DOI:10.32604/cmc.2022.026205
| Computers, Materials & Continua DOI:10.32604/cmc.2022.026205 | |
| Article |
Mutation Prediction for Coronaviruses Using Genome Sequence and Recurrent Neural Networks
1Bharati Vidyapeeth’s College of Engineering, New Delhi, 110063, India
2Department of Natural and Exact Sciences, Samarkand State University, Samarkand, Uzbekistan
3Graduate School, Duy Tan University, Da Nang, 550000, Viet Nam
4Faculty of Information Technology, Duy Tan University, Da Nang, 550000, Viet Nam
5Department of Information Technology, College of Computers and Information Technology, Taif University, Taif, 21944, Saudi Arabia
*Corresponding Author: Anand Nayyar. Email: anandnayyar@duytan.edu.vn
Received: 18 December 2021; Accepted: 08 April 2022
Abstract: The study of viruses and their genetics has been an opportunity as well as a challenge for the scientific community. The recent ongoing SARS-Cov2 (Severe Acute Respiratory Syndrome) pandemic proved the unpreparedness for these situations. Not only the countermeasures for the effect caused by virus need to be tackled but the mutation taking place in the very genome of the virus is needed to be kept in check frequently. One major way to find out more information about such pathogens is by extracting the genetic data of such viruses. Though genetic data of viruses have been cultured and stored as well as isolated in form of their genome sequences, there is still limited methods on what new viruses can be generated in future due to mutation. This research proposes a deep learning model to predict the genome sequences of the SARS-Cov2 virus using only the previous viruses of the coronaviridae family with the help of RNN-LSTM (Recurrent Neural Network-Long Short-Term Memory) and RNN-GRU (Gated Recurrent Unit) so that in the future, several counter measures can be taken by predicting possible changes in the genome with the help of existing mutations in the virus. After the process of testing the model, the F1-recall came out to be more than 0.95. The mutation detection’s accuracy of both the models come out about 98.5% which shows the capability of the recurrent neural network to predict future changes in the genome of virus.
Keywords: COVID-19; genome sequence; coronaviridae; RNN-LSTM; RNN-GRU
COVID-19 is a contagious and extremely infectious disease that is caused by SARS-CoV-2 virus [1]. The virus originated from the Wuhan province of China when the first case was reported on 17 November 2019 in Hubei [2]. Since then, the virus is spreading at an alarming rate that has resulted into a global pandemic as declared by World Health Organization (WHO) on 31 January 2020 [3]. More than 15.2 million cases have been identified across 188 countries and territories out of which 623,000 have resulted in death as of 23 July 2020 [4]. Apart from this, the pandemic has caused a lot of economic and social disruption [5] including the largest global recession [6] and global famines [7]. Some of the common visible symptoms also include cough, fever, shortness of breath and lack of smell as well as taste [8–10]. Although most of the cases end with meek symptoms, some gradually develop into acute respiratory distress syndrome known as ARDS that can precipitate through a cytokine storm [10,11], blood clots and multi-organ failure [12]. The virus spreads through tiny droplets released during coughing, sneezing, and talking [13,14]. Therefore, the virus is transmitted more frequently amongst individuals in close contact.
COVID-19 virus medically termed as SARS-CoV-2 is a positively single-stranded Ribonucleic Acid (RNA) coronavirus that comes under the Betacoronavirus genus [15,16]. The first genomic sequence was discovered in China on 10 January 2020 and was kept in the National Center for Biotechnology Information GenBank (NCBI). The gene sequence is Deoxyribose Nucleic Acid (DNA) type that is Uracil (U) is substituted by Thymine (T) although the virus is an RNA type for understanding. The virus mutates during replication of genomic information which is caused because of some errors while duplicating the RNA into a new cell [17,18]. Therefore, it becomes necessary to study the genome sequence of the virus as the mutation takes place with every replication. Artificial intelligence subfields such as machine learning (ML) and deep learning are playing very important role in health care sector recently [19–22]. Various Machine Learning and Deep Learning (DL) are being used to analyze the data on COVID-19 to help in preparing the vaccines, evaluating the drug responses, genome sequencing and predicting the proximity of the disease in the patients. In this paper, LSTM-RNN and GRU-RNN based models are proposed and implemented to predict the genome sequence of viruses in the future caused by mutation in the genome sequence of the coronavirus so that necessary preparation and treatment are taken care of in advance. Such studies can help not only study changes in the genetic nature of these viruses but also help in knowing what effect a vaccine or a medicine cause on their genome and hence can be used to run different simulations before live testing.
The research paper addresses following research objectives:
Objective 1: In-depth study of research work done within the fields of genome sequencing and Neural Networks for predicting sequences.
Objective 2: Design and Development done for RNN-LSTM and RNN-GRU models according to shape of pre-processed & time-sequenced genomic data collected and combined from NCBI repository.
Objective 3: Using Training data collected for respective models to compare their performance in terms of accuracy and F1-score, as well as use the models to predict an existing genome sequence and compare with to find mutation accuracy for respective models.
Objective 4: The results of mutation accuracy were analyzed as well as compared with contemporary works over its performance and algorithm used to predict genome sequence.
Organization of the Paper
Section 2 covers related works and studies done in genome sequencing of coronavirus. Section 3 gives an introduction on genome sequencing and discusses all the methodologies used in both the models. Section 4 describes the flow and implementation process of the proposed model including data description and data preprocessing. Section 5 analyses the results of both the models and their comparison. Section 6 concludes the paper with future scope.
Several researchers have worked on the genome sequence of the SARS-CoV-2 virus to achieve fruitful results. This section discusses such existing works in detail.
Alejandro Lopez-Rincon et al. [23] proposed an assisted detection approach to solve this issue by integrating the molecular analysis with machine learning and Artificial Intelligence (AI). The method uses a deep convolution neural network that extracts features from the genome sequence.
Studies with Novel Coronavirus Tool (2019nCoVR) have shown that the proposed system is sufficiently capable of correctly classifying SARS-CoV-2, differentiating it from other coronavirus mutants, such as Middle East Respiratory Syndrome (MERS-CoV), Human Coronavirus (HCoV-229E), HCoV-NL63, HCoV-OC43, HCoV-HKU1 and the virus’s predecessor SARS-CoV, regardless of insufficient description as well as sequential noises or errors.
Biswas [24] (2020) presented a phylogenetic analysis of the SARS-CoV-2 virus. In the study, complete genomic sequence of the virus was mentioned. The authors established the endemicity component of the virus and then worked on discovering the next SARS-CoV-2 source and disclosed that all sequences of this virus were formed in a single group with no branching but did not support the results with a comprehensive mathematical analysis.
Mooney [25] (2014) and Roach [26] (2010) addressed the assembly, stoichiometry, and composition of RNA synthesizing complexes. One practical outcome of reverse genetics, according to the authors is the development of stable coronavirus-based replicated reservoirs for vaccines and other biomedical uses.
The development of an in vitro method of replication of viruses such as the one used for poliovirus in which complete replication of viruses in cell lysates can be achieved is still ongoing. This approach will allow for a far more in-depth analysis of the requirements for gene replication, beginning with the transcript of an infectious gene.
Lauber [27] (2012) optimized genome design conservation to segment of all the genome into five non-overlapping regions: 59 untranslated regions (UTRs) as well as open reading frames (ORFs) 1a, ORF1b, 39 ORFs (including 39 proximal ORFs) and 39 UTRs. Under different models, each area was examined for its contribution to shifts in genome scale. Statistically, the non-linear solution outperformed the linear model and obtained 0.92% of the data variance.
Examination of the SARS-CoV-2 gene signature was conducted by Das and Ghate [28] (2020). They measured the ancestry rate of the European genome using the qpAdm statistical tool. Then, with the help of GraphPad Prism v8.4.0, GraphPad Program, Pearson applied the coefficient of association between different ethnicity levels of the European genome and conducted a statistical study of the death to recovery ratio.
Yadav [29] (2020) researched on the SARS-CoV-2 genome sequence of three cases that had a positive record of travel from Wuhan, China. Almost complete genomes of case 1, case 3 (29,851 nucleotides) as well as a partial genome of case 2 were found. It has been noted that the Indian SARS-CoV-2 sequences shared almost 99 percent identification with the pneumonia virus contained in the Wuhan seafood industry. They proposed that genome sequencing of cases from India will be performed to establish whether the virus is developing.
Ye [30] (2013) proposed inclusion of AI in solving the problems proposed by the coronavirus and did an extensive literature review on the models such as extreme machine learning, generative adversarial networks etc. For monitoring patients, diagnosing the disease, and predicting the spread of the virus in different stages.
Based on the above literature survey, it is quite evident that there is extensive research work done on genome structure of coronavirus or related viruses and genome sequencing. With the use of prediction power of recurrent Neural Networks., change and nature of genome of virus can be detected and analyzed. It can be concluded that the development of a model that could the predict the new mutated genome sequence of SARS-CoV-2 would benefit the world to avoid the situation such as the ongoing pandemic soon by developing potential treatments for example vaccines and medicinal drugs after medically examining the genome sequence of the predicted virus.
In this section, a general overview of Deep Learning algorithms is performed.
Genome sequence is a full series of nucleotides which composes all chromosomes of an organism. In a population, the huge percentage of nucleotides are similar among organisms, although the analysis of several organisms is important to explain genetic diversity.
The method of establishing the full DNA sequence of the genome of the organism at a particular time that includes the decoding of all the chromosomal DNA of the individual, and the DNA found in the mitochondria and in the chloroplast for plants is known as Genome Sequencing [31]. Genome is an organism’s genetic code which contains DNA and RNA for viruses. It contains both mitochondrial DNA as well as chloroplast DNA. In general, gene sequences that are completely total are often considered entire genome sequences [32]. Genome sequencing is primarily used as a testing method but has been extended to clinics in 2014 [33]. In the succeeding phase of precision medicine, full genome sequence data can be an essential resource to direct clinical involvement [34]. It can therefore lay the groundwork for predicting disease severity and proximity as well as drug reaction.
The SARS-CoV-2 genome contains between 26,000 to 31,500 base pairs, which sound like a long sequence, with 31,500 positions filled with one nucleotide. As per the analysis performed by Woo et al. [34], in this virus, the number of Gs & C’s single nucleotide polymorphism (SNPs) ranges up to 32 percent. Fig. 1 shows around 43 percent of the specific numbers of ORFs are found in genes such as ORF1ab, shell, membrane, spike, and nucleocapsid [35]. In this paper, genome sequence of future viruses is predicted using the existing genome sequence of the coronavirus and applying the RNN models on it.
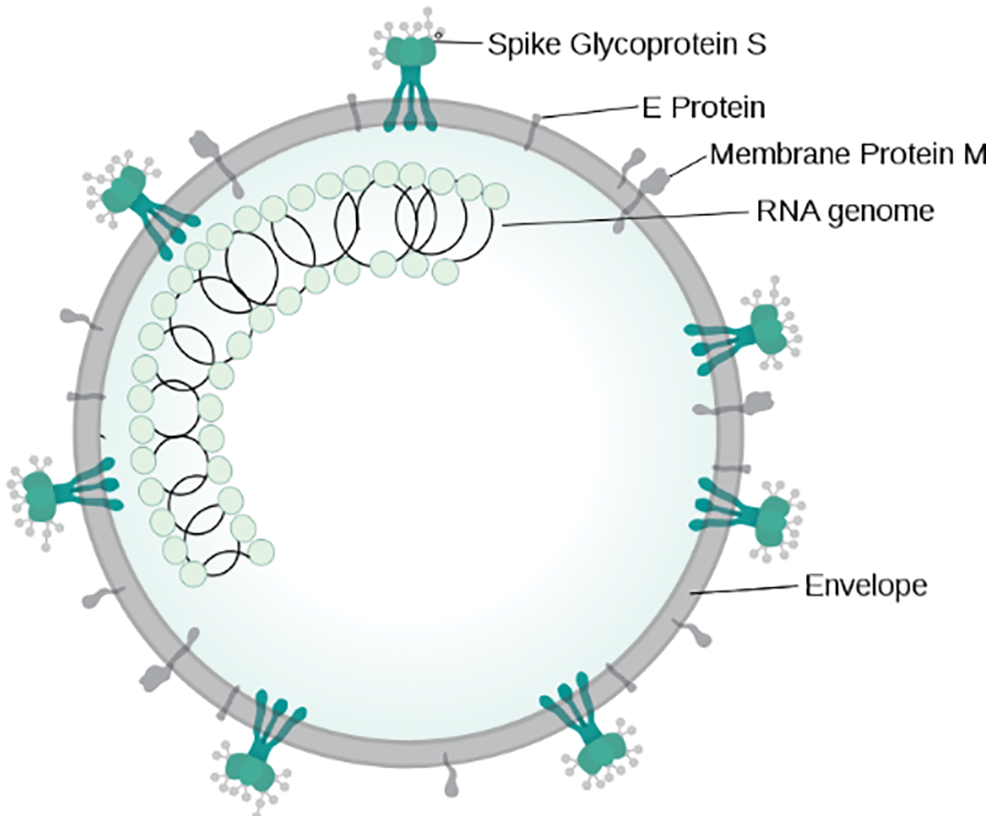
Figure 1: Illustration of SARS-CoV virion
3.2 Recurrent Neural Network (RNN)
A recurrent neural network (RNN) is a feedforward neural network where the relationship between nodes forms a directed graph along a time series or sequence that enables temporal contextual actions to be seen. RNNs are based on feedforward neural networks [36]. Therefore, they can process variable duration sequence of inputs using their internal state or memory.
The word “recurrent neural network” is known to point to two large groups of neural networks with a common framework, one which is a finite i.e., known impulse and the other is an infinite i.e., unknown impulse. All network groups view time sensitive behavior [37]. The finite impulse recurring network is a supervised learning model that could be unfolded and replaced by a purely feed-forward neural network, while the infinite impulse recurring network is a cyclical nature graph that cannot be unfolded. These impulses have specific storage states that are directly controlled by the network. These form the basis of the LSTMs and GRUs.
Each data element is taken as a token after tokenization. During the forward propagation as being shown in Fig. 2 in which, at specific time(t), the output at each hidden layer (h) is calculated using an activation function by multiplying the input (x) with the weights (U and V) initialized. However, in this contrary to other neural networks, one more term included in the function that is the output of the previous layer multiplied by some different weights (W) initialized due to which after each subsequent layer, the next output of one layer depends on the output of the previous at a time, therefore, maintaining the sequence in which the input is fed to the network [38]. The final output is calculated using a different activation function (mostly SoftMax) and to get the loss function. Similarly, in the backward propagation, the weights are updated according to the outputs of the previous layer to get a global minimum during gradient descent to reduce the loss in each iteration [39].
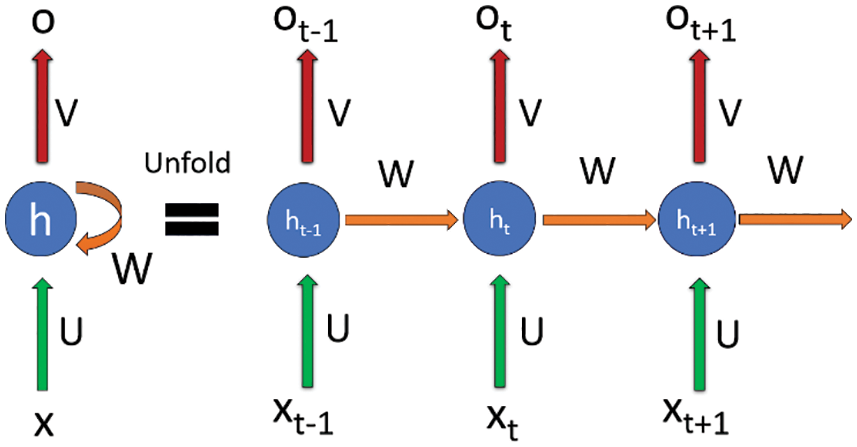
Figure 2: Illustration of forward propagation in recurrent neural network (RNN)
3.3 Long-Short Term Memory (LSTM)
RNNs are not particularly effective if the input sequences are long as it is difficult to preserve and carry the information from the previous steps to the next ones due to which they might not include the information from the start. Moreover, RNNs suffer from vanishing gradient problem during backpropagation i.e., if the gradient value goes on to become lesser and lesser, it cannot help in training and learning of the model. LSTM tends to solve all the shortcomings of the typical recurrent neural network.
Long short-term memory is a recurrent neural network (RNN) based architecture [40–42]. Unlike normal neural feedforward networks, LSTM has regulated feedback loops. It does not only read singular data points like images, even applying to whole data sequences like audio or video stream. For instance, it is useful for applications like handwriting recognition, speech recognition, time series prediction [43], sign language translation [44,45] and many more. Some of the applications in healthcare sector include predicting subcellular localization of proteins [46] and various prediction in medical care pathways [47–50].
LSTM based RNN model is based on supervised learning that trains by utilizing gradient descent algorithm which is an optimization technique on a series of training sequences over time in order to measure the gradients required to optimize the model so that the weights of LSTM model are revised in proportion to the error derivative with reference to the corresponding weight using backpropagation. The issue of vanishing gradient with RNNs is resolved by LSTM as when the erroneous values are carried from specific output layer, they persist in the cell due to which it simultaneously returns error until the model is trained to cut off the value in all the gates. In this paper, LSTM based RNN model is implemented.
3.4 Gated Recurrent Unit (GRU)
Gated recurrent unit (GRU) uses a gating function in RNNs as it avoids usage of cell state and uses hidden state as a tool to transmit information [51]. It has only namely two gates i.e., a reset gate and an update gate. It is like LSTM which has less parameters in forget gate as it requires an entry gate [52]. The performance of GRU in some activities of natural language processing and polyphonic music modeling came out to be close to that of LSTM [53,54]. Nevertheless, it has shown stronger performance on relatively small and less regular datasets [55].
Like LSTM, it solves the issue of vanishing gradient in standard RNN by using update gate and reset gate. There are two primary sequences that determine which data will be carried on to the output. The remarkable aspect about GRU is that it can be learned to retain information even for a large amount of time, without wiping it over time or deleting information that is unrelated to the prediction.
However according to Weiss [56], the LSTM is better and stronger as compared to GRU because it performs unbounded counting whereas the GRU cannot be due to which it cannot learn common languages that are learned easily by the LSTM.
Fig. 3 shows the complete flow of our research study, and each block is separately explained. We first start with collecting our data and implementing suitable data preparation techniques to convert the raw data into the form that can be passed through the deep learning model. The steps include tokenization, feature extraction and feature selection of the data. After that the data is passed through the models and then they are optimized and evaluated with suitable metrics such as accuracy and F1 score. After getting the best performances of these models their results are compared and analyzed for to get further insights about our study.
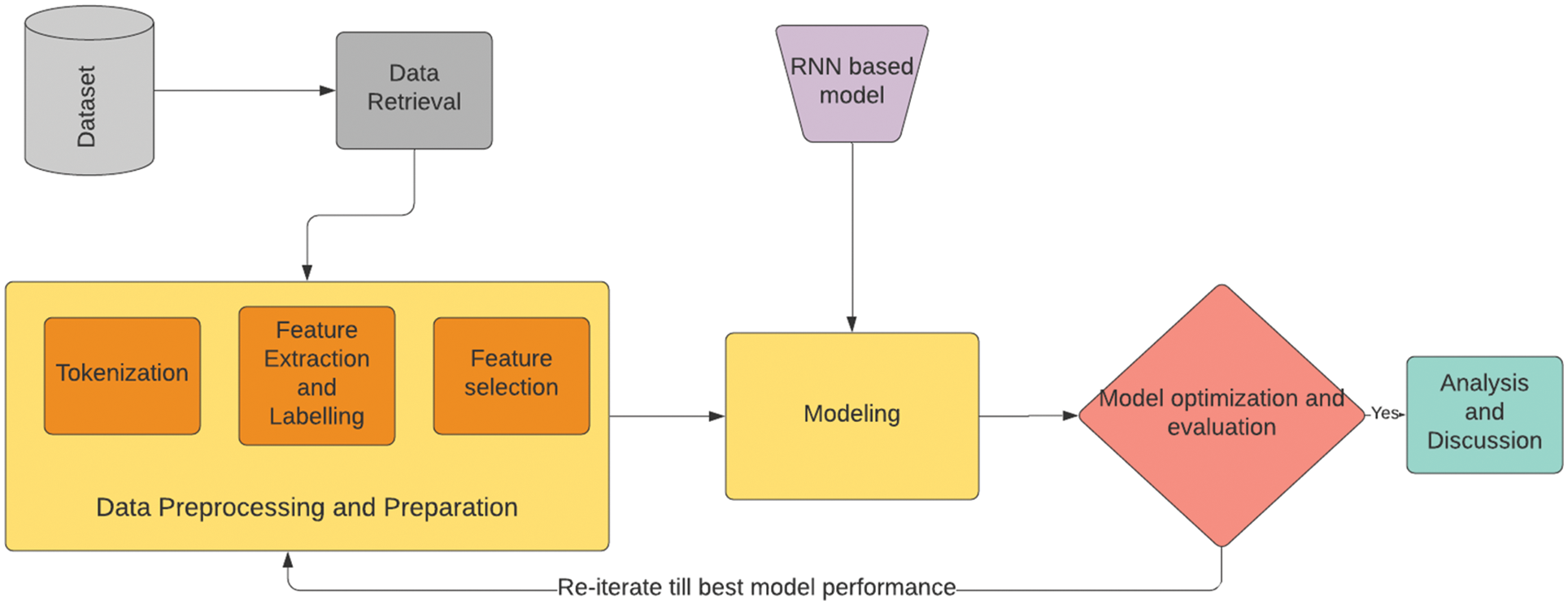
Figure 3: Flow of the research
In this paper, LSTM-RNN and GRU-RNN models are used for training and learning. Figs. 4 and 5 displays the model architecture of LSTM and GRU models respectively with the input and output dimension after each layer. The first layer is input layer in which all shaped data sequences are fed to the model. The second layer is the LSTM recurrent layer in the case of the LSTM model and GRU recurrent layer in the case of the GRU model with the activation function, tan h. The next layer is Dropout layer which is used for regularization initialized using dropout rate, 0.2, followed by a Dense layer which is the standard fully connected neural network layer with activation function, sigmoid. The next two layers are also Dropout and Dense layers. However, the activation function is SoftMax in the last Dense layer which is the output layer.
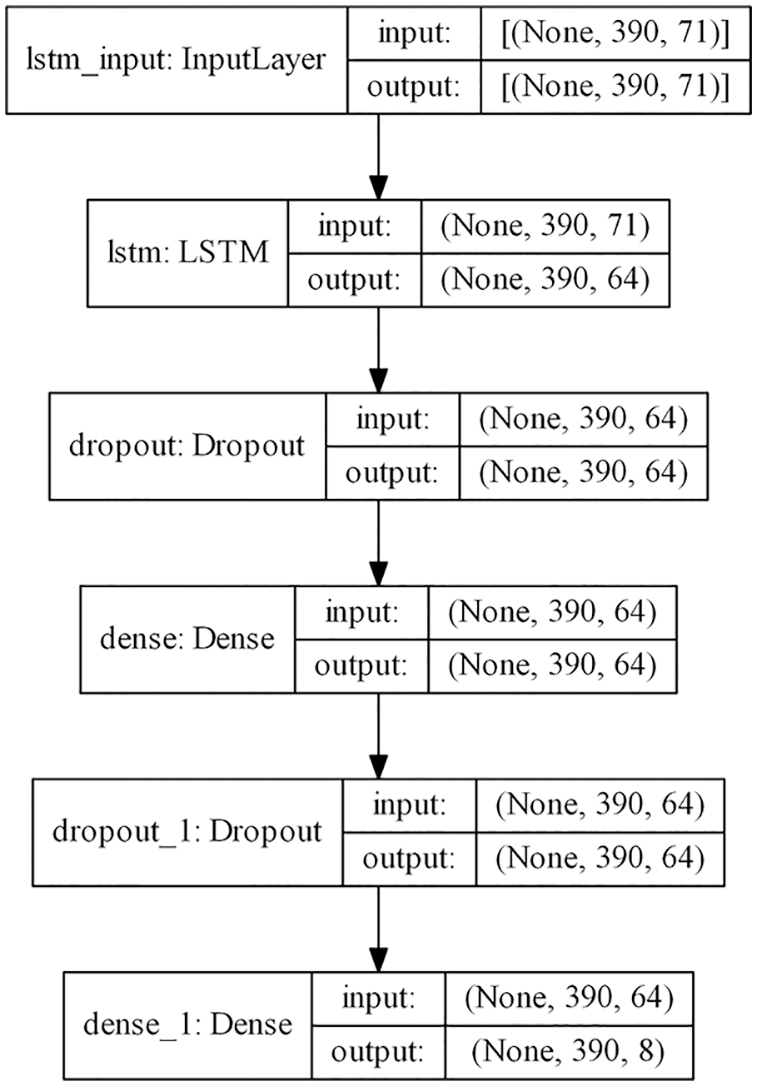
Figure 4: Model architecture of LSTM-RNN model

Figure 5: Model architecture of GRU-RNN model
4.2 Data Collection and Processing
The data used for training the model is collected from six different genome sequences of coronavirus family. The first is the complete genome sequence of coronavirus HKU1 (CoV-HKU1) cultured from a 71-year-aged male person with pneumonia who recently returned from Shenzhen province, China [57–60]. The second is the genome sequence of novel HCoV-229E that was isolated from a man diagnosed with case of acute pneumonia along with failure of renal functions in 2012. The third is the genome sequence of a fourth HCoV-NL63 which was isolated from a 7-month-old suffering from both conjunctivitis as well as bronchiolitis [61–64]. The viral genome sequence consisted of unique characteristics that included a distinctive N-terminal fragment. The fourth is the genome sequence of SARS Coronavirus (SARS-CoV). The fifth is the complete genome sequence of MERS-CoV. The last one is the genome sequence of SARS-CoV-2 i.e., COVID-19 responsible for the ongoing pandemic [64–66]. It was isolated from a patient who used to work at the Huanan seafood marketplace in Wuhan and had to be admit in the Wuhan Central Hospital on 26 December 2020. All these six genome sequences were collected and compiled into one dataset for the further learning and training of the model. Tab. 1 gives the complete Dataset description.

After the data was collected and compiled, the next step was to preprocess the raw text data so that it can be used as an input to the model for training and learning. The data included the genome sequences as a data stream of letters. Using TensorFlow and Keras library [67], firstly the genome sequences were tokenized into a series of integers where each integer was the index of the token. In the second step, the letters present in the genome sequences were tokenized. Finally, all the data was cleaned and checked for any null or duplicate values present in the final dataset.
4.3 Feature Extraction and Labelling
First, tokenization of the genome sequences was done. The converted tokens of the characters in the genome sequence are in the form of integers ranging from 1 to 5. Furthermore, the integer 5 always occurs at the end of the token array and hence has no greater significance in the feature extraction process.
The process of extracting the features from the preprocessed dataset was done by iterating over the token list of the sequences. A fixed length of the token was taken at a time and combined with the next sequence of tokens of length T till the end of the token list was reached using the append method. It was later converted into an array. At the end of the process, the feature array is created with shape (y, T) where y is represented in Eq. (1).
During the feature extraction process, an index was given to each feature array, and a label array of the length of dataset size was created which was then multiplied by the indices of unique character that came out to be 8 in this case. This array was enumerated or defined by iterating over its indices and giving each element value of either 0 for all features not matching the label array indices or 1 for all features matching the label array indices giving us a label array of shape (y*8,8).
The fitting of the RNN-LSTM and RNN-GRU model on the dataset was done by using the Adam optimizer which uses stochastic gradient descent algorithm. The learning rate and decay rate were manually set and adjusted to avoid overfitting in case of extremely small learning rate or underfitting in case of very high learning rate. Instead of using gradients, partial derivatives were used which are very helpful since in the dataset comprised of multiple tokens that were being propagated in the RNN based models.
The model is generated using a TensorFlow v2.0 Environment, with the system using NVIDIA GeForce MX110 GPU, 16GB Ram and Python version 3.7. Although the genome sequences of viruses are of same length, their width is different, so they need to be trimmed to same dimension before pre-processing to make them uniform i.e., 72 X 395 so that it easier to give dimensions in neural network layers.
For Metrics we have Chosen Accuracy and F1 score which require true positives (TP), true negatives (TN), false positives (FP) and false negatives (FN) need to be defined.
TP: When actual sequence is “ABCD” (in sequence) and predicted sequence is also “ABCD”.
TN: When actual sequence is “CBAD” (not in sequence) and predicted sequence is also “CBAD”.
FP: When actual sequence is “ABCD” (in sequence) and predicted sequence is “CBAD” (not in sequence).
FN: When actual sequence is “CBAD” (not in sequence) and predicted sequence is “ABCD” (in sequence).
The dataset containing the genome sequences after preprocessing is split into training and validation datasets to prevent overfitting and for evaluating the performance of respective LSTM and GRU models. The metrics of evaluation used in this paper are accuracy and F1 score. F1 score is an indicator of the precision of the dataset’s accuracy. The more the number of true positives, the greater would be the F1 score.
Tab. 2 highlights the accuracy and F1 score for both the models after epoch intervals of 5, 10, and 15. The accuracy in the case of both the models is approximately equal but in the case of the F1 score, LSTM-RNN performs better than the GRU-RNN model implying that the former gives a greater number of positive results as compared to the latter. Further insights can be drawn after plotting the training and validation graphs for both the modes.

For further analysis, the training and validation accuracy as well as loss per epoch is plotted. Figs. 6 and 7 displays the training and validation accuracy of both the models. Although the validation accuracy is almost the same in both graphs, the training accuracy differs as in the LSTM-RNN model it achieves high accuracy after five epochs which is much earlier than the GRU-RNN model in which it achieves high accuracy after 10–12 epochs. Similarly, Figs. 8 and 9 display the training and validation loss per epoch. From both the graphs, it is visible that the loss in the LSTM-RNN model is less than the loss in the GRU-RNN model.
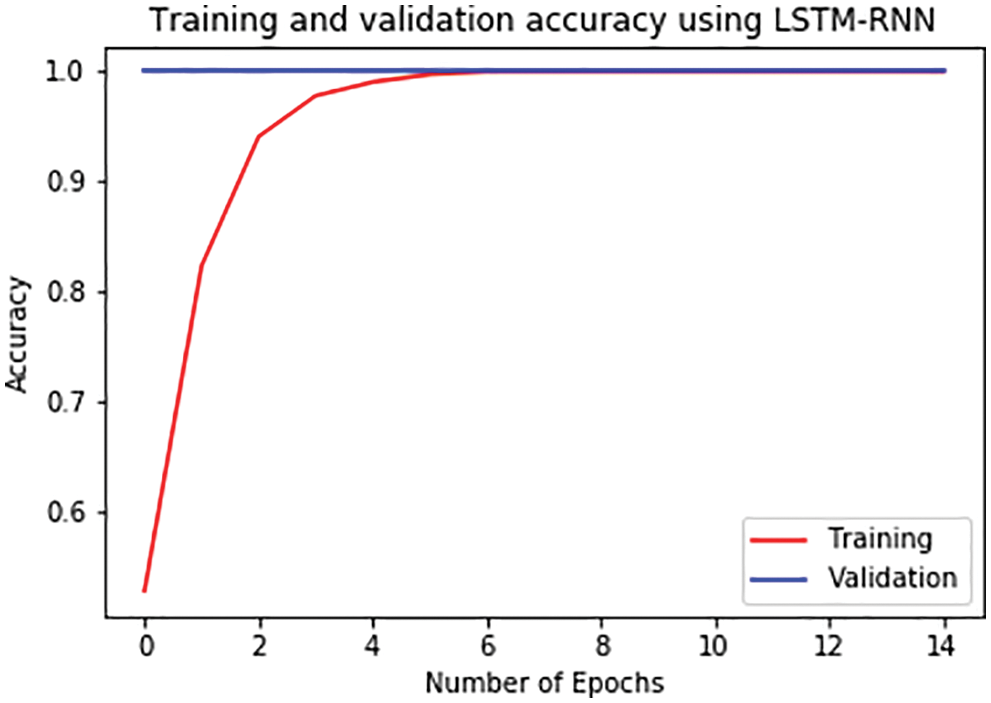
Figure 6: Training and validation accuracy per epoch in LSTM-RNN
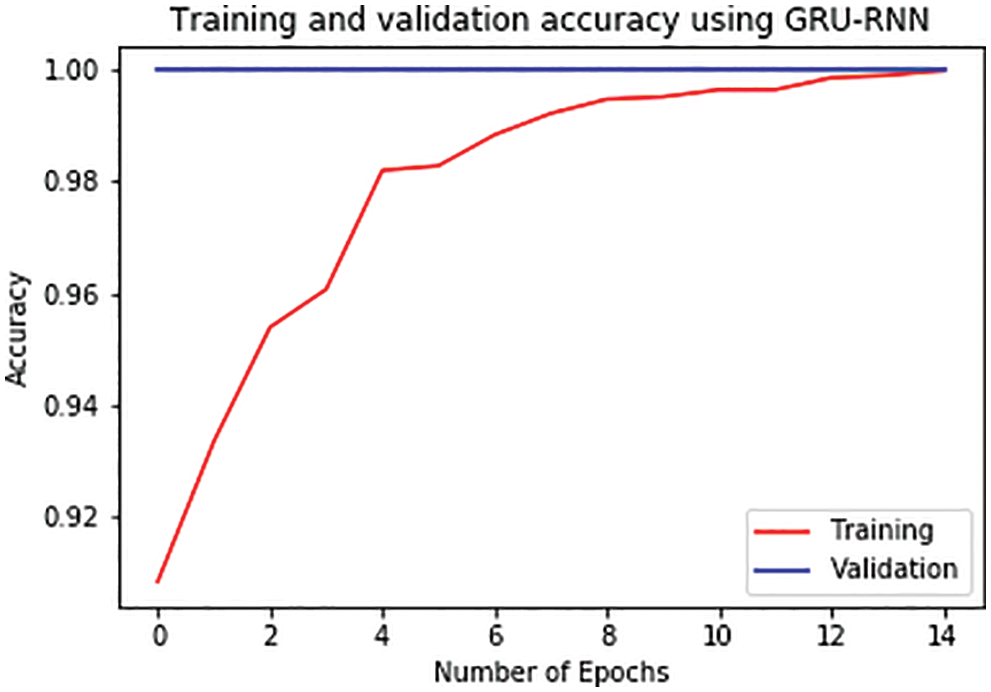
Figure 7: Training and validation accuracy per epoch in GRU-RNN
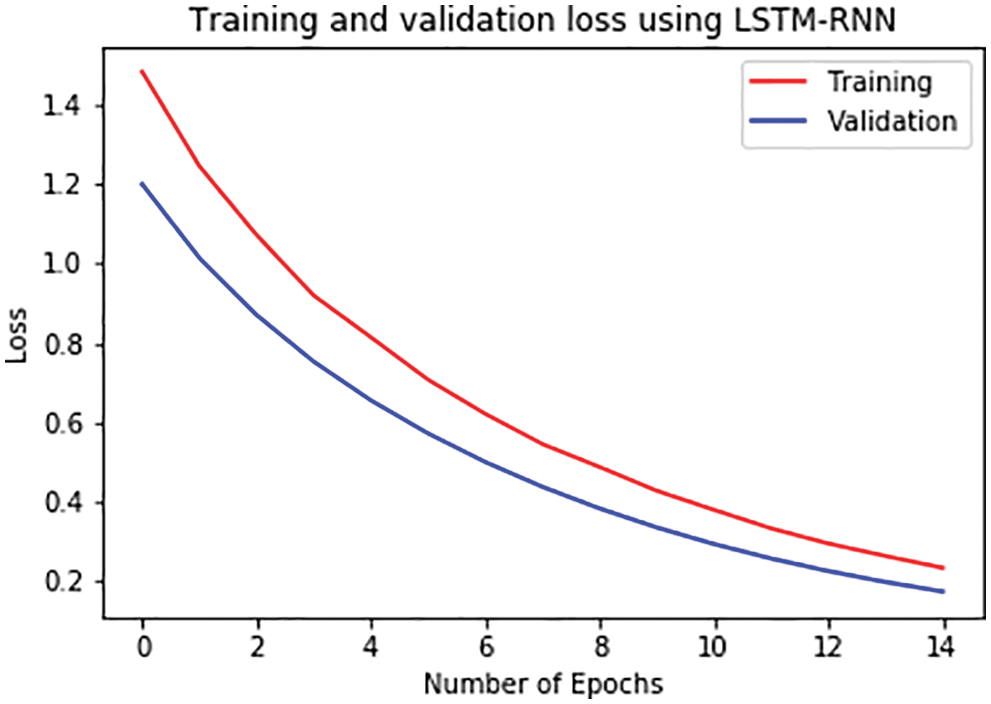
Figure 8: Training and validation loss per epoch in LSTM-RNN
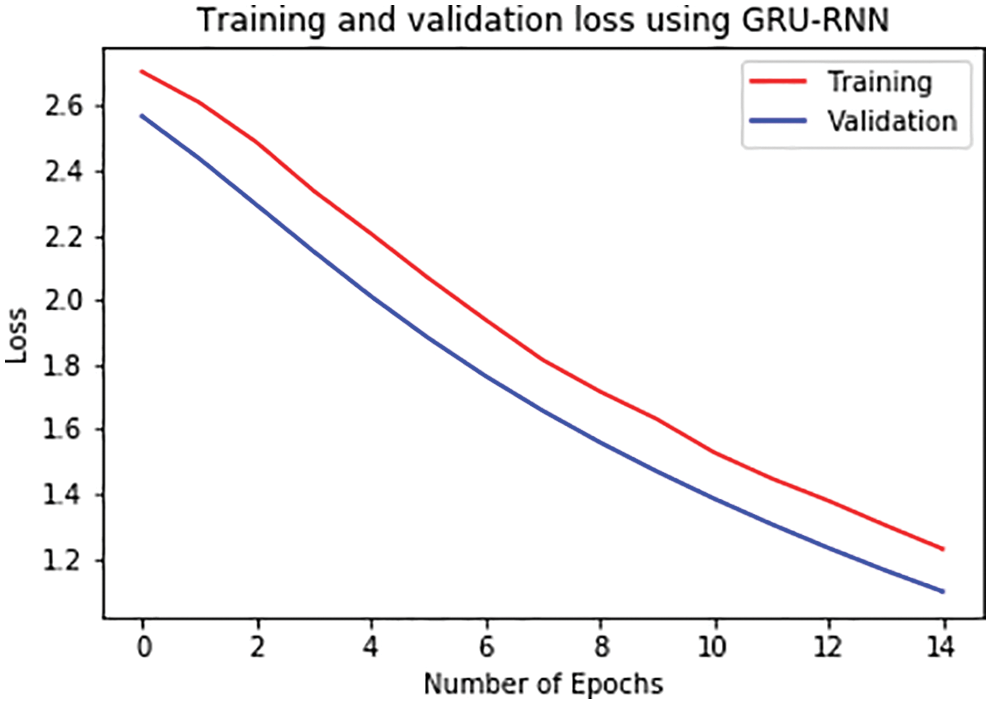
Figure 9: Training and validation loss per epoch in GRU-RNN
Now we try to implement our model on an existing piece of genome sequence and try to predict it mutations, i.e., change in its genome and calculate mutation rate and compare it with the mutated genome to calculate the mutation accuracy. Fig. 10 demonstrates the genome sequences of the virus.
It was then passed through the same data preprocessing procedure as in the training of the two models Fi.

Figure 10: Part of genome sequence of a virus

Figure 11: Token form of the output
The data we get is in the adjusted vector form which we change to the genome sequence by following the data preprocessing in reverse direction. Fig. 12 highlights the output coversion from token.

Figure 12: Output after conversion from token
When both the models are applied on the test set shown above following results regarding the mutation rate and mutation detection accuracy were observed.
Tab. 3 gives the mutation percentage as well as the mutation accuracy for both models. The mutation percentage in the GRU-RNN model is greater than the LSTM-RNN which could be due to larger number of true negative results. From all the analysis and comparison of the results of both the models, it can be concluded that LSTM-RNN works better than the GRU-RNN model for this research.

For Comparison analysis two contemporary works have been taken into consideration.
Pipek [68] uses a filtering algorithm using IsoMut tool and identifies the changes in the genome structure using changes in the filtering parameters in those sequences. Its advantage over conventional statistical approaches is its visualization power of the changes such as False Positive Rate and True Positive Rate threshold is highly informational. On comparison with other tools based on single core performance and time taken to complete the task being 7 min, it performed better than other tools with the next best one being 1 h 20 min.
Thireou [69] in their work used bi-directional LSTM for predicting subcellular localization in eukaryotic proteins and were able to get an accuracy of 93% on plant proteins and 88% on non-plant proteins. Their work showed the ability of deep learning networks in predicting and analyzing bioinformatics. Our work has been an approach to learn patterns in the sequences of family of coronaviruses using LSTM and GRU also helped us to treat the genome sequences as time series data due to their different instances of identification and giving us the opportunity to predict possible changes in their genome sequence [70].
Viral genome structures provide an opportunity as well as a challenge to dig deeper into the biology of the viruses. This research was a simple attempt at creating a model to learn the pattern changes of genome sequences in these viruses and predict the mutation that took place in the most recent virus of concern that is SARS-CoV2. A neural network especially an RNN based network suits best for these types of studies as they keep on learning and improving their results on increasing the training cycles. In this paper, two models were implemented: RNN-LSTM and RNN-GRU. Although RNN-GRU provided slightly higher accuracy for the initial cycles, RNN-LSTM provided a considerably higher F1 score, hence making the model more suitable for the prediction.
Although the two models performed very well on the sequenced dataset, in case of very huge datasets of viruses which can contain significant outliers and hence, may end up getting overfitted by these models. Therefore, in future well designed neural networks like modular neural networks can help in these cases where multiple neural networks work simultaneously and can avoid overfitting. Furthermore, not only nucleotide data but ribosome data can also be included for not only predicting the mutation but also predicting the complete genome structure of the virus that can be possibly encountered soon.
Also, these models can be used to run simulations on change in genetic data of viruses with respect to any change in surrounding condition, or any chemical compound from any under-development vaccine or medicine to get additional insight on the change in biology of the virus.
Acknowledgement: Authors would like to thank for the support of Taif University Researchers Supporting Project number (TURSP-2020/211), Taif University, Taif, Saudi Arabia.
Funding Statement: Taif University Researchers are supporting project number (TURSP-2020/211), Taif University, Taif, Saudi Arabia.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
1. M. Grant, L. Geoghegan, M. Arbyn, Z. Mohammed, L. McGuiness et al., “The prevalence of symptoms in 24,410 adults infected by the novel coronavirus (SARS-CoV-2; COVID-19A systematic review and meta-analysis of 148 studies from 9 countries,” PLoS one, vol. 15, no. 6, pp. e0234765, 2020. [Google Scholar]
2. “COVID-19 dashboard by the center for systems science and engineering (CSSE) at Johns Hopkins University (JHU),” ArcGIS. Johns Hopkins University, Feb, 2020. [Google Scholar]
3. “Here comes the coronavirus pandemic: Now, after many fire drills, the World may be facing a real fire,” Editorial, The New York Times, Feb, 2020. [Google Scholar]
4. K. Sharma, H. Singh, D. Sharma, A. Kumar, A. Nayyar et al., “Dynamic models and control techniques for drone delivery of medications and other healthcare items in COVID-19 hotspots,” Emerging Technologies for Battling Covid-19: Applications and Innovations, vol. 1, pp. 1–34, 2021. [Google Scholar]
5. “The great lockdown: Worst economic downturn since the great depression,” IMF blog, April, 2020. [Google Scholar]
6. “As famines of ‘biblical proportion’ loom, Security Council urged to ‘act fast,’” UN News, April, 2020. [Google Scholar]
7. “Symptoms of coronavirus,” U.S. centers for disease control and prevention (CDCMay, 2020. [Google Scholar]
8. Q. Ye, B. Wang and J. Mao, “The pathogenesis and treatment of the `Cytokine storm’ in COVID-19,” The Journal of Infection, vol. 80, no. 6, pp. 607–613, 2020. [Google Scholar]
9. S. Murthy, C. Gomersall and R. Fowler, “Care for critically Ill patients with COVID-19,” Journal of American Medical Association, vol. 323, no. 15, pp. 1499–1500, 2020. [Google Scholar]
10. M. Cascella, M. Rajnik, A. Aleem, S. Dulebohn and R. Napoli, “Features, evaluation and treatment coronavirus (COVID-19),” StatPearls, StatPearls Publishing, 2020. [Google Scholar]
11. V. Stadnytskyi, C. Bax, A. Bax and P. Anfinrud, “The airborne lifetime of small speech droplets and their potential importance in SARS-CoV-2 transmission,” Proceedings of the National Academy of Sciences of the United States of America, vol. 117, no. 22, pp. 11875–11877, 2020. [Google Scholar]
12. “Guidance on social distancing for everyone in the UK,”. GOV.UK. May, 2020. [Google Scholar]
13. A. Gorbalenya, S. Baker, R. Baric, R. Groot, C. Drosten et al., “The species severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2,” Nature Microbiology, vol. 5, no. 4, pp. 536–544, 2020. [Google Scholar]
14. S. Taneja, A. Nayyar and P. Nagrath, “Face mask detection using deep learning during COVID-19,” in Proc. of Second Int. Conf. on Computing, Communications, and Cyber-Security, Springer, Singapore. pp. 39–51, 2021. [Google Scholar]
15. C. Zimmer, “DNA linked to COVID-19 was inherited from neanderthals, study finds - The stretch of six genes seems to increase the risk of severe illness from the coronavirus,” NY Times, 2020. [Google Scholar]
16. C. Campbell, J. Chong, M. Malig, A. Ko, B. Dumont et al., “Estimating the human mutation rate using auto zygosity in a founder population,” Nature Genetics, vol. 44, no. 11, pp. 1277–1281, 2012. [Google Scholar]
17. D. Bhowmik, S. Pal, A. Lahiri, A. Talukdar and S. Paul, “Emergence of multiple variants of SARS-CoV-2 with signature structural changes,” BioRxiv, 2020. [Google Scholar]
18. R. He, F. Dobie, M. Ballantine, A. Leeson, Y. Li et al., “Analysis of multimerization of the SARS coronavirus nucleocapsid protein,” Biochemical and Biophysical Research Communications, vol. 316, no. 2, pp. 476–483, 2004. [Google Scholar]
19. M. Ridley, “Genome: The autobiography of a species in 23 chapters (PDF),” New York Harper Perennial, 2006. [Google Scholar]
20. “Definition of whole-genome sequencing - NCI dictionary of cancer terms,”. National Cancer Institute. [Google Scholar]
21. C. Gilissen, J. Hehir-kwa, D. Thung, M. Vorst, B. Bon et al., “Genome sequencing identifies major causes of severe intellectual disability,” Nature, vol. 511, no. 7509, pp. 344–347, 2014. [Google Scholar]
22. C. Van, M. Cornel, P. Borry, R. Hastings, F. Fellmann et al., “Whole-genome sequencing in health care. recommendations of the european society of human genetics,” European Journal of Human Genetics, vol. 21, no. 6, pp. 580–584, 2013. [Google Scholar]
23. A. Rincon, A. Tonda, L. Maldonado, D. Mulders, R. Molenkamp et al., “Accurate identification of SARS-CoV-2 from viral genome sequences using deep learning,” BioRxiv, 2020. [Google Scholar]
24. A. Biswas, U. Bhattacharjee, A. Chakrabarti, D. Tewari, H. Banu et al., “Emergence of novel coronavirus and COVID-19: Whether to stay or die out?,” Critical Reviews Microbiology, vol. 46, no. 2, pp. 182–193, 2020. [Google Scholar]
25. S. Mooney, “Progress towards the integration of pharmacogenomics in practice,” Human Genetics, vol. 134, no. 5, pp. 459–465, 2014. [Google Scholar]
26. J. Roach, G. Glusman A. Smit, D. Chad, R. Hubley et al., “Analysis of genetic inheritance in a family quartet by whole-genome sequencing,” Science, vol. 328, no. 5978, pp. 636–639, 2010. [Google Scholar]
27. M. Boheemen, M. Graaf, C. Lauber, T. Bestebroer, V. Raj et al., “Genomic characterization of a newly discovered coronavirus associated with acute respiratory distress syndrome in humans,” mBio, vol. 3, no. 6, pp. e00473–12, 2012. [Google Scholar]
28. R. Das and S. Ghate, “Investigating the likely association between genetic ancestry and COVID-19 manifestation,” medRxiv, 2020. [Google Scholar]
29. P. Yadav, V. Potdar, M. Choudhary, D. Nyayanit, M. Agrawal et al., “Full-genome sequences of the first two SARS-CoV-2 viruses from India,” Indian Journal of Medical Research, vol. 151, no. 2, pp. 200–209, 2020. [Google Scholar]
30. K. Ye, M. Beekman, E. Lameijer, Y. Zhang, M. Moed et al., “Aging as accelerated accumulation of somatic variants: Whole-genome sequencing of centenarian and middle-aged monozygotic twin pairs,” Twin Research and Human Genetics, vol. 16, no. 6, pp. 1026–1032, 2013. [Google Scholar]
31. Towards data science, an article on machine learning for biology: How will COVID-19 mutate next? By Andrew Ye, https://towardsdatascience.com/machine-learning-forbiology-how-will-COVID-19-mutate-next-4df93cfaf544, accessed on April 11, 2020. [Google Scholar]
32. L. Hoek, K. Pyrc, M. Jebbink, W. Oost, R. Berkhout et al., “Identification of a new human coronavirus,” Nature Medicine, vol. 10, no. 4, pp. 368–373, 2004. [Google Scholar]
33. R. Pathan, M. Biswas and M. Khandaker, “Time series prediction of COVID-19 by mutation rate analysis using recurrent neural network-based LSTM model,” Chaos, Solitons, and Fractals, vol. 138, pp. 110018, 2020. [Google Scholar]
34. F. Wu, S. Zhao, B. Yu, Y. Chen, W. Wang et al., “A new coronavirus associated with human respiratory disease in China,” Published Correction Appears in Nature, vol. 579, no. 7798, pp. 265–269, 2020. [Google Scholar]
35. Figure 1: By SPQR10Binte altaf - Own work, CC BY-SA 4.0, https://commons.wikimedia.org/w/index.php?curid=88349537. 2020. [Google Scholar]
36. S. Dupond, “A thorough review on the current advance of neural network structures,” Annual Reviews in Control, vol. 14, pp. 200–230, 2019. [Google Scholar]
37. M. Miljanovic, “Comparative analysis of recurrent and finite impulse response neural networks in time series prediction,” Indian Journal of Computer and Engineering, vol. 3, no. 1, pp. 180–191, 2012. [Google Scholar]
38. K. Cho, B. Merrienboer, C. Gulcehre, D. Bahdanau, F. Bougares et al., “Learning phrase representations using rnn encoder-decoder for statistical machine translation,” arXiv, pp. 1724–1734, 2014. [Google Scholar]
39. A. Kumar, K. Sharma, H. Singh, P. Srikanth, R. Krishnamurthi et al., “Drone-based social distancing, sanitization, inspection, monitoring, and control room for COVID-19,” Artificial Intelligence and Machine Learning for COVID-19, vol. 924, pp. 153–173, 2021. [Google Scholar]
40. H. Sak, A. Senior and F. Beaufays, “Long short-term memory recurrent neural network architectures for large scale acoustic modeling,” Interspeech, pp. 3368–342, 2014. [Google Scholar]
41. J. Alzubi, A. Nayyar and A. Kumar, “Machine learning from theory to algorithms: An overview,” Journal of Physics: Conference Series, vol. 1142, no. 1, pp. 012012, 2018. [Google Scholar]
42. X. Li and X. Wu, “Constructing long short-term memory based deep recurrent neural networks for large vocabulary speech recognition,” IEEE ICASSP, pp. 4520–4524, 2015. [Google Scholar]
43. A. Graves, M. Liwicki, S. Fernandez, R. Bertolami, H. Bunke et al., “A novel connectionist system for improved unconstrained handwriting recognition,” IEEE Transactions on Pattern Analysis and Machine Intelligence, vol. 31, no. 5, pp. 855–868, 2009. [Google Scholar]
44. J. Huang, W. Zhou, Q. Zhang, H. Li and W. Li, “Video-based sign language recognition without temporal segmentation,” arXiv, pp. 2257–2264, 2018. [Google Scholar]
45. S. Hochreiter and J. Schmidhuber, “Long short-term memory,” Neural Computation, vol. 9, no. 8, pp. 1735–1780, 1997. [Google Scholar]
46. E. Choi, M. Bahadori, E. Schuetz, W. Stewart and J. Sun, “Doctor AI: Predicting clinical events via recurrent neural networks,” in Proc. of the 1st Machine Learning for Healthcare Conf., Northeastern University, Boston, USA, vol. 56, pp. 301–318, 2016. [Google Scholar]
47. D. Wierstra, J. Schmidhuber and F. Gomez, “Evolino: Hybrid neuroevolution/optimal linear search for sequence learning,” in Proc. of the 19th Int. Joint Conf. on Artificial Intelligence (IJCAI), Edinburgh, Scotland, pp. 853–858, 2005. [Google Scholar]
48. G. Felix, J. Schmidhuber and F. Cummins, “Learning to forget: Continual prediction with LSTM,” Neural Computation, vol. 12, no. 10, pp. 2451–2471, 2000. [Google Scholar]
49. “Recurrent neural network tutorial, Part 4 – Implementing a GRU/LSTM RNN with python and theano – WildML,” Wildml.com, 2015. [Google Scholar]
50. Y. Su and J. Kuo, “On extended long short-term memory and dependent bidirectional recurrent neural network,” NeuroComputing, vol. 356, pp. 151–161, 2019. [Google Scholar]
51. M. Ravanelli, P. Brakel, M. Omologo and Y. Bengio, “Light gated recurrent units for speech recognition,” IEEE Transactions on Emerging Topics in Computing, vol. 2, pp. 92–102, 2018. [Google Scholar]
52. Y. Su and J. Kuo, “Empirical evaluation of gated recurrent neural networks on sequence modeling,” arXiv, 2014. [Google Scholar]
53. N. Gruber and A. Jockisch, “Are GRU cells more specific and LSTM cells more sensitive in motive classification of text?,” Frontiers in Artificial Intelligence, vol. 3, 2020. [Google Scholar]
54. A. Devi and A. Nayyar, “Perspectives on the definition of data visualization: A mapping study and discussion on coronavirus (COVID-19) dataset,” Emerging Technologies for Battling Covid-19: Applications and Innovations, vol. 424, pp. 223–240, 2021. [Google Scholar]
55. A. Devi and A. Nayyar, “Evaluation of geotagging twitter data using sentiment analysis during COVID-19,” in Proc. of the Second Int. Conf. on Information Management and Machine Intelligence, Poornima Institute of Engineering and Technology, Jaipur, India: Springer, vol. 166, pp. 601–608, 2021. [Google Scholar]
56. G. Weiss, Y. Goldberg and E. Yahav, “On the practical computational power of finite precision RNNs for language recognition,” in Proc. of the 56th Annual Meeting of the Association of Computational, Melbourne, Australia, Linguistics, vol. 2, pp. 740–745, 2018. [Google Scholar]
57. N. Tayarani and H. Mohammed, “Applications of artificial intelligence in battling against COVID-19: A literature review,” Chaos, Solitons, and Fractals, vol. 142, pp. 110338. 2021. [Google Scholar]
58. M. Zivkovic, N. Bacanin, K. Venkatachalam, A. Nayyar, A. Djordjevic et al., “COVID-19 cases prediction by using hybrid machine learning and beetle antennae search approach,” Sustainable Cities and Society, vol. 66, pp. 102669, 2021. [Google Scholar]
59. C. Sara, D. Wim, F. Vagner, K. Wasim, G. Marta et al., “Genome detective coronavirus typing tool for rapid identification and characterization of novel coronavirus genomes,” Bioinformatics, vol. 36, no. 11, pp. 3552–3555, 2020. [Google Scholar]
60. D. Stone, A. Furthmann, V. Sandig and A. Lieber, “The complete nucleotide sequence, genome organization, and origin of human adenovirus type 11,” Virology, vol. 309, no. 1, pp. 152–165, 2003. [Google Scholar]
61. F. Turjman, A. Devi and A. Nayyar, “Emerging technologies for battling COVID-19,” 2021. [Google Scholar]
62. P. Woo, S. Lau, C. Chu, K. Chan, H. Tsoi et al., “Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia.” Journal of Virology, vol. 79, no. 2, pp. 884–895, 2005. [Google Scholar]
63. T. Jabeen, H. Ashraf, N. Jhanjhi, H. Mamoona, M. Mehedi et al., “A monte carlo based COVID-19 detection framework for smart healthcare,” Computers, Materials, & Continua,” vol. 70, no. 2, pp. 2365–2380, 2022. [Google Scholar]
64. J. Ma, “Coronavirus: China’s first confirmed COVID-19 case traced back to November 17,” South China Morning Post, 2020. [Google Scholar]
65. M. Pachetti, B. Marini, F. Benedetti, F. Giudici, E. Mauro et al., “Emerging SARS-CoV-2 mutation hot spots include a novel RNA-dependent-RNA polymerase variant.” Journal of Translational Medicine, vol. 18, no. 1, pp. 179–194, 2020. [Google Scholar]
66. M. Awal, M. Masud, M. Hossain, A. Bulbul, S. Mahmud et al., “A novel Bayesian optimization-based machine learning framework for COVID-19 detection from inpatient facility data,” IEEE Access, vol. 9, pp. 10263–10281, 2021. [Google Scholar]
67. S. Pruthi, “Coronavirus disease (COVID-19)—Symptoms and causes,” Mayo Clinic. 2019. [Google Scholar]
68. T. Thireou and M. Reczko, “Bidirectional long short-term memory networks for predicting the subcellular localization of eukaryotic proteins,” IEEE/ACM Transactions on Computational Biology and Bioinformatics, vol. 4, no. 3, pp. 441–446, 2007. [Google Scholar]
69. O. Pipek, D. Ribli, J. Molnár, A. Póti, M. Krzystanek et al., “Fast and accurate mutation detection in whole genome sequences of multiple isogenic samples with IsoMut,” BMC Bioinformatics, vol. 18, no. 1, pp. 73–83, 2017. [Google Scholar]
70. T. Jasarevice, C. Lindmeier and F. Chaib, “Statement on the second meeting of the International Health Regulations Emergency Committee regarding the outbreak of novel coronavirus (2019-nCoV),” World Health Organization (WHO) Statement, Geneva, Switzerland, 2020. [Google Scholar]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |