DOI:10.32604/cmc.2022.028958
| Computers, Materials & Continua DOI:10.32604/cmc.2022.028958 | |
| Article |
Single and Mitochondrial Gene Inheritance Disorder Prediction Using Machine Learning
1Riphah School of Computing & Innovation, Faculty of Computing, Riphah International University Lahore Campus, Lahore, 54000, Pakistan
2Pattern Recognition and Machine Learning Lab, Department of Software, Gachon University, Seongnam, 13120, Gyeonggido, Korea
3Faculty of Computing, Riphah International University, Islamabad, 45000, Pakistan
4School of Information Technology, Skyline University College, Sharjah, 1797, UAE
5Network and Communication Technology Lab, Center for Cyber Security, Faculty of Information Science and Technology Universiti Kebangsaan Malaysia, 43600, Malaysia
6Canadian University, Dubai, 00000, UAE
7Cyber-Physical Systems, Khalifa University, Abu Dhabi, 127788, UAE
*Corresponding Author: Hussam Al Hamadi. Email: hussam.alhamadi@ku.ac.ae
Received: 22 February 2022; Accepted: 24 March 2022
Abstract: One of the most difficult jobs in the post-genomic age is identifying a genetic disease from a massive amount of genetic data. Furthermore, the complicated genetic disease has a very diverse genotype, making it challenging to find genetic markers. This is a challenging process since it must be completed effectively and efficiently. This research article focuses largely on which patients are more likely to have a genetic disorder based on numerous medical parameters. Using the patient’s medical history, we used a genetic disease prediction algorithm that predicts if the patient is likely to be diagnosed with a genetic disorder. To predict and categorize the patient with a genetic disease, we utilize several deep and machine learning techniques such as Artificial neural network (ANN), K-nearest neighbors (KNN), and Support vector machine (SVM). To enhance the accuracy of predicting the genetic disease in any patient, a highly efficient approach was utilized to control how the model can be used. To predict genetic disease, deep and machine learning approaches are performed. The most productive tool model provides more precise efficiency. The simulation results demonstrate that by using the proposed model with the ANN, we achieve the highest model performance of 85.7%, 84.9%, 84.3% accuracy of training, testing and validation respectively. This approach will undoubtedly transform genetic disorder prediction and give a real competitive strategy to save patients’ lives.
Keywords: Genetic disorder; machine learning; deep learning; single gene inheritance gene disorder; mitochondrial gene inheritance disorder
Complicated disorders with a significant genetic effect, such as Single gene inheritance disorder (SGID) and Mitochondrial gene inheritance disorder (MGID), may have numerous syndromes involving the number of genes. Latest developments in genetic technology have resulted in more accurate genetic data acquisition. Various major genetic investigations for SGID and MID, for example, have found hundreds of people with diseases [1,2]. However, given the large volumes of data collected by this large-scale research, finding the actual genes causing diseases has become a difficult challenge. Worldwide child mortality rates have fallen rapidly in recent years, owing mostly to fewer fatalities from pathogens, diarrhea, and immunization illnesses. As a result, child mortality is now very reduced in many contexts, and program targets are moving to noncommunicable diseases, which now account for a higher share of all below five fatalities [3].
In situations with very low acute disease mortality, genetically determined disorders account for a large fraction of stillbirths, infant mortality, and ongoing handicap. Genetically determined illnesses are classified into two categories: “single gene inheritance disorders” produced by strong gene variations and “genetic risk factors e-g mitochondrial gene inheritance disorders” caused by feeble genetic variations that cause disease only when associated with other genetic and/or environmental parameters.
A single gene disease begins with a genetic change in one gene. Because this may happen in any gene, single-gene diseases can impact every element of functioning and are astonishingly varied [4]. Despite their clinical differences, single-gene diseases all share the same biological foundation, have the ability to be carried down to children, and demand the same fundamental genetic and advisory services. Accurate diagnosis, risk assessment, and information for the afflicted individuals and their families, as well as access to risk management choices and assistance for sick adults and children.
Mitochondrial gene inheritance disorders are by far the most common type of inborn metabolic mistake [5], accounting for 1.6 out of every 5 k people [6]. The vast majority of organ involvement is multisystemic, with a preference for cells that require a lot of energy. These cells rely on the preservation of an efficient energy balance, and patients’ symptoms are often moderate to severe and regressed during periods of metabolic stress.
Deep learning and machine learning have been applied effectively in a variety of biological situations in latest years. Deep learning and machine learning-based algorithms are effective enough to tackle enormous data sets with high levels of noise, complexity, and/or imperfection while making just a few guesses probability distributions and data creation techniques. Prediction is the central objective of deep learning and machine learning methods, as opposed to the inferential approach of traditional statistical methodologies [7].
People are 99.9% genetically related; we all have the same code of 6 billion letters of chemical compositions (A, T, C, and G), which join in base pairs to form our Deoxyribonucleic acid (DNA). What distinguishes us is the >1% of information that changes from individual to individual; these variances are known as genetic mutations. At least 4 M of these genetic variations is distinct from others [8]. Researchers are divided on how to predict disorder. Some argue that most disorders are neither genetic or that there aren’t enough genetic differences to predict risk. Strokes and cardiovascular disease, for example, are not caused by a single or numerous mutations, but rather by genetic and behavioral variables.
Greater genotyping and testing methods have resulted in an increase in genetic data collection. Although this expansion, the methods of action by which genetic variations cause disease progression to remain unknown. Despite the fact that genomic alleles and malignant variants are continually mapped, the majority of them still lack genomic information [9]. The initial attempts to discovering non-experimental illness gene connections relied on association studies, which calculates the likelihood of seeing genotypes in an organism against chance.
Previous researchers have also revealed that illnesses with contiguous sections have strong phenotypic and comorbidity characteristics [10]. It has been proposed that genetic data are especially informative because distinct perturbations in a single disorder module frequently achieve similar phenotypes [11], and networks of phenomenon (where genes are endpoints that are attached if they indicate associated phenotypic statuses) are highly linked with proteins. Relationships between proteins and transcription factor networks [12]. Furthermore, disorders located in the interactome remote neighbors cause distinct phenotypes [10]. Several approaches for predicting genes disorder that incorporate these various forms of data have been presented [13]. A collection of techniques combines the information into a single graph, which is subsequently utilized for prediction.
Similar approaches have been used to predict disorder modules, a comparable challenge; disordered genes can be discovered within groups of these modules. Liu et al. [14] recover disease components by evaluating genetic data and expression network partitions; Ghiassian et al. [15] continuously add genes to categories using immediate neighbor analysis in nutrient interaction nets. It has been established that genetic risk prediction may have an influence on individuals and populations, for a certain period [16], but it is only significant developments in high-density genotyping technology that have brought genetic risk prediction within reach. Genes linked to cardiovascular disease may also be implicated in intermediate outcomes such as dyslipidemia, hypertension, or even smoking [17]. Genetic variations implicated in intermediate variables will no more be relevant when put into a dependent variable with these intermediate factors, according to the fundamental principles of scientific studies. When genetic variations are engaged in undiscovered pathways or processes with immeasurable intermediary components, they can enhance illness prediction beyond established risk factors. Some diseases may be more prone to have new yet undiscovered pathways than others. A crucial but not improbable point is that gene findings may uncover novel etiological networks and intermediate biomarkers, which may be better predictors of disease than the genetic variant that brought to their discovery.
As per previous researches, most genome disorders work based on genome sequencing. Major limitations in genome sequencing have stated below:
• Analytical and validity problem because it is possible during the prediction mostly genome segments could be read below the minimum coverage path of DNA sequence if this depth is not read sufficiently so it is possible that the base will not predict actual genome disorder in a person.
• Clinical interpretation problem, because with development of genetic technologies total prediction process has automated. So, without an individual, there is no way to predict DNA sequencing on automated algorithms.
• Clinical legitimacy.
So, in our proposed model covered most of the limitations to improve the prediction of genetic disorders with the help of patients’ medical history.
The dataset is downloaded from Kaggle. The total patient records are 22083 with 35 features that are used to predict genetic disorders. In data pre-processing replaced the null values with the help of different data normalization techniques and for the best feature apply the linear regression technique to choose the best 14 features from 35 features with the help of a mean square error.
Early detection of genetic disorders helps the patient to improve their health before any major consequences. Early detection of genetic disorders helps in health improvement and changes in lifestyle for patients. In our research article, we present the model of neural network using deep learning, SVM, and KNN model using machine learning for detection of a genetic disorder. After analysis, we will use the highest accurate model for genetic disorder prediction. Fig. 1 shows our prediction framework.
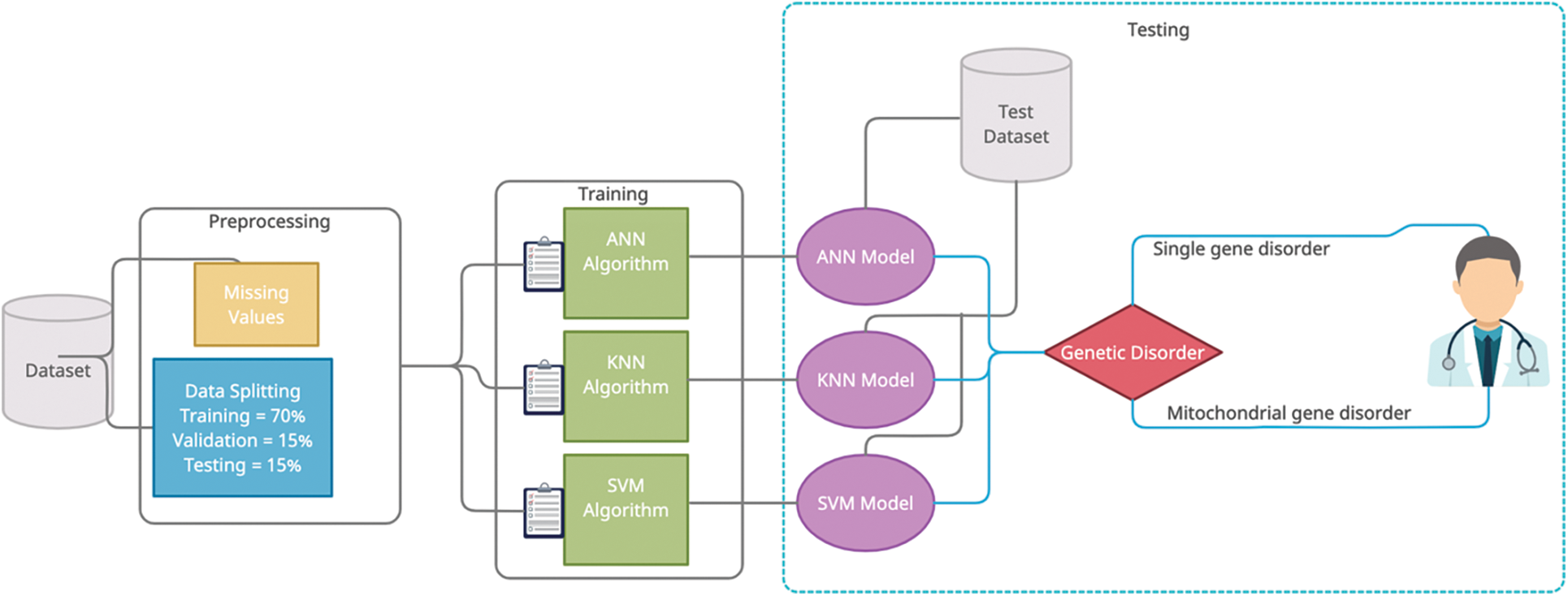
Figure 1: Proposed model of genetic disorder prediction
The proper dataset selection before the training and preprocessing phase. In this study, we selected a labeled dataset for the use of our prediction framework. This dataset consists of 22083 instances with 35 features in which 34 features are independent and one feature (output class) is dependent. In pre-processing phase involves two steps first is data cleaning in this phase we replace missing values with the help of the normalization technique right after pre-processing, we used the linear regressing technique to choose the best fourteen independent features and the second step is data splitting which is done randomly in ratio, training data is 70%, testing data is 15% and validation data is 15%. After pre-processing phase, the training phase is a takeover and, in this phase, proposed model used three supervised classification techniques i-e ANN, KNN, and SVM. The training phase receives input from the pre-processing phase. In the Artificial Neural Network technique, model used five hidden layers and one hundred neurons for each layer and the backpropagation technique (Scaled Conjugate Gradient) to tune the weights. Every neuron in the hidden layer has an activation function which is the sigmoid function. After the testing phase, we choose the best prediction model based on testing parameters and present it in the result section.
In the artificial neural network technique, proposed model divides pre-processed data into three parts: 70% for training, 15% for validation, and 15% for testing the total pre-processed dataset. Pre-processed data is running on five hidden layers of neurons and train the model. Scaled conjugate gradient backpropagation activation function used in the training phase of ANN. In the artificial neural network, there are one hundred neurons for each layer and two neurons for the output layer, which contain two classes single gene inheritance disorder and mitochondrial gene inheritance disorder. The mathematical interpretation of artificial neural network is given below:
There are fourteen input neurons which are represented as ῠ1, ῠ2, ῠ3, . . ., ῠ14 and in the hidden layer there are one hundred neurons on each layer (five hidden layers) which is represented as ñ1, ñ2, ñ3, …, ñ5 and the output layer is represented as out and the biases are signified as ƃ1 and ƃ2 respectively.
By using above mentioned Eqs. (2)–(5), we can calculate outð, netƍ, and outƍ.
The sigmoid function of the proposed prediction model can be interpreted as:
Input derived from the output layer is
The output layer activation function is
6 Scaled Conjugate Gradient Algorithm
Moller’s scaled conjugate gradient (SCG) method is based on conjugate gradients, but unlike other conjugate gradient techniques that need a linear search at every repetition, this approach somehow doesn’t execute a linear search at each iterative process. Scaled Conjugate Gradient was created to eliminate the need for time-consuming linear searches.
In MATLAB, ‘trainscg’ is a network training function that modifies the weight and bias variables using the scaled conjugate gradient technique. Any network may be trained as long as its weight, net-input, and backpropagation contain derivatives. The phase margin in the SCG method is a quadratic estimate function of the error function, making it more resilient and independent of the user-defined parameters.
A different method is used to estimate step size. The second order term is computed as follows:
where λk is a scalar and is adjusted each time according to the sign of δk.
Step size,
Artificial Neural Network algorithm is more efficient in calculating the output of large datasets. So, the efficiency of the artificial neural network algorithm is analyzed as to its accuracy, miss classification rate, recall, precision, and F1 score. After transferring the dataset into the training phase in which data is trained by the artificial neural network, support vector machine, and KNN algorithm. After that trained data is transfer into the testing phase, in this phase data is tested from all trained models individually after this we selected the best prediction model based on prediction accuracy which is the artificial neural network. We explained the results of ANN because this proposed model gained the highest prediction accuracy as compared to the other models. Simulation results of ANN from the proposed model are explained below,
The dataset of 22083 instances was into three-phase, first, model trained 70% data, second, model validate 15% data and at the end, and tested 15% data than model applied ANN on this data division, and all ANN simulation results obtained are shown and justified in graphical and tabular form.
The simulation results in Fig. 2 provide the training accuracy which is 85.7% and its miss classification rate is 14.3%. The recall value of the ANN training phase is 85.8% and precision is 99.7%. The F1 score of this simulation is 92.2%. In this simulation, model applied and explain the ANN algorithm because it gained the highest accuracy above all. The blue line of this simulation shows class 1 which is a single gene inheritance disorder and the lime green line represents class 2 which is mitochondrial gene inheritance disorder.
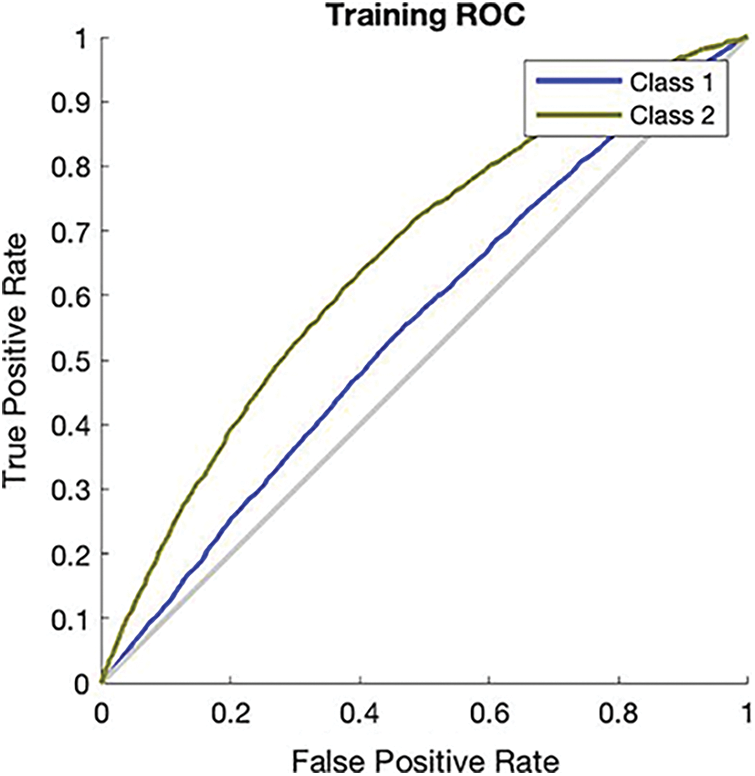
Figure 2: Receiver operating characteristics curve of ANN training phase
The simulation results in Fig. 3 provide the validation accuracy which is 84.3% and its miss classification rate is 15.7%. The recall value of the ANN validation phase is 84.5% and precision is 99.6%. The F1 score of this simulation is 91.3%. The simulation results in Fig. 4 provide the testing accuracy which is 84.9% and its miss classification rate is 15.1%. The recall value of the ANN testing phase is 85% and precision is 99.7%. The F1 score of this simulation is 92%.
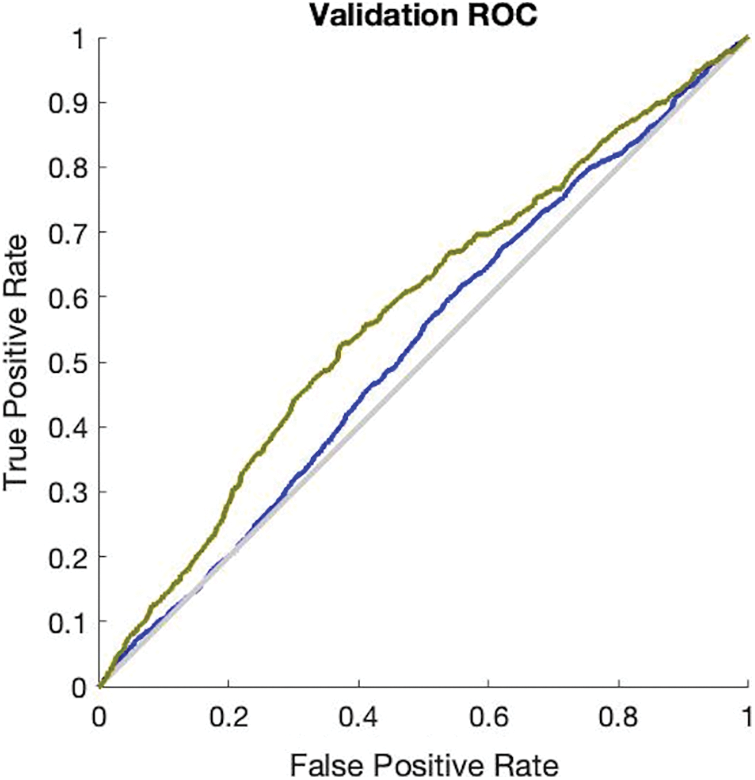
Figure 3: Receiver operating characteristics curve of ANN validation phase
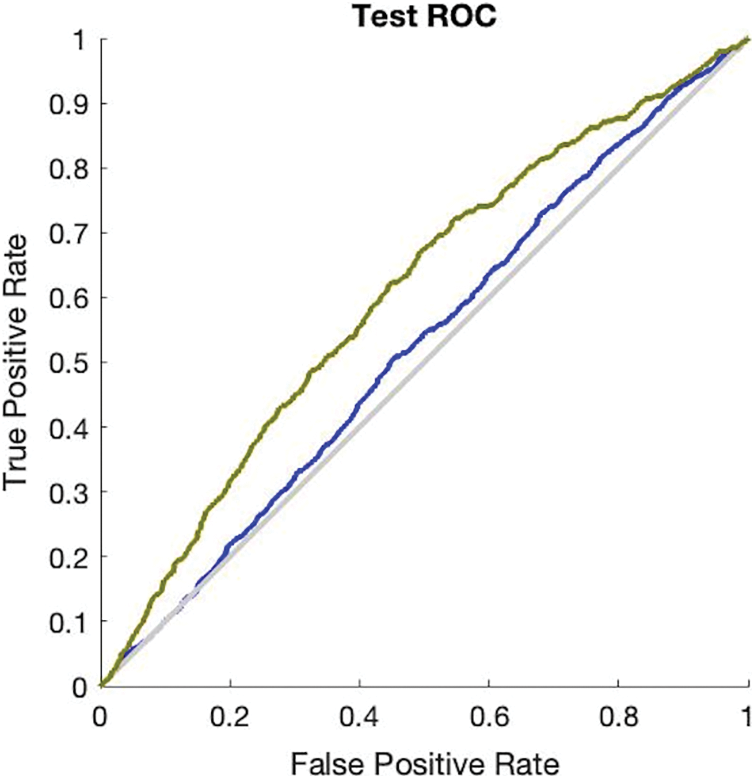
Figure 4: Receiver operating characteristics curve of ANN test phase
The simulation results in Fig. 5 provide the detail about the best validation mean squared error which is 0.22, it means the prediction accuracy of ANN is outstanding, the lower the MSE the higher the predicted value. At 24 epoch regressions lines are equal to train, validation, and testing.
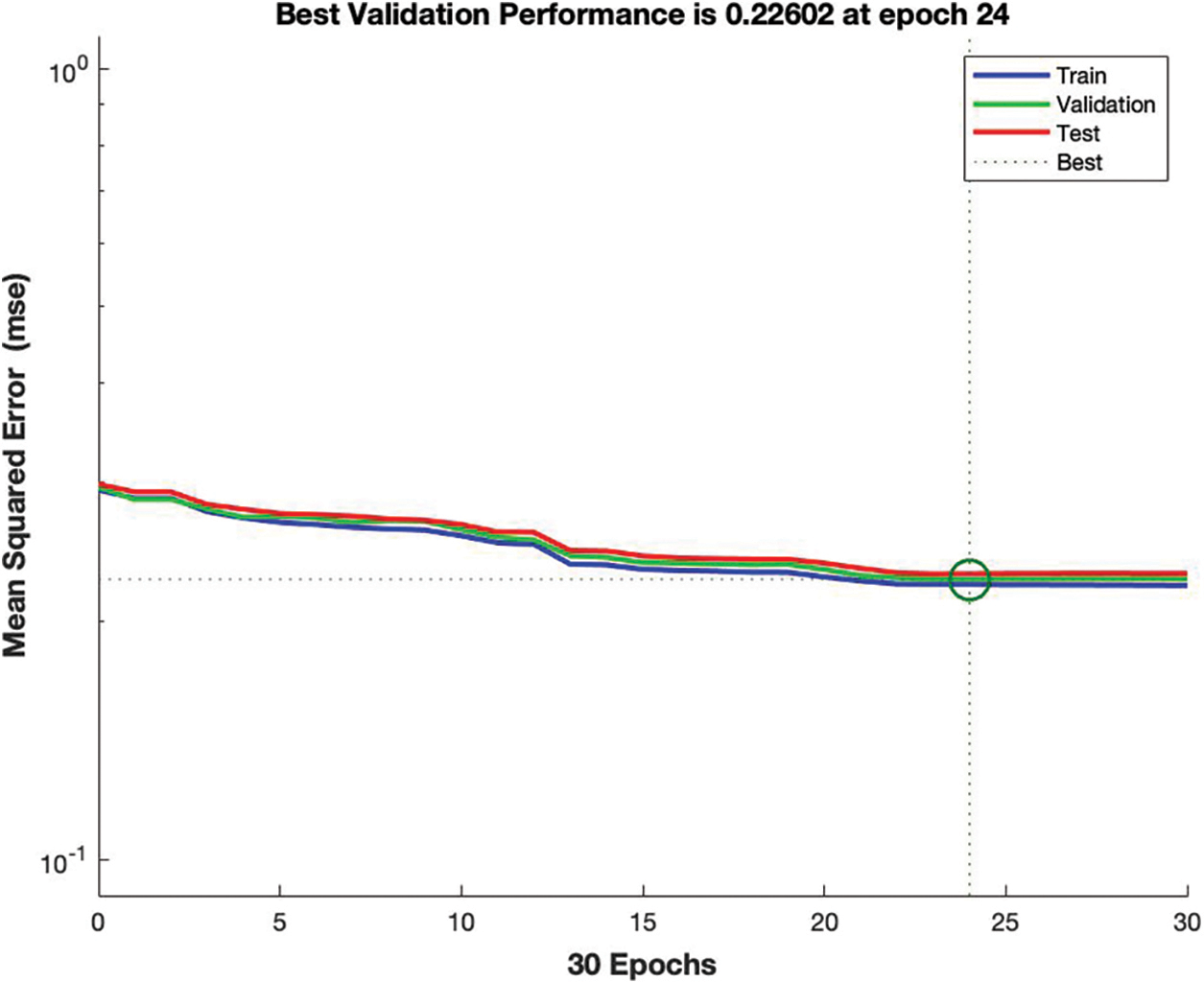
Figure 5: Mean squared error of ANN
In Tab. 1 the accuracy, miss classification rate, recall, precision, and F1 score values are calculated by using the formulas mentioned below.
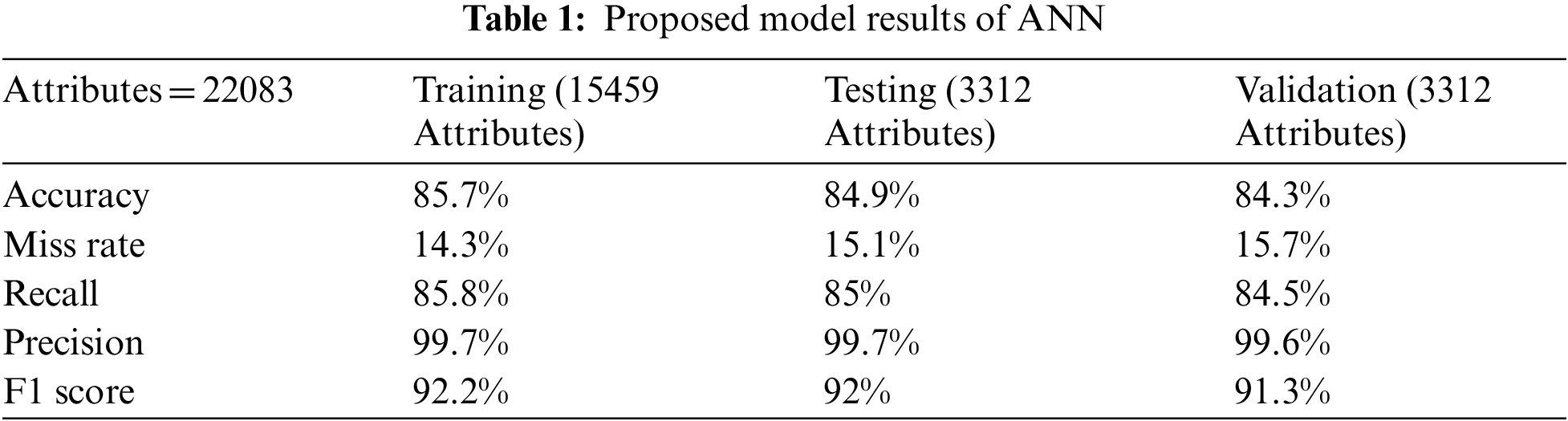
Tab. 1 shown the proposed model ANN results of single and mitochondrial gene inheritance disorder in training, testing and validation phase. Proposed model divides total 22083 attributes into training, testing, validation of 15459, 3312, 3312 respectively. During the training phase proposed model predict 13229, 15, 29, 2129 attributes of true positive, true negative, false positive and false negative respectively. Furthermore, during the testing phase proposed model predict 2812, 1, 6, 493 attributes of true positive, true negative, false positive and false negative respectively and in validation phase proposed model achieved 2793 true positive attributes and 1, 8, 510 attributes of true negative, false positive and false negative respectively.
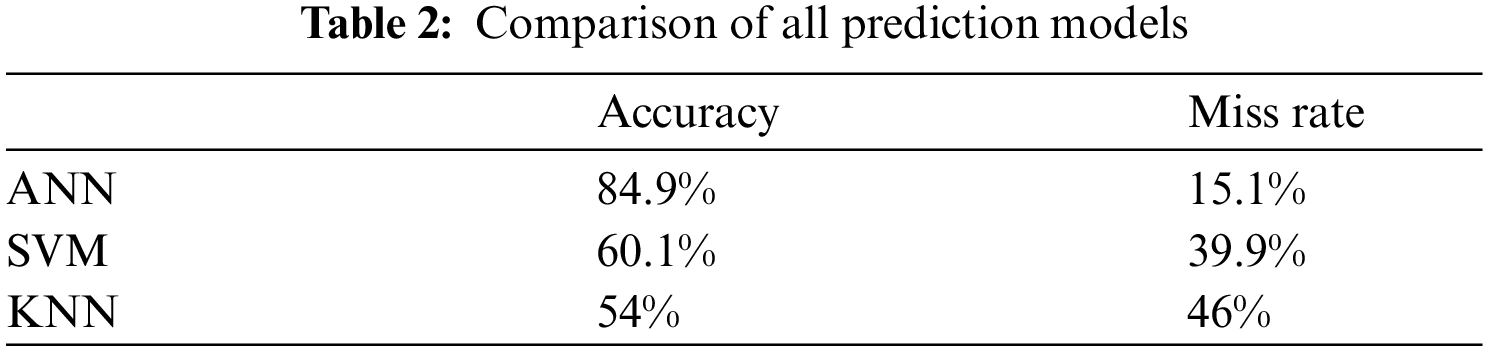
Tab. 2 shown the comparative results of all model’s accuracy and miss classification value. It clearly observed that proposed model achieved accuracy 84.9%, 60.1%, 54% from ANN, SVM and KNN respectively and proposed model miss rate 15.1%, 39.9%, 46% of ANN, SVM and KNN respectively.
The machine and deep learning approach usually uses to predict gene disorders in the medical field. In this study, proposed model doing binary classification of genetic disorders by using different experimental techniques of supervised learning and their comparison. Proposed model assessed the stability of these experimental techniques with respect to their testing accuracy. Prediction results showed the artificial neural network performed best based on accuracy, miss classification rate, and validation mean squared error. As we used the medical history of patient data which easily overcome the genetic disorder prediction limitation on genetic sequence data for prediction. So, to remove this prediction uncertainty proposed model performed binary classification of genetic disorder prediction on patient medical history which gives best whether patient present on time or not. Therefore, this study will be helpful to predict the genetic disorder before time on basis of medical history, and with the help of this process, we can easily save many adult and pre-mature lives. In the future, we will do genetic disorder classification by using multifactor gene inheritance disorder based on vast medical history.
Acknowledgement: Thanks to our families & colleagues who supported us morally.
Funding Statement: The authors received no specific funding for this study.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
1. S. J. Sanders, “First glimpses of the neurobiology of autism spectrum disorder,” Genetics and Development, vol. 33, pp. 80–92, 2015. [Google Scholar]
2. Schizophrenia working group of the psychiatric genomics consortium, “Biological insights from 108 schizophrenia-associated genetic loci,” Nature, vol. 511, pp. 421–427, 2014. [Google Scholar]
3. L. Liu, S. Oza, D. Hugan, Y. Chu, J. Perin et al., “Global, regional, and national causes of under-5 mortality in 2000–15: An updated systematic analysis with implications for the sustainable development goals,” Lancet, vol. 388, pp. 3027–3035, 2017. [Google Scholar]
4. JH. Medicine, “Research news article,” 3 May 2017. [Online]. Available: https://www.hopkinsmedicine.org/news/media/releases/media_advisory_online_mendelian_inheritance_in_man_omim_hosts_symposium_celebrating_50_years. [Accessed 5 December 2021]. [Google Scholar]
5. C. R. Ferreira, C. D. M. van Karnebeek, J. Vockley and N. Blaue, “A proposed nosology of inborn errors of metabolism,” Genetic Medicine, vol. 21, no. 1, pp. 102–106, 2019. [Google Scholar]
6. J. Tan, M. Wagner, S. L. Stenton, T. M. Storm, S. B. Wortmaan et al., “Lifetime risk of autosomal recessive mitochondrial disorders calculated from genetic databases,” Lancet, vol. 54, pp. 111–119, 2019. [Google Scholar]
7. D. Bzdok, N. Altman and M. Krzywinski, “Statistics versus machine learning,” Nature, vol. 15, pp. 233–234, 2018. [Google Scholar]
8. Psomagen, “How can genetics help predict diseases?” Psomagen, 31 August 2020. [Online]. Available: https://psomagen.com/how-can-genetics-help-predict-diseases-psomagen/. [Accessed 11 December 2021]. [Google Scholar]
9. J. Das, J. Muhammed and H. Yu, “Genome-scale analysis of interaction dynamics reveals organization of biological networks,” Bioinformatics, vol. 28, no. 14, pp. 1873–1878, 2012. [Google Scholar]
10. J. Menche, A. Sharma, M. Kitsak, S. Ghiassian, M. Vidal et al., “Uncovering disease-disease relationships through the incomplete human interactome,” Science, vol. 347, no. 6224, pp. 1257601, 2015. [Google Scholar]
11. A. L. Barabási, N. Gulbahce and J. Loscalzo, “Network medicine: A network-based approach to human disease,” Nature, vol. 12, pp. 56–68, 2011. [Google Scholar]
12. M. Vidal, M. E. Cusik and A. L. Barabási, “Interactome networks and human disease,” Cell, vol. 144, no. 6, pp. 986–998, 2011. [Google Scholar]
13. L. Madeddu, G. Stillo and P. Vilardi, “Network-based methods for human disease gene prediction,” Brief Functional Genomics, vol. 10, no. 5, pp. 280–293, 2011. [Google Scholar]
14. Y. liu, D. A. Tennant, Z. Zhu, J. K. Health, X. Yao et al., “Dime: A scalable disease module identification algorithm with application to glioma progression,” Plos One, vol. 9, no. 2, pp. 866–876, 2014. [Google Scholar]
15. S. D. Ghiassian, J. Menche and A. L. Barabasi, “A disease module detection (diamond) algorithm derived from a systematic analysis of connectivity patterns of disease proteins in the human interactome,” Plos Computational Biology, vol. 11, no. 4, pp. 1004120, 2015. [Google Scholar]
16. M. T. Scheuner and J. I. Rotter, “Quantifying the health benefits of genetic tests: The importance of a population perspective,” Nature, vol. 8, pp. 141–142, 2006. [Google Scholar]
17. S. Katherisan, O. Millander, D. Anaveski and C. Guiducci, “Polymorphisms associated with cholesterol and risk of cardiovascular events,” New England Journal of Medicine, vol. 358, no. 12, pp. 1240–9, 2008. [Google Scholar]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |