DOI:10.32604/iasc.2021.015205
| Intelligent Automation & Soft Computing DOI:10.32604/iasc.2021.015205 | |
| Article |
Multifactorial Disease Detection Using Regressive Multi-Array Deep Neural Classifier
1Department of Electronics and Communication Engineering, KPR Institute of Engineering and Technology, Coimbatore, India
2Department of Electronics and Communication Engineering, University College of Engineering, BIT Campus, Anna University, Tiruchirappalli, India
3School of Computing, SRM Institute of Science and Technology, Kattankulathur, 603203, India
4School of Computer Science and Engineering, Vellore Institute of Technology, Chennai, India
5Department of Electrical and Electronics Engineering, National Engineering College, Kovilpatti, India
*Corresponding Author: T. Jayasankar. Email: jayasankar27681@gmail.com
Received: 10 November 2020; Accepted: 27 December 2020
Abstract: Comprehensive evaluation of common complex diseases associated with common gene mutations is currently a hot area of human genome research into causative new developments. A multi-fractal analysis of the genome is performed by placing the entire DNA sequence into smaller fragments and using the chaotic game representation and systematic methods to calculate the general dimensional spectrum of each fragment. This is a time consuming process as it uses floating point to represent large data sets and requires processing time. The proposed Regressive Multi-Array Deep Neural Classifier (RMDNC) system is implemented to reduce the computation time, it is called a polymorphic processor, the system design a dedicated processor, based on a hardware-oriented algorithm that we have proposed to efficiently compute the general dimensional spectrum of DNA sequences. The proposed Regressive Multi-Array Deep Neural Classifier (RMDNC) system concept of the biology information is classified as follows. Protein-Protein Interaction (PPI) networks explain the understanding of organisms in coronary arteries, genetics, gender studies, cardiovascular risk factors and atherosclerosis, the development and identification of carotid intimal media thickness Pay particular attention to arterial calcification, which is an important factor in improving. Also, multiple biological activities of the human body are responsible for these interactions. In this work, computational studies have been completed to understand the PPI network of obstructive sleep apnea, cardiovascular disease, stroke and epilepsy.
Keywords: Regressive Multi-Array Deep Neural Classifier (RMDNC); cardiovascular disease; epilepsy; sleep apnea; DNA protein ring
Genetic and environmental factors understand the central multi-factors of common disease processes. A key challenge facing molecular assembly research in the post-genome era is understanding the network interconnection of media and in-house products through the onset of various environmental changes. In biology information of system in polymorphic genes that interact with each other as well as with environmental sensitivities and consequences of complex diseases. The development of common complex diseases interacts non-linearly, hierarchically with multiple genes and epistasis effects and is effected by various genes and different environmental condition. For complex diseases, the combination of genes can be susceptible to the disease, and environmental factors exacerbate the effects of these genes. If these continuation are neglected, the relative risk of self-genetic variation is expected to be low. Considering the interaction between genes, it does not weaken the relationship between exposure to the environment, diseases and genetic diseases. Therefore, there is increasing interest in optimizing treatments and identifying prognostic genetic elements of the disease. However, sex chromosomes not included in a genome-wide association study (GWAS) to consider gene analysis variables targeting men and women investigating genes for cardiovascular disease in men and women, and to examine hormonal gene interactions. The environmental factors that appear to have a slight effect at the most useful level can have a certain relative risk of a certain genetic predisposition. For instance, the danger of significant basic ailments, for example, malignant growth, cardiovascular sickness, dysfunctional behavior and diabetes is relied upon to be influenced by extreme DNA designs.
Obstructive Sleep Apnea (OSA) is a turmoil that happens when the tongue and delicate sense of taste breakdown (hinders) onto the rear of the throat weakening during rest. This issue causes a recoil in the aviation route which can prompt shallow breathing or even a stoppage in relaxing for brief timeframes extraordinary the night. OSA finding is a short-term demonstrating redundant obstructive apneas and hypopneas during rest, which can prompt drops in oxygen impregnation levels, rest feelings of excitement, and hemodynamic changes. Therefore, a method of calculating reliability is being developed where automatic prediction is based on structures derived from single sequences of binding residues generic information DNA and also possible derived from them. There are also computational challenges in studying DNA interactions to predict genes in key methods and common diseases, combinations of environment and gene-gene interactions. What is needed is an effective method for analyzing large amounts of DNA data that goes beyond traditional statistical methods for complex diseases.
In the post-genome era, many problems in bioinformatics are caused by the generation of large amounts of unbalanced data. Specifically, there is a high imbalance in the computational class of precursor microRNAs (premiRNAs) [1]. High-throughput technology screens have provided a large amount of drug susceptibility data for cancer cell lines and groups of hundreds of compounds. Identifying Computational Drug-Sensitive Genome-Molecule Determinants Can Obtain Anti-Cancer Therapy, Methods for Analyzing These Data, and Development of New Anti-Cancer Agents [2]. Interpretation factor restricting locales (TFBSs) are fundamental for displaying essential requirement instruments and ensuing cell work. Convolutional neural systems (CNN) are already superior to methods for predicting TFBSs from main DNA sequences [3]. DNA methylation assumes a significant job in the guideline of certain organic procedures. As of recently, the improvement of machine learning models and deep learning models based on several sequences used to predict the methylation status of DNA have been used to generate traditional learning methods such as random forests and support vector machines. May get better performance than the method [4].
The identification of couple rehashes in DNA arrangements is a troublesome issue for researchers and specialists of the current time. Identification of couple duplications is likewise a significant piece of quality comment, which is useful for identifying different genetic diseases and human identities [5]. The advances in DNA methylation investigation have prepared for comprehension the epigenetic components basic different maladies, for example of disease [6]. In spite of the fact that this strategy has demonstrated helpful for recognizing ailment defenseless SNPs, there is proof that huge numbers of these are, actually, bogus positives. In this manner, there are a few ambiguities about the most proper limit for guaranteeing entire genome suggestions [7]. Deep sequencing technology reveals new insights, different transcriptional subtypes and accurate expression measurements to determine the level of transcription and shows clear advantages in sequence coverage analysis [8].
Deep learning algorithms work better than traditional methods of predicting specific DNA-protein sequence binding. They lack the consideration of the dependence between different transcription factors (TF) [9] and nucleotides of different bond lengths. It proposes a method for the diagnosis of genetic diseases. To achieve this goal, we need to select the most informative people who can accurately distinguish different types of genes [10]. Non-B DNA structures can contend with nucleosomes for genomic positions [11], regardless of whether just B-DNA can be folded over the nucleosome. The logic circuit of DNA is a hot research theme of biological computing. First, DNA was introduced as a simple logic gate. After that, a simple circuit that proposed a DNA logic circuit model was successfully proved [12].
In numerous unpredictable maladies, the progress from a sound stage to a cataclysmic stage doesn’t happen bit by bit. Late examinations have demonstrated that the turn of events and improvement of these illnesses includes three stages, including a solid stage, a pre-stage, and an infection stage [13]. Multi-fractal investigation of the genome is performed by combining the total DNA grouping into littler sections and utilizing disorderly game portrayal and the case strategy to calculate the general dimensional spectra of each fragment [14]. The reason for this article is to build up a program that can consequently address these hazy spots. This application includes a shiny online program accessible on the R stage and runs essentially [15]. Surface plasmon reverberation (SPR) is one of the most alluring and ongoing strategies for surface-delicate bio atomic collaboration investigation (BIA) and location of different marvels. Its primary focal points incorporate surface delicate responses, no imprint recognition, continuous estimation capacities, and even the capacity to recognize little changes [16].
DNA has become a very useful tool for predicting disease. By allowing a health care professional to recognize a DNA gene as a marker of a disease, one can help make the appropriate lifestyle and similar modifications that help reduce the risk of the disease [17]. Repetitive identification plays an important role in DNA sequence analysis because most DNA sequences contain specific repeating patterns of specific cycles [18].Numerous attributes that group different diseases and disorders based on a particular common. Our goal is to study any pattern that can extract information based on the evaluation of those diseases and genes in a large group [19]. Current microarray classification algorithms yield false positives at high speed, which is typically an unacceptable diagnostic application [20]. Human disease involves a complex array of interactions between different organic procedures. Specifically, different biologic information, for example, single nucleotide polymorphisms (SNPs), duplicate number polymorphisms (CNVs), DNA methylations (DMs), and their communications all the while assume a significant job in human sickness [21].
Despite the popularity of DNA in human body fluids, the origin of its molecule is still unknown. Except for the exogenous source of DNA, mechanisms of homology with several possible sources have been proposed. First, initial studies showed that DNA enters cells under circulation at the interface between the tumor and the circulation. However, this reversal indicates that the concentration of DNA in the blood of cancer patients is greater than can be explained by the quality of the cells. The proposed Regressive Multi-Array Deep Neural Classifier (RMDNC) shown in Fig. 1 is implemented that DNA can be derived from the destruction of tumor micro and circulating tumor cells. However, this turned out to be incorrect, because certain mutations in the DNA from CRC patients may not be associated with mutations in the cell layer where micro metastases should be present. The gene database collects significant genes. The main selection part of the gene set, the total number of genes that carry 127 groups is collected as OSA and 1598532 times 1292 genes collected for epilepsy, CVD and stroke. All major collected genes do not belong to human tissues. It represents a major gene collection of human only biological gene extraction technology. The proposed system is generate the pattern and check the similarity sequence of this DNA pattern. The input sequence identification of the system is identify the DNA protein ring to calculate the sequence. The gene is filtered to significant changes in the disease like Obstructive sleep apnea the gene value is 121, epilepsy gene value is 143 and cardiovascular disease gene for 463, this the gene values are collected in the human gene set of this system.
The unique biological properties of DNA include the specificity of double-stranded base pairing that together keep the double helix together. The protein ring of DNA is shown in Fig. 2. The DNA double helix, which usually results from complementary interactions, is a linear molecule. However, branched-chain DNA molecules have emerged as key intermediates in DNA metabolism, especially during replication, recombination, and repair. The physical properties of the DNA double helix are different from any other natural or synthetic polymer. The proposed system
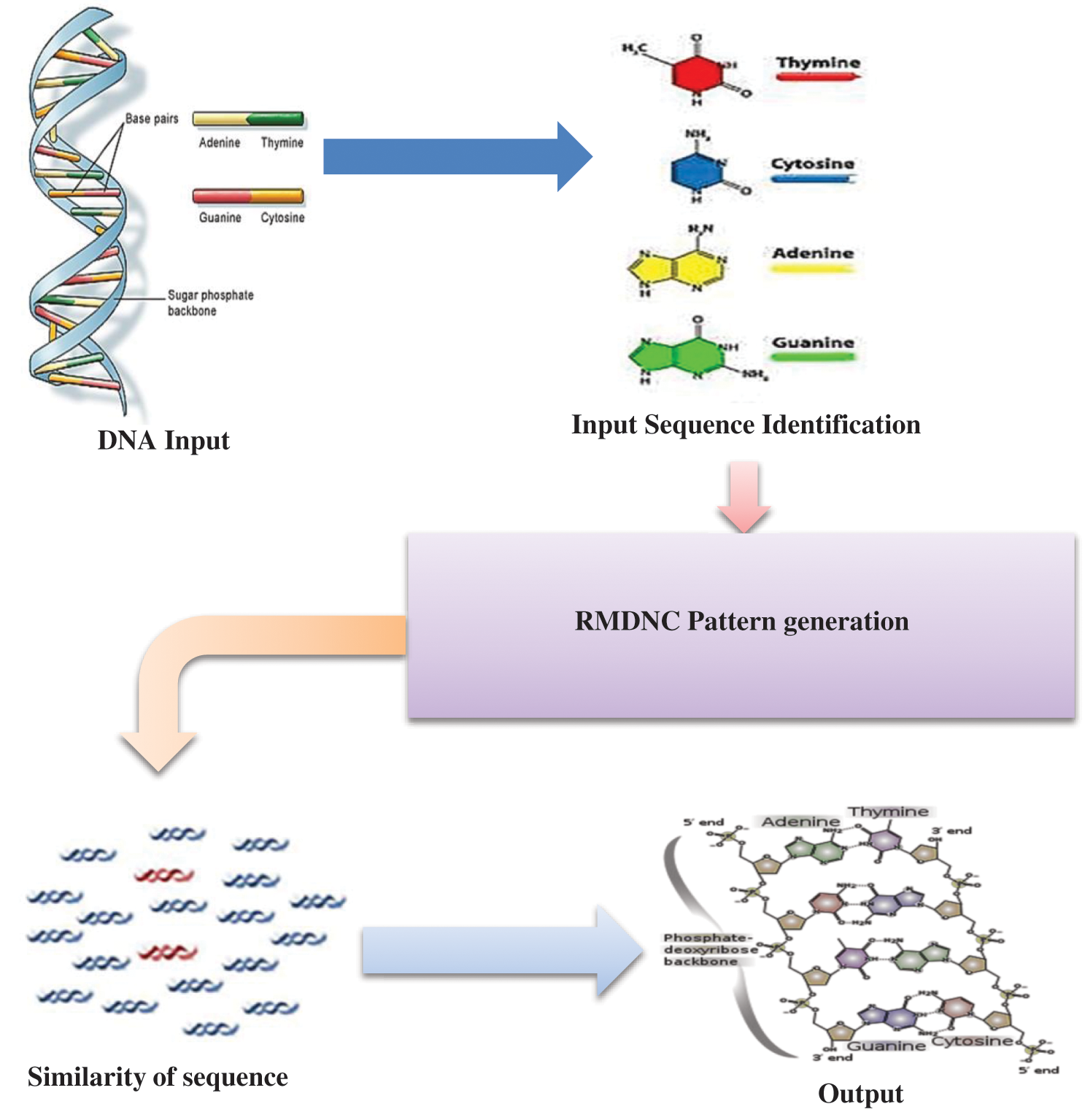
Figure 1: Proposed system block diagram
The characteristic base-stacking and braided structure of the molecule lends it extraordinary rigidity: it bends double-stranded DNA molecules into loops approximately 50 times, rather than performing the same action on single-stranded DNA. It requires more energy. Also, double-stranded DNA molecules are very stable. These properties make double-stranded DNA molecules, ideal candidates for scaffolding other molecules.
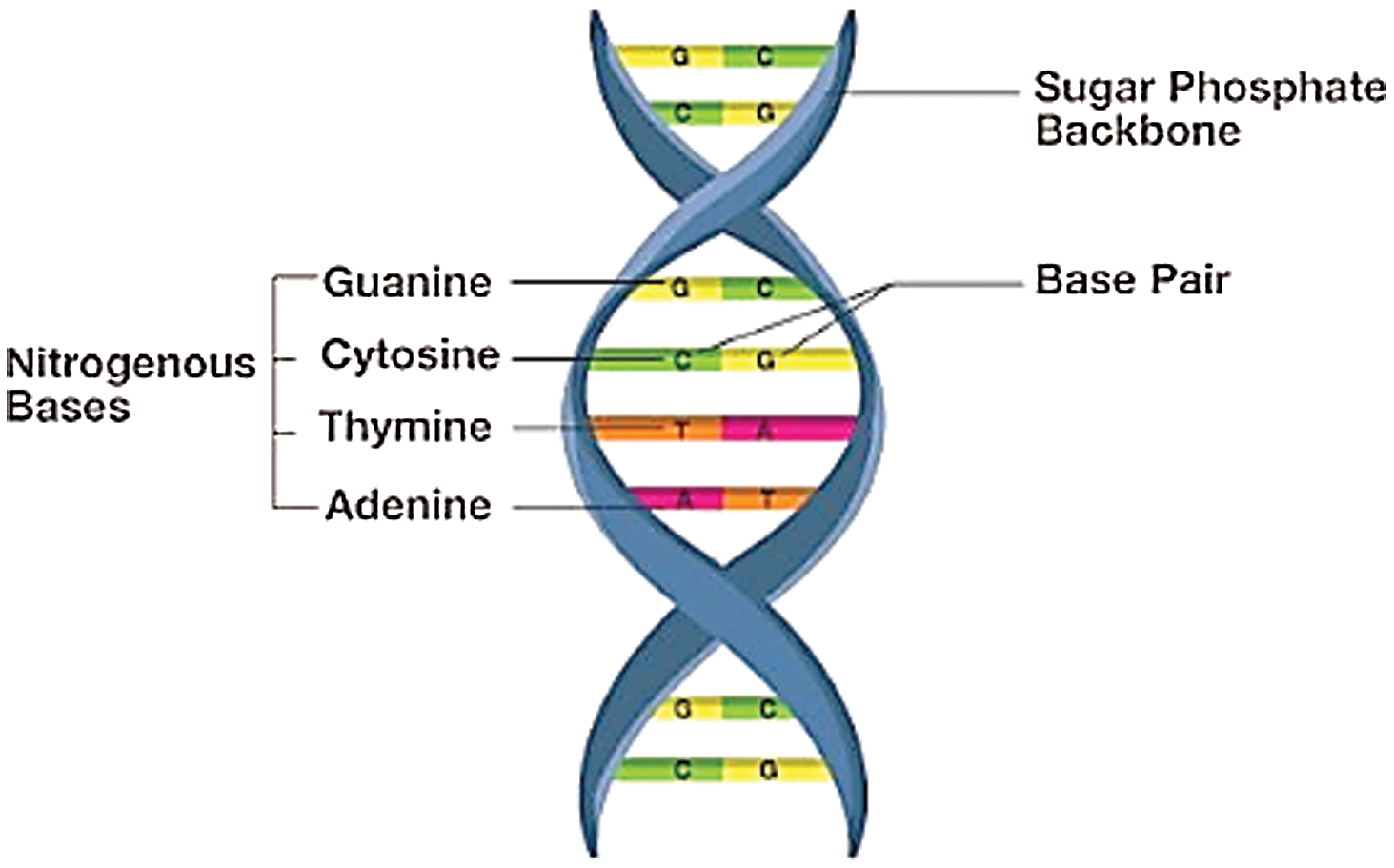
Figure 2: Protein ring of DNA
Adenine (A), Guanine (G), Cytosine (C) and Thymine (T) this is a four protein ring consist in the DNA. A gene is a fragment of DNA that contains the code for the function of a particular protein used in many types of human cells. A chromosome is a structure that contains human genes inside a cell. This gene is contained on a chromosome in the nucleus. The function of the chromosome is to carry the basic genetic material, DNA. DNA provides the type of genetic information for various cellular functions.
3.2 Risk Factors of Cardiovascular
Cardiovascular disease is the leading cause of death and disability in the Western world. Cardiovascular illness is a mind boggling sickness with natural and hereditary elements. Hazard Factors that create coronary illness known to improve the probability of cardiovascular coronary illness contain diabetes, hypertension, weight, hypercholesterolemia, and diet. Additionally, a positive family ancestry of coronary illness in first-degree family members is a solid autonomous hazard factor for cardiovascular coronary illness. The mutation of PTPNII is the pattern of congenital heart disease is one of the form of cardiovascular disease, genes is important one to analyzing the disease above 50% of cases. The studies of gene mutation pathway is related to analysis spectrum disease.
3.3 Identification of DNA Sequence
This technique delivers a succession set of given groupings, and this strategy produces staggered arrangement similitude dependent on design sets. The DNA sequence is depict in Fig. 3. For each degree of DNA arrangement, succession comparability is determined by the total grouping similarity is measure and determined by this technique and the last strategy. The technique utilized for every class keeps up an alternate succession, and this strategy computes different degrees of arrangement comparability for every classification. Class selection and distinguish the most comparable successions dependent on how you measure arrangement similitude.
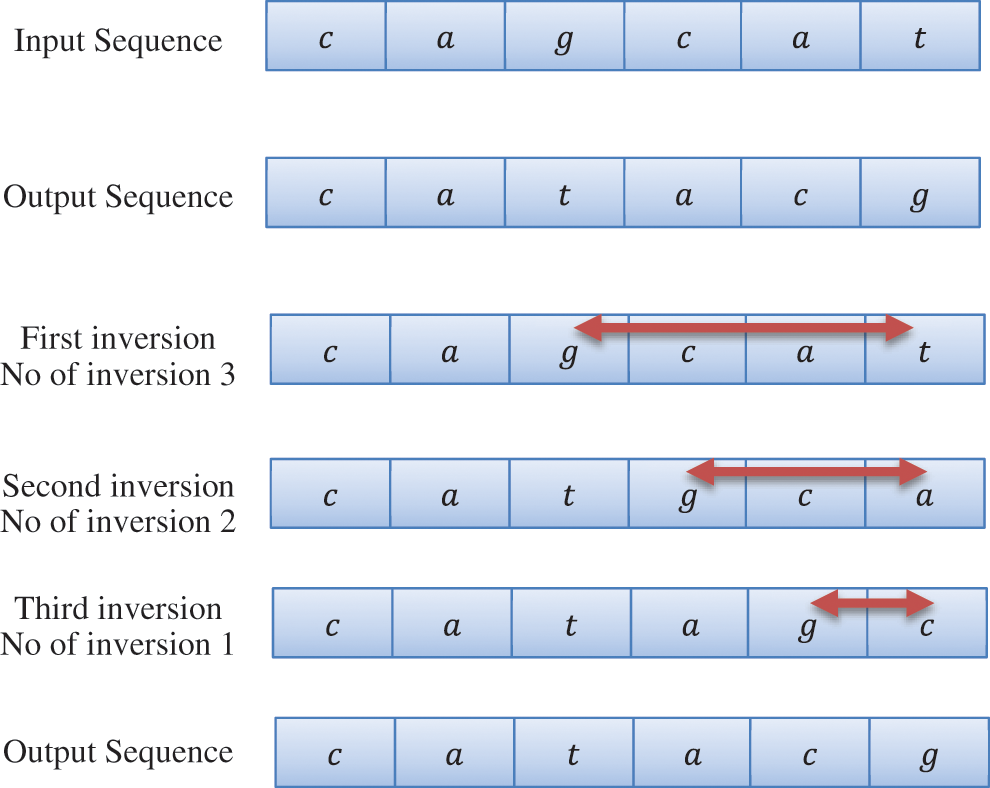
Figure 3: DNA Sequence
The examples are utilized for distinguishing the ailment by means of various sizes and diverse example blends. The DNA pattern generation is shown in Fig. 4. For each sort of DNA succession, the technique creates various degrees of various arrangement designs. The pattern create from the various measurements and the example will be utilized to compute the grouping to analysis. The contribution to the framework is a quality database grouping, which has the questions database is found.
Figure 4: DNA Pattern generation
The yield of the framework is the likeness between the two strings at the area of the locale with these comparability scores. Arrangement Represented as a quality score for each pair, the scores are arranged. Scoring grid utilizes bases (DNA) to compute the base score for adjustment. The arrangement score is the whole of the scores at each position.
Regressive Multi-Array Deep Neural Classifier (RMDNC)
Step 1: Initialize the input data value.
Step 2: Analysis the input data values and store in the system further operation.
Step 3: The collected data is trained and computed to the system and analysis the DNA sequence.
Step 4: This system classified input and the output neuron and the equ is
where,
S is the system neuron output
Step 5: The order execution of the framework is given the component esteem and to figure the information wellness esteem in the framework.
where,
S is training samples
T is steps time.
Step 6: TheRegressive Multi-Array Deep Neural Classifier (RMDNC) is used to identifying the output signal value.
Step 7: The Regressive Multi-Array Deep Neural Classifier (RMDNC) is used to analysis and changing the tested value of the system parameter.
Step 8: End.
The neural system is a significant strategy to remove the component in the info sign, and its created depends on the Regressive Multi-Array Deep Neural Classifier (RMDNC) model is shown in Fig. 5; it has the three-layer system.
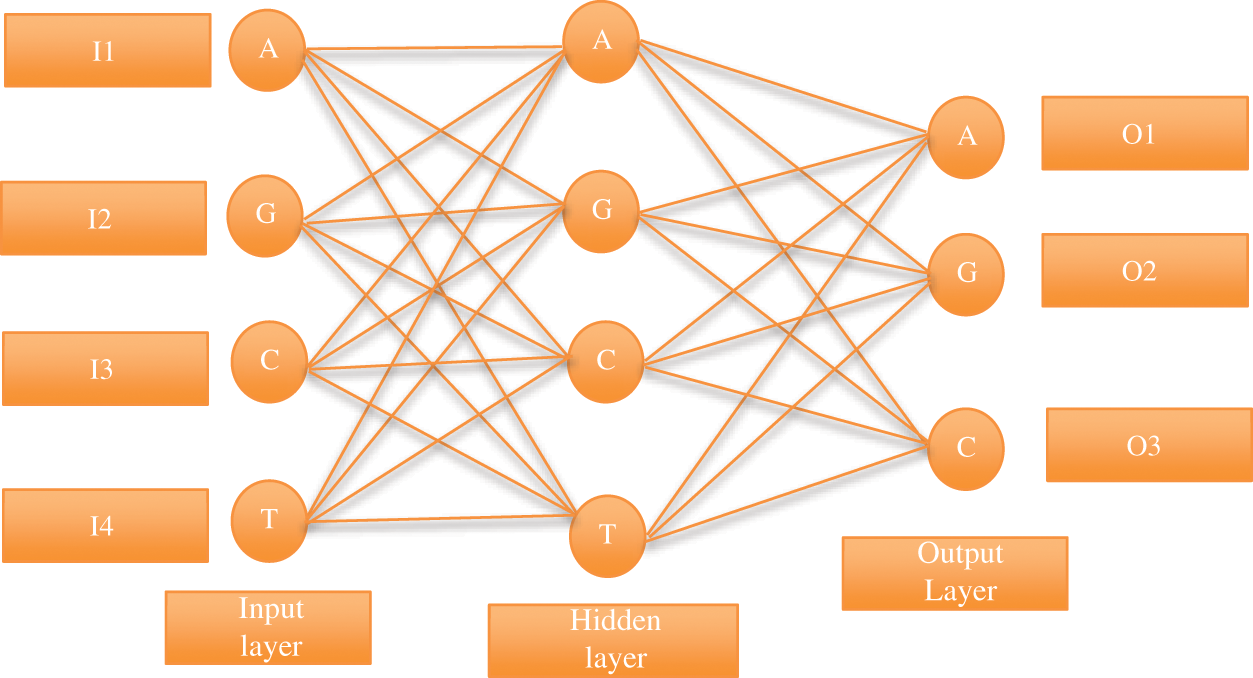
Figure 5: The proposed Regressive Multi-Array Deep Neural Classifier (RMDNC) system
Systems ordinarily utilize the sigmoid yield include. The quantity of hubs in every one of these layers is controlled by understanding in addition to relative yield signal quality. At the point when the shrouded number of layers in hubs is huge, the capacity to gain proficiency with the connection between the solid information and the yield is solid. Be that as it may, the hubs with such a large number of layers center on the relationship among learning and learning and not the system of preparing tests.
Application of an innovative Regressive Multi-Array Deep Neural Classifier (RMDNC) sorting method used in Regressive Multi-Array Deep Neural Classifier (RMDNC). In traditional broadcasting systems, it is close to the typical and midpoints. The notable hub qualities are broke down dependent on the Regressive Multi-Array Deep Neural Classifier (RMDNC) geography investigation. This technique has been demonstrated to utilize amino corrosive arrangements for various DNA successions and their productivity. The effectiveness of this technique has been affirmed by computing the time multifaceted nature of the succession discovery exactness got.
Table 1: Details of data set used
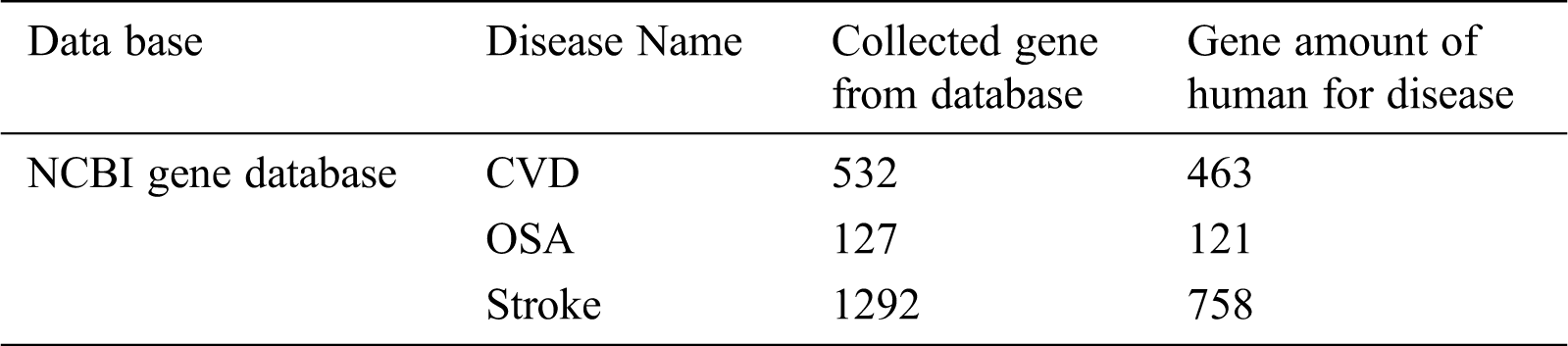
The Tab. 1, shows the subtleties of informational index being utilized to assess the presentation of the proposed approach. The technique has been approved for its productivity utilizing various informational collections and the strategy has been approved for its effectiveness in arrangement recognizable proof and its time intricacy.
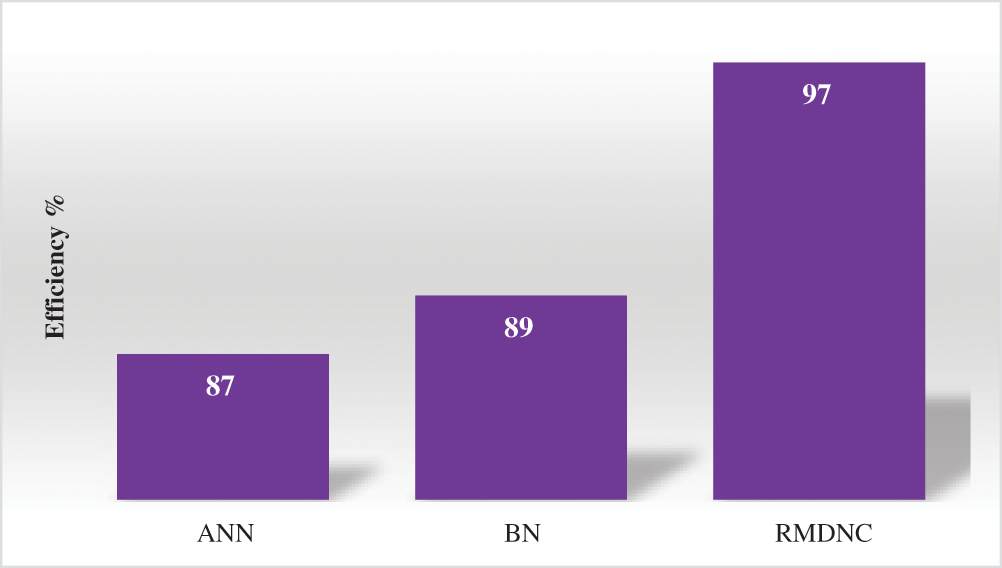
Figure 6: Comparison of efficiency
The Fig. 6 gives the correlation of arrangement recognizable proof effectiveness created by various existing techniques is Artificial Neural Network, Bayesian Network and it compared to the proposed Regressive Multi-Array Deep Neural Classifier (RMDNC) method has produces higher efficiency than other methods.
Table 2: Proposed system performance comparison

Tab. 2 is given the sensitivity, accuracy, and specificity of the proposed Regressive Multi-Array Deep Neural Classifier (RMDNC) strategy is higher than the other existing framework like SVM and Fuzzy and the correlation graph is given beneath.
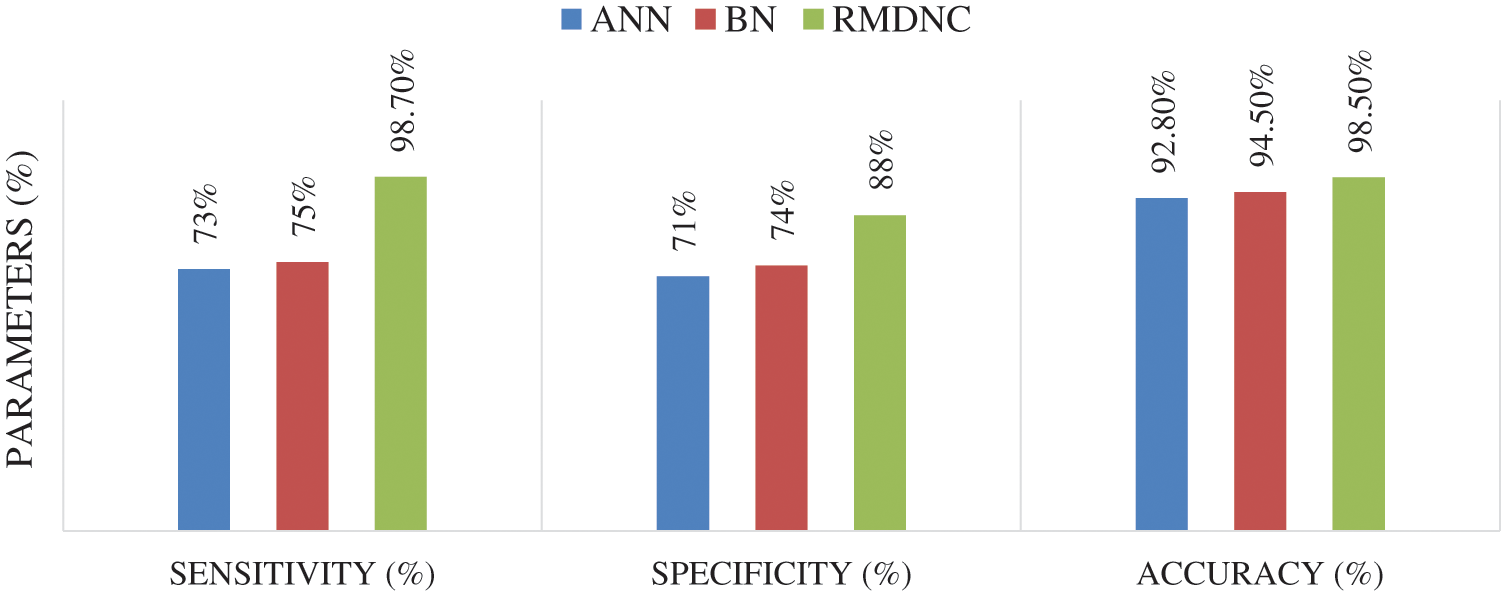
Figure 7: Comparison of proposed RPDLC system
Fig. 7 gives the performance examination of the proposed RPDLC framework with the regular technique. As contrasted and traditional Artificial Neural Network, and Bayesian Network methods, the proposed Regressive Multi-Array Deep Neural Classifier (RMDNC) method was delivered significant results for every working condition.
Table 3: Comparison analysis of variation of FAR and FRR

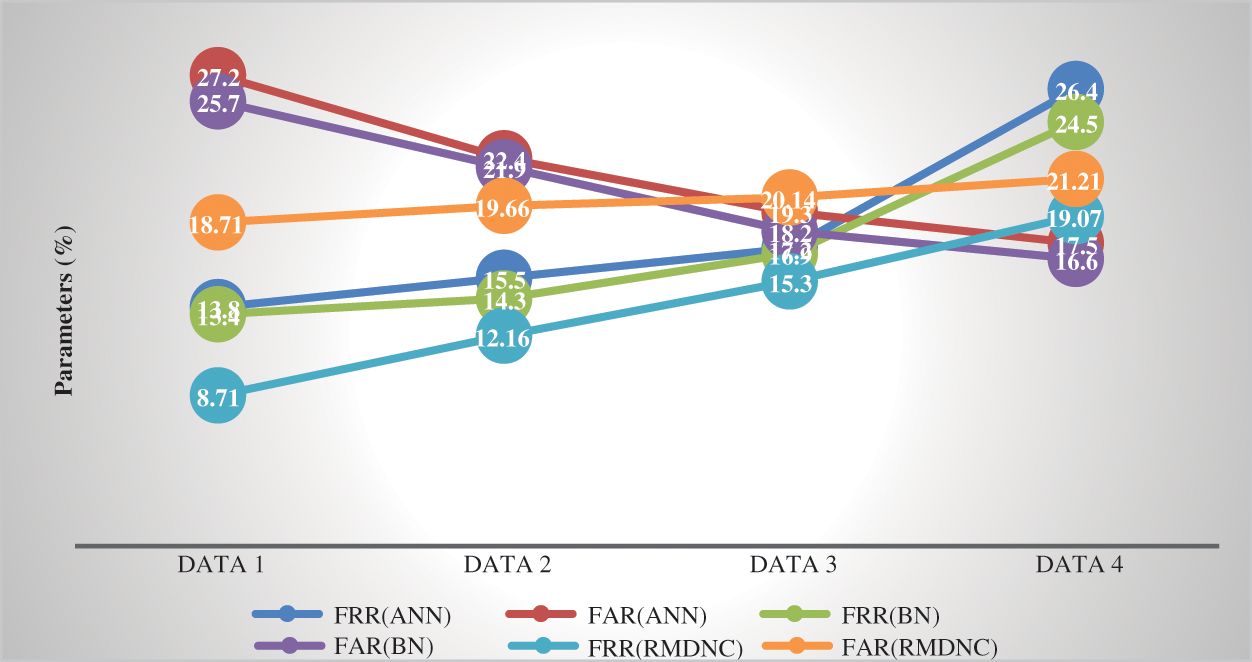
Figure 8: Comparison chart false ratio
Fig. 8 show the correlation outline of the mean proportion, and it’s contrasted with the current technique. Tab. 3 represents the comparision analysis of variation in FAR and FRR with absolute data with existing techniques. Besides, the proposed bogus technique proportion is decreased, which makes our framework productive. The proposed Regressive Multi-Array Deep Neural Classifier (RMDNC) strategies accomplish the best outcomes.
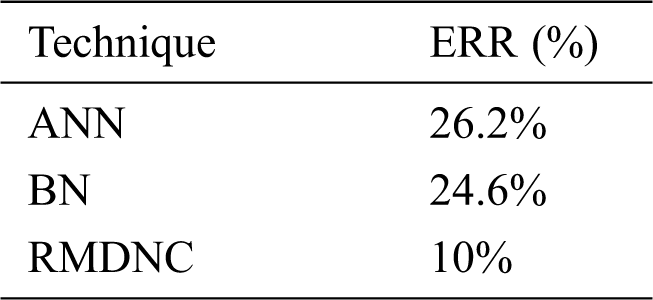
Tab. 4 gives the error rejection ratio of the Regressive Multi-Array Deep Neural Classifier (RMDNC) system.
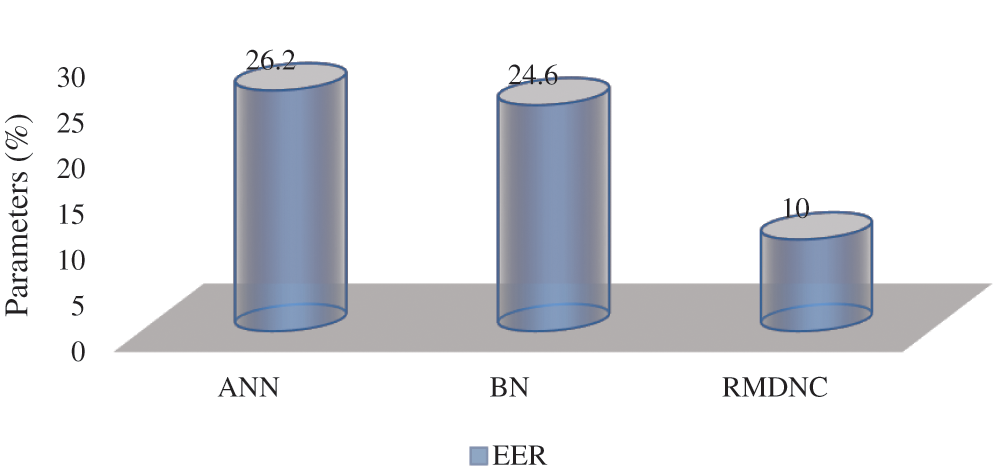
Figure 9: Average EER
Fig. 9 gives the proposed Regressive Multi-Array Deep Neural Classifier (RMDNC) system Error Rejection Rate.
The genes that have to be collected in this system are jointly responsible for genetic selection, they come from filters from gene databases from authoritative sources, just to get genes based on human organisms. Find all illnesses. It clusters subnet networks and ultimately determines the key gene ontologies for embedded PPI network analysis topology analysis. Our results show that they are jointly responsible for the nature of the interactions between genes. These types of studies are likely to be useful in producing recommendations for genetically-based evidence in the field of network biology. Our results show that they are jointly responsible for the nature of the interactions between genes. These types of studies are likely to be useful in producing gene-based evidence recommendations for use in the field of network biology. They also demonstrated that gene behavior must be displayed in a regulatory network environment to determine gene interactions and their roles and effects on disease development. The proposed regressive multi-array deep neural classifier (RMDNC) approach has allowed them to identify candidate genes and confirm their association with the importance of wet-lab for encoded proteins or rejection. Under this expectation, the study integrates several data mining techniques to identify potential novel and gene signaling-related multisource in female cardiovascular disease.
Funding Statement: The authors received no specific funding for this study.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
1. L. Cristian. (2019). “Deep neural architectures for highly imbalanced data in bioinformatics,” in IEEE Transactions on Neural Networks and Learning Systems, vol. 31, no. 8, pp. 2857–2867. [Google Scholar]
2. Y. Wang and M. Li. (2019). “Deep dsc: A deep learning method to predict drug sensitivity of cancer cell lines,” in IEEE/ACM Transactions on Computational Biology and Bioinformatics, pp. 1–11. [Google Scholar]
3. F. Jing and S. W. Zhang. (2019). “An integrative framework for combining sequence and epigenomic data to predict transcription factor binding sites using deep learning,” in IEEE Transactions on Computational Biology and Bioinformatics Class Files, vol. 18, no. 1, pp. 355–364. [Google Scholar]
4. L. Fu and Q. Peng. (2019). “Predicting DNA methylation states with hybrid information based deep-learning model,” in IEEE/ACM Transactions on Computational Biology and Bioinformatics, vol. 17, no. 5, pp. 1721–1728. [Google Scholar]
5. P. Garg and S. D. Sharma. (2019). “MGWT based algorithm for tandem repeats detection in dna sequences,” in Int. Conf. on Signal Processing, Computing and Control, Solan, India. [Google Scholar]
6. H. Damgacioglu and E. Celik. (2020). “Intra-cluster distance minimization in DNA methylation analysis using an advanced tabu-based iterative k-medoids clustering algorithm (T-CLUST),” in IEEE/ACM Transactions on Computational Biology and Bioinformatics, vol. 17, no. 4, pp. 1241–1252. [Google Scholar]
7. P. Fergus and R. Casimiro. (2018). “Utilising deep learning and genome wide association studies for epistatic-driven preterm birth classification in african- american women,” in IEEE/ACM Transactions on Computational Biology and Bioinformatics, vol. 17, no. 2, pp. 668–678. [Google Scholar]
8. B. Aniruddha. (2018). “Deep sequencing data analysis,” in IEEE/ACM Transactions on Computational Biology and Bioinformatics, vol. 15, no. 2, pp. 482–483. [Google Scholar]
9. Q. H. Zhang and L. Zhu. (2018). “High order convolutional neural network architecture for predicting DNA-protein binding sites,” in IEEE/ACM Transactions on Computational Biology and Bioinformatics, vol. 16, no. 4, pp. 1184–1192. [Google Scholar]
10. F. Francis and A. Namitha. (2018). “Ensemble approach for predicting genetic disease through case-control study,” in Int. Conf. on Inventive Communication and Computational Technologies, Coimbatore, India. [Google Scholar]
11. E. Tevanyan and M. Poptsova. (2018). “Recognizing patterns of nucleosome and DNA structures positioning,” in Int. Conf. on Bioinformatics and Biomedicine, Madrid, Spain. [Google Scholar]
12. B. Yimwadsana and P. Artiwet. (2018). “On Optimizing DNA Sequence Design for DNA Logic AND Circuit,” in proc. TENCON 2018 - 2018 IEEE Region 10 Conf., Jeju, Korea, pp. 1828–1833. [Google Scholar]
13. A. D. Torshizi and L. Petzold. (2018). “Sparse pathway-induced dynamic network biomarker discovery for early warning signal detection in complex diseases,” IEEE/ACM Transactions on Computational Biology and Bioinformatics, vol. 15, no. 3, pp. 1028–1034. [Google Scholar]
14. J. E. Duarte and S. Jaime. (2017). “Hardware accelerator for the multi-fractal analysis of DNA sequences,” IEEE/ACM Transactions on Computational Biology and Bioinformatics, vol. 15, no. 1, pp. 1611–1624. [Google Scholar]
15. Z. Elyazghi and L. E. Yazouli. (2017). “ABI base recall: Automatic correction and ends trimming of DNA sequences,” IEEE Transactions on NanoBioscience, vol. 16, no. 8, pp. 682–686. [Google Scholar]
16. S. Shafayet and S. Mohammad. (2017). “Detection of DNA mutation, urinary diseases and blood diseases using surface plasmon resonance biosensors based on kretschmann configuration,” in Int. Conf. on Electrical, Computer and Communication Engineering, Cox’s Bazar, Bangladesh. [Google Scholar]
17. M. Y. Sikkandar, T. Jayasankar, K. R. Kavitha, N. B. Prakash, N. M. Sudharsan et al. (2020). , “Three factor nonnegative matrix factorization based he stain unmixing in histopathological images,” Journal of Ambient Intelligence and Humanized Computing, pp. 1–9. [Google Scholar]
18. P. Garg, S. Sharma and S. N. Sharma. (2017). “Tandem repeats detection in DNA sequences using p-spectrum based algorithm,” in proc. 2017 Conf. on Information and Communication Technology (CICTGwalior, India, pp. 1–5. [Google Scholar]
19. J. Jayanthi, E. Laxmi Lydia, N. Krishnaraj, T. Jayasankar, R. Lenin Babu et al. (2020). , “An effective deep learning features based integrated framework for iris detection and recognition,” Journal of Ambient Intelligence and Humanized Computing, pp. 1–11. [Google Scholar]
20. S. Hsieh and Y. Chou. (2016). “A faster cDNA microarray gene expression data classifier for diagnosing diseases,” IEEE/ACM Transactions on Computational Biology and Bioinformatics, vol. 13, no. 1, pp. 43–54. [Google Scholar]
21. M. Kang, J. Park, D. Kim, A. K. Biswas, C. Liu et al. (2017). , “Multi-Block bipartite graph for integrative genomic analysis,” IEEE/ACM Transactions on Computational Biology and Bioinformatics, vol. 14, no. 6, pp. 1350–1358. [Google Scholar]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |