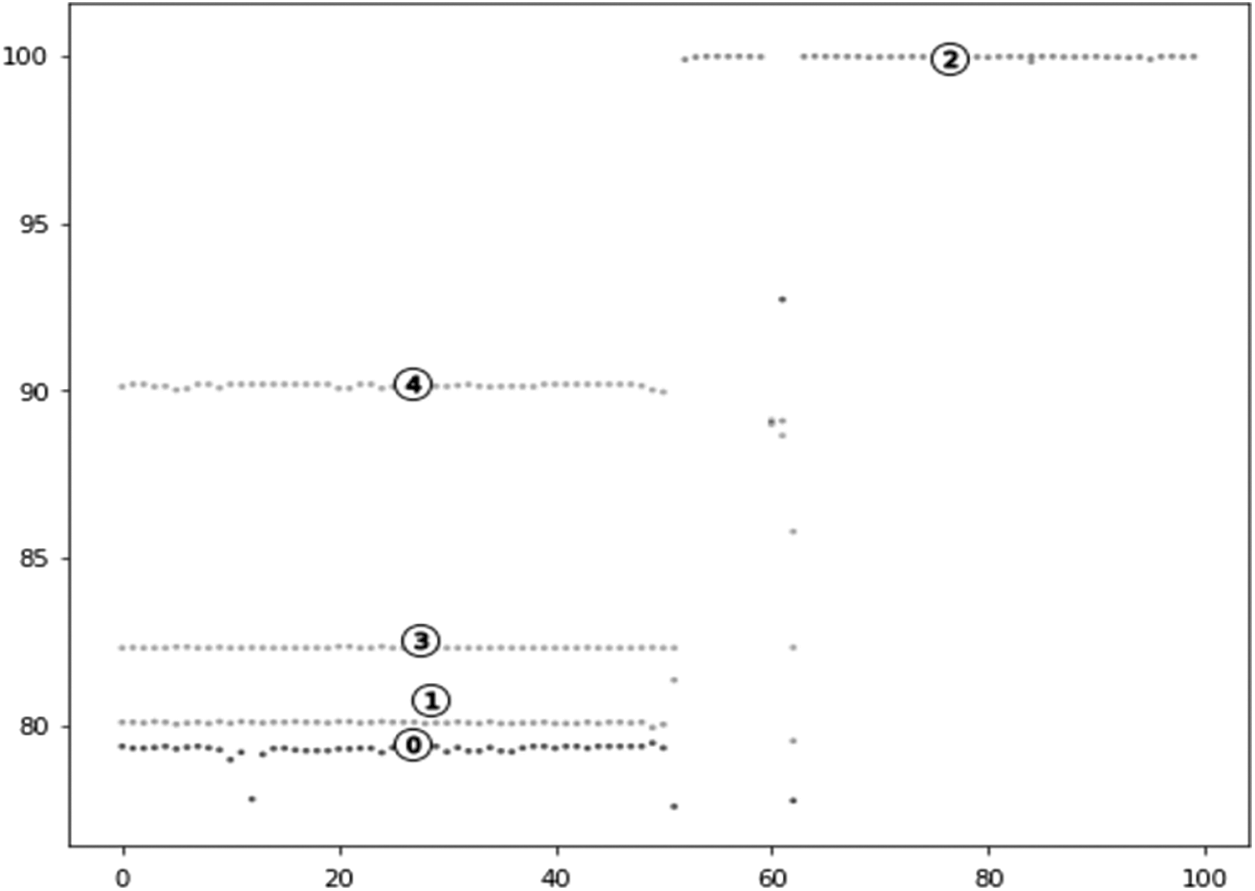
Figure 7: Clustered data visualization
Fig. 7 represents five clusters of virus mutations that are indicated with numerical numbers. Higher cluster value represents higher alignment length which represents cluster closer to parent virus whereas lower cluster value represents the genome of a genetically clustered genome from the parent virus. Most of the cluster of viruses differs from the original virus and hence scientists are attempting to create of vaccine which is focused on virus mutation.
Here the results of the experiment are treated as heat maps that appear above in the Results and Discussion section. This heat-map used correlations based on Spearman’s rank statistics to generate proximity to genome sequences of n-CoV traits from the genus Betacoronavirus.
The correlation metric performed by Spearman’s rank statistics provides the similarity between a pair of positional factors related to each other. This correlation metric is superior to the Carl Pearson correlation statistic because it does not require explicit boundaries to find relationships between information. Furthermore, it works admirably on all information that remains constant during analysis (e.g., age, length, mass). The dataset objective in this research is to represent the association between SARS-CoV2 infection and pneumonia infection and other SARS variations found in the Wuhan Seafood Market virus. It is a complete dataset because it includes variables, for example, BP length, open-close interval, rate of similarity, etc. The heat map of the data in Fig. 8 shows that the data are highly correlated with each other. The alignment length of the array is highly correlated with the bit score. Now preprocess the data and apply k-mean clustering with a total of 5 clusters that best fit the data. These clusters represent the numerical evaluation of the 5 main types of mutations. The heat map of the correlation matrix between the variables in the data is as follows:

Figure 8: Heat map of the complex matrix of 11 variables of the COVID-19 genome mutation data
The heatmap in Fig. 9 represents the characteristics of each cluster by column. Because the scores were scaled up, the actual annotated values have nothing to do with quantitative meaning. Scaled properties can be considered in each segment. If scientists somehow managed to develop a vaccine, it should address these major groups of viruses.
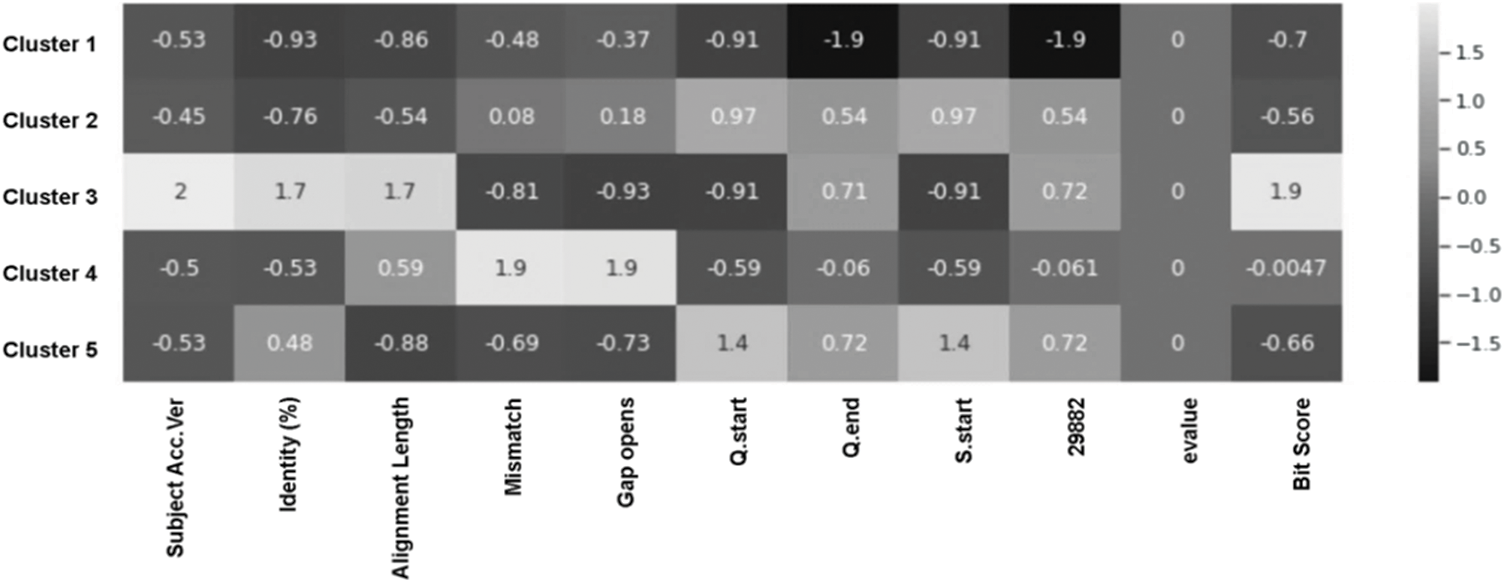
Figure 9: Heat map of the complex matrix of 11 variables of the COVID-19 genome mutation data and k-Means computed clusters
This research provides an analysis-based scientific approach to the issue of COVID-19 by dissecting the genomic clustering of infections using the machine learning cluster process, for example, k-means. Using the k-means method will enable the identification of five main clusters of mutations (
Acknowledgement: This project was funded by the Deanship of Scientific Research (DSR), King Abdulaziz University, Jeddah, under Grant No. (D-111-611-1443). The authors, therefore, gratefully acknowledge DSR technical and financial support.
Funding Statement: This project was funded by the Deanship of Scientific Research (DSR), King Abdulaziz University, Jeddah, under Grant No. (D-111-611-1443). The authors, therefore, gratefully acknowledge DSR technical and financial support.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
1. World Health Organization. (2020). Coronavirus disease 2019 (COVID-19Situation report, 72. World Health Organization. https://apps.who.int/iris/handle/10665/331685. [Google Scholar]
2. N. Zhu, D. Zhang, W. Wang, X. Li, B. Yang et al., “A novel coronavirus from patients with pneumonia in China, 2019,” New England Journal of Medicine, vol. 382, no. 8, pp. 727–733, 2020. [Google Scholar]
3. A. Fehr, R., Channappanavar, R., and S. Perlman, “Middle East respiratory syndrome: Emergence of a pathogenic human coronavirus,” Annual Eeview of Medicine, vol. 68, pp. 387–399, 2017. [Google Scholar]
4. H. Mohd. A., J. Al-Tawfiq, A., and Z. A. Memish, “Middle East respiratory syndrome coronavirus (MERS-CoV) origin and animal reservoir,” Virology Journal, vol. 13, no. 1, pp. 87, 2016. [Google Scholar]
5. P. Zhou, X. Yang., X. Wang, B. Hu, L. Zhang et al., “A pneumonia outbreak associated with a new coronavirus of probable bat origin,” Nature, vol. 579, no. 7798, pp. 270–273, 2020. [Google Scholar]
6. X. Tang, C. Wu, X. Li, Y. Song, X. Yao et al., “On the origin and continuing evolution of SARS-CoV-2,” National Science Review, vol. 7, no. 6, pp. 1012–1023, 2020. [Google Scholar]
7. S. van Boheemen., M. de Graaf, C. Lauber, T. M. Bestebroer, V. stalin Raj et al., “Genomic characterization of a newly discovered coronavirus associated with acute respiratory distress syndrome in humans,” MBio, vol. 3, no. 6, pp. e00473–12, 2012. [Google Scholar]
8. Y. Ding, L. He, Q. Zhang, Z. Huang, X. Che et al., “Organ distribution of severe acute respiratory syndrome (SARS) associated coronavirus (SARS-CoV) in SARS patients: Implications for pathogenesis and virus transmission pathways,” The Journal of Pathology: A Journal of the Pathological Society of Great Britain and Ireland, vol. 2, no. 203, pp. 622–630, 2004. [Google Scholar]
9. V. M. Corman, O. Landt, M. Kaiser, R. Molenkamp, A. Meijer et al., “Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR,” Eurosurveillance, vol. 25, no. 3, pp. 2000045, 2020. [Google Scholar]
10. Chen, Y., Liu, Q., and Guo, D., “Emerging coronaviruses: Genome structure, replication, and pathogenesis,” Journal of Medical Virology, vol. 92, no. 4, pp. 418–423, 2020. [Google Scholar]
11. K. Stadler, V. Masignani, M. Eickmann, S. Becker, S. Abrignani et al., “SARS—beginning to understand a new virus,” Nature Reviews Microbiology, vol. 1, no. 3, pp. 209–218, 2003. [Google Scholar]
12. T. Li, Y. Zhang, L. Fu, C. Yu, Y. Li et al., “siRNA targeting the leader sequence of SARS-CoV inhibits virus replication,” Gene Therapy, vol. 12, no. 9, pp. 751–761, 2005. [Google Scholar]
13. J. Lei, Y. Kusov, and R. Hilgenfeld, “Nsp3 of coronaviruses: Structures and functions of a large multi-domain protein,” Antiviral Research, vol. 149, pp. 58–74, 2018. [Google Scholar]
14. J. Chen, “Pathogenicity and transmissibility of 2019-nCoV—a quick overview and comparison with other emerging viruses,” Microbes and Infection, vol. 22, no. 2, pp. 69–71, 2020. [Google Scholar]
15. P. Serrano, M. A. Johnson, A. Chatterjee, B. W. Neuman, J. S. Joseph et al., “Nuclear magnetic resonance structure of the nucleic acid-binding domain of severe acute respiratory syndrome coronavirus nonstructural protein 3,” Journal of Virology, vol. 83, no. 24, pp. 12998–13008, 2009. [Google Scholar]
16. K. Anand, J. Ziebuhr, P. Wadhwani, J. R. Mesters, R. Hilgenfeld et al., “Coronavirus main proteinase (3CLpro) structure: Basis for design of anti-SARS drugs,” Science, vol. 300, no. 5626, pp. 1763–1767, 2003. [Google Scholar]
17. M. M. Angelini, M. Akhlaghpour, B. J. Neuman, M. J. Buchmeier, “Severe acute respiratory syndrome coronavirus nonstructural proteins 3, 4, and 6 induce double-membrane vesicles,” MBio, vol. 4, no. 4, pp. e00524–13, 2013. [Google Scholar]
18. B. H. Harcourt, D. Jukneliene, A. Kanjanahaluethai, J. Bechill, K. M. Severson et al., “Identification of severe acute respiratory syndrome coronavirus replicase products and characterization of papain-like protease activity,” Journal of Virology, vol. 78, no. 24, pp. 13600–13612, 2004. [Google Scholar]
19. G. Sutton, E. Fry, L. Carter, S. Sainsbury, T. Walter et al., “The nsp9 replicase protein of SARS-coronavirus, structure and functional insights,” Structure, vol. 12, no. 2, pp. 341–353, 2004. [Google Scholar]
20. S. K. Wong, W. Li, M. J. Moore, H. Choe, M. Farzan et al., “A 193-amino acid fragment of the SARS coronavirus S protein efficiently binds angiotensin-converting enzyme 2,” Journal of Biological Chemistry, vol. 279, no. 5, pp. 3197–3201, 2004. [Google Scholar]
21. B. Coutard, C. Valle, X. de Lamballerie, B. Canard, N. G. Seidah et al., “The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade,” Antiviral Research, vol. 176, pp. 104742, 2020. [Google Scholar]
22. I. Glowacka, S. Bertram, M. A. Müller, P. Allen, E. Soillux et al., “Evidence that TMPRSS2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response,” Journal of Virology, vol. 85, no. 9, pp. 4122–4134, 2011. [Google Scholar]
23. R. He, A. Leeson, M. Ballantine, A. Andonov, L. Baker et al., “Characterization of protein–protein interactions between the nucleocapsid protein and membrane protein of the SARS coronavirus,” Virus Research, vol. 105, no. 2, pp. 121–125, 2004. [Google Scholar]
24. W. Guan, Z. Ni, Y. Hu, W. Liang, C. Ou et al., “Clinical characteristics of coronavirus disease 2019 in China,” New England Journal of Medicine, vol. 382, no. 18, pp. 1708–1720, 2020. [Google Scholar]
25. J. Wang, and X. Su, “An improved K-means clustering algorithm,” in 2011 IEEE 3rd Int. Conf. on Communication Software and Networks, Xi'an, China, IEEE, pp. 44–46, 2011. [Google Scholar]
26. T. M. Ghazal, M. Z. Hussain, R. A. Said, A. Nadeem, M. K. Hassan et al., “Performances of K-means clustering algorithm with different distance metrics,” Intelligent Automation & Soft Computing, vol. 30, no. 2, pp. 735–742, 2021. [Google Scholar]
27. M. J. Rezaee, M. Eshkevari, M. Saberi, O. Hussain et al., “GBK-Means clustering algorithm: An improvement to the K-means algorithm based on the bargaining game,” Knowledge-Based Systems, vol. 213, pp. 106672, 2021. [Google Scholar]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |