| Journal of Renewable Materials |
DOI: 10.32604/jrm.2022.016365
ARTICLE
Feasibility Study of the Synthesis of Isocyanate-Free Polyurethanes from Catechin
1Faculté des Sciences et Technologies, Laboratoire d’Etudes et de Recherche sur le Matériau Bois, EA 4370-USC INRAE, Université de Lorraine, Nancy, 54506, France
2Laboratoire d’Etudes et de Recherche sur le Matériau Bois, EA 4370-USC INRAE, Université de Lorraine, Epinal, 88000, France
*Corresponding Author: Christine Gérardin-Charbonnier. Email: Christine.Gerardin@univ-lorraine.fr
Received: 01 March 2021; Accepted: 26 July 2021
Abstract: With the current trend of increasing efforts to develop non-isocyanate-based polyurethanes (NIPUs), this study aimed to check the feasibility of the development of a method using cyclic carbonate modified catechin and amine to synthesis non-isocyanate urethane with the objective to further extend these results to polyurethane synthesis. The methods used in this study consist of four steps: glycidilation of catechin, hydrolysis of epoxide, cyclic carbonate synthesis, and carbamate synthesis through condensation of butylamine. The resulting products were analyzed using FTIR (Fourier transform infrared) spectroscopy and NMR (nuclear magnetic resonance) spectroscopy. The results showed that carbamate could be successfully obtained through this four-steps synthesis, opening the possibility to further developments for the synthesis of polyurethanes starting from catechin and condensed tannins.
Keywords: Catechin; cyclic carbonate; isocyanate-free; glycidilation; carbamate
Polyurethanes are organic polymers linked by carbamate junctions. They are primarily used in the manufacture of seals, foams, high-performance adhesives, and coatings, as well as in a multitude of other fields [1]. Conventional polyurethanes are prepared through a polyaddition reaction using isocyanates and polyols. The production of polyurethanes is a major concern, as it generally involves the use of toxic petrochemical diisocyanates [2]. Therefore, many studies aiming to create bio-based, environmentally friendly, and non-toxic green polyurethanes have been conducted. One example is the development of non-isocyanate polyurethanes (NIPUs) by minimizing the utilization of isocyanates in polyurethane synthesis to reduce the severe toxicity issues [3–5]. The NIPUs have also gained much attention because of their improvements in porosity, water absorption, and thermal and chemical resistance compared to conventional polyurethanes [6].
In this context, numerous studies have been conducted to develop isocyanate-free polyurethane from natural compounds. Some of the chemical compounds used in these studies are polyphenols, which can be found naturally in wood and bark [7]. The chemical modification of bio-sourced polyphenol is interesting because of its environmentally friendly traits [8,9]. These syntheses have been conducted by replacing bisphenol A and isocyanate by natural polyphenol or sugar [10–14]. Recently, different studies reported modification of polyphenolic compounds such as catechin and tannins, two flavonoid-type polyphenol compounds, for the synthesis of different polymers [15–18] presenting antioxidant properties [19]. Benyahya et al. [15] described the synthesis of epoxide through glycidilation starting from catechin, while Thébault et al. [16] described the synthesis of urethane from tannins. On the basis of these results, it seems interesting to modify hydroxyl group of catechin used as model of polyphenolic compounds to glycidyl derivative, which can be subsequently hydrolyzed to diol [20], and transformed into cyclic carbonate [21,22] allowing urethane synthesis through reaction with amine. The aim of this study is to carried out a preliminary study to show the feasibility of urethane formation through reaction of mono amine with cyclic carbonate obtained from catechin used as model compound of flavan-3-ol involved in condensed tannins.
(+)-Catechin hydrate, epichlorohydrin (99.0%), benzyltriethylammonium chloride (≥98.0%), sodium hydroxide (≥98.0%), potassium carbonate, cesium carbonate and butylamine were obtained from Sigma-Aldrich Chimie, France.
2.1.1 Procedure for Glycidilation of Catechin
A 100-mL two-necked flask equipped with a condenser, a septum cap, and a magnetic stirring bar was charged with 1 g of catechin (3.445 mmol). Catechin was mixed with epichlorohydrin (5 mEq./OH) and heated under reflux at 100°C, after which benzyltriethylammonium chloride (0.05 mEq./catechin) was added. After one hour, the resulting solution was cooled down to 30°C and an aqueous solution of NaOH 20 wt% (2 mEq./OH) was added dropwise with additional amount of benzyltriethylammonium chloride (0.05 mEq./catechin). The mixture was stirred vigorously to prevent precipitation of the reaction mixture for 90 min. The organic layer was separated, dried over MgSO4, and concentrated under vacuum.
2.1.2 Procedure of Hydrolysis of Epoxide in Hot Water
In a 100-mL round-bottom flask equipped with a condenser, glycidyl catechin (1.03 g, 2 mmol) was added to 12 mL of distilled water; then, 6 mL of tetrahydrofuran (THF) was added. The solution was stirred at 80°C and monitored using thin-layer chromatography (TLC). Increase of the temperature to 80°C allowed reaction of water with epoxide and homogenization of the mixture. The reaction monitored by TLC was completed after 4 to 5 h. After completion, the mixture was concentrated under vacuum to remove THF. The remaining water was then removed using a freeze dryer.
2.1.3 Procedure of Carbonate Synthesis from the Diol Derivative
In a 100-mL three-neck flask fitted with a condenser and a CaCl2 guard tube, the diol was stirred with 24 eq. of dimethyl carbonate (3.64 mL, 43.18 mmol) and 0.03 eq. of K2CO3 (0.07 g, 0.054 mmol). The mixture was refluxed at 80°C for 24 h. The excess of dimethyl carbonate was then evaporated under reduced pressure.
2.1.4 Procedure of One-Pot Carbamate Synthesis from Cyclic Carbonate
In a 100-mL three-neck flask fitted with a condenser and a CaCl2 guard tube, butyl amine and cyclic carbonate catechin derivative were stirred, and then 5 to 10 mL of methanol was added to lower the viscosity of the reaction mixture. The reaction medium was subsequently, vigorously stirred using magnetic stirrer and heated at 80°C for 8 h. The progress of the reaction was monitored by Fourier transform infrared (FTIR) analysis. After completion, methanol was removed under vacuum using a rotary evaporator. The reaction mixture was then extracted using 150 mL of ethyl acetate. The organic phase was washed with 2 × 35 mL of 1 N HCl and 35 mL of a saturated NaCl solution, dried over MgSO4, filtered and then concentrated under reduced pressure.
2.2 Nuclear Magnetic Resonance (NMR) Analysis
The products were identified by NMR performed on a Bruker Avance DRX 400 (Bruker, Germany) at 400 MHz. Samples were dissolved either in methanol-d4, or in chloroform-d, or in DMSO-d6 for 1H NMR analysis. Chemical shifts were expressed in parts per million (ppm).
2.3 Fourier Transform Infrared (FTIR) Analysis
FTIR spectrums were recorded with an ATR cell on a Perkin Elmer Spectrum 2000 instrument in the range of 4000 to 600 cm−1 at a resolution of 4 cm−1.
The glycidylation of catechin was conducted in the presence of a phase transfer catalyst [15]. The catechin was initially reacted with epichlorohydrin (5 mEq./aromatic hydroxyl group) in the presence of benzyltriethylammonium chloride used as phase transfer catalyst. Afterwards, the reaction mixture was subjected to an alkaline treatment with NaOH (27.6 mmol, 1.105 g) at 30°C without removing the epichlorohydrin (Fig. 1). Purification using column chromatography was not conducted to avoid the opening of epoxide ring by water on the silica under acidic conditions [23]. The yield of the crude product obtained as a colourless oily product after extraction was of 74%.

Figure 1: Reaction of catechin and epichlorohydrin
Chemical structure of the tetraglycidylcatechin was assigned by 1H NMR and FTIR analysis (Figs. 2 and 3).

Figure 2: 1H NMR spectrum of tetraglycidylcatechin
NMR spectrum of catechin after glycidylation indicated aliphatic signals arising from methyl oxirane groups. Signal of hydrogens of CH2 group of epoxide ring (2 × Ha), overlapping with hydrogens of benzylic group belonging to the flavonoid skeleton (2 × Hj), appeared as a massif between 2.75 and 2.95 ppm. A multiplet between 3.20 and 3.40 ppm corresponds to the hydrogen of CH of epoxide (Hb). The two massifs between 3.68 and 3.85 ppm and 4.05 and 4.35 ppm correspond each to one hydrogen of the methylene in α of the epoxide group (2 × Hc). H2 of catechin (Hh) appeared as a doublet at 4.5 ppm, while H3 (Hi) was masked under the signals of epoxide. H6 (He) and H8 (Hd) signals of catechin moiety appeared as a massif around 6.03 ppm, while those of H2′, H5′ and H6′ appeared as a massif between 6.75 and 7.25 ppm.

Figure 3: FTIR spectra of catechin and after glycidylation
Comparison of the FTIR spectrum of catechin before and after glycidylation shows the presence epoxide ring at 908.5 cm−1 in the reaction product. Intensity of the stretching vibration bands of hydroxyl groups in the 3000 to 3500 cm−1 area also differ showing a decrease of hydroxyl band in the tertraglycidylated product ascribable to the substitution of phenolic groups.
3.2 Hydrolysis of Epoxide in Hot Water
Hydrolysis of epoxide was conducted by heating the crude product in hot water at a temperature comprised between 60 and 80°C for 16 h (Fig. 4). Wang et al. [20] reported that the ring opening reactions of epoxides can be achieved in hot water with a good yield, water acting as a weak acid catalyst. THF was added to increase the solubility in the water of the tetraglycidylcatechin derivative. The final product from the reaction was obtained as a colourless oily product.

Figure 4: Hydrolysis of epoxide
FTIR analysis (Fig. 5) clearly indicated the formation of the diol resulting from the hydrolysis of epoxide. An important absorption band appeared at 3347 cm−1, while the oxirane band at 908 cm−1 disappeared quite totally.
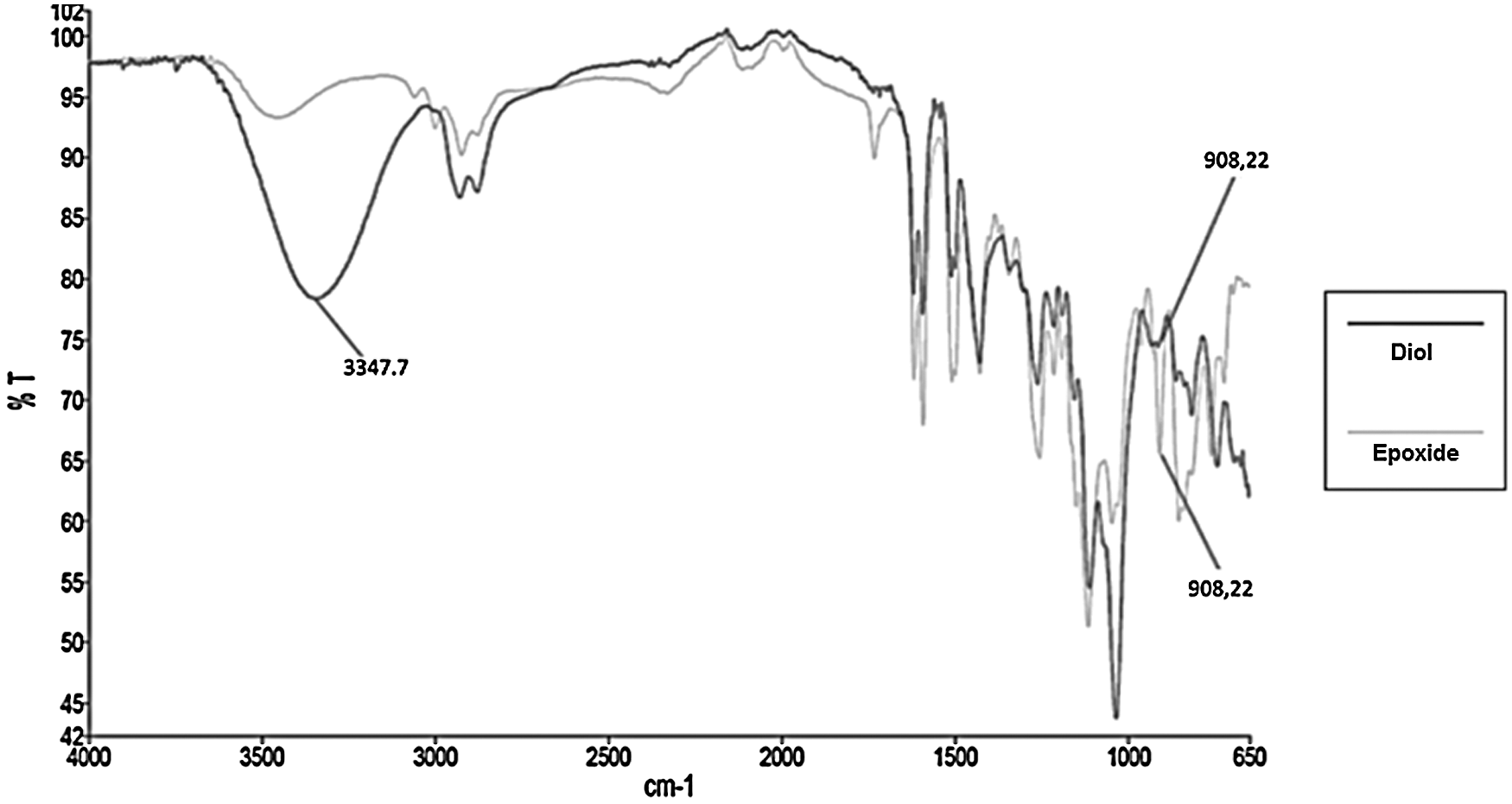
Figure 5: FTIR spectrum of tetraglycidylcatechin derivative after hydrolysis
To confirm the formation of diol, NMR analysis was conducted. Fig. 6 presents the NMR spectrum of the crude hydrolysis product after evaporation of solvents. The multiplet à 3.20 ppm correspond to hydrogen of CH2OH group (2 × Ha), while those of CHOH group (Hb) and CH2O (2 × Hc) appeared between 3.30 and 3.70 ppm.
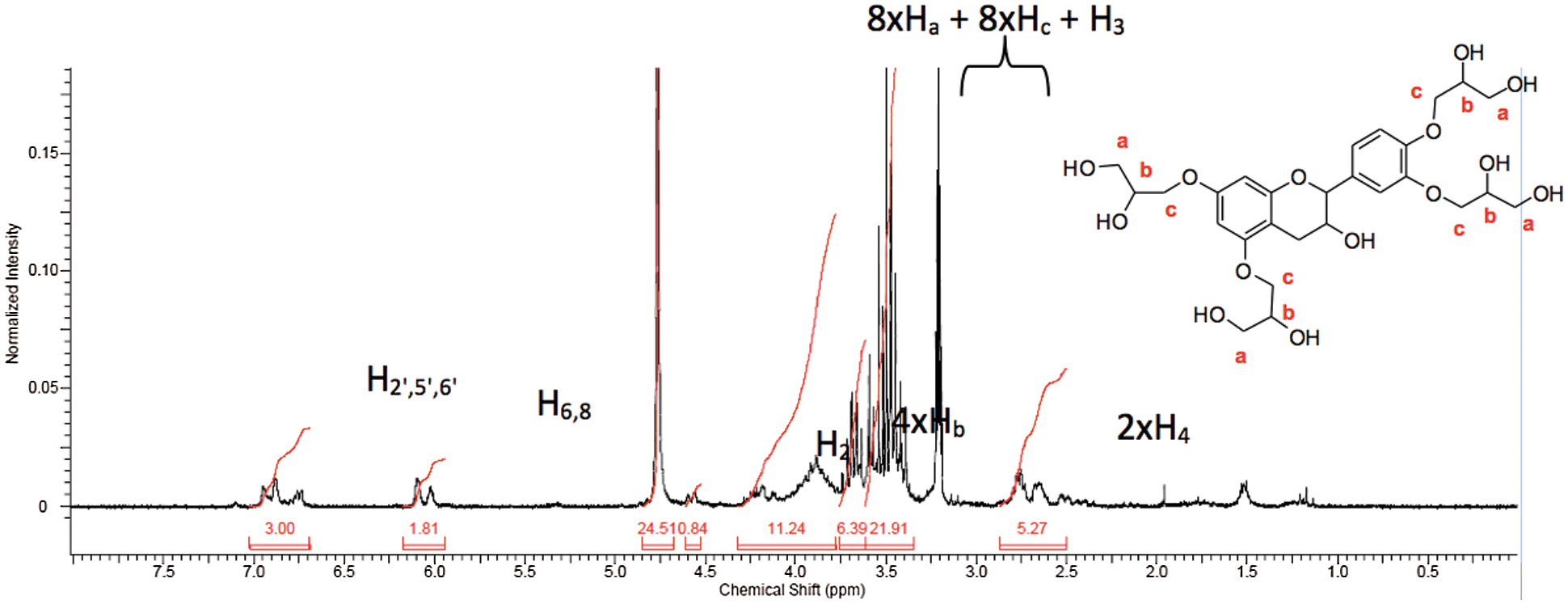
Figure 6: 1H NMR spectrum after epoxide hydrolysis
3.3 Cyclic Carbonate Synthesis
Carbonate synthesis was conducted to obtain cyclic carbonate, which afterwards can be reacted with amines to provide carbamate function. The reaction with dimethyl carbonate was carried out at 80°C for 24 h as shown in Fig. 7. Dimethyl carbonate was used in excess (24 eq.) to allow solubilization of the diol.
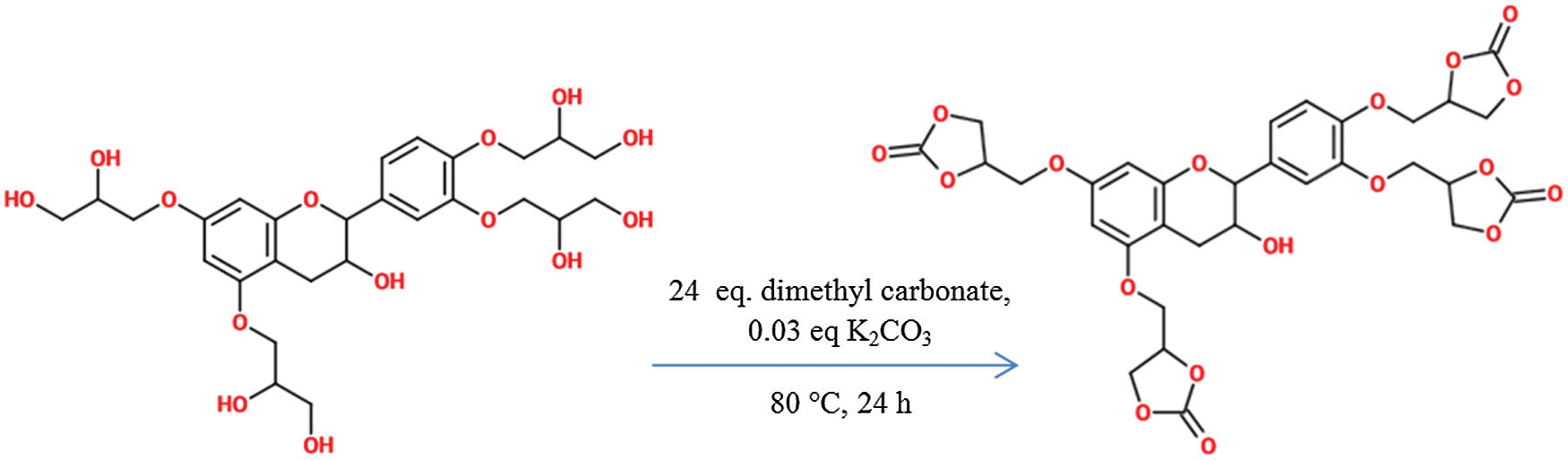
Figure 7: Synthesis of cyclic carbonate derivative
Formation of cyclic carbonate was investigated by FTIR (Fig. 8). The presence of expected cyclic carbonate was confirmed by typical absorption band at 1793 cm−1 in infrared spectra. The decrease of the alcohol wide band at 3445 cm−1 indicated the carbonatation of the diol moieties. The signal at 1748 cm−1 may be due to the formation of non-cyclic carbonate or to the presence of residual dimethyl carbonate. The total raw yield after carbonation was 69%.
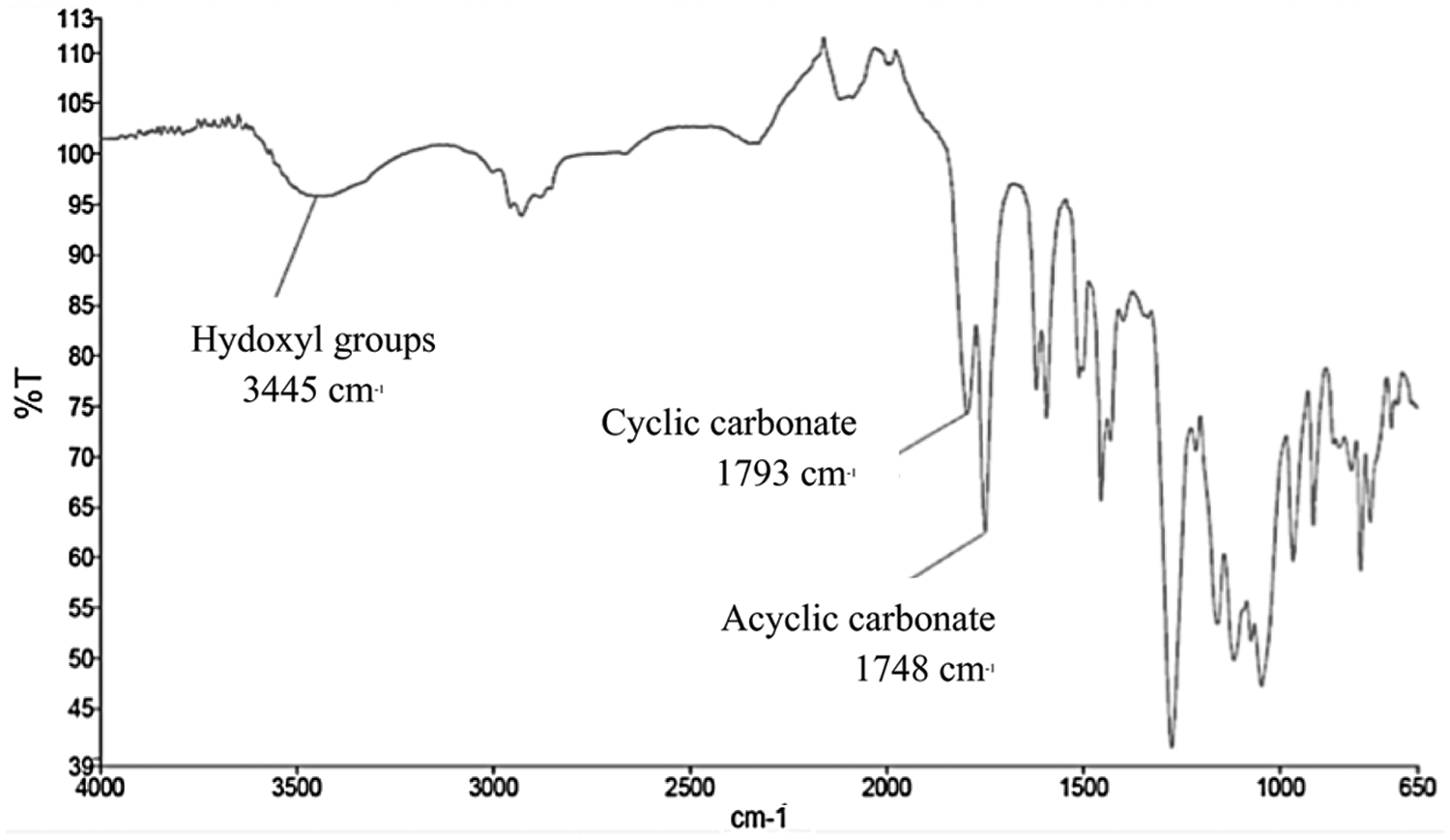
Figure 8: FTIR spectrum of carbonated catechin
To confirm the chemical structure of the cyclic carbonate derivative, NMR analysis was conducted. Fig. 9 presents the NMR spectrum. Protons of the original glycerol part were significantly shifted downfield with cyclic carbonate formation. Indeed, introduction of the carbonate moiety deshields the neighbouring protons Ha, Hb, and Hc by 0.75 to 1 ppm. The rest of the signals referring to the flavonoid block were very little affected by insertion of the carbonate function.
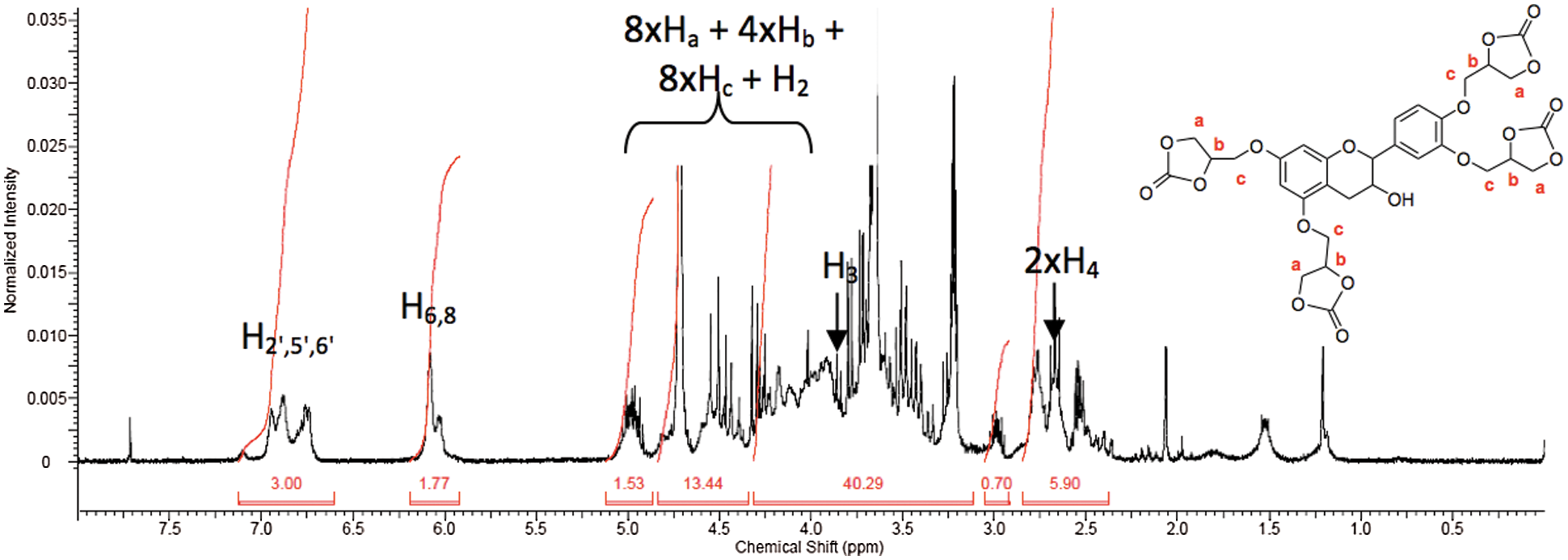
Figure 9: 1H NMR spectrum after carbonation reaction
To evaluate the feasibility of carbamate formation, butylamine was used as an model amine to test reactivity of carbonate moiety. The reaction was carried out by heating the cyclic carbonate with 10 eq. of butylamine at 80°C for 16 h (Fig. 10).
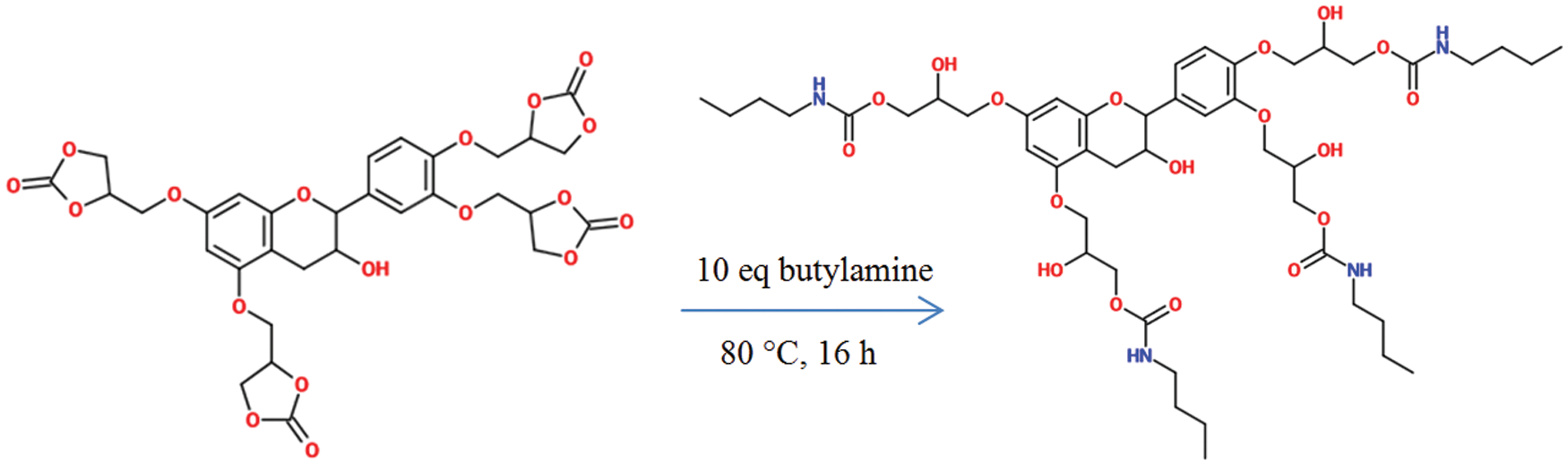
Figure 10: Reaction of cyclic carbamate modified catechin with butylamine
To confirm the formation of urethane bond, the product was then examined using FTIR (Fig. 11). The appearance of the strong absorption band at 1696 cm−1 indicated the formation of the carbamate group as in previous study conducted by Thébault et al. [21].
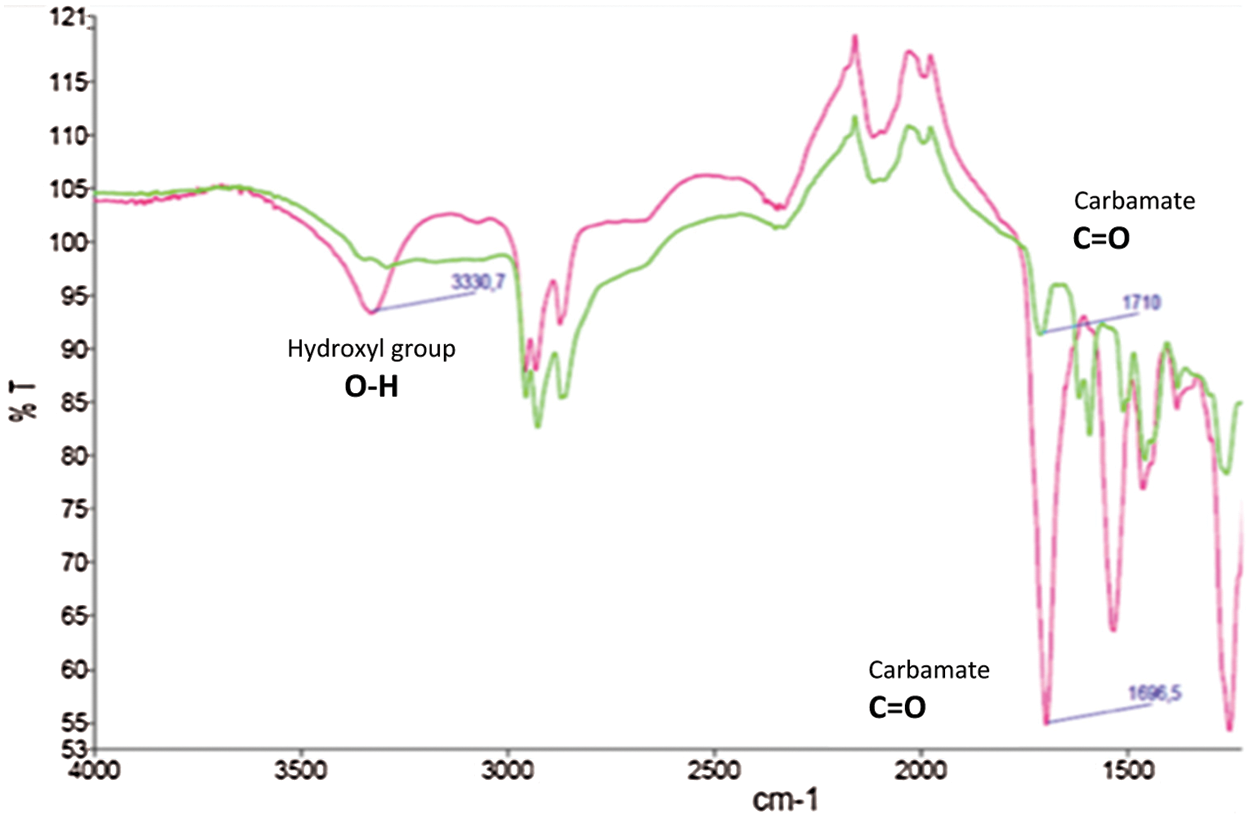
Figure 11: FTIR spectra showing the urethane bond formation through reaction of cyclic carbonate modified catechin with butylamine
Measurements on crude product using 1H NMR were difficult to interpret due to the presence of residual butyl amine masking partially signals of butyl moiety in the urethane (Fig. 12). However, evidence of urethane formation was confirmed by the appearance at 3.1 ppm of a signal characteristic of CH2 in α position of the carbamate compared to the corresponding CH2 at 2.68 ppm in the residual butylamine.
.

Figure 12: 1H NMR spectrum after carbamatation reaction with butylamine
The synthesis of carbamate starting from catechin carbonate derivative has successfully been achieved using butylamine chose as model amine. All the fourth steps of the synthesis involving glycidilation of catechin, hydrolysis of epoxyde, carbonatation with dimethylcarbonate and reaction of cyclic carbonate with amine have been completed and characterized using FTIR showing the formation of the carbamate at the end of the synthesis. Further works are under investigation to extend these preliminary results to the formation of polyurethanes using diamine instead of butylamine, but also to extend the procedure to tannins instead of catechin used in this study.
Acknowledgement: Febrina Boer would like to express her gratitude to the Ministry of Education and Culture of Indonesia (Kementerian Pendidikan dan Kebudayaan Indonesia) for their scholarship, “Beasiswa Unggulan”, which supports her double degree Master education both in Indonesia and France, and Campus France with French Ministry of Foreign Affairs for their social coverage scholarship. The authors gratefully acknowledge the “Ministère des Affaires étrangères et du développement international” (MAEDI)” and the “Ministère de l’Education nationale, de l’Enseignement supérieur et de la recherche (MENESR)” for the financial support through the Bio-Asie Program (2015–2016). LERMAB is supported by a grant overseen by the French National Research Agency (ANR) as part of the “Investissements d’Avenir” Program (ANR-11-LABX-0002-01. Lab of Excellence ARBRE) and by the “Impact Biomolecules” Project of the “Lorraine Université d’Excellence” “Investissements d’avenir–ANR 15-004” and by the French Ministry of Agriculture and the Lorraine-FEDER for the support of “EXTRAFOREST” Project.
Funding Statement: The authors received no specific funding for this study.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
1. Rokicki, G., Parzuchowski, P. G., Mazurek, M. (2015). Non-isocyanate polyurethanes: Synthesis, properties, and applications. Polymers Advanced Technologies, 26(7), 707–761. DOI 10.1002/pat.3522. [Google Scholar] [CrossRef]
2. Bayer, O. (1947). Das di-isocyanat-polyadditions verfahren (polyurethane). Angewandte Chemie, 59(9), 257–272. DOI 10.1002/(ISSN)1521-3757. [Google Scholar] [CrossRef]
3. Gao, Z. H., Li, D. (2007). Chemical modification of poplar wood with foaming polyurethane resins. Journal of Applied Polymer Science, 104(5), 2980–2985. DOI 10.1002/(ISSN)1097-4628. [Google Scholar] [CrossRef]
4. Maisonneuve, L., Lamarzelle, O., Rix, E., Grau, E., Cramail, H. (2015). Isocyanate-free routes to polyurethanes and poly(hydroxyurethane)s. Chemical Reviews, 115(22), 12407–12439. DOI 10.1021/acs.chemrev.5b00355. [Google Scholar] [CrossRef]
5. Datta, J., Włoch, M. (2016). Progress in non-isocyanate polyurethanes synthesized from cyclic carbonate intermediates and di- or polyamines in the context of structure-properties relationship and from an environmental point of view. Polymer Bulletin, 73(5), 1459–1496. DOI 10.1007/s00289-015-1546-6. [Google Scholar] [CrossRef]
6. Guan, J., Song, Y., Lin, Y., Yin, X., Zuo, M. et al. (2011). Progress in study of non-isocyanate polyurethane. Industrial & Engineering Chemistry Research, 50(11), 6517–6527. DOI 10.1021/ie101995j. [Google Scholar] [CrossRef]
7. Arbenz, A., Avérous, L. (2015). Chemical modification of tannins to elaborate aromatic biobased macromolecular architectures. Green Chemistry, 17(5), 2626–2646. DOI 10.1039/C5GC00282F. [Google Scholar] [CrossRef]
8. Peng, Y., Zheng, Z., Sun, P., Wang, X., Zhang, T. (2013). Synthesis and characterization of polyphenol-based polyurethane. New Journal of Chemistry, 37(3), 729–734. DOI 10.1039/c2nj41079f. [Google Scholar] [CrossRef]
9. Pizzi, A. (2016). Wood products and green chemistry. Annals of Forest Science, 73(1), 185–203. DOI 10.1007/s13595-014-0448-3. [Google Scholar] [CrossRef]
10. Aouf, C., Nouailhas, H., Fache, M., Caillol, S., Boutevin, B. et al. (2013). Multi-functionalization of gallic acid. Synthesis of a novel bio-based epoxy resin. European Polymer Journal, 49(6), 1185–1195. DOI 10.1016/j.eurpolymj.2012.11.025. [Google Scholar] [CrossRef]
11. Nouailhas, H., Aouf, C., Le Guerneve, C., Caillol, S., Boutevin, B. et al. (2011). Synthesis and properties of biobased epoxy resins. Part 1. Glycidylation of flavonoids by epichlorohydrin. Journal of Polymer Science Part A: Polymer Chemistry, 49(14), 2261–2270. DOI 10.1002/pola.24659. [Google Scholar] [CrossRef]
12. Xi, X., Pizzi, A., Gérardin, C., Du, G. (2019). Glucose-biobased non-isocyanate polyurethane rigid foams. Journal of Renewable Material, 7(3), 301–312. DOI 10.32604/jrm.2019.04174. [Google Scholar] [CrossRef]
13. Xi, X., Wu, Z., Pizzi, A., Gérardin, C., Lei, H. et al. (2019). Non-isocyanate polyurethane adhesive from sucrose used for particleboard. Wood Science and Technology, 53(2), 393–405. DOI 10.1007/s00226-019-01083-2. [Google Scholar] [CrossRef]
14. Chen, X., Pizzi, A., Essawy, H., Fredon, E., Gerardin, C. et al. (2021). Non-furanic humins-based non-isocyanate polyurethane (NIPU) thermoset wood adhesives. Polymers, 13(3), 372. DOI 10.3390/polym13030372. [Google Scholar] [CrossRef]
15. Benyahya, S., Aouf, C., Caillol, S., Boutevin, B., Pascault, J. P. et al. (2014). Functionalized green tea tannins as phenolic prepolymers for bio-based epoxy resins. Industrial Crops and Products, 53(6), 296–307. DOI 10.1016/j.indcrop.2013.12.045. [Google Scholar] [CrossRef]
16. Thébault, M., Pizzi, A., Dumarçay, S., Gérardin, P., Fredon, E. et al. (2014). Polyurethanes from hydrolysable tannins obtained without using isocyanates. Industrial Crops and Products, 59(3), 329–336. DOI 10.1016/j.indcrop.2014.05.036. [Google Scholar] [CrossRef]
17. Delgado-Sanchez, C., Letellier, M., Fierro, V., Chapuis, H., Gérardin, C. et al. (2016). Hydrophobisation of tannin-based foams by covalent grafting of silanes. Industrial Crops and Products, 92, 116–126. DOI 10.1016/j.indcrop.2016.08.002. [Google Scholar] [CrossRef]
18. Rangel, G., Chapuis, H., Basso, M.C., Pizzi, A., Delgado-Sanchez, C. et al. (2016). Improving water repellence and friability of tannin-furanic foams by oil-grafted flavonoid tannins. BioResources, 11(3), 7754–7768. DOI 10.15376/biores.11.3.7754-7768. [Google Scholar] [CrossRef]
19. Gadkari, P. V., Balaraman, M. (2015). Catechins: Sources, extraction and encapsulation: A review. Food and Bioproducts Processing, 93, 122–138. DOI 10.1016/j.fbp.2013.12.004. [Google Scholar] [CrossRef]
20. Wang Z., Cui Y. T., Xu Z. B., Qu J. (2008). Hot water-promoted ring-opening of epoxides and aziridines by water and other nucleophiles. Journal of Organic Chemistry, 73(6), 2270–2274. DOI 10.1021/jo702401t. [Google Scholar] [CrossRef]
21. Thébault, M., Pizzi, A., Essawy, H. A., Barhoum, A., van Assche, G. (2015). Isocyanate free condensed tannin-based polyurethanes. European Polymer Journal, 67(3), 513–526. DOI 10.1016/j.eurpolymj.2014.10.022. [Google Scholar] [CrossRef]
22. Panchgalle, S. P., Kunte, S. S., Chavan, S. P., Kalkote, U. R. (2011). Exploration of L-proline-catalyzed α-aminoxylation of aldehyde to (S)-guaifenesin-related drug molecules. Synthetic Communications, 41(13), 1938–1946. DOI 10.1080/00397911.2010.493631. [Google Scholar] [CrossRef]
23. Mohan, P. (2013). A critical review: The modification, properties, and applications of epoxy resins. Polymer-Plastics Technology and Engineering, 52(2), 107–125. DOI 10.1080/03602559.2012.727057. [Google Scholar] [CrossRef]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |