DOI:10.32604/iasc.2022.017683
| Intelligent Automation & Soft Computing DOI:10.32604/iasc.2022.017683 | |
| Article |
Hybrid Approach for Taxonomic Classification Based on Deep Learning
1Department of Information Technology, College of Computer and Information Sciences, Princess Nourah Bint Abdulrahman University, Riyadh, Saudi Arabia
2Department of Electronics and Electrical Communications Engineering, Faculty of Electronic Engineering, Menoufia University, Menoufia, 32952, Egypt
3Department of Computer Science and Engineering, Faculty of Electronic Engineering, Menoufia University, Menoufia, 32952, Egypt
4Ministry of Education, Riyadh, Saudi Arabia
5Department of Computer Sciences, College of Computer and Information Sciences, Princess Nourah Bint Abdulrahman University, Riyadh, Saudi Arabia
*Corresponding Author: Naglaa. F. Soliman. Email: nfsoliman@pnu.edu.sa
Received: 07 February 2021; Accepted: 01 September 2021
Abstract: Recently, deep learning has opened a remarkable research direction in the track of bioinformatics, especially for the applications that need classification and regression. With deep learning techniques, DNA sequences can be classified with high accuracy. Firstly, a DNA sequence should be represented, numerically. After that, DNA features are extracted from the numerical representations based on deep learning techniques to improve the classification process. Recently, several architectures have been developed based on deep learning for DNA sequence classification. Convolutional Neural Network (CNN) and Recurrent Neural Network (RNN) are the default deep learning architectures used for this task. This paper presents a hybrid module that combines a CNN with an RNN for DNA classification. The CNN is used for feature extraction, and this is followed by a subsampling layer, while the RNN is trained for classifying bacteria into taxonomic levels. Besides, a wavelet-based pooling strategy is adopted in the subsampling layer, because the wavelet transform with down-sampling allows signal compression, while maintaining the most discriminative features of the signal. The proposed hybrid module is compared with a CNN based on Random Projection (RP) and an RNN based on histogram-oriented gradient features. The simulation results show that the hybrid module has the best performance among other ones.
Keywords: Deep learning; CNN; RNN; DNA; random projection; wavelet transform; taxonomic classification
Deoxyribonucleic acid (DNA) molecules contain the essential inheritable information. Such information is represented as a long sequence of nucleotides, which can be represented into four alphabets (nucleotides) {A, C, T, G} [1–3] as shown in Fig. 1. The genome sequence is the complete list of sequences (nucleotides) that makes up the DNA. These nucleotides are closely related among individuals of the same species. They differ only in small subsets. More than 34,000 species [4] have their genomes sequenced, the bacterial being one of them. Living things can be classified based on the similarity between DNA sequences. According to the hierarchy, classification can be performed to Kingdom, Phylum, Class, Order, Family, Genus and Species [5]. The main objective of this work is the classification of bacteria based on DNA sequences. Hence, the DNA sequences with similar structures also have similar functions. The problem here is finding regularities (repetitions) in DNA sequences to classify them into different groups with the same regularities. Taxonomy classification allows to recognize and classify the discovered and undiscovered species and other taxa based on DNA sequences. Sequence similarity is traditionally estimated using sequence alignment methods [6,7]. These sequence alignment methods involve a feature selection stage. Spectral representation of DNA sequences can be used to determine the sequence similarity in order to enhance the classification performance [8,9]. The time computational complexity remains the reason for restricting the use of alignment approaches.
Figure 1: DNA structure
Machine learning has emerged as a successful technique in classification and regression applications. Classification is the activity of examining the features of an object, and assigning it to a predefined set of classes based on supervised learning [10]. The selection of which features are more suitable to face the given target remains a crucial and challenging step in machine learning. Deep learning has recently emerged as a successful paradigm for big data processing because of the technological advances in the low-level cost of parallel computing architectures. Hence, deep learning has given significant contributions to several basic, but arduous artificial intelligence tasks.
Deep learning [11] is a relatively new artificial intelligence field that achieves remarkable results in image recognition, text interpretation, translation, and other domains, such as drug and genome detection. Deep learning reveals complex structures in vast databases by using back-propagation. The model adjusts the internal weights to calculate data in the current layer based on the previous layer output data. Classification models using deep learning have a multi-layered architecture. They consist of input, hidden, and output layers. Each layer is a relatively simple computer unit that tries to learn a certain level of representation; nevertheless, the layers are interconnected with non-linear functions. The output of a lower layer is thus the input to a higher layer of the model.
The Convolutional Neural Networks (CNNs) and Recurrent Neural Networks (RNNs) are supreme models in deep learning that can be used for DNA signal classification. The CNNs are types of discriminative connectionist models. They are originally designed to work directly on observed images without pre-processing [12]. The CNNs comprise one or more convolutional layers, and subsampling layers, followed by one or more fully-connected layers as in standard neural networks. The RNNs are good in modeling of dynamic data characteristics. They can remember the context information due to their recurrent schematic [13]. For tasks that include input data in the form of sequences such as speech recognition and natural language processing, it is often better to use RNNs [14,15] than CNNs. A CNN can be combined with an RNN to improve the classification accuracy in several applications. The output of the final fully-connected layer of a CNN model is used as the RNN input [16]. The high-dimensional data can be reduced to a low-dimensional data with the most significant features in the form of vectors using different down-sampling layers. There are different kinds of down-sampling layers, such as pooling and RP layers, to reduce the dimensionality of feature maps associated with multi-layer CNNs [17,18].
This paper presents a hybrid module consisting of a CNN followed by a wavelet-based pooling and an RNN to improve the classifier accuracy. Since deep learning models can only work with numerical values, we need to transform the DNA characters into numbers. The one-hot coding and Frequency Chaos Game Representation (FCGR) are usually used for DNA classification in most research as sequence mapping [19–23]. In the one hot coding, the DNA nucleotides are mapped as binary vectors with all elements set to zero and one as A = (1 0 0 0), G = (0 1 0 0), C = (0 0 1 0) and T = (0 0 0 1). This representation scheme was used efficiently for promoter recognition in imbalanced DNA sequence datasets using support vector machines [24]. It was used for Ecoli promoter prediction using neural networks [25]. Furthermore, it was extended for gene identification in human, Drosophila melanogaster, and Arabidopsis thaliana using a neural network-based multi-classifier [26]. The FCGR is a kind of DNA sequence mapping that keeps up the patterns in the arrangement and changes picture highlights [27]. It is considered a graphical and numerical representation.
The Chaos Game Representation (CGR) is a scale-independent representation developed by Jeffrey [27] that explains the oligonucleotides frequencies as an image. The main physiognomies of the whole genome using CGR can be shown [28]. The CGR pattern of the same genome nucleotide sequences is similar, but differs from those of other species, quantitatively. This biological characteristic makes the unique genomic signature that is suitable for classification and clustering. Besides, the CGR technique has the advantage of presenting the abundance of all k-mers (a group of successive k nucleotides) in a given sequence. To estimate the sub-sequence frequency occurrence, we should transfer from the Chaos Game Representation to the FCGR [29]. An image can be constructed from the FCGR representation matrix as indicated in Fig. 2 and the dimensions of this image are a function of the dimension k. For example, for k = 6, the size of the input image is

Figure 2: Distribution of K-mers in CGR
The main objective is to test whether the methods can classify the correct taxonomic class sequences, even if we have only the 500 bp long part of all sequences available. There are several deep learning (DL) techniques such as CNNs, RNNs, and the proposed hybrid module for achieving this objective. The rest of this paper is organized as follows. Sections 2 and 3 present the related work and dataset, respectively. The proposed module is explained in Section 4. The experimental results are explained in Section 5. Finally, the concluding remarks are provided in Section 6.
2 Related Work to DNA Classification
There are several methods that have been used in the classification of DNA sequences such as alignment methods and DL models [19,21,30]. The alignment methods depend on positioning of the biological sequences to identify regions of similarity. These methods may be alignment-based or alignment-free methods [30]. Although the alignment methods are very effective in several applications, the key issue that seriously limits the performance remains their time computational complexity. For this reason, it is necessary to have sequence classification methods that do not depend on alignment. Recently, DL methods have been used in bioinformatics. Angermueller et al. [31] presented a review study that discusses the applications of DL approaches in regulatory genomics and cellular imaging. In [32], the authors added a dropout layer to the deep neural network. This layer results in an improved performance of Gene Expression Classification (GEC).
The CNN and RNN are the default DL architectures that are mainly used in recognition tasks and DNA classification [21–23,33]. Collobert et al. [34] have firstly shown that CNNs can be used effectively for sequence analysis, in the case of a generic text. Fig. 3 demonstrates the structure of a simple CNN. The network begins with an input layer. Then, an initial layer of convolutional filters is used, followed by a nonlinearity, and a pooling layer. The network ends with a fully-associated layer and a softmax layer to forecast set labels. With the introduction of convolutional layers, the complexity of learning increases. Hence, we adopt a pooling method or an RP method [16]. These methods reduce the number of parameters. Therefore, the speed of the algorithm is increased. Recently, the CNNs have given effective training on DNA sequences without using feature extraction [35,36]. The RP and wavelet-domain pooling can be used as subsampling layers, for reducing the original CNN feature high dimension. Johnson et al. [17] provided evidence that the RP has distance preserving properties in reducing dimensions, so that the loss of information is well controlled. In addition, wavelet pooling contains a subsampling stage in its structure, while giving more valuable features [18].
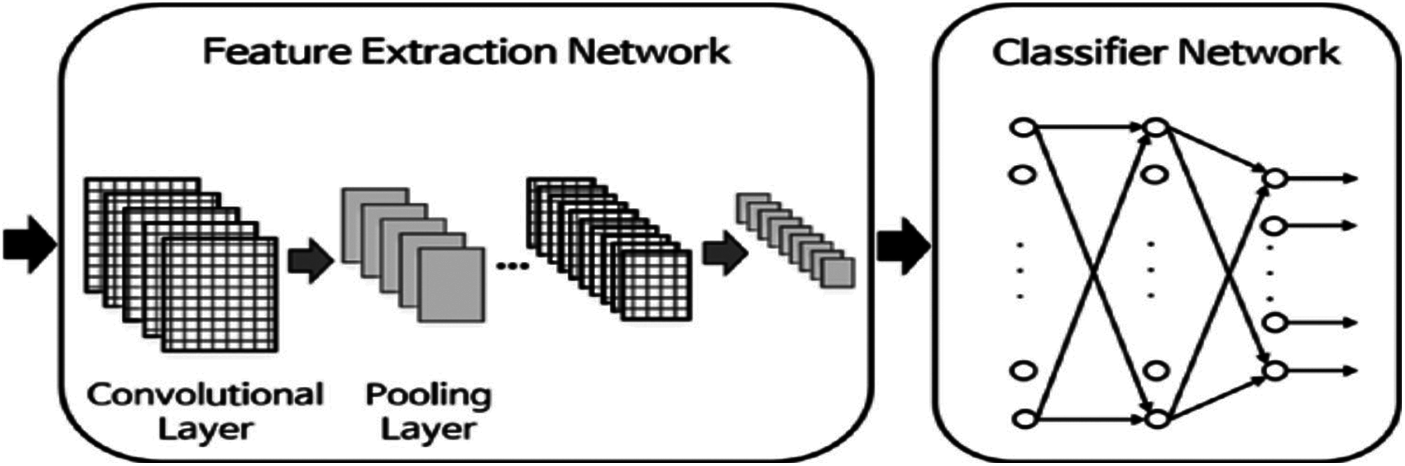
Figure 3: Typical architecture of a CNN
Recurrence networks process the input data one by one, one at a time, and store information about the history of all previous states in their hidden layers. The simplified version of an RNN has an internal status ht, which is a summary of the sequence seen before at (t−1), and is used in conjunction with the new input xt as follows [23]:
where
In recent years, the RNN has been used to classify DNA sequences without providing a priori information (feature extraction) [23], where the authors used character embedding after mapping of the DNA sequence by one-hot coding. In [39], the authors combined the histogram of oriented gradient for feature extraction with an RNN used as a classifier in scene text recognition. The CNN has a powerful feature representation ability compared to the hand-crafted features in the recognition task. The authors of [40] used the CNN features with an RNN classifier in scene text word image recognition.
The Wavelet Transform (WT) is presented as a subsampling layer in the proposed hybrid module. The basic idea of the WT is to select a certain sub-band after implementing the transformation [41]. The wavelet transform can be implemented and a certain sub-band can be used to represent the DNA sequence, especially the low-frequency sub-band. This process achieves the data reduction, while most of the signal energy is kept.
Datasets were obtained from the Ribosomal Database Project (RDP) repository [42], Release 11. Two different sequences were used for comparison: (a) full-length sequences with a length of approximately 1200–1500 nucleotides and (b) 500 bp DNA sequence fragments. The complete set of data includes sequences of the 16SrRNA gene of bacteria belonging to 3 different phylum, 5 different classes, 19 different orders, 65 different families, and 100 different genus.
The DNA databases have been mapped using one-hot coding or FCGR. Since more data with learned features usually result in the best performance, effort should be spent on cleaning and normalizing data. The mapped sequences are converted to a number of feature maps using Histogram of Oriented Gradient (HOGs) or features extracted from CNN model. Then wavelet pooling layer or RP is used as subsampling layer. Finally, the RNN with BLSTM is trained and compared with the CNN based on RP for choosing the best performance. The performance is evaluated by different metrics such as Accuracy, Precision, Recall, and F1 score. They can be defined as follows [43]:
where TP represents true positives, FP represents false positives, TN represents true negatives, and FN represents false negatives.
The DNA datasets were mapped using FCGR with k-mers equal to 6. The mapped sequences are converted to feature maps extracted from trained multi-layer CNNs. Then, 2D DWT or 2D RP is used as a down-sampling layer. Finally, the RNN with BLSTM is trained. The block diagram of the proposed model is depicted in Fig. 4. This model consists of five layers, whose input is in the form of FCGR images. The first four layers are composed of two convolutional layers, each followed by a down-sampling layer (RP or DWT). These convolutional layers use filters of size 5 × 5, to give feature maps that are converted to sequences. These sequences are fed to the BLSTM with 100 hidden layers (recurrent layer). The architecture of the hybrid model is shown in Fig. 4b. Besides, the HOG features of the BLSTM network have the same structure of the previous CNN features based on RNN with BLSTM network except at the first layer, where it consists of feature maps extracted from HOGs followed by max-pooling layer.

Figure 4: The proposed module. (a) CNN based on RP model, (b) Architecture of the hybrid model
Simulation experiments have been carried out to evaluate the encoded bacterial DNA sequence classification based on different approaches for achieving high performance. The DNA sequences have been encoded using the FCGR algorithm or by one-hot coding. The parameters used in the simulation are the k-mers of the FCGR algorithm equal to 6. A batch size of 128 training samples is employed to depict the performance of the hybrid model. Five classification models have been adopted as follows:
a) Model 1: Classification of mapped DNA sequences using a classical CNN and an RP layer (sub-sampling layer).
b) Model 2: Classification of feature maps extracted from HOGs using RNN with BLSTM.
c) Model 3: Classification of feature maps extracted with CNN followed by max-pooling using RNN with BLSTM.
d) Model 4: Classification of feature maps extracted from CNN followed by wavelet pooling using RNN with BLSTM.
e) Model 5: Classification of feature maps extracted from CNN followed by RP using RNN with BLSTM.
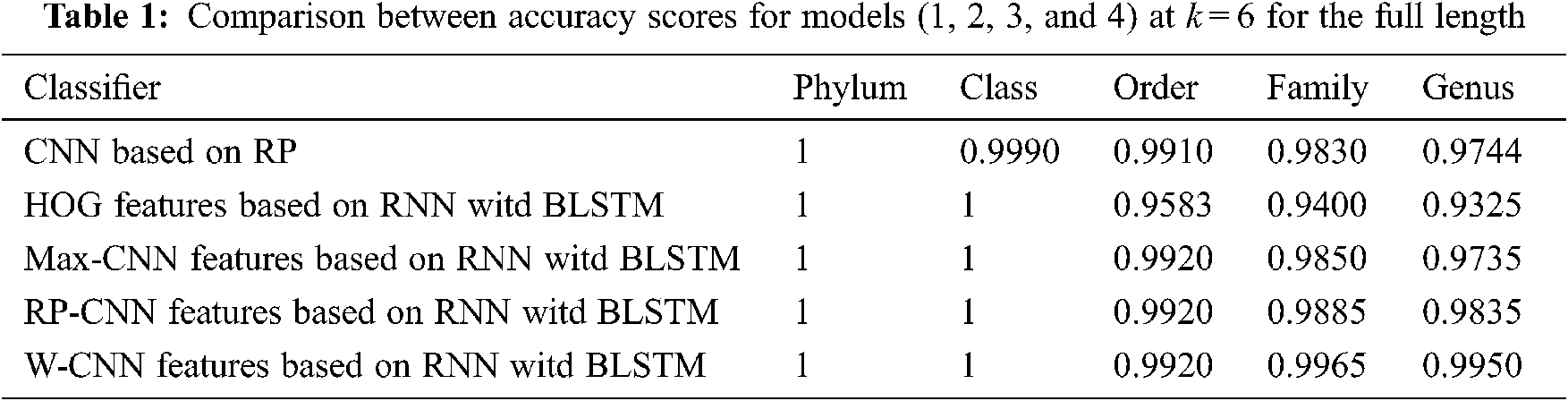
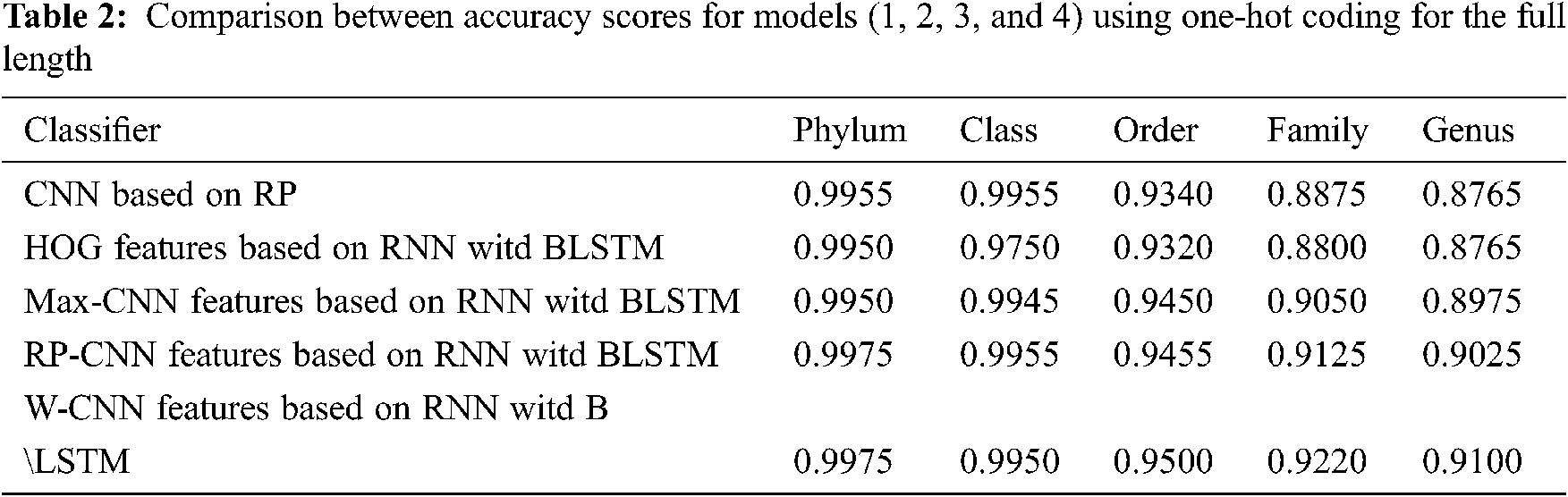
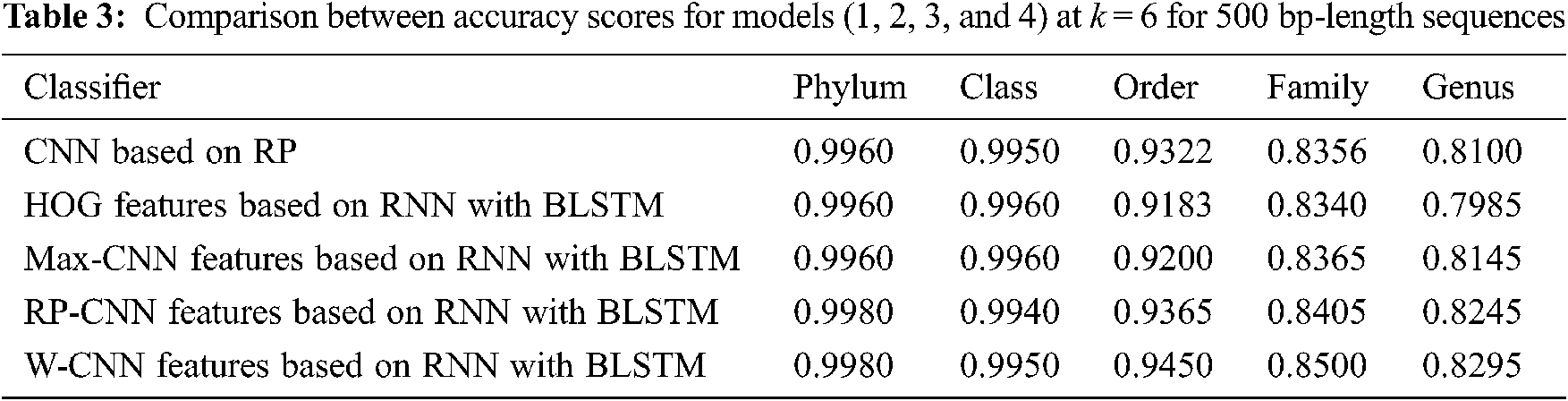
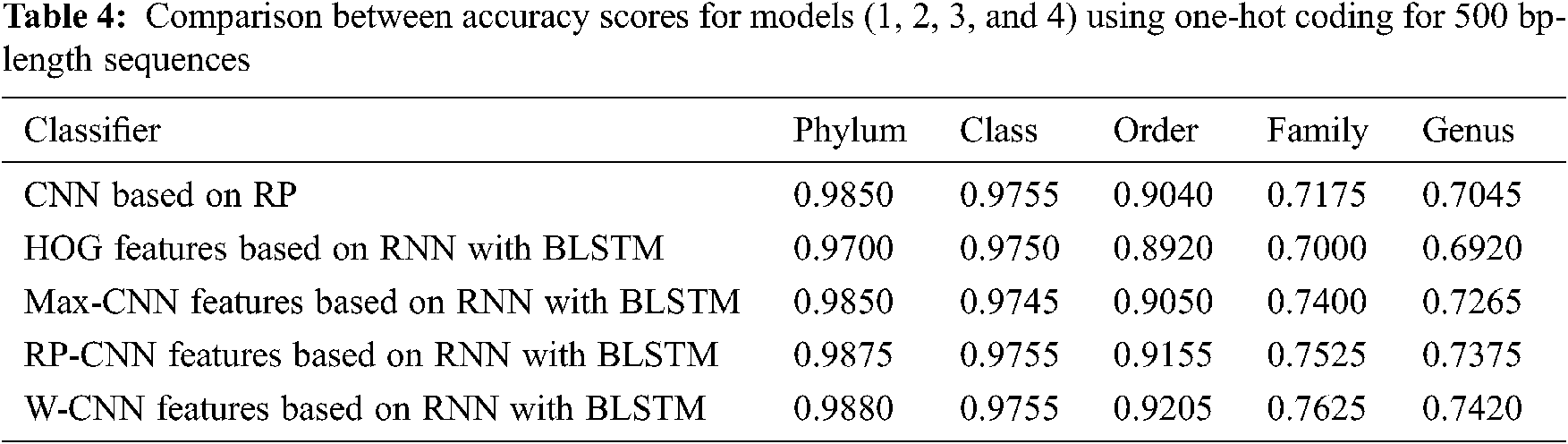
The proposed models have been trained using 70% of the input data and tested using the remaining 30%. A comparison of the accuracy performance among the five models is demonstrated in Tabs. 1–4. The resultant full-length DNA sequence is specified in Tabs. 1 and 2. Tabs. 3 and 4 are obtained according to 500 bp-length sequences. Figs. 5 and 6 show a comparison of the F1 score performance among the five models. According to the previous results, the W-CNN features of BLSTM (model 4) have the best accuracy among all the other models, especially on the genus and family levels. Additionally, the FCGR mapping is more suitable for encoding. Nevertheless, the proposed classical CNN based on RP consumes less running time.
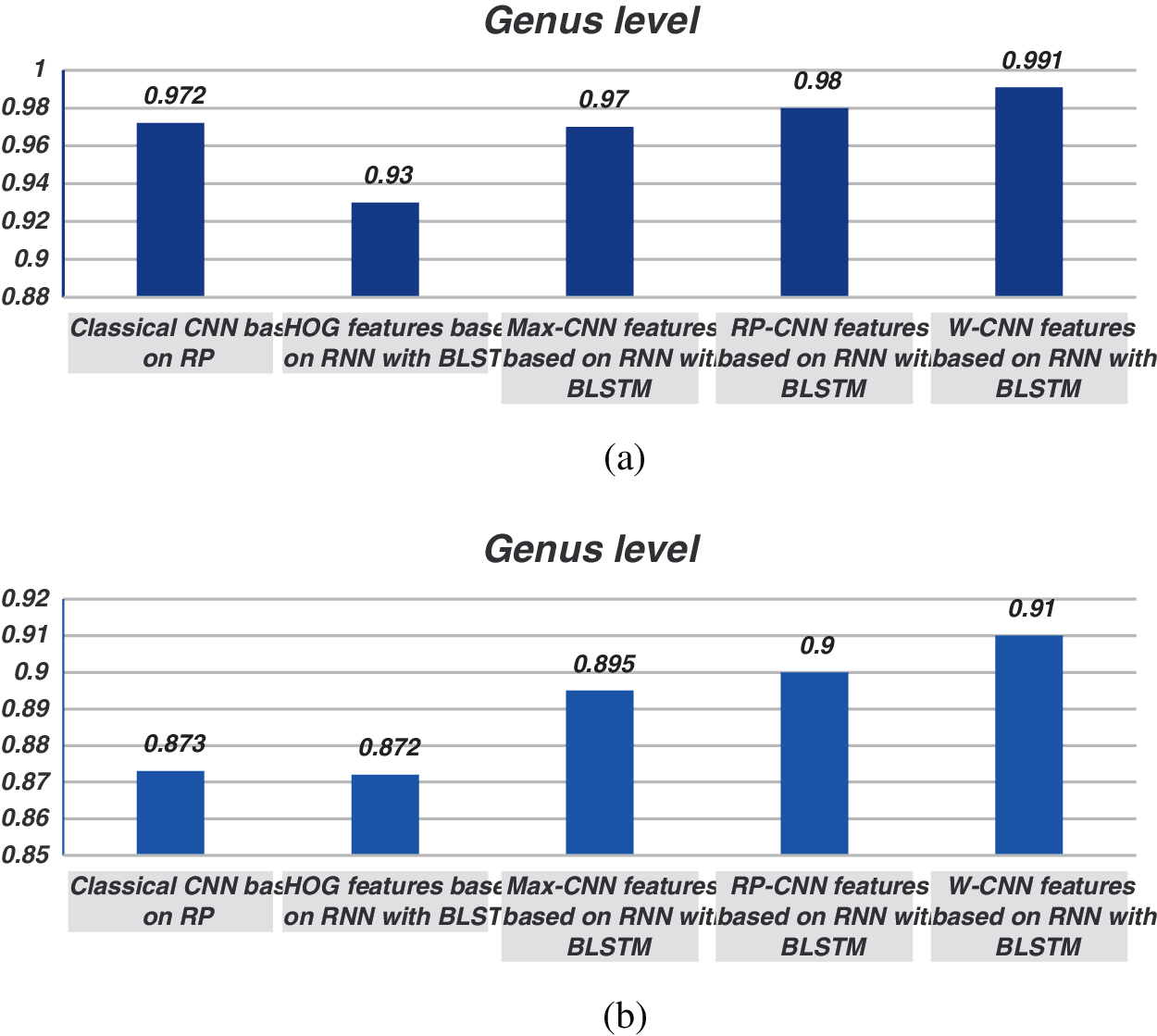
Figure 5: Comparison between F1 scores for models (1, 2, 3, and 4) at the genus level for the full length. (a) At k = 6, (b) Using one-hot coding 6
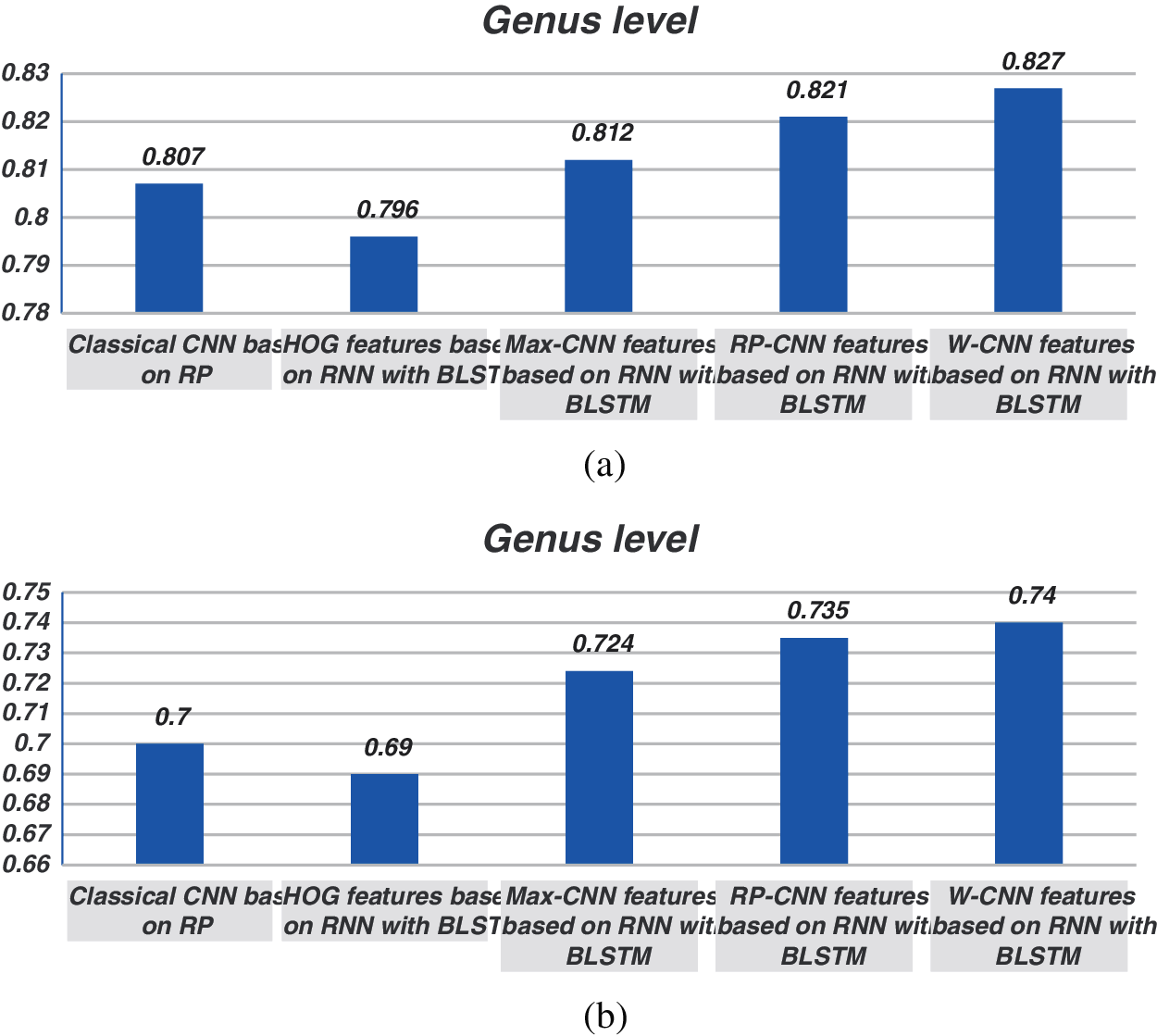
Figure 6: Comparison between F1 scores for models (1, 2, 3, and 4) at the genus level for 500 bp-length sequences. (a) At k = 6, (b) Using one-hot coding
A hybrid approach has been proposed for bacterial classification to achieve taxonomic-rank improvement with efficient encoding. This approach consists of multi-layer CNN followed by wavelet transform that is used to give the input to the BLSTM classifier. Multi-layer CNN is used for extracting features due to its powerful representation ability compared with that of the hand-crafted features. Wavelet transform is supposed to reduce the dimensionality problem associated with the multi-layer CNN and add more features to it. According to the obtained results, the accuracy and F1score in the hybrid module are the best compared to those of other models, but it has a longer processing time compared with other models. Besides, the FCGR images are more suitable than other mappings.
Acknowledgement: The authors would like to thank the support of the Deanship of Scientific Research at Princess Nourah bint Abdulrahman University.
Funding Statement: This research was funded by the Deanship of Scientific Research at Princess Nourah Bint Abdulrahman University through the Fast-track Research Funding Program.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
1. B. Alberts, “Molecular Biology of the Cell,” 4th ed, Chapter 4. DNA and Chromosomes, New York: Garland Science, 2002. [Google Scholar]
2. D. Moore, “The Developing Genome: An Introduction to Behavioral Epigenetics,” United Kingdom: Oxford University Press, 2015. [Google Scholar]
3. B. Tropp and D. Freifelder, “Molecular Biology: Genes to Proteins,” Chapter 4 Nucleic Acid Structure, 3rd ed, Sudbury, Mass: Jones and Bartlett Publishers, 2008. [Google Scholar]
4. H. Tettelin, D. Riley, C. Cattuto and D. Medini, “Comparative genomics: The bacterial pan-genome,” Current Opinion in Microbiology, vol. 11, no. 5, pp. 472–477, 2008. [Google Scholar]
5. Homology Concepts, [Online]. Available: http://en.wikipedia.org/wiki/homology_(biology), last access on 11-07-2020. [Google Scholar]
6. S. Altschul, W. Gish, W. Miller, E. W. Myers and D. J. Lipman, “Basic local alignment search tool,” J. Mol. Biol, vol. 215, no. 3, pp. 403–410, 1990. [Google Scholar]
7. D. Lipman and W. Pearson, “Rapid and sensitive protein similarity searches,” Science, vol. 227, no. 4693, pp. 1435–1441, 1985. [Google Scholar]
8. G. Bosco and L. Pinello, “A new feature selection methodology for k-mers representation of DNA sequences,” CIBB, LNCS, Springer, Heidelberg, vol. 8623, no. 4, pp. 99–108, 2015. [Google Scholar]
9. G. Bosco, “Alignment free dissimilarities for nucleosome classification,” CIBB, LNCS, Springer, Heidelberg, vol. 9874, no. 7, pp. 114–128, 2016. [Google Scholar]
10. S. Fernando and S. Perera, “Empirical analysis of data mining techniques for social network,” COMPUSOFT, An International Journal of Advanced Computer Technology, vol. 3, no. 2, pp. 201–223, 2014. [Google Scholar]
11. Y. LeCun, Y. Bengio and G. Hinton, “Deep learning,” Nature, vol. 521, no. 2, pp. 436–444, 2015. [Google Scholar]
12. K. Shea and R. Nash, “An introduction to convolutional neural networks,” ArXiv Preprint ArXiv:1511.08458, vol. 4, pp. 1–13, 2015. [Google Scholar]
13. J. Hochreiter, “Long short-term memory,” Neural Computation, vol. 9, no. 8, pp. 1735–1780, 1997. [Google Scholar]
14. A. Graves, A. Mohamed and G. Hinton, “Speech recognition with deep recurrent neural networks,” in Proc. IEEE Int. Conf. on Acoustics, Speech and Signal Processing, Vancouver, BC, Canada, pp. 6645–6649, 2013. [Google Scholar]
15. T. Mikolov, M. Karaat, L. Burget, J. Cernocky and S. Khudanpur, “Recurrent neural network-based language model,” InInterspeech, vol. 2, no. 6, pp. 1045–1048, 2010. [Google Scholar]
16. R. Wu, S. Yang, D. Leng, Z. Luo and Y. Wang, “Random projected convolutional feature for scene text recognition,” in Proc. 15th IEEE Int. Conf. on Frontiers in Handwriting Recognition, Shenzhen, China, pp. 132–137, 2016. [Google Scholar]
17. W. Johnson and J. Lindenstrauss, “Extensions of lipchitz mapping into hilbert space,” in Proc. Conf. in Modern Analysis and Probability, Amer. Math. Soc., of Contemporary Mathematics, Jerusalem, Israel, pp. 189–206, 1984. [Google Scholar]
18. W. El-Shafai, F. Khallaf, E. El-Rabaie and F. Abd El-Samie, “Robust medical image encryption based on DNA-chaos cryptosystem for secure telemedicine and healthcare applications,” Journal of Ambient Intelligence and Humanized Computing, vol. 3, no. 2, pp. 1–29, 2021. [Google Scholar]
19. G. Sakakibara, “Convolutional neural networks for classification of alignments of non-coding RNA sequences,” Bioinformatics, vol. 34, no. 13, pp. i237–i244, 2018. [Google Scholar]
20. Y. Wang, K. Hill, S. Singh and L. Kari, “The spectrum of genomic signatures: From dinucleotides to chaos game representation,” Gene, vol. 346, no. 2, pp. 173–185, 2005. [Google Scholar]
21. R. Rizzo, A. Fiannaca, M. Rosa and A. Urso, “Classification experiments of DNA sequences by using a deep neural network and chaos game representation,” in Proc. IEEE Int. Conf. on Computer Systems and Technologies CompSysTech’16, Palermo, Italy, pp. 222–228, 2016. [Google Scholar]
22. C. Angermueller, T. Pärnamaa, L. Parts and O. Stegle1, “Deep learning for computational biology,” Molecular Systems Biology, vol. 12, no. 7, pp. 207–211, 2016. [Google Scholar]
23. G. Bosco and M. Gangi, “Deep learning architectures for DNA sequence classification,” in Proc. 11th Int. Workshop of Fuzzy Logic and Soft Computing Applications, Naples, Italy, pp. 162–171, 2017. [Google Scholar]
24. R. Damasevicius, “Analysis of binary feature mapping rules for promoter recognition in imbalanced DNA sequence datasets using support vector machine, intelligent systems,” in Proc. 4th Int. IEEE Conf. Intelligent Systems, Varna, Bulgaria, pp. 11–25, 2008. [Google Scholar]
25. R. Ranawana and V. Palade, “A neural network based multi-classifier system for gene identification in DNA sequences,” Neural Computing & Applications, vol. 14, no. 2, pp. 122–131, 2005. [Google Scholar]
26. S. Arniker, H. Kwan, N. Law and D. Lun, “Promoter prediction using DNA numerical representation and neural network,” in Proc. IEEE Annual Case Study with Three Organisms, India Conf. (INDICONHyderabad, India, pp. 1–4, 2011. [Google Scholar]
27. H. Jeffrey, “Chaos game visualization of sequences,” Computers & Graphics, vol. 16, no. 1, pp. 25–33, 1990. [Google Scholar]
28. I. Messaoudi, A. Oueslati and Z. Lachiri, “Building specific signals from frequency chaos game and revealing periodicities using a smoothed Fourier analysis,” IEEE/ACM Trans Comput Biol Bioinform, vol. 11, no. 5, pp. 863–77, 2014. [Google Scholar]
29. J. Almeida, J. Carrico, A. Noble and M. Fletcher, “Analysis of genomic sequences by chaos game representationl,” Bioinformatics, vol. 17, no. 3, pp. 429–437, 2001. [Google Scholar]
30. A. Zielezinski, S. Vinga, J. Almeida and W. Karlowski, “Alignment-free sequence comparison: Benefits, applications, and tools,” Genome Biol, vol. 18, no. 1, pp. 1–19, 2017. [Google Scholar]
31. C. Angermueller, T. Pärnamaa, L. Parts and O. Stegle1, “Deep learning for computational biology,” Molecular Systems Biology, vol. 12, no. 7, pp. 107–115, 2016. [Google Scholar]
32. O. Ahmed and A. Brifcani, “Gene expression classification based on deep learning,” in Proc. 4th IEEE Scientific Int. Conf. Najaf (SICN),” Al-Najef, Iraq, pp. 145–149, 2019. [Google Scholar]
33. B. Alipanahi, A. Delong, M. Weirauch and B. Frey, “Predicting the sequence specificities of DNA-and RNA-binding proteins by deep learning,” NatBiotechnol, vol. 33, no. 2, pp. 831–838, 2015. [Google Scholar]
34. R. Collobert, J. Weston, L. Bottou, M. Karlen and P. Kuksa, “Natural language processing (Almost) from scratch,” Journal of Machine Learning Research, vol. 12, no. 3, pp. 2493–2537, 2011. [Google Scholar]
35. J. Zhou and O. Troyanskaya, “Predicting effects of noncoding variants with deep learning-based sequence model,” Nat Methods, vol. 12, no. 4, pp. 931–934, 2015. [Google Scholar]
36. C. Angermueller, H. Lee and W. Reik, “Accurate prediction of single cell DNA methylation states using deep learning,” Genome Biology, vol. 8, no. 1, pp. 1–17, 2016. [Google Scholar]
37. J. Chung, C. Gulcehre, K. Cho and Y. Bengio, “Empirical evaluation of gated recurrent neural networks on sequence modeling,” ArXiv Preprint ArXiv:1412.3555, vol. 7, pp. 1–9, 2014. [Google Scholar]
38. G. Li, X. Du, X. Li L. Zou and G. Zhang, “Prediction of DNA binding proteins using local features and long-term dependencies with primary sequences based on deep learning,” Bioinformatics and Genomics, vol. 4, no. 2, pp. 1–17, pp. 2021. [Google Scholar]
39. S. Su, “Accurate scene text recognition based on recurrent neural network,” in Proc. Asian Conf. on Computer Vision, Singapore, Singapore, pp. 345–48, 2014. [Google Scholar]
40. P. He, W. Huang, Y. Qiao, C. C. Loy and X. Tang, “Reading scene text in deep convolutional sequences,” in Proc. Thirtieth AAAI Conf. on Artificial Intelligence, Arizona, USA, pp. 3501–3508, 2016. [Google Scholar]
41. K. Abdelwahab, S. Abd El-atty, W. El-Shafai, S. El-Rabaie and F. Abd El-Samie, “Efficient SVD-based audio watermarking technique in FRT domain,” Multimedia Tools and Applications, vol. 79, no. 9, pp. 5617–5648, 2020. [Google Scholar]
42. Sequence Analysis Tools, [Online]. Available: Https://rdp.cme.msu.edu, last access on 11-05-2018. [Google Scholar]
43. Y. Jiao and P. Du, “Performance measures in evaluating machine learning based bioinformatics predictors for classifications,” Quantitative Biology, vol. 4, no. 4, pp. 320–330, 2016. [Google Scholar]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |