DOI:10.32604/iasc.2023.027670
| Intelligent Automation & Soft Computing DOI:10.32604/iasc.2023.027670 | |
| Article |
Drug–Target Interaction Prediction Model Using Optimal Recurrent Neural Network
Department of Computer Science and Engineering, Anna University, Chennai, 600025, India
*Corresponding Author: G. Kavipriya. Email: kavipriya112@gmail.com
Received: 23 January 2022; Accepted: 21 March 2022
Abstract: Drug-target interactions prediction (DTIP) remains an important requirement in the field of drug discovery and human medicine. The identification of interaction among the drug compound and target protein plays an essential process in the drug discovery process. It is a lengthier and complex process for predicting the drug target interaction (DTI) utilizing experimental approaches. To resolve these issues, computational intelligence based DTIP techniques were developed to offer an efficient predictive model with low cost. The recently developed deep learning (DL) models can be employed for the design of effective predictive approaches for DTIP. With this motivation, this paper presents a new drug target interaction prediction using optimal recurrent neural network (DTIP-ORNN) technique. The goal of the DTIP-ORNN technique is to predict the DTIs in a semi-supervised way, i.e., inclusion of both labelled and unlabelled instances. Initially, the DTIP-ORNN technique performs data preparation process and also includes class labelling process, where the target interactions from the database are used to determine the final label of the unlabelled instances. Besides, drug-to-drug (D-D) and target-to-target (T-T) interactions are used for the weight initiation of the RNN based bidirectional long short term memory (BiLSTM) model which is then utilized to the prediction of DTIs. Since hyperparameters significantly affect the prediction performance of the BiLSTM technique, the Adam optimizer is used which mainly helps to improve the DTI prediction outcomes. In order to ensure the enhanced predictive outcomes of the DTIP-ORNN technique, a series of simulations are implemented on four benchmark datasets. The comparative result analysis shows the promising performance of the DTIP-ORNN method on the recent approaches.
Keywords: Drug target interaction; deep learning; recurrent neural network; parameter tuning; semi-supervised learning
The evolution of new drugs is a time-consuming and cost-effective method. A per the US Food and Drug Administrations’ (FDA) static data, the costs of novel molecular entity detection are around $1.8 billion and it takes typically thirteen years [1]. Additionally, twenty new molecular entities are permitted by FDA annually. Hence, it is a major challenge in minimizing these costs in drug finding. The computation method provides a powerful tool to solve the problem [2]. With the growth of higher-throughput models, a large amount of drug–target interaction (DTI) information was established [3]. Some data bases have been generated for providing relevant retrieval servers and storing interaction data. As experimental approach for potential DTI remains a challenge, computation prediction method is required to resolve the issue. So far, several silico models were introduced for predicting interactions between their targets and drugs. The computation models are classified into receptor-based method, literature text mining method, and ligand-based method [4].
In recent years, various statistical models were introduced to gather DTI data under the assumption that similar ligand is possible to have interacted with similar protein [5]. The prediction can be performed by incorporating biological data, like drug target protein sequence, compound-protein interaction, and chemical structures. Machine learning (ML), a computer technique for data analysis developed to construct prediction methods utilizing data sets, which become an essential tool of current biological study. It became a conventional model for solving and analyzing difficulties included in drug–target interaction prediction studies. Earlier supervised methods [6] considered the unknown DTI as negative samples that will mainly impact the predictive performance. The major drawback of supervised learning method is that the dataset must be hand-labelled whether by a data scientist or a machine learning engineer.
To address the problem, the concepts of Semi-Supervised Learning have been proposed. In this kind of learning, the model is trained on integration of labelled and unlabelled information. Generally, both combinations have smaller amount of labelled data and larger amount of unlabelled data [7]. The fundamental process included is that firstly, the programmer cluster similar data using an unsupervised learning method and later use the current labelled data for labelling remaining unlabelled data. According to the complex network model, Chen et al. [8] introduced a network-related inference model, NBI, to DTI predictions that used known DTI data. Xia et al. [9] developed a semi-supervised learning model, NetLapRLS, to forecast drug-protein interaction through labelled and unlabelled data. Cheng et al. [10] presented an inference model, NRWRH, by random walk on heterogeneous networks, involving drug-drug similarity networks, known DTI system, and protein-protein similarity network. The common issues of the above-mentioned models are that they cannot be used for the drugs without the knowledge of target data.
This paper introduces a novel drug target interaction prediction using optimal recurrent neural network (DTIP-ORNN) model in a semi-supervised way. The DTIP-ORNN technique involves data preparation and class labelling process, where the target interactions from the database are used to determine the final label of the unlabelled instances. Moreover, drug-to-drug (D-D) and target-to-target (T-T) interactions are used for the weight initiation of the RNN based bidirectional long short term memory (BiLSTM) technique which is then utilized to the prediction of DTIs. Furthermore, the Adam optimizer is used for the hyperparameter tuning of the BiLSTM model. For examining the improved performance of the DTIP-ORNN technique, a wide range of experiments were performed on four benchmark datasets.
The rest of the paper is orgnanized as follows. Section 2 offers related works and Section 3 provides proposed model. Next, Section 4 provides experimental validation and Section 5 concludes the work.
Kumar et al. [11] presented a new approach to predict DDI based similarity of drugs involving distance-based similarity, side effects, chemical similarity, ligand similarity, and so on., using FCN models. Xie et al. [12] modelled the DTI prediction as binary classification method. By employing transcriptome data in the L1000 dataset of LINCS projects, we proposed an architecture-based DL technique to predict drug target interaction. The experiment result shows that this method discovers more reliable DTI when compared to other models. Feng et al. [13] contrived PADME (Protein and Drug Molecule interaction prediction), an architecture based DNN model, to forecast real-time interaction robustness among proteins and compounds without the need of feature engineering. PADME takes protein and compound data as input; hence it is able to solve cold-target (and cold-drug) issues.
Lim et al. [14] presented a DL model to forecast DTI using a graph NN model. Also, present a distance-aware graph attention process for differentiating different kinds of intermolecular interaction. Moreover, extracts the graph feature of molecular interaction straightaway from the three-dimensional data on the protein–ligand binding pose. Therefore, the algorithm learns key features for precise prediction of drug–target interactions instead of memorizing some pattern of ligand molecule. Lee et al. [15] introduced a DL based DTI predictive method to capture local residue pattern of protein participating in DTI. While employing a CNN on raw protein sequence, we performed convolution on different lengths of amino acid subsequence for capturing local residue pattern of generalization protein class. It is trained the model with largescale DTI data and demonstrates the efficiency of the presented technique.
Islam et al. [16] presented DTI-SNNFRA, an architecture to predict DTI, on the basis of fuzzy-rough approximation (FRA) and shared nearest neighbour (SNN). It applies sampling models to jointly decrease the searching space covering the available targets, drugs, and millions of interactions amongst others. Ye et al. [17] developed a DTI predictive method called AdvB-DTI. Regarding this, the feature of DTI expression profiles is related to Adversarial Bayesian Personalized Ranking via matrix factorization. First, based on the known drug-target relationship, a collection of ternary partial order relationships was created. At last, the score of drug-target pair can be attained by the inner products of latent factor, as well as the DTI predictions can be implemented on the basis of score ranking.
Peng et al. [18] proposed an ‘end-to-end’ learning-based architecture based heterogeneous ‘graph’ convolution network for predicting ‘DTI’ named end-to-end graph (EEG)-DTI. Assumed a heterogeneous network comprising different kinds of biological entities (that is, protein, drug, side-effect, disease), the presented method learns lower-dimension feature representations of targets and drugs through a graph convolution network-based method and predicts DTI on the basis of learned features.
In this study, a novel DTIP-ORNN technique has been developed to predict the DTIs in a semi-supervised way, i.e., inclusion of both labelled and unlabelled instances. The DTIP-ORNN technique encompasses a series of subprocesses namely data preparation, class labelling, D-D and T-T based weight initialization, BiLSTM based prediction, and Adam optimizer based hyperparameter tuning. The application of class labelling, weight initiation, and hyperparameter tuning processes helps to considerably improve the DTIP predictive outcomes. Fig. 1 illustrates the working process of DTIP-ORNN technique.
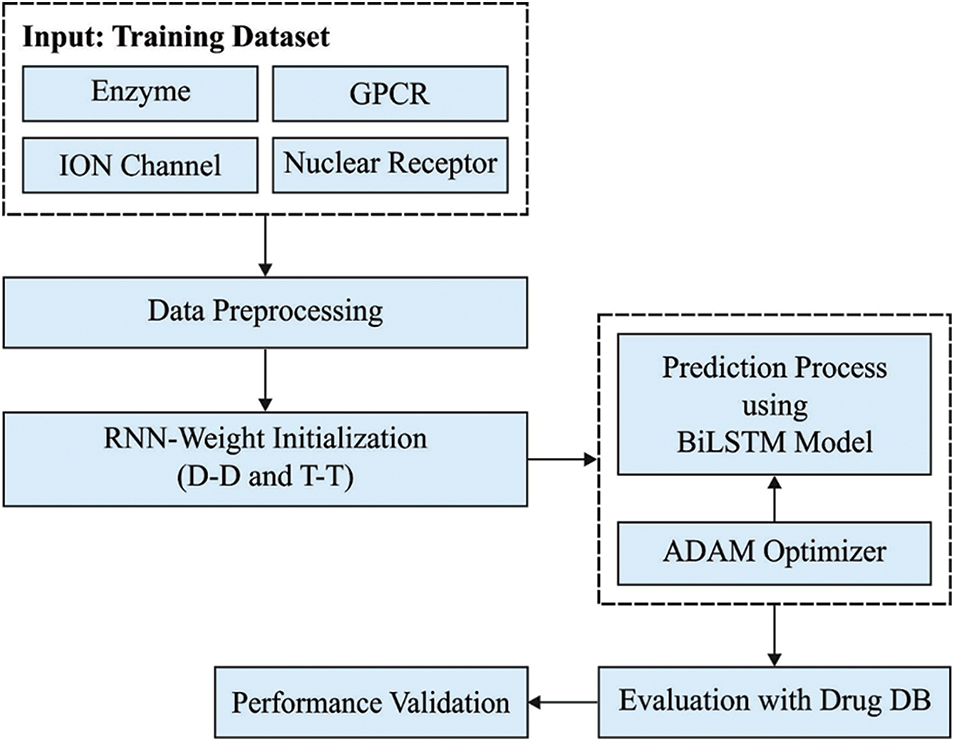
Figure 1: Working process of DTIP-ORNN technique
3.1 Data Preparation and Class Labeling
In this study, we have employed four DTIP datasets namely Enzyme, Ion Channel, GPCR, and Nuclear Receptor, available at http://web.kuicr.kyoto-u.ac.jp/supp/yoshi/drugtarget/. These datasets are gathered in the KEGG BRITE, BRENDA, SuperTarget, and DrugBank. Drug chemical structure data can be obtained in DRUG AND COMPOUND Sections in the KEGG LIGAND [19]. The chemical formation resemblance among the components can be determined by SIMCOMP that offers a scoring value depending upon the size of usual substructures with graph alignments. The sequence similarity among the target can be determined using the normalized Smith–Waterman technique, depending upon the data of amino acid order of target protein derived from KEGG GENE database. Consider a pair of proteins Ai and Aj, the sequence similarity among them can be determined using Eq. (1):
where SW(Ai, Aj) refers the score of Smith–Waterman technique.
Then, the DTIP-ORNN technique undergoes class labelling process where the target interactions that exist in the database are used to label the unknown instances.
3.2 Design of RNN Based BiLSTM Model for Predictive Process
RNN is a neural network developed to examine the data stream interms of hidden layers. In several application areas as text processing, speech detection, and DNA sequence, the final outcome is mainly based on the earlier computation [20]. As the RNN has the ability to deal with sequential data, it finds helpful for biomedical informatics domain where massive quantities of sequential data are available for processing [21].
The simple RNN is designed by altering the simple network to hold latent data from time step to time step. Initially, the single examination case is clearly defined, which relates to sequence of length 1:
Then, the linear transformation is utilized in the hidden state processing, which is based on the input x and also previous information which is held by the hidden state. The earlier hidden state is considered as 0 that can be comprehended by holding over no details related to the past:
In the modeling preview, the RNN is almost identical to the classical NN. However, it varies not only in the notations and operation counts, it can be extended to various instances.
This RNN method can able to process sequence of length, e.g., length of T = 1 and T = 7, since the transition function and parameter is shared oνer time.
The network training is performed i.e., almost equal to the procedure defined above for feedforward network. Now, all the patients correspond to sequences of examination, x1, …, xT, and label γT. They formed computational graph by unrolling the RNN over time step, and by adding operations to calculate the possibility of loss and malignancy. Next, attains gradient through backpropagation, and enhances through stochastic gradient descent.
where the hidden layer is neglected, and frequently consider as h0 = 0. Fig. 2 illustrates the framework of RNN.
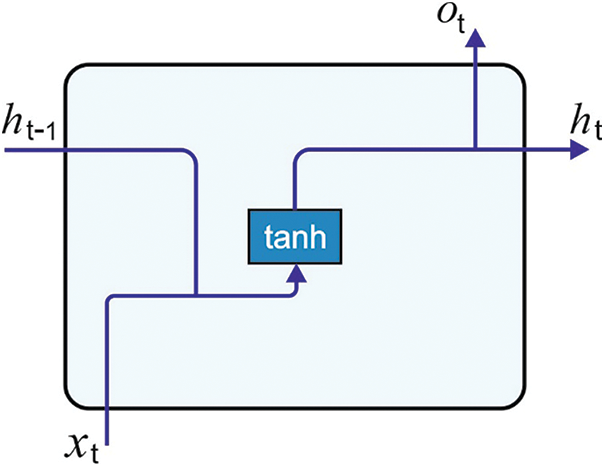
Figure 2: RNN structure
At this stage, the D-D and T-T interactions are used to initialize the weights of the RNN based BiLSTM model which helps to improve the predictive efficiency. LSTM [22] is a different version of RNN. The LSTM is particularly supposed to avoid the long-term dependency problem. The LSTM has many gates like output gate, input gate, forget gate, and constant memory cell. In the LSTM based sequential method, it, ft, and ot signify the input, forget, and output gates correspondingly in Eqs. (13)–(15).
When the input order
This point
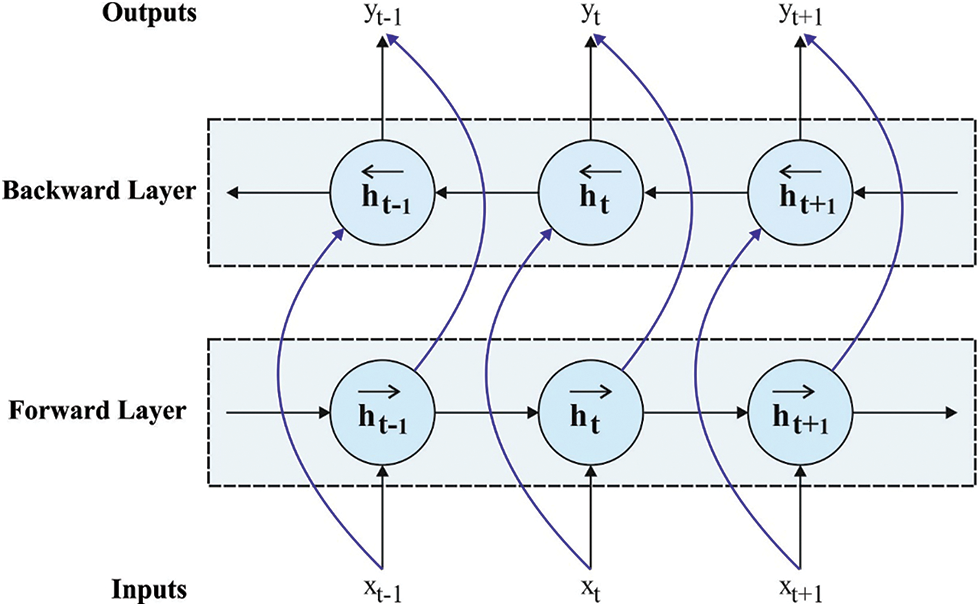
Figure 3: BiLSTM structure
For optimally tuning the hyperparameters of the BiLSTM model, the Adam optimizer is applied. Adam is another widely employed technique which alters the learning rate adoptively for all the parameters. Adam is an integration of distinct gradient optimization models. It is exponentially decaying average of previous squared gradient calculated, such as RMSprop and Adadelta, as well as Adam take an exponentially decaying average of previous gradient that is analogous to Momentum.
In which β1 and β2 represent the decay rate that is recommended to follow the default value. Mt and Gt represent the mean and uncentered variance of historical gradient, correspondingly.
Since the decaying rate generally causes some bias problems, it is needed to perform the bias-correction work.
Hence, the updated values of Adam are determined by the following equation [23]:
The gradient part of
It is found that each operation is based on historical gradient of the existing parameter that has no relationship to learning rate. Therefore, Adam has an effective performance with the help of learning rate method.
The performance validation of the DTIP-ORNN technique takes place using four datasets, available at http://web.kuicr.kyoto-u.ac.jp/supp/yoshi/drugtarget/. The first enzyme dataset includes 445 instances into drug, 664 instances into target, and 2926 instances into interactions. The second Ion channel dataset comprises 210 instances into drug, 204 instances into target and 1467 instances into interactions. The third GPCR dataset contains 223 instances into drug, 95 instances into target and 635 instances into interactions. The final Nuclear Receptor dataset encompasses 54 instances into drug, 26 instances into target and 90 instances into interactions as shown in Tab. 1 and Fig. 4. All these datasets have three combinations namely D-D, D-T, and T-T. In this study, the D-D and D-T are used for the weight initiation process of the BiLSTM model. The remaining D-T instances are used for the prediction process.

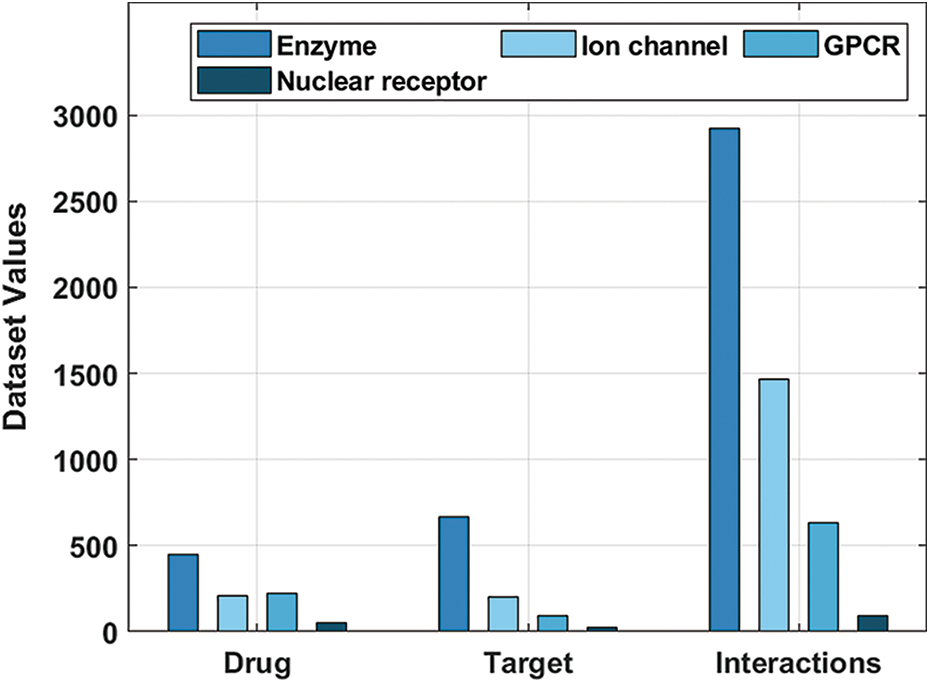
Figure 4: Dataset descriptions
Tab. 2 and Fig. 5 provide a brief result analysis of the proposed DTIP-ORNN technique interms of labeled and unlabeled instances in the test datasets. Besides, the results are reported interms of top k(%) instances. The results show that the DTIP-ORNN technique has effectively labeled the unknown instances based on the interactions that exist in the known instances.

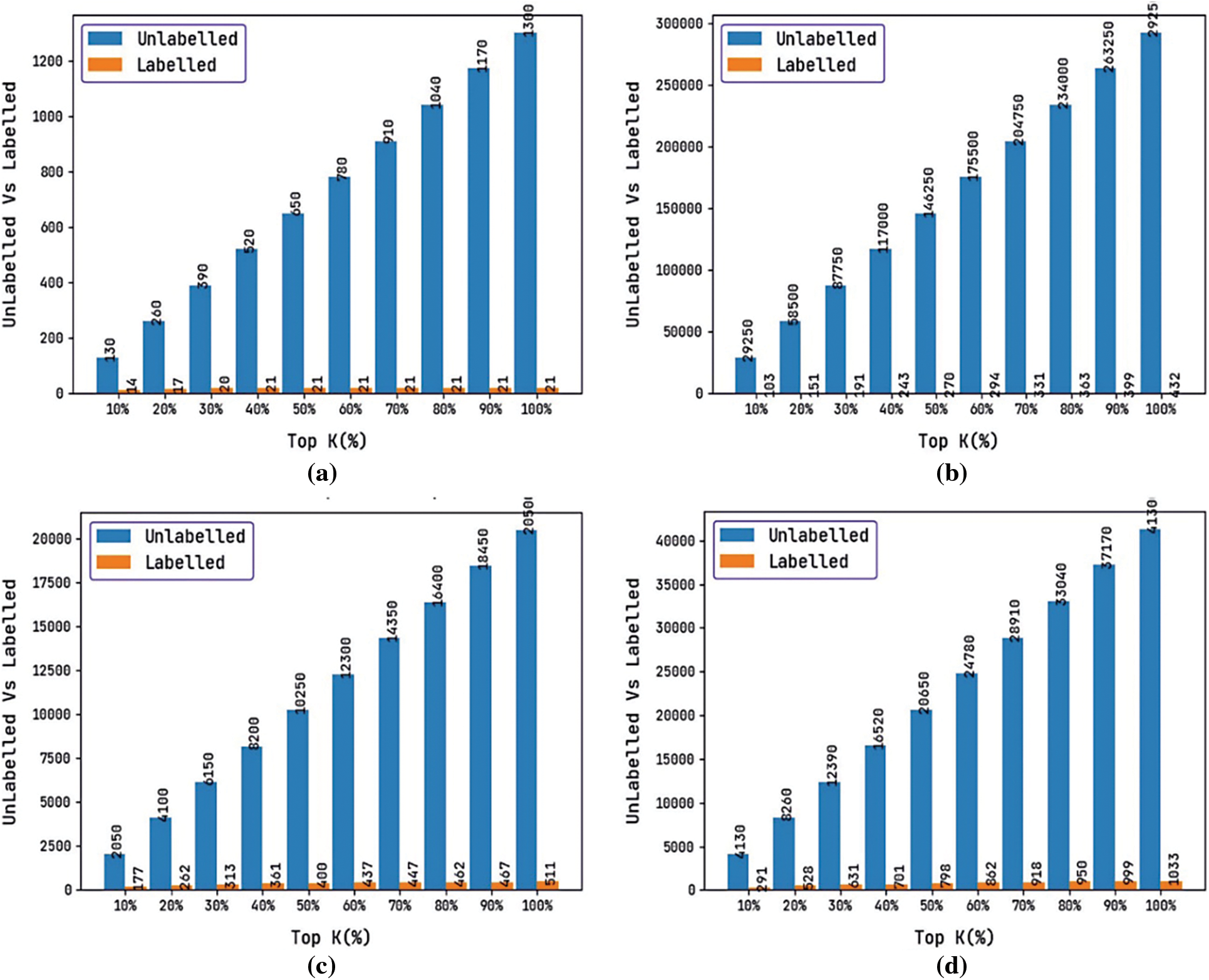
Figure 5: Labeled data analysis of DTIP-ORNN technique
For instance, on the enzyme dataset with top 10% instances, the count of unlabeled instances is 29250 and the count of labeled instances is 103. In addition, on the GPCR dataset with top 10% instances, the count of unlabeled instances is 2050 and the count of labeled instances is 177. Similarly, on the ION Channel dataset with top 10% instances, the count of unlabeled instances is 4130 and the count of labeled instances is 291. Lastly, on the nuclear receptor dataset with top 10% instances, the count of unlabeled instances is 130 and the count of labeled instances is 14.
Tab. 3 and Fig. 6 reports the performance validation of the DTIP-ORNN technique under distinct SEED values on the applied datasets. The experimental values indicated that the DTIP-ORNN technique has obtained effective predictive outcomes with the maximum AUC and AUPR values on the test datasets. For instance, on the enzyme dataset with CV_SEED value of 3201, the DTIP-ORNN technique has obtained higher AUC and AUPR values of 91.81% and 53.25% correspondingly. Meanwhile, on the GPCR dataset with CV_SEED value of 3201, the DTIP-ORNN technique has attained increased AUC and AUPR values of 85.20% and 61.50% correspondingly. Eventually, on the ION Channel with CV_SEED value of 3201, the DTIP-ORNN technique has resulted in improved AUC and AUPR values of 81.57% and 64.95% respectively. Lastly, on the Nuclear Receptor dataset with CV_SEED value of 3201, the DTIP-ORNN technique has offered higher AUC and AUPR values of 89.84% and 73.21% respectively.
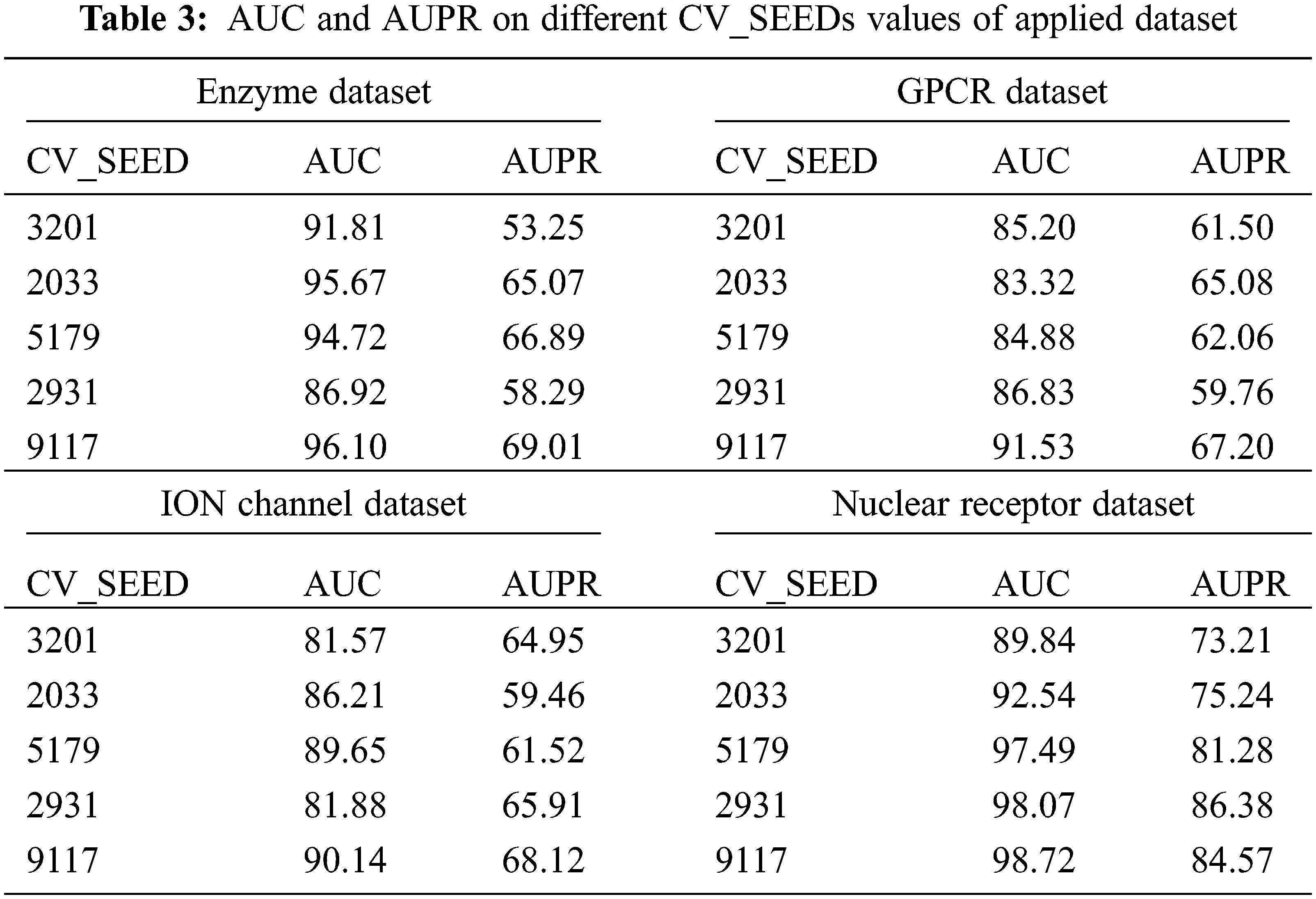
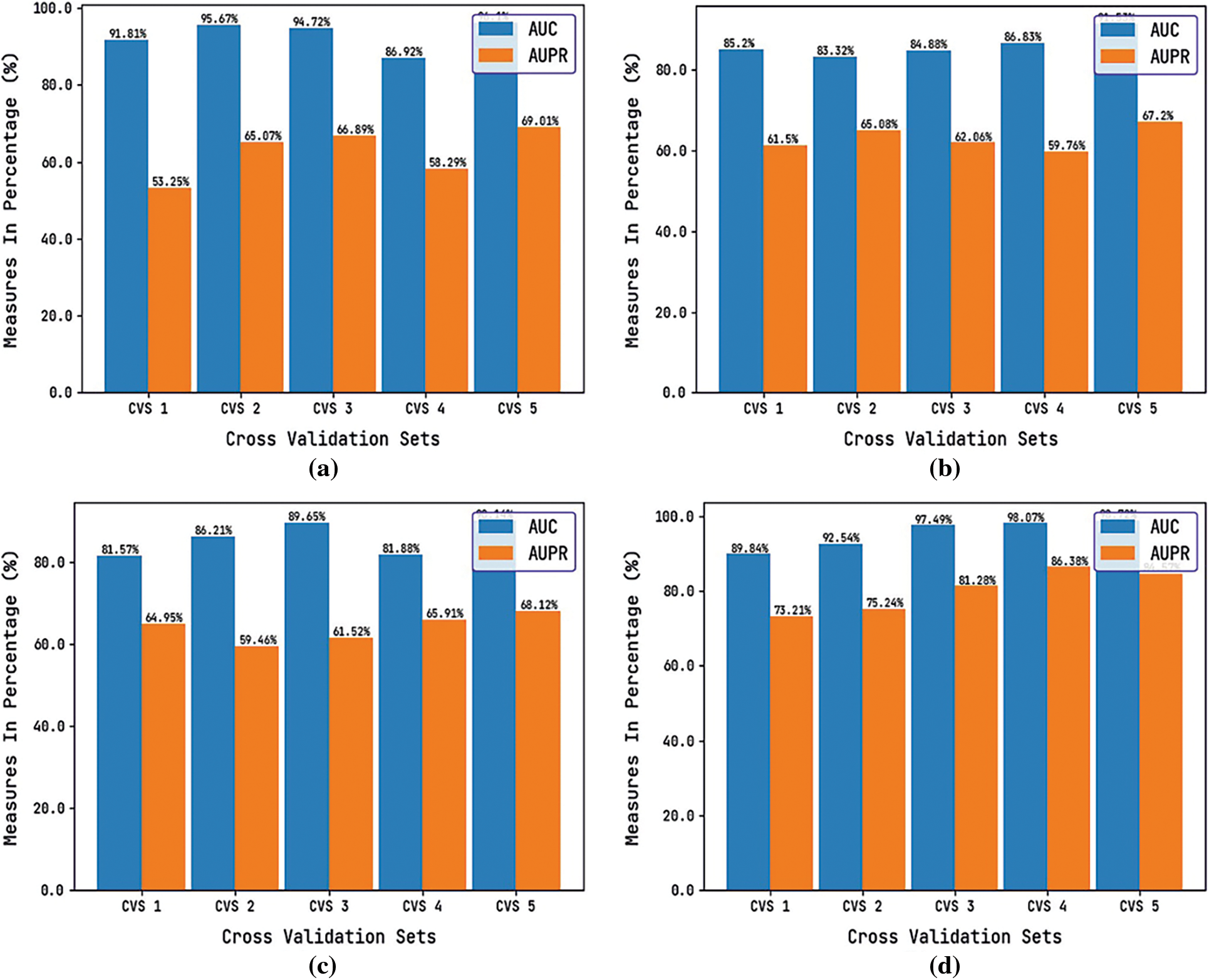
Figure 6: AUC and AUPR analysis of DTIP-ORNN technique on different CV_SEEDs value
Tab. 4 provides a comparative AUC analysis of the DTIP-ORNN technique with recent approaches on four datasets. Fig. 7 inspects the AUC analysis of the DTIP-ORNN technique on the Enzyme and GPCR datasets. The figure reported that the DBSI technique has offered lower AUC values of 80.6% and 80.3% on the test Enzyme and GPCR datasets respectively. Followed by, the KBMF2K, IFB Model, UDTPP, and Nearest Neighbour techniques have resulted in moderately closer values of AUC. Though the Bi-gram PSSM model has reached near optimal AUC of 94.80% and 88.90%, the presented DTIP-ORNN technique has accomplished higher AUC values of 96.10% and 91.53% respectively.
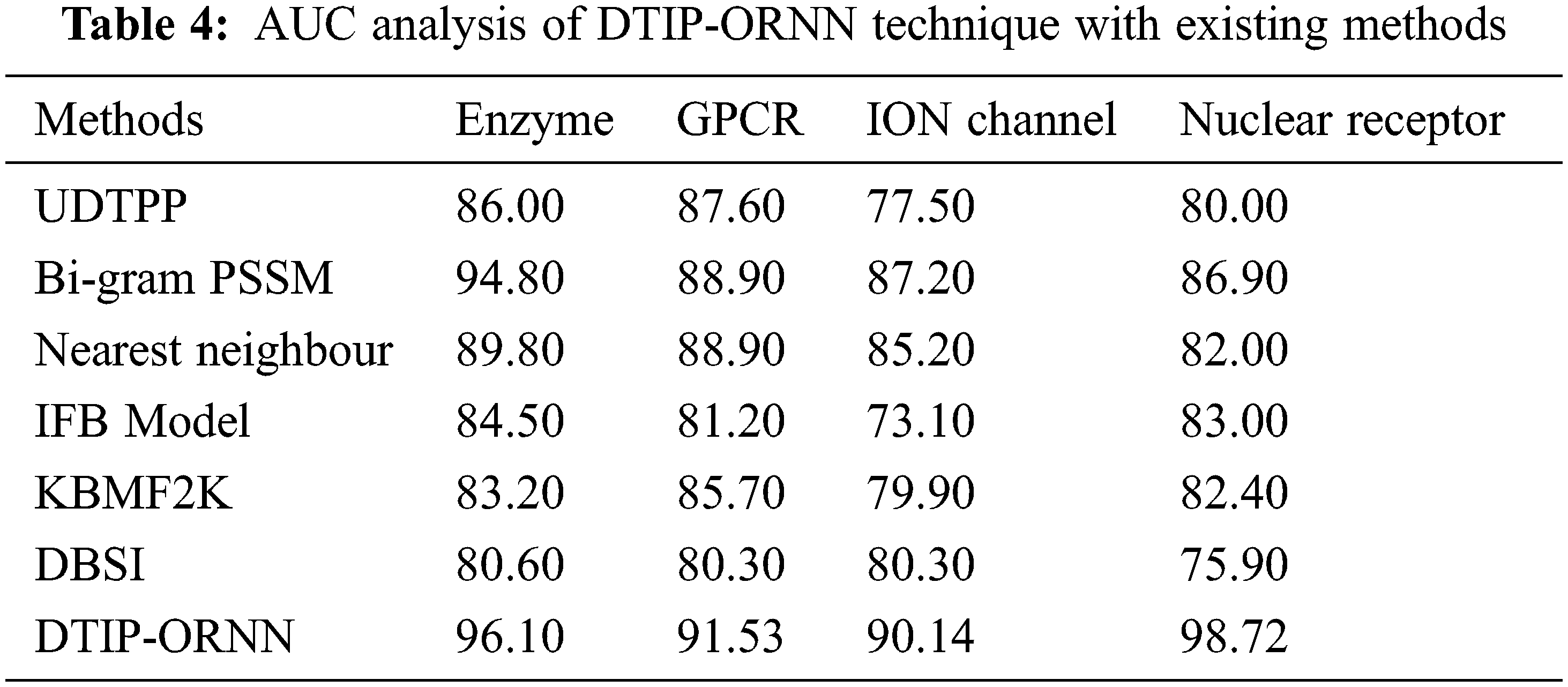
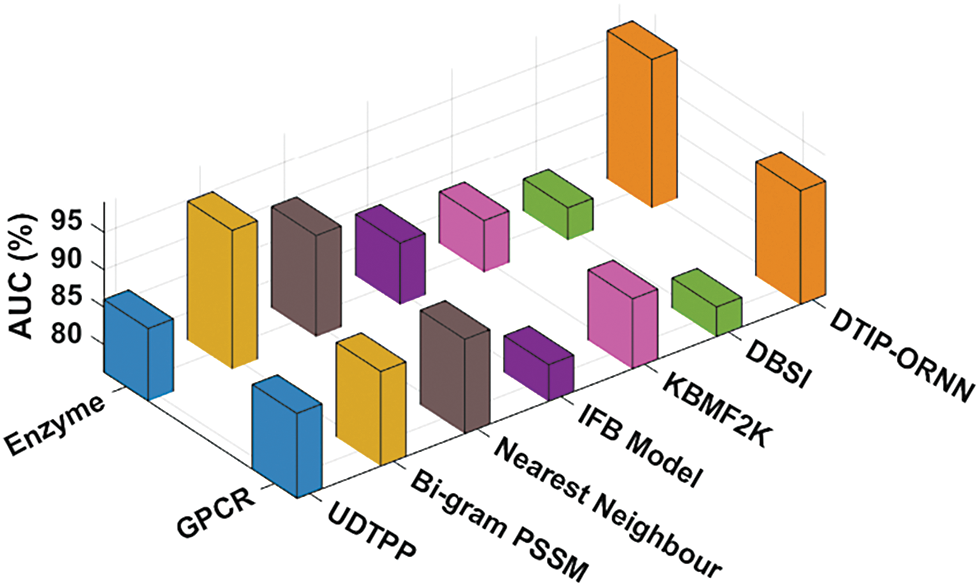
Figure 7: AUC analysis of DTIP-ORNN technique on enzyme and GPCR datasets
Fig. 8 demonstrates the AUC analysis of the DTIP-ORNN approach on the ION channel and nuclear receptor datasets. The figure reported that the DBSI methodology has accessible minimal AUC values of 80.30% and 75.90% on the test ION channel and nuclear receptor datasets correspondingly. Then, the KBMF2K, IFB Model, UDTPP, and Nearest Neighbour methodologies have resulted in moderately closer values of AUC. However, the Bi-gram PSSM system has reached to near optimal AUC of 87.20% and 86.90%, the projected DTIP-ORNN approach has accomplished maximum AUC values of 90.14% and 98.72% correspondingly.
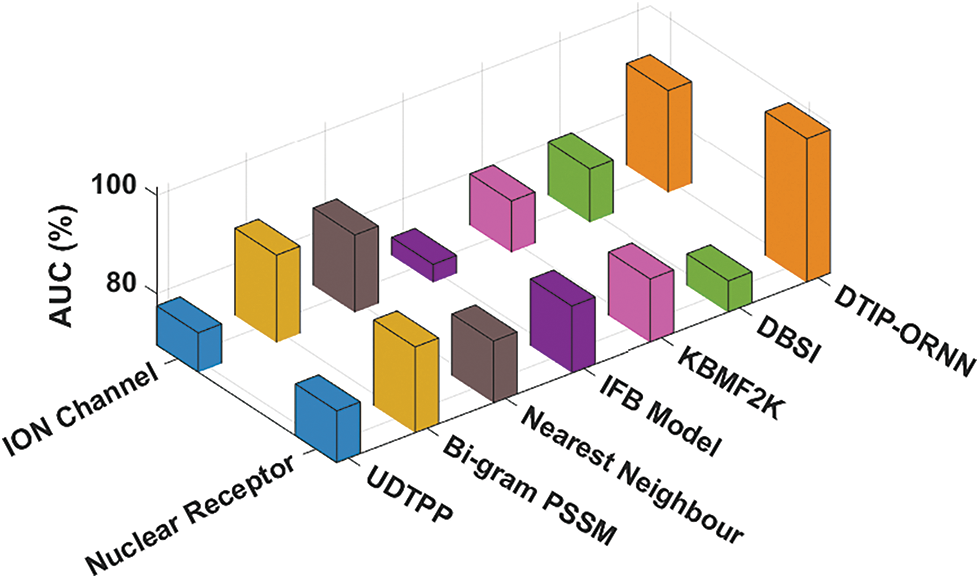
Figure 8: AUC analysis of DTIP-ORNN technique on ION channel and nuclear receptor datasets
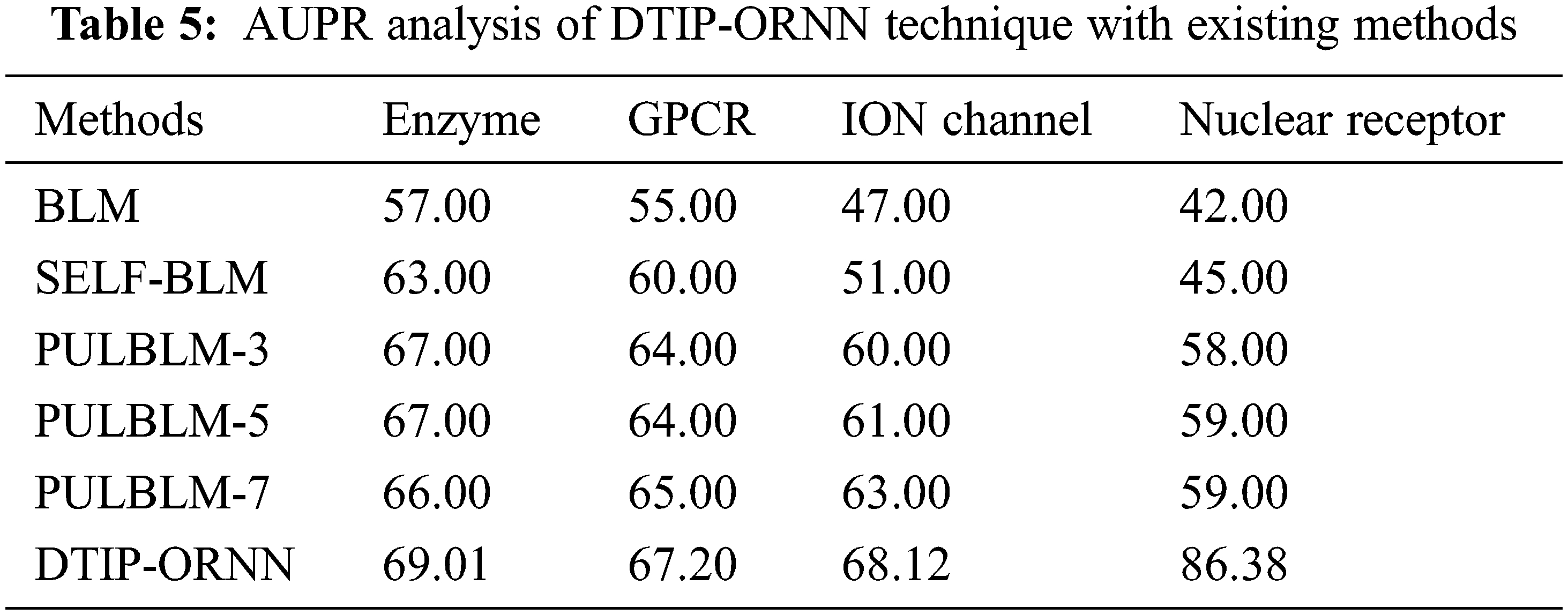
Tab. 5 offers a comparative AUPR analysis of the DTIP-ORNN approach with existing methods on four datasets. The results stated that the BLM technique has offered lower AUPR values of 57% and 55% on the test Enzyme and GPCR datasets correspondingly. Afterward, the SELF-BLM, PULBLM-7, and PULBLM-3 approaches have resulted in moderately closer values of AUPR. Then, the PULBLM-5 algorithm has reached near optimal AUPR of 67% and 64%, the presented DTIP-ORNN methodology has accomplished maximum AUPR values of 69.01% and 67.20% correspondingly. The table values indicated that the BLM system has offered lesser AUPR values of 47% and 42% on the test ION channel and nuclear receptor datasets correspondingly. Similarly, the SELF-BLM, PULBLM-7, and PULBLM-3 techniques have resulted in moderately closer values of AUPR. Though the PULBLM-5 technique has achieved near optimal AUPR of 61% and 59%, the projected DTIP-ORNN methodology has accomplished superior AUPR values of 68.12% and 86.38% correspondingly.
In order to further ensure the predictive outcomes of the DTIP-ORNN technique, a validation process of the predicted interactions takes place with four databases namely chembl (C), Drug bank (D), Kegg (K), and Metador (M). The interaction confidence obtained by the DTIP-ORNN technique with the matching databases is listed out in Tab. 6 [24,25].
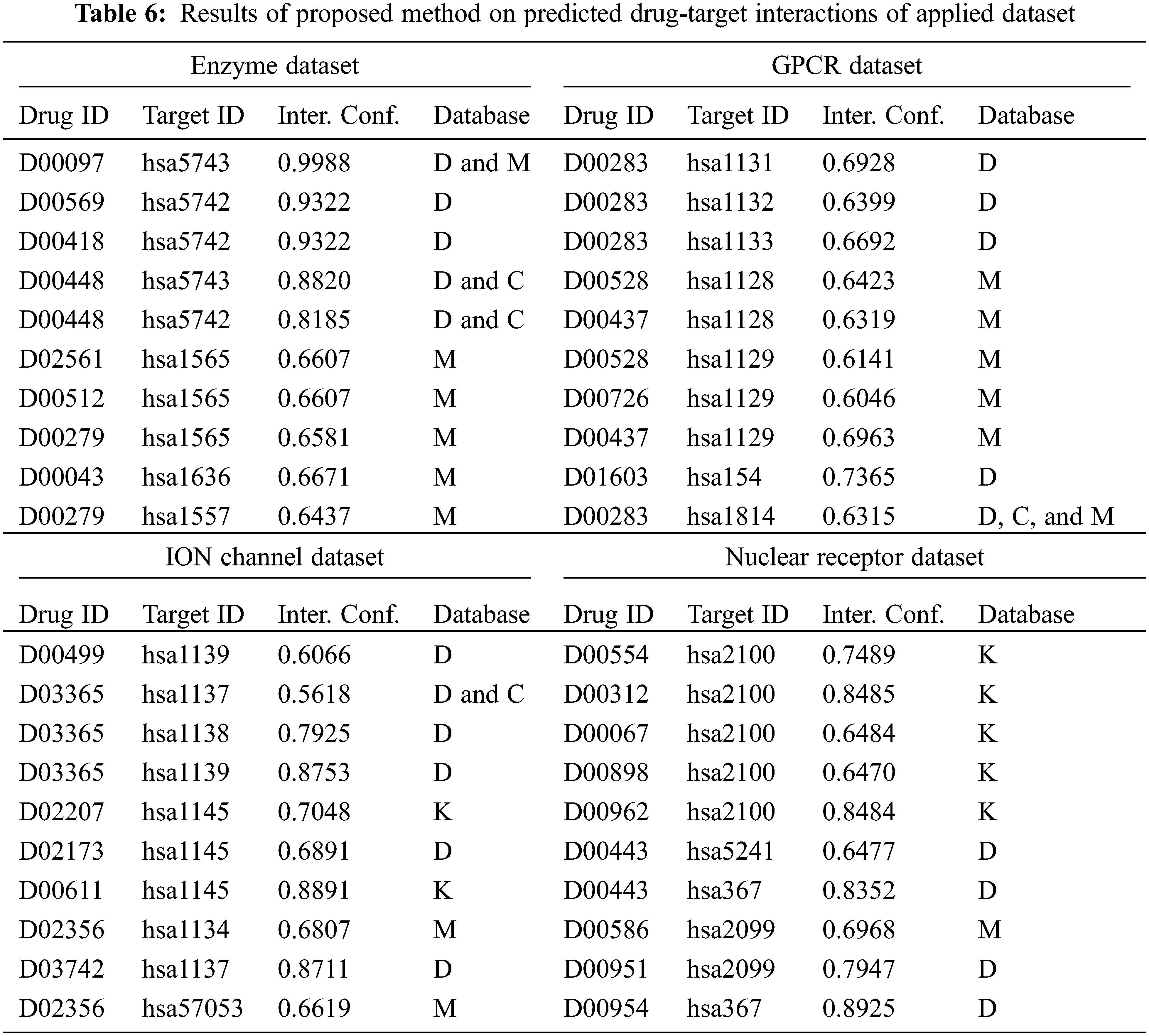
The drug id, target id, predicted interaction confidence, and matching database details are provided. On the enzyme dataset, the interaction confidence of 0.9988 has been obtained for the drug id D00097 and target id hsa5743, which is matched with the D and M database. By looking into the above mentioned tables and figures, it is ensured that the DTIP-ORNN technique has accomplished effective predictive outcomes over the other approaches.
This paper has developed a novel DTIP-ORNN method for predicting the DTIs in a semi-supervised way, i.e., inclusion of both labelled and unlabelled instances. The DTIP-ORNN technique encompasses a series of subprocesses namely data preparation, class labelling, D-D and T-T based weight initialization, BiLSTM based prediction, and Adam optimizer based hyperparameter tuning. The application of class labelling, weight initiation, and hyperparameter tuning processes helps to considerably improve the DTIP predictive outcomes. For examining the improved performance of the DTIP-ORNN technique, a wide range of experiments are performed on four benchmark datasets. The comparative result analysis shows the promising performance of the DTIP-ORNN technique over the recent approaches. Therefore, the DTIP-ORNN technique can appear as an effective tool for DTIP. In future, fusion of DL models can be designed to boost the predictive outcomes of the DTIP-ORNN technique.
Funding Statement: The authors received no specific funding for this study.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
1. S. S. Sadeghi and M. R. Keyvanpour, “An analytical review of computational drug repurposing,” IEEE/ACM Transactions on Computational Biology and Bioinformatics, vol. 18, no. 2, pp. 472–488, 2019. [Google Scholar]
2. H. Ding, I. Takigawa, H. Mamitsuka and S. Zhu, “Similarity based machine learning methods for predicting drug-target interactions: A brief review,” Briefings in Bioinformatics, vol. 15, no. 5, pp. 734–747, 2013. [Google Scholar]
3. F. Rayhan, S. Ahmed, S. Shatabda, D. M. Farid, Z. Mousavian et al., “iDTI-ESBoost: Identification of drug target interaction using evolutionary and structural features with boosting,” Scientific Reports, vol. 7, no. 1, pp. 1–18, 2017. [Google Scholar]
4. P. Xuan, C. Sun, T. Zhang, Y. Ye, T. Shen et al., “Gradient boosting decision tree-based method for predicting interactions between target genes and drugs,” Frontiers in Genetics, vol. 10, no. 459, pp. 1–11, 2019. [Google Scholar]
5. L. Peng, W. Zhu, B. Liao, Y. Duan, M. Chen et al., “Screening drug-target interactions with positive-unlabeled learning,” Scientific Reports, vol. 7, no. 1, pp. 1–17, 2017. [Google Scholar]
6. A. Ezzat, M. Wu, X. Li and C. K. Kwoh, “Computational prediction of drug-target interactions using chemogenomic approaches: An empirical survey,” Briefings in Bioinformatics, vol. 20, no. 8, pp. 1337–1357, 2018. [Google Scholar]
7. Y. Luo, X. Zhao, J. Zhou, J. Yang, Y. Zhang et al., “A network integration approach for drug-target interaction prediction and computational drug repositioning from heterogeneous information,” Nature Communications, vol. 8, pp. 1–13, 2017. [Google Scholar]
8. X. Chen, M. -X. Liu and G. -Y. Yan, “Drug–target interaction prediction by random walk on the heterogeneous network,” Molecular Biosystems, vol. 8, no. 7, pp. 1970–1978, 2012. [Google Scholar]
9. Z. Xia, L. -Y. Wu, X. Zhou and S. T. Wong, “Semi-supervised drug-protein interaction prediction from heterogeneous biological spaces,” BMC Systems Biology, vol. 4, no. S2, pp. 1–16, 2010. [Google Scholar]
10. F. Cheng, C. Liu, J. Jiang, W. Lu, W. Li et al., “Prediction of drug-target interactions and drug repositioning via network-based inference,” PLoS Computational Biology, vol. 8, no. 5, pp. 1–12, 2012. [Google Scholar]
11. A. Kumar and M. Sharma, “Drug-drug interaction prediction based on drug similarity matrix using a fully connected neural network,” in Proc. of Second Doctoral Symp. on Computational Intelligence, Singapore, pp. 911–919, 2022. [Google Scholar]
12. L. Xie, S. He, X. Song, X. Bo and Z. Zhang, “Deep learning-based transcriptome data classification for drug-target interaction prediction,” BMC Genomics, vol. 19, no. S7, pp. 667, 2018. [Google Scholar]
13. Q. Feng, E. Dueva, A. Cherkasov and M. Ester, “Padme: A deep learning-based framework for drug-target interaction prediction,” arXiv Preprint arXiv:1807.09741, vol. 2018, pp. 1–29, 2018. [Google Scholar]
14. J. Lim, S. Ryu, K. Park, Y. J. Choe, J. Ham et al., “Predicting drug–Target interaction using a novel graph neural network with 3D structure-embedded graph representation,” Journal of Chemical Information and Modeling, vol. 59, no. 9, pp. 3981–3988, 2019. [Google Scholar]
15. I. Lee, J. Keum and H. Nam, “DeepConv-DTI: Prediction of drug-target interactions via deep learning with convolution on protein sequences,” PLoS Computation Biology, vol. 15, no. 6, pp. e1007129, 2019. [Google Scholar]
16. S. M. Islam, S. M. M. Hossain and S. Ray, “DTI-SNNFRA: Drug-target interaction prediction by shared nearest neighbors and fuzzy-rough approximation,” PLoS ONE, vol. 16, no. 2, pp. e0246920, 2021. [Google Scholar]
17. Y. Ye, Y. Wen, Z. Zhang, S. He and X. Bo, “Drug-target interaction prediction based on adversarial Bayesian personalized ranking,” BioMed Research International, vol. 2021, pp. 1–16, 2021. [Google Scholar]
18. J. Peng, Y. Wang, J. Guan, J. Li, R. Han et al., “An end-to-end heterogeneous graph representation learning-based framework for drug–target interaction prediction,” Briefings in Bioinformatics, vol. 22, no. 5, pp. 1–9, 2021. [Google Scholar]
19. M. Kanehisa, “From genomics to chemical genomics: New developments in KEGG,” Nucleic Acids Research, vol. 34, no. 90001, pp. D354–D357, 2006. [Google Scholar]
20. Y. LeCun, Y. Bengio and G. Hinton, “Deep learning,” Nature, vol. 521, no. 7553, pp. 436–444, 2015. [Google Scholar]
21. S. Khan and T. Yairi, “A review on the application of deep learning in system health management,” Mechanical Systems and Signal Processing, vol. 107, pp. 241–265, 2018. [Google Scholar]
22. S. Siami-Namini, N. Tavakoli and A. S. Namin, “The performance of LSTM and BiLSTM in forecasting time series,” in 2019 IEEE Int. Conf. on Big Data (Big Data), Los Angeles, CA, USA, pp. 3285–3292, 2019. [Google Scholar]
23. Z. Zhang, “Improved adam optimizer for deep neural networks,” in 2018 IEEE/ACM 26th Int. Symp. on Quality of Service (IWQoS), Banff, AB, Canada, pp. 1–2, 2018. [Google Scholar]
24. H. R. Rajpura and A. Ngom, “Drug target interaction predictions using PU- leaming under different experimental setting for four formulations namely known drug target pair prediction, drug prediction, target prediction and unknown drug target pair prediction,” in 2018 IEEE Conf. on Computational Intelligence in Bioinformatics and Computational Biology (CIBCB), St. Louis, MO, pp. 1–7, 2018. [Google Scholar]
25. W. Lan, J. Wang, M. Li, J. Liu, Y. Li et al., “Predicting drug–target interaction using positive-unlabeled learning,” Neurocomputing, vol. 206, pp. 50–57, 2016. [Google Scholar]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |