| Journal of Renewable Materials |
DOI: 10.32604/jrm.2022.019071
ARTICLE
Effects of Porous Graphene on LiOH Based Composite Materials for Low Temperature Thermochemical Heat Storage
1Thermal and Environmental Engineering Institute, School of Mechanical Engineering, Tongji University, Shanghai, 200092, China
2Guangzhou Institute of Energy Conversion, Chinese Academy of Sciences, Guangzhou, 510640, China
3Department of Chemical Engineering, Graduate School of Engineering, Nagoya University, Nagoya, 464-8603, Japan
4National Institute of Clean-and-Low-Carbon Energy, Beijing, 102200, China
*Corresponding Authors: Hongyu Huang. Email: huanghy@ms.giec.ac.cn; You Zhou. Email: you.zhou.c@chnenergy.com.cn
Received: 01 September 2021; Accepted: 22 December 2021
Abstract: Thermochemical heat storage material inorganic hydrate LiOH is selected as a promising candidate material for storing low-temperature heat energy because of its high energy density (1440 kJ/kg) and mild reaction process. However, the low hydration rate of LiOH limits the performance of low temperature thermochemical heat storage system as well as the thermal conductivity. In this study, porous-graphene/LiOH composite thermochemical heat storage materials with strong water sorption property and higher thermal conductivity were synthesized by hydrothermal process. The experimental results show that the hydration rate of the composites was greatly improved. The heat storage density of the composite materials was increased by 47% (from 661 kJ/kg to 974 kJ/kg). By combing the porous graphene, the thermal conductivity of composites with different contents were highly increased by 21.1% to 78.7%, but the increase of heat storage density is opposite to that of thermal conductivity. The development of high-performance materials for thermochemical heat storage should consider the relationship between the heat storage density and thermal conductivity of the material, and the thermal conductivity of the supporter needs to be further improved.
Keywords: Thermochemical; heat storage; hydration; thermal conductivity; porous graphene
Low-grade heat energy (below 100°C) [1] such as industrial waste heat and solar energy has the characteristics of great potential utilization and wide distribution. However, how to solve the problem of time-space mismatch between supply and demand in low-grade heat energy utilization and further improve energy utilization has become very important [2]. Thermal energy storage technology can be divided into sensible heat [3,4], latent heat [5,6] and thermochemical heat storage (TCS) system [7–9], all of which are potential means to solve the problems by storing the low-grade heat energy [10].
Among the above technologies, TCS is more suitable for efficient utilization of thermal energy due to their high heat storage densities [11–14] and probably no energy loss [15]. Many literatures have described the adsorption process of metal salts with water [16,17], ammonia [18–21], methanol [22,23] or metal alloys [24,25] in order to utilize low-grade heat energy. Because of its high energy density (1440 kJ/kg) and mild reaction process, the inorganic hydrate LiOH has been selected as the promising candidate for storing low-temperature heat energy efficiently. However, the low hydration rate of thermochemical heat storage materials as an important influence factor still limits the performance of low temperature thermochemical heat storage systems [26–28].
In order to improve the hydration rate, hydrophilic inorganic salt as an additive was prepared to composite, which can react with H2O stably because of good water adsorption performance of salt. For example, Posern et al. [28] compounded MgSO4 with MgCl2. Ishitobi et al. [29] compounded Mg(OH)2 with LiCl and explored the mechanism of hydration hydrolysis reaction of the material. The previous studies show that the materials mixing hydrophilic inorganic salts can improve the hydration rate and also reduce the regeneration temperature. However, the inorganic salt materials have no pores, and this modification method cannot solve the problems of swelling and caking of the materials. In addition, Kim et al. [30] compounded CaCl2 with Mg(OH)2 and expanded graphite, effectively improving the hydration reaction rate of the composite, and the porosity of expanded graphite can help the composite solve the selling and caking problem. But the triple compound composite will decrease the content of the reactant. In the case of limited heat transfer enhancement by materials, the optimization of reactor structure is also an effective means to improve the heat storage and release performance of materials [31]. However, the low thermal conductivity leads to the huge volume of the reactor, which is not conducive to its popularization and application.
The preparation and application of porous nano-carbon materials have become one of the advanced researches and hot spots in the field of materials [32,33]. From previous studies [25–27], nano-carbon materials can increase the hydration rate and thermal conductivity of the thermochemical heat storage materials owing to the high thermal conductivity and high surface area of the nano-carbon supporter. Porous graphene denoted by PG is an excellent nano-carbon material, which is composed of graphene and forms a porous structure. Therefore, PG exhibits large surface area, low bulk density, and high thermal conductivity. The particle size of LiOH·H2O is greatly reduced by the high specific surface area and porous structure of PG, so as to reduce the activation energy and diffusion resistance of the material. In this study, PG/LiOH·H2O composite thermochemical heat storage materials with strong water sorption property and higher hydration rate are of great significance to be synthesized. Furthermore, the heat storage performance study on dehydration and hydration transformations of composite materials with different LiOH·H2O content (30%, 47%, 63%) was carried out on a thermogravimetric and differential scanning calorimetric analyzer after 60 min hydration. The hydration behaviors and thermal conductivities of composite were also studied experimentally.
To prepare PG, the precursor graphene oxide was first prepared by a modified Hummers method [34]. The subsequent PG was synthesized by hydrothermal method as follows. The prepared 8 mg/mL graphene oxide solution was placed at 150 mL with a capacity of 200 mL Teflon lined stainless steel autoclave. The hydrothermal temperature and time were set at 180°C and 12 h, respectively. After the end of the reaction, the reactor was naturally cooled to room temperature and the reaction product was taken out for freeze-drying. Thus, the preparation process of PG was completed.
The PG and LiOH·H2O composite was also prepared by hydrothermal method. Firstly, LiOH·H2O and PG supporter in different mass ratios of 30%, 47%, 63% are added to a 150 mL Teflon lined stainless steel autoclave and fully mixed with adding a small amount of deionized water (just immersed sample). Then, the reactor was heated to 105°C for hydrothermal reaction for 12 h, and then the reactor was naturally cooled to room temperature. The composite samples were taken out for freeze-drying. After freeze-drying, put the final sample into a tubular furnace and hydrolyze it at 150°C in argon atmosphere for 3 h. The hydrolyzed product is cooled to 30°C, and the nitrogen flow at 120 mL/min is used as supporter gas to carry water vapor. It is introduced into a tubular furnace for hydration reaction at 30°C The partial pressure of water vapor is 2.97 kPa. After the hydration reaction, the required composite PG/LiOH·H2O chemical heat storage materials with LiOH·H2O content of 30%, 47% and 63% were obtained and denoted as PG/LiOH·H2O-30%, PG/LiOH·H2O-47% and PG/LiOH·H2O-63%.
2.2 The Characterization of Composites
X-ray diffraction (XRD) analysis was performed on PG and composites using an X-ray diffractometer (D8-advance X, Bruker Co., Ltd. (Karlsruhe, Baden-Württemberg, Germany)) with Cu target (40 kV, 40 mA) at a scanning step of 0.0167° and a counting time of 10.160 s. The pore structure of the composite was calculated by nitrogen adsorption isotherms at 77 K and relative pressure of 0.1–1.0 using a gas adsorption apparatus (QDS-30, Quantachrome Instruments Co. Ltd. (Florida, USA)). The weight of material is about 70–100 mg. The material degassing temperature was 180°C for 8 h. Surface topography was measured by scanning electron microscopy (SEM, S-4800, Hitachi Co., Ltd. (Tokyo, Japan)) and trans-mission electron micrographs (TEM, JEM-2100F, JOEL Co., Ltd. (Tokyo, Japan)) with the controlling voltage of 200 kV.
2.3 Hydration Behaviors Experiments
Hydration experiments on the PG/LiOH composites were conducted with an electronic balance analyzer (Ohaus Corp. (Florham Park, NJ), AX124ZH) with a temperature and humidity control system. Firstly, the composite material was dehydrated in the vacuum drying oven at 150°C for 180 min. Then, the samples were taken out to the temperature and humidity control system with a relative humidity of 80% at 30°C. The weight change of the composite material was recorded for 60 min. In this experiment, the LiOH hydration reaction of the composite was carried out simultaneously with the adsorption of water vapor by PG. Therefore, the hydration process of PG/LiOH composite materials is defined as hydration reaction and water adsorption.
2.4 Heat Storage Performance and Thermal Conductivity Evaluation Experiments
The heat storage performance test was conducted as follows. Firstly, the sample is put into a tube furnace to hydrolyze at 150°C for 180 min in argon atmosphere. Then, the dehydrated material is cooled to 30°C in argon atmosphere. And the water vapor partial pressure was set to 2.97 kPa and flowed into the sample tank by flowing nitrogen at 30°C and the hydration reaction maintained for 60 min. After hydration, the heat storage performances of the samples were carried out on a thermogravimetric and differential scanning calorimetric analyzer (TG-DSC, STA-200, Nanjingdazhan Co., Ltd. Nanjing, China). The dehydration TG-DSC curves of the materials were obtained at a heating temperature range of 30°C–200°C and a heating rate of 10 °C/min under nitrogen atmosphere.
The thermal conductivity of the sample was measured by steady heat flux method instrument (DRL-II system, Xiangtan xiangqi instrument Co., Ltd. (Xiangtan, China)). The testing sheet samples with a thickness range of 2–3 mm were pressed by using cylindrical mold with diameter of 30 mm and 20 MPa pressure. The effective thermal conductivities of samples could be obtained by Fourier’s law.
Fig. 1 shows the XRD patterns of PG and 63% content of LiOH·H2O on PG supporter. It can be seen that there are obvious diffraction peaks near 15°, 20°, 30°, 32.19°, 34.84°, 38.83°, 50°, 56° and 60°, all of which are attributed to LiOH·H2O (JCPDS 25-0486). The diffraction peaks at 25° and 43° belong to graphite carbon of PG (JCPDS 24-0619). The peak dispersion diffraction intensity of LiOH·H2O of composite is low, indicating that LiOH·H2O is highly dispersed on the surface of supporter. It also proves that PG/LiOH was successfully synthesized after hydrothermal treatment.
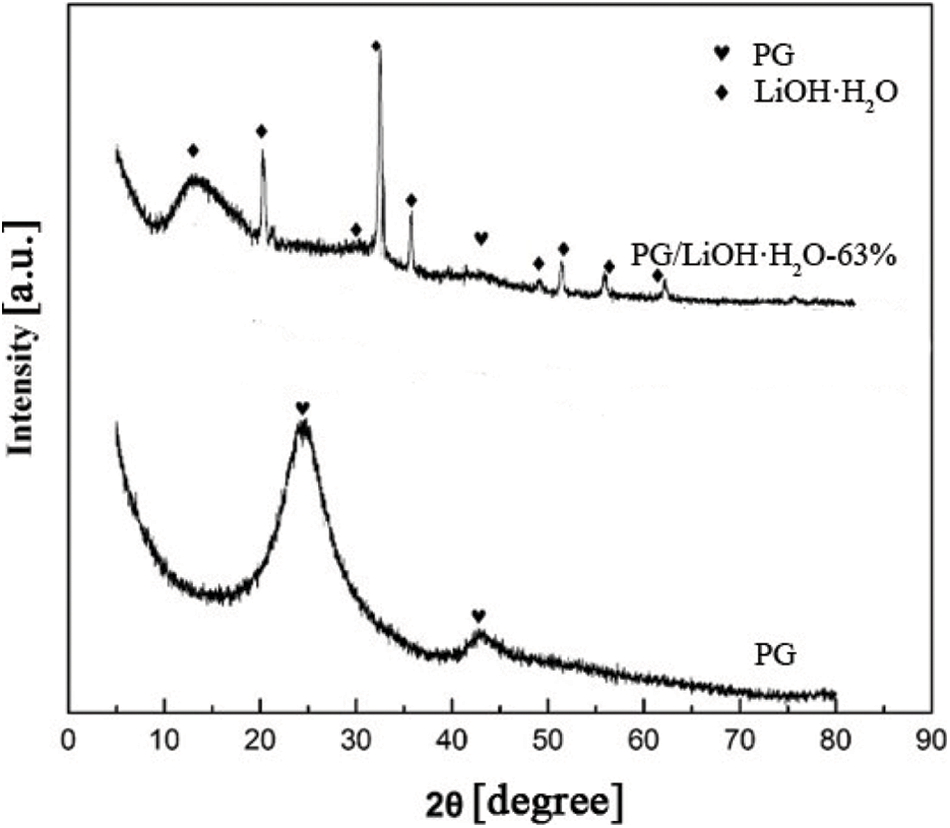
Figure 1: XRD patterns of PG and PG/LiOH·H2O composites
Table 1 shows the pore structure of PG and PG/LiOH composites. The specific surface area of PG is much higher than LiOH. Fig. 2 shows the pore size distribution of composites. It can be seen from figure that all samples have nano channels, and the pore diameter of pure LiOH is mainly concentrated near 45 nm (Fig. 2a). It can be seen from results that the average pore diameter of pure LiOH is near 45 nm, and the composite has nano channels (average pore diameter of 4 nm). The smaller pore diameter size of PG/LiOH changed from 45 to 4 nm, indicating that the pore size of the composite material modified by PG has been redistributed at nanoscale.

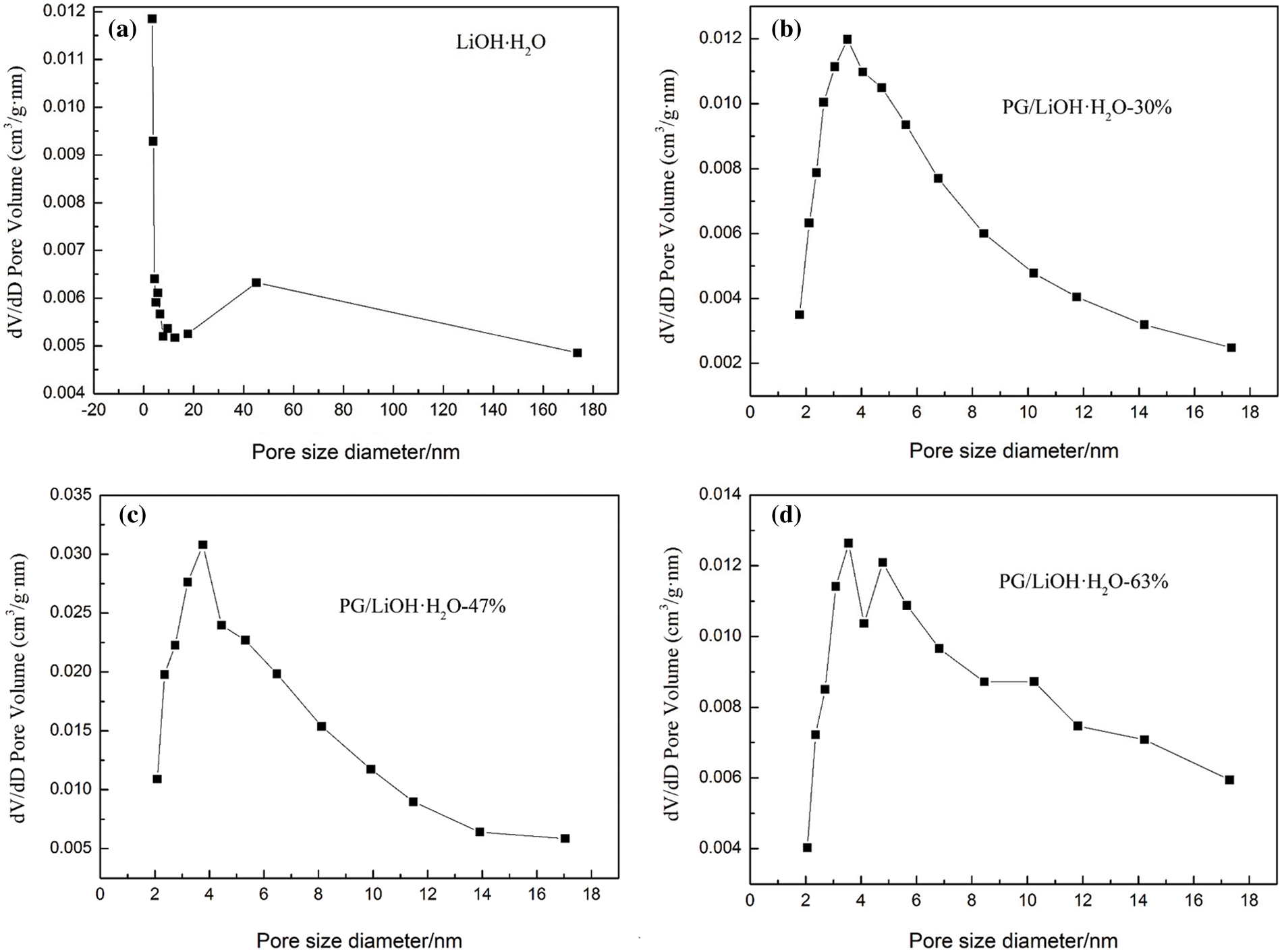
Figure 2: Pore size distribution of (a) pure LiOH·H2O, (b) PG/LiOH·H2O-30% composite, (c) PG/LiOH·H2O-47%, (d) PG/LiOH·H2O-63%. (a) Adapted with permission from reference [35], Copyright© 2021, Wilkey-VCH GmbH
The SEM and TEM images of the composite with different LiOH·H2O content are shown in the Figs. 3 and 4, respectively. As shown in from Figs. 3a–3c, the SEM images of LiOH·H2O were loaded content of 30%, 47% and 63% on the PG, respectively. When the content of LiOH·H2O was less than 30%, the structure of the PG supporter in the composite did not change significantly (Fig. 3a). With the increase of LiOH·H2O content, there are no obvious particles on the surface of the supporter, but a few large particles of LiOH·H2O are difficult to locate on the surface of PG (as shown in Figs. 3b and 3c). The particle size of pure LiOH·H2O ranges from 300 nm to 2 μm without the addition of PG, thus it can be explained laterally that LiOH·H2O particles exist in PG supporter at smaller nanoscale. The results indicated that LiOH·H2O added into the composites did not lead to the structural damage of the whole materials.

Figure 3: SEM images of (a) PG/LiOH·H2O-30%, (b) PG/LiOH·H2O-47%, (c) PG/LiOH·H2O-63%
The TEM images of PG/LiOH·H2O-30%, PG/LiOH·H2O-47% and PG/LiOH·H2O-63% are shown in Fig. 4 to observe more microscopic structures of the composites. Obvious particles have been pointed out in the figures, which indicate that about 5–10 nm LiOH·H2O nanoparticles have been successfully dispersed on PG supporter. By comparing with Fig. 4a, the diameter of LiOH·H2O nanoparticles shows an increasing trend with the increase of its content loading on the PG supporter (shown in Figs. 4b and 4c). When the content of LiOH·H2O reaches 63% (Fig. 4c), the particle size of LiOH·H2O nanoparticles can be increased to about 10 nm, which is higher than that of LiOH·H2O nanoparticles with the lower LiOH·H2O content. Because the nanoparticles easily agglomerated on the surface during the preparation of composites when the mass ratio of LiOH·H2O increased. However, the pure LiOH·H2O particles with a size range of 300 nm–2 μm exist in laminated bulk form. The large particle LiOH·H2O can be effectively induced to form small-sized nanoparticles dispersing on PG supporter. Combining the results of micromorphology characterization shows that high specific surface and nanopore size are the important factors for LiOH·H2O particles achieving nanoscale dispersion.
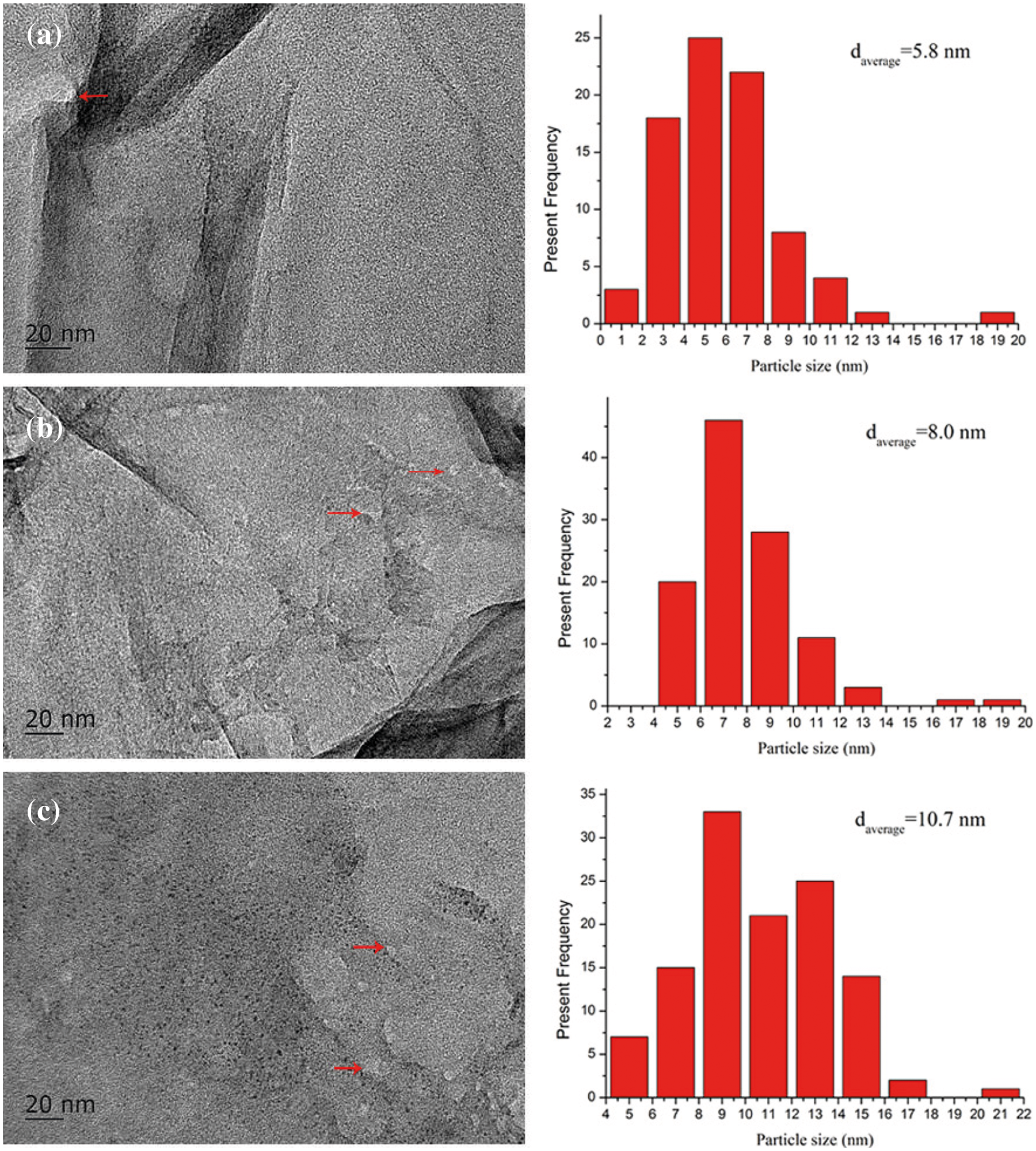
Figure 4: TEM images of (a) PG/LiOH·H2O-30%, (b) TEMPG/LiOH·H2O-47%, (c) PG/LiOH·H2O-63%
Fig. 5 shows the temporal change in amount of hydration of PG, LiOH and PG/LiOH-63% when each sample was hydrated at temperature of 30°C and relative humidity of 80%. LiOH gradually reacts with water vapor, and the amount of hydration increases monotonously with time. The amount of hydration of LiOH is 0.24 g/g at 60 min, while PG rapidly adsorbs water vapor and almost reaches equilibrium state within 20 min. The equilibrium amount of hydration was 0.1 g/g at 60 min. Meanwhile, the hydration rate of PG/LiOH·H2O-63% composite was faster than that of LiOH and PG. The PG/LiOH·H2O-63% amount of hydration was 0.48 g/g at 60 min, and equilibrium amount of hydration was 0.58 g/g at 120 min. According to the results of hydration behaviors, the average hydration rate of the composite is 2 times that of the pure material at 60 min, so that the composite can store and release more thermal energy at the same time.
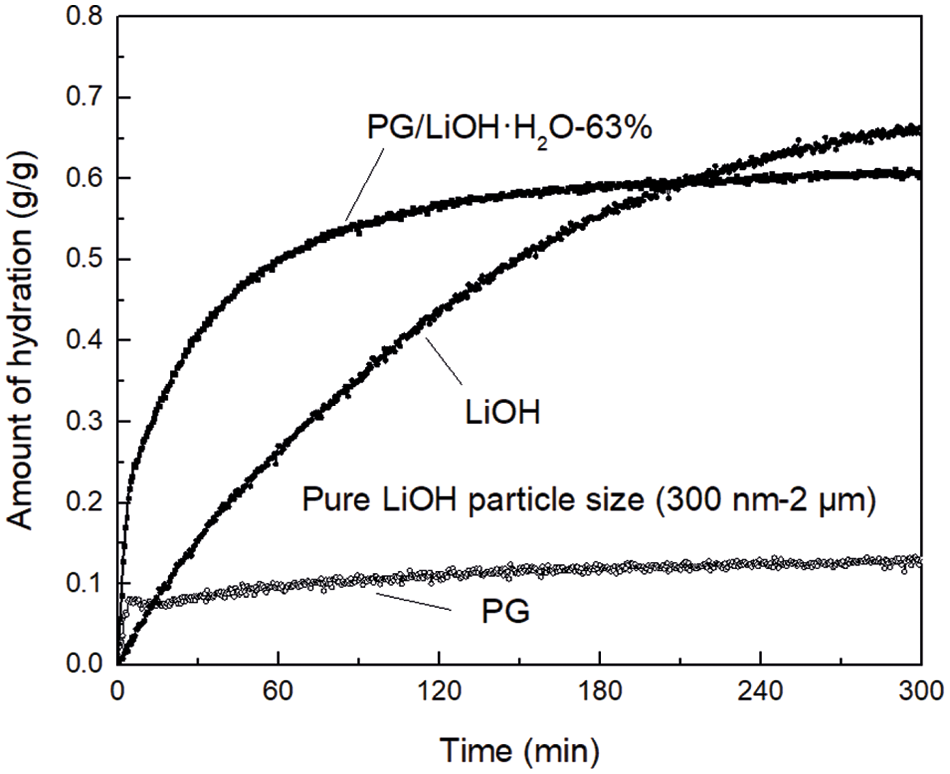
Figure 5: Temporal changes in amount of hydration for pure LiOH·H2O, PG/LiOH·H2O-63% and PG at 30°C with 80% relative humidity
Fig. 6 is the TG-DSC curve of hydration reaction with different content of LiOH·H2O loaded on PG for 60 min. The content of LiOH·H2O was 30%, 47% and 63%, respectively (Figs. 6b–6d) and Fig. 6a is the TG-DSC curve of pure LiOH·H2O. Because the hydration rate of pure LiOH is slow, only 43.7% of LiOH is hydrated after 60 min of hydration, so its heat storage density is only about 661 kJ/kg (Fig. 6a). Compared with pure LiOH, the hydration rate of LiOH is greatly increased by the addition of PG, so the LiOH monomer can react completely after 60 min hydration. By changing the content of LiOH·H2O, the overall thermal storage density of composites can reach 549 kJ/kg (30%), 752 kJ/kg (47%) and 974 kJ/kg (63%), respectively, and the highest thermal storage density of LiOH·H2O monomer can be increased to 1830 kJ/kg (the heat storage density of PG is only 59 kJ/kg). The reason why the hydration rate and heat storage density of PG/LiOH·H2O composites have been greatly increased is that on the one hand, there are a large number of hydrophilic groups such as hydroxy, carbonyl and carboxyl groups on the surface of the PG supporters, which provides an efficient hydrophilic interface for the LiOH hydration process, and enhances the adsorption capacity of materials on water molecules to improve the hydration reaction rate, on the other hand, when PG is compounded with LiOH·H2O, the size of LiOH·H2O particles reaches nano-scale and the surface atomic quantity increases significantly. Moreover, the lattice field and binding energy of the internal atoms are significantly different from that of the surface atoms, and the surface atoms have many suspended bonds due to the lack of adjacent atoms. Under these conditions, the thermodynamic properties of nanoparticles will be enhanced, which makes LiOH·H2O after hydration reaction have higher heat storage density. The evaluation results of thermal storage performance of PG/LiOH·H2O composites show that the hydration rate of LiOH monomer is greatly increased by the compounding of PG supporter, and the temperature range of LiOH monomer is significantly expanded compared with that of pure LiOH·H2O (Fig. 6a). The initial temperature range (60°C–110°C) is extended to 30°C–200°C, and the energy storage density is increased to varying degrees, which will greatly expand the range of available heat energy and effectively improve the energy utilization rate.
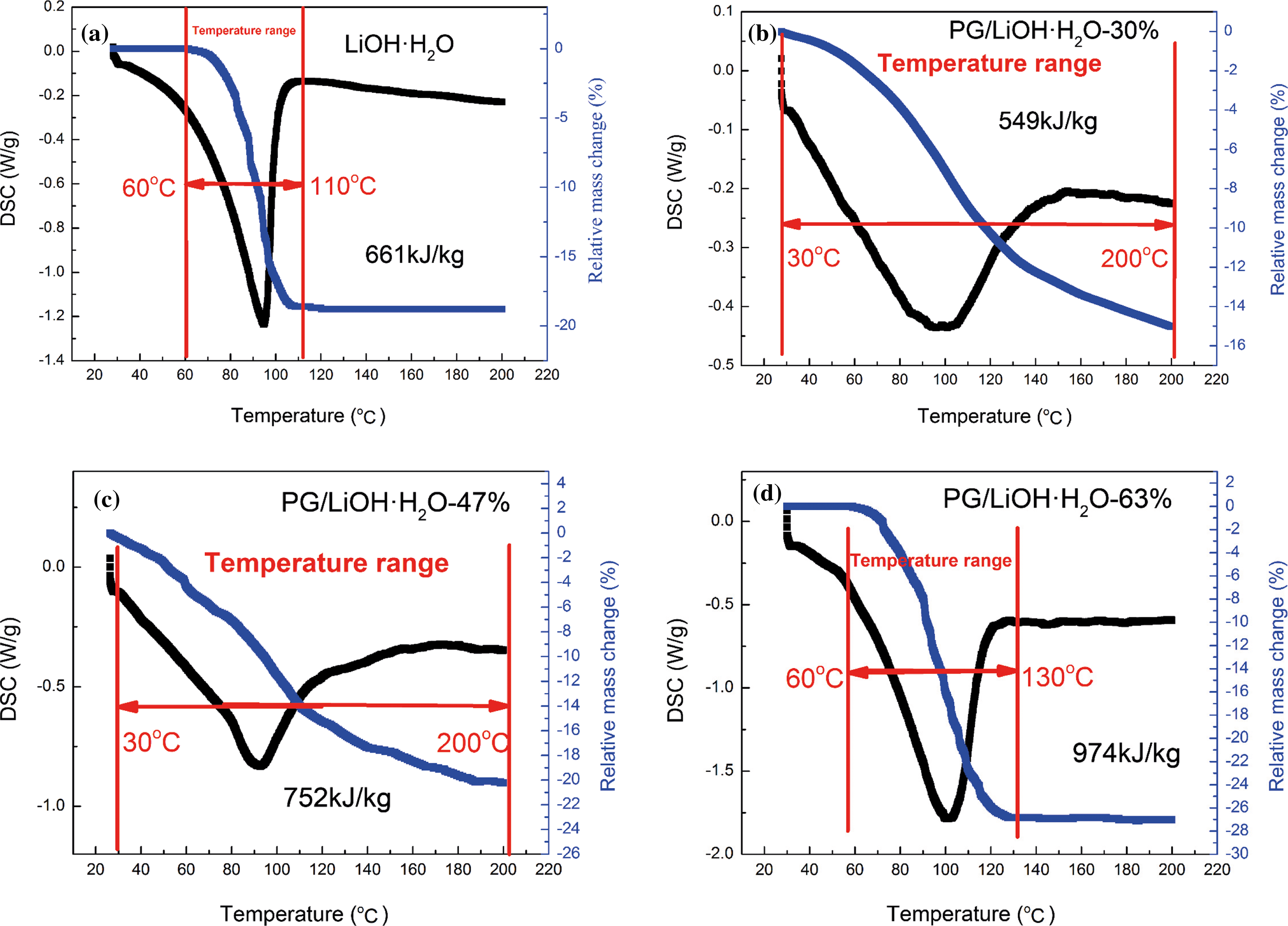
Figure 6: TG-DSC curves of (a) pure LiOH·H2O, (b) PG/LiOH·H2O-30%, (c) PG/LiOH·H2O-47% and (d) PG/LiOH·H2O-63%. (a) Adapted with permission from reference [35], Copyright© 2021, Wilkey-VCH GmbH
Fig. 7 shows the thermal conductivities of pure LiOH·H2O and the composites with different LiOH·H2O content. As shown in the figure, the thermal conductivity of the composites decreased with the increase of LiOH content. With the addition of PG supporters, the thermal conductivity of composite with different LiOH·H2O content increased from 1.69 W/(m·°C) to 2.05, 2.54 and 3.02 W/(m·°C), respectively. In order to better analyze the role of the supporter, the thermal conductivity model of porous composite heat conduction model is used for analysis. From the previous microscopic characterization analysis, it can be assumed that the PG in composite belongs to the continuous phase and the LiOH·H2O belongs to the dispersed phase. Thus, Maxwell–Eucken model is used as a famous theoretical calculation model of thermal conductivity of porous composites [36]. The thermal conductivity calculation equation was as Eq. (1).
where K is the effective thermal conductivity, k1 is the thermal conductivity of continuous phase and k2 is the thermal conductivity of dispersed phase, and
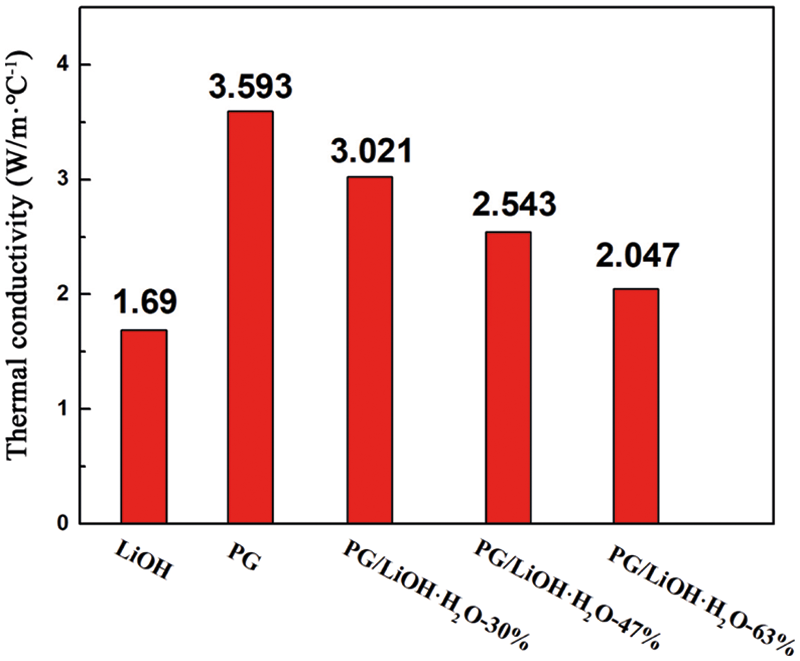
Figure 7: Thermal conductivities of LiOH·H2O and PG/LiOH·H2O composites
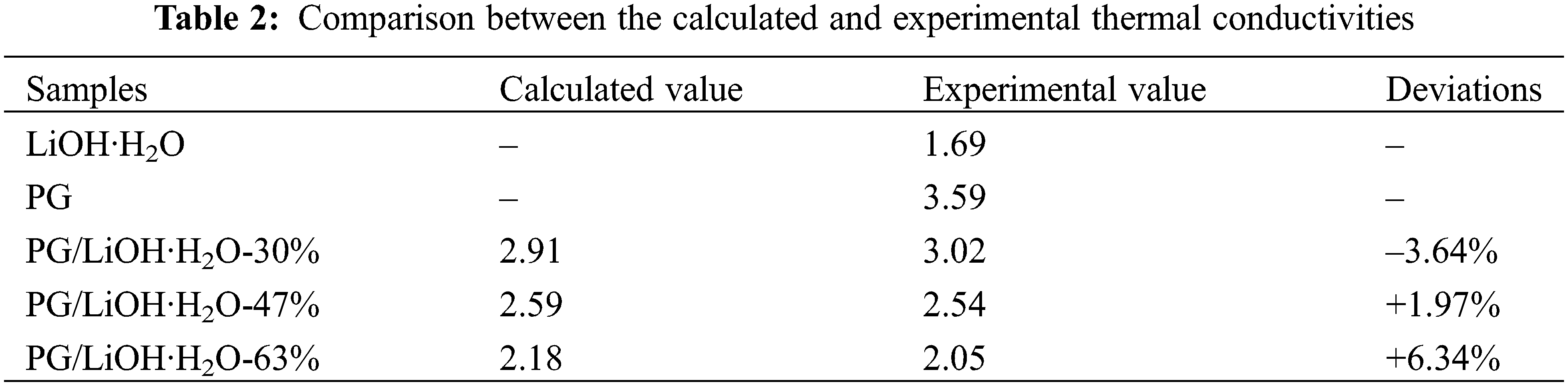
According to Eq. (1), the thermal conductivity of the composites was predicted by using the thermal conductivity of LiOH·H2O and PG. The comparison between the calculated and the experimental results is shown in Table 2. The deviations of the theoretically calculated values from the experimental values were all less than 10%. The deviation may be caused by uniform material distribution and experimental errors when theoretical model is used. Therefore, it can be assumed that the thermal conductivity of PG/LiOH·H2O composites is affected by the LiOH·H2O content. Because the thermal conductivity of pure LiOH·H2O is lower than PG, the thermal conductivity of the composite shows a decreasing trend with the increase of LiOH·H2O content. The results show that the thermal conductivity of the composites increased by 78.7%, 50.4% and 21.1% when the content of LiOH·H2O is 30%, 47% and 63%, respectively. Therefore, when preparing high-performance composites, it is necessary to comprehensively consider the heat storage density and thermal conductivity.
We mainly study on the thermochemical heat storage materials hydrate LiOH for storing low temperature heat. However, its application is limited by the low hydration rate and thermal conductivity. In order to improve its hydration rate and thermal conductivity, porous graphene combing LiOH·H2O was synthesized to composite PG/LiOH·H2O by hydrothermal method. The main conclusions are summarized as follows:
1. The addition of PG caused the composites to have higher specific surface, nanopores distribution and nanoparticle size of LiOH·H2O. The composite materials exhibited improved hydration rate. The hydration rate of PG/LiOH·H2O-63% was twice as high as that of pure LiOH·H2O at 60 min.
2. Compared with the pure LiOH·H2O, the heat storage density of PG/LiOH·H2O composite is greatly increased to 974 kJ/kg from 661 kJ/kg. The heat storage density of PG/LiOH·H2O-63% is 47.3% higher than that of pure LiOH·H2O.
3. By combing with PG, the thermal conductivity of the composite is greatly improved. With the LiOH·H2O content of 30%, 47% and 63%, the thermal conductivities of composite were increased by 78.7%, 50.4% and 21.1%, respectively. According to theoretical analysis, the key factor affects the thermal conductivity of composites is the proportion of components.
4. With the increase of LiOH·H2O content, the heat storage density and thermal conductivity of composite show the opposite trend. The development of high-performance materials for thermochemical heat storage should consider the relationship between the heat storage density and thermal conductivity of the material, and the thermal conductivity of the supporter needs to be further improved.
Funding Statement: This work was supported by the Key Area Research and Development Program of Guangdong Province (2019B110209003), Key Research Program of Frontier Sciences, Chinese Academy of Sciences, China (QYZDY-SSW-JSC038), Natural Science Foundation of Guangdong Province (2017A030310185) and Science and Technology Project of China Energy Investment Corporation (GJNY-20-121).
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
1. Xiong, H. (2015). Policies for utilization of industrial waste heat and its current situation and potential in China. China: Energy Research Institute, National Development and Reform Commission. [Google Scholar]
2. Jouhara, H., Khordehgah, N., Almahmoud, S., Delpech, B., Chauhan, A. et al. (2018). Waste heat recovery technologies and applications. Thermal Science and Engineering Progress, 6(1), 268–289. DOI 10.1016/j.tsep.2018.04.017. [Google Scholar] [CrossRef]
3. Prasad, L., Muthukumar, P. (2013). Design and optimization of lab-scale sensible heat storage prototype for solar thermal power plant application. Solar Energy, 97(5), 217–229. DOI 10.1016/j.solener.2013.08.022. [Google Scholar] [CrossRef]
4. Salomoni, V. A., Majorana, C. E., Giannuzzi, G. M., Miliozzi, A., Maggio, R. D. et al. (2014). Thermal storage of sensible heat using concrete modules in solar power plants. Solar Energy, 103(4), 303–315. DOI 10.1016/j.solener.2014.02.022. [Google Scholar] [CrossRef]
5. Pinel, P., Cruickshank, C. A., Beausoleil-Morrison, I., Wills, A. (2011). A review of available methods for seasonal storage of solar thermal energy in residential applications. Renewable and Sustainable Energy Reviews, 15(7), 3341–3359. DOI 10.1016/j.rser.2011.04.013. [Google Scholar] [CrossRef]
6. Kenisarin, M. M. (2010). High-temperature phase change materials for thermal energy storage. Renewable and Sustainable Energy Reviews, 14(3), 955–970. DOI 10.1016/j.rser.2009.11.011. [Google Scholar] [CrossRef]
7. Rosato, A., Sibilio, S. (2013). Preliminary experimental characterization of a three-phase absorption heat pump. International Journal of Refrigeration, 36(3), 717–729. DOI 10.1016/j.ijrefrig.2012.11.015. [Google Scholar] [CrossRef]
8. Pardoa, P., Deydier, A., Anxionnaz-Minvielle, Z., Rougé, S., Cabassud, M. et al. (2014). A review on high temperature thermochemical heat energy storage. Renewable and Sustainable Energy Reviews, 32, 591–610. DOI 10.1016/j.rser.2013.12.014. [Google Scholar] [CrossRef]
9. Abedin, A. H., Rosen, M. A. (2011). A critical review of thermochemical energy storage systems. Open Renewable Energy Journal, 4, 42–46. DOI 10.2174/1876387101004010042. [Google Scholar] [CrossRef]
10. N’Tsoukpoe, K. E., Restuccia, G., Schmidt, T., Py, X. (2014). The size of sorbents in low pressure sorption or thermochemical energy storage processes. Energy, 77, 983–998. DOI 10.1016/j.energy.2014.10.013. [Google Scholar] [CrossRef]
11. Zhang, Y., Wang, R. Z. (2020). Sorption thermal energy storage: Concept, process, applications and perspectives. Energy Storage Materials, 27, 352–369. DOI 10.1016/j.ensm.2020.02.024. [Google Scholar] [CrossRef]
12. Balasubramanian, G., Ghommem, M., Hajj, M. R., Wong, W. P., Tomlin, J. A. et al. (2010). Modeling of thermochemical energy storage by salt hydrates. International Journal of Heat and Mass Transfer, 53(25–26), 5700–5706. DOI 10.1016/j.ijheatmasstransfer.2010.08.012. [Google Scholar] [CrossRef]
13. Farcot, L., Pierrès, N. L., Michel, B., Fourmigué, J. F., Papillon, P. (2018). Numerical investigations of a continuous thermochemical heat storage reactor. Journal of Energy Storage, 20(7), 109–119. DOI 10.1016/j.est.2018.08.020. [Google Scholar] [CrossRef]
14. Malley-Ernewein, A., Lorente, S. (2019). Constructal design of thermochemical energy storage. International Journal of Heat and Mass Transfer, 130, 1299–1306. DOI 10.1016/j.ijheatmasstransfer.2018.10.097. [Google Scholar] [CrossRef]
15. Yu, N., Wang, R. Z., Lu, Z. S., Wang, L. W. (2014). Development and characterization of silica gel-LiCl composite sorbents for thermal energy storage. Chemical Engineering Science, 111, 73–84. DOI 10.1016/j.ces.2014.02.012. [Google Scholar] [CrossRef]
16. Dawoud, B., Vedder, U., Amer, E. H., Dunne, S. (2007). Non-isothermal adsorption kinetics of water vapour into a consolidated zeolite layer. International Journal of Heat and Mass Transfer, 50(11–12), 2190–2199. DOI 10.1016/j.ijheatmasstransfer.2006.10.052. [Google Scholar] [CrossRef]
17. Wu, S., Li, T. X., Yan, T., Wang, R. Z. (2019). Advanced thermochemical resorption heat transformer for high-efficiency energy storage and heat transformation. Energy, 175(14), 1222–1233. DOI 10.1016/j.energy.2019.03.159. [Google Scholar] [CrossRef]
18. Yana, T., Wanga, C. Y., Li, D. (2019). Performance analysis of a solid-gas thermochemical composite sorption system for thermal energy storage and energy upgrade. Applied Thermal Engineering, 150, 512–521. DOI 10.1016/j.applthermaleng.2019.01.004. [Google Scholar] [CrossRef]
19. Yan, T., Wang, R. Z., Li, T. X. (2018). Experimental investigation on thermochemical heat storage using manganese chloride/ammonia. Energy, 143, 562–574. DOI 10.1016/j.energy.2017.11.030. [Google Scholar] [CrossRef]
20. Fitóa, J., Coronas, A., Mauran, S., Mazeta, N., Perier-Muzeta, M. et al. (2019). Hybrid system combining mechanical compression and thermochemical storage of ammonia vapor for cold production. Energy Conversion and Management, 180, 709–723. DOI 10.1016/j.enconman.2018.11.019. [Google Scholar] [CrossRef]
21. Grekova, A., Strelova, S., Gordeeva, L., Aristov, Y. (2019). “LiCl/vermiculite—Methanol” as working pair for adsorption heat storage: Adsorption equilibrium and dynamics. Energy, 186, 115775. DOI 10.1016/j.energy.2019.07.105. [Google Scholar] [CrossRef]
22. Gordeeva, L. G., Aristov, Y. I. (2011). Composite sorbent of methanol “LiCl in mesoporous silica gel” for adsorption cooling: Dynamic optimization. Energy, 36(2), 1273–1279. DOI 10.1016/j.energy.2010.11.016. [Google Scholar] [CrossRef]
23. Calabrese, L., Brancato, V., Paolomba, V., Proverbio, E. (2019). An experimental study on the corrosion sensitivity of metal alloys for usage in PCM thermal energy storages. Renewable Energy, 138, 1018–1027. DOI 10.1016/j.renene.2019.02.013. [Google Scholar] [CrossRef]
24. Wanga, G., Xua, C., Wei, G., Du, X. (2019). Numerical study of a novel dual-PCM thermal energy storage structure filled with inorganic salts and metal alloy as the PCMs. Energy Procedia, 158(1), 4423–4428. DOI 10.1016/j.egypro.2019.01.774. [Google Scholar] [CrossRef]
25. Li, S., Huang, H., Yang, X., Bai, Y., Li, J. et al. (2018). Hydrophilic substance assisted low temperature LiOH·H2O based composite thermochemical materials for thermal energy storage. Applied Thermal Engineering, 128, 706–711. DOI 10.1016/j.applthermaleng.2017.09.050. [Google Scholar] [CrossRef]
26. Kubota, M., Matsumoto, S., Matsuda, H. (2019). Enhancement of hydration rate of LiOH by combining with mesoporous carbon for low-temperature chemical heat storage. Applied Thermal Engineering, 150, 858–863. DOI 10.1016/j.applthermaleng.2019.01.049. [Google Scholar] [CrossRef]
27. Yang, X., Huang, H., Wang, Z., Kubota, M., He, Z. et al. (2016). Facile synthesis of graphene oxide-modified lithium hydroxide for low-temperature chemical heat storage. Chemical Physics Letters, 644, 31–34. DOI 10.1016/j.cplett.2015.11.033. [Google Scholar] [CrossRef]
28. Posern, K., Kaps, C. (2010). Calorimetric studies of thermochemical heat storage materials based on mixtures of MgSO4 and MgCl2. Thermochimica Acta, 502(1–2), 73–76. DOI 10.1016/j.tca.2010.02.009. [Google Scholar] [CrossRef]
29. Ishitobi, H., Uruma, K., Takeuchi, M., Ryu, J., Kato, Y. (2013). Dehydration and hydration behavior of metal-salt-modified materials for chemical heat pumps. Applied Thermal Engineering, 50(2), 1639–1644. DOI 10.1016/j.applthermaleng.2011.07.020. [Google Scholar] [CrossRef]
30. Kim, S. T., Ryu, J., Kato, Y. (2011). Reactivity enhancement of chemical materials used in packed bed reactor of chemical heat pump. Progress in Nuclear Energy, 53(7), 1027–1033. DOI 10.1016/j.pnucene.2011.05.013. [Google Scholar] [CrossRef]
31. Shi, T., Xu, H., Qi, C., Lei, B., Wu, Y. et al. (2021). Multi-physics modeling of thermochemical heat storage with enhance heat transfer. Applied Thermal Engineering, 198, 117508. DOI 10.1016/j.applthermaleng.2021.117508. [Google Scholar] [CrossRef]
32. Suh, D., Moon, C. M., Kim, D., Baik, S. (2016). Ultrahigh thermal conductivity of interface materials by silver-functionalized carbon nanotube phonon conduits. Advanced. Materials, 28, 7220–7227. DOI 10.1002/adma.201600642. [Google Scholar] [CrossRef]
33. Feng, W., Qin, M., Feng, Y. (2016). Toward highly thermally conductive all-carbon composites: Structure control. Carbon, 109, 575–597. DOI 10.1016/j.carbon.2016.08.059. [Google Scholar] [CrossRef]
34. Leila, S., Athawale, A. (2014). Graphene oxide synthesized by using modified hummers approach. International Journal of Renewable Energy and Environmental Engineering, 2(1), 58–63. [Google Scholar]
35. Li, L., Li, S. J., Zeng, T., Deng, L. S., Huang, H. Y. et al. (2021). Comparative analysis of 3D graphene-LiOH·H2O composites by modification with phytic acid and ascorbic acid for low-grade thermal energy Storages. Energy Technology, 9, 2001086. DOI 10.1002/ente.202001086. [Google Scholar] [CrossRef]
36. Wang, J. F., Carson, J. K., North, M. F., Cleland, D. J. (2006). A new approach to modelling the effective thermal conductivity of heterogeneous materials. International Journal of Heat and Mass Transfer, 49(17–18), 3075–3083. DOI 10.1016/j.ijheatmasstransfer.2006.02.007. [Google Scholar] [CrossRef]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |