| Journal of Renewable Materials |
DOI: 10.32604/jrm.2022.018935
ARTICLE
Preparation and Characterization of Waterborne Polyurethane with Unique Nanoparticles by Controlling Water
Faculty of Printing, Packing Engineering and Digital Media Technology, Xi’an University of Technology, Xi’an, 710048, China
*Corresponding Author: Xing Zhou. Emails: zdxnlxaut@163.com, zdxnl@xaut.edu.cn
Received: 26 August 2021; Accepted: 15 October 2021
Abstract: A facile method, focusing on emulsification, chain extension and dispersion process in preparing waterborne polyurethane, was developed to prepare emulsion with rod-like nanoparticles. The facile method involves a water addition procedure by in situ generated water to react with polyurethane prepolymer instead of the external water addition process. As a comparison, waterborne polyurethane was synthesized through the external water addition process. According to the characterization methods including FTIR, 1H-NMR, TEM and water swelling experiments, it is suggested there are two kinds of hydrogen bonds interactions in hard/soft domain of the novel polyurethane, and the phase separation of hard/soft domains increases significantly. The morphology of the two polyurethanes was quite different (nanorods and spherical particles, respectively), presenting a different surface property. In addition, the water swelling of the novel polyurethane indicates that it holds significant potential application as degradable material.
Keywords: Polyurethane; nanoparticles; coatings; degradation; swelling
Water has been devoted to organic reactions due to the unusual properties in pure form and as a solvent, and even the induction of unique reactivity and selectivity, reduction of harmful organic solvents, and environmentally friendly chemical processes [1–4]. One of the most important applications of water in organic reactions is the synthesis of waterborne polyurethane (WPU) dispersion, in which water plays a key role due to the aqueous synthetic environment [5,6]. The most widely used synthesis route of WPU is to add deionized water into WPU precursor to emulsify and disperse the hydrophilic WPU prepolymer, with the process of chain extension [7–9], which was called as external water addition synthesis process. However, because of high viscosity of the WPU prepolymer, it is commonly hard to disperse WPU prepolymer into deionized water or adding deionized water into the prepolymer [10,11].
Actually, water, playing the role of chain extender, emulsifier and solvent, or maybe more, is one of the most important components to decide whether WPU can be obtained. Williams et al. [10] reported that water acts as a plasticizer and significantly influences the mechanical performance due to the expected hydrophilic nature of the ionic domains. Nasrullah et al. [12] incorporated an automated parallel reactor system to synthesize WPU dispersions. They controlled the addition of neutralizer, water, and chain extender to study the impact of their amount and addition rate on properties of WPU dispersions. They found that the rate of water addition determines the final WPU dispersions properties and made a conclusion that a shorter time is better according to the results of viscosity, particle size and percent solids of dispersions. Asplund et al. [13] investigated the siginificant and interesting polyurethane synthesis process by adding water in vapor phase. The addition of water is the most important step, which should be done slowly and continuously. It is stated in this paper that this synthesis method is able to eliminate the tedious control to approach stoichiometry, is less sensitive to impurities, involves no intermediate isolation steps, and does not involve any chain extender. Still, a relatively large amount of organic solvent was needed to decrease the viscosity. Shahrousvand et al. [14] had realized early that water addition rate is a critical parameter to obtain stable dispersion. Therefore, they used a tubing pump to add water for 6 min at a constant flow rate to the WPU prepolymer. Kim et al. [15] also studied phenomena which occurred during the water emulsification process, such as phase inversion, viscosity, conductivity, and isocyanate value change. Madbouly et al. [16] and Dieterich [17] both summarized the importance of water addition. They stated that WPU dispersion is mainly affected by the slow addition of water to the polymer solution. Meanwhile, the water emulsification function was well documented by Dieterich. Therefore, the time and rate for water addition are critical to determine whether good WPU is formed or not [18]. However, the rate of water addition and time is hardly controlled without a special experiment device and the function of water on WPU prepolymer has seldom been investigated [19–21].
This work proposed a facilely controlled water addition procedure for the WPU prepolymer chain extending, emulsifying and dispersing. To prepare homogeneous (relatively narrow molecular weight distribution) WPU dispersions avoiding the prepolymer with high viscosity, the new strategy for preparation of WPU dispersions in situ water generating process (in situ WGP) was developed (as is shown in Fig. 1), including emulsification, chain extension, and dispersion processes for WPU prepolymer by in situ water generation [9,22]. The main advantages of the novel process are as follows, firstly, the control of water addition rate and time can be easily accomplished without special pump and addition equipment. Secondly, the in situ WGP can react with all the prepolymer chains simultaneously, avoiding the influence of prepolymer high viscosity, and decreasing the amount of organic solvent, especially for acetone. Furthermore, the in situ WGP makes it possible to control the water generating rate and amount, indicating that the kinetics of emulsification, chain extension and dispersion of water to WPU prepolymer can be investigated, of which procedure can also be applied to the ractions involving water. This study investigated the bulk structures of the polyurethane chains and nanoparticles, and water swelling property of WPU dispersion prepared through in situ WGP. For comparative purposes, another WPU dispersion was prepared through the external water addition process.
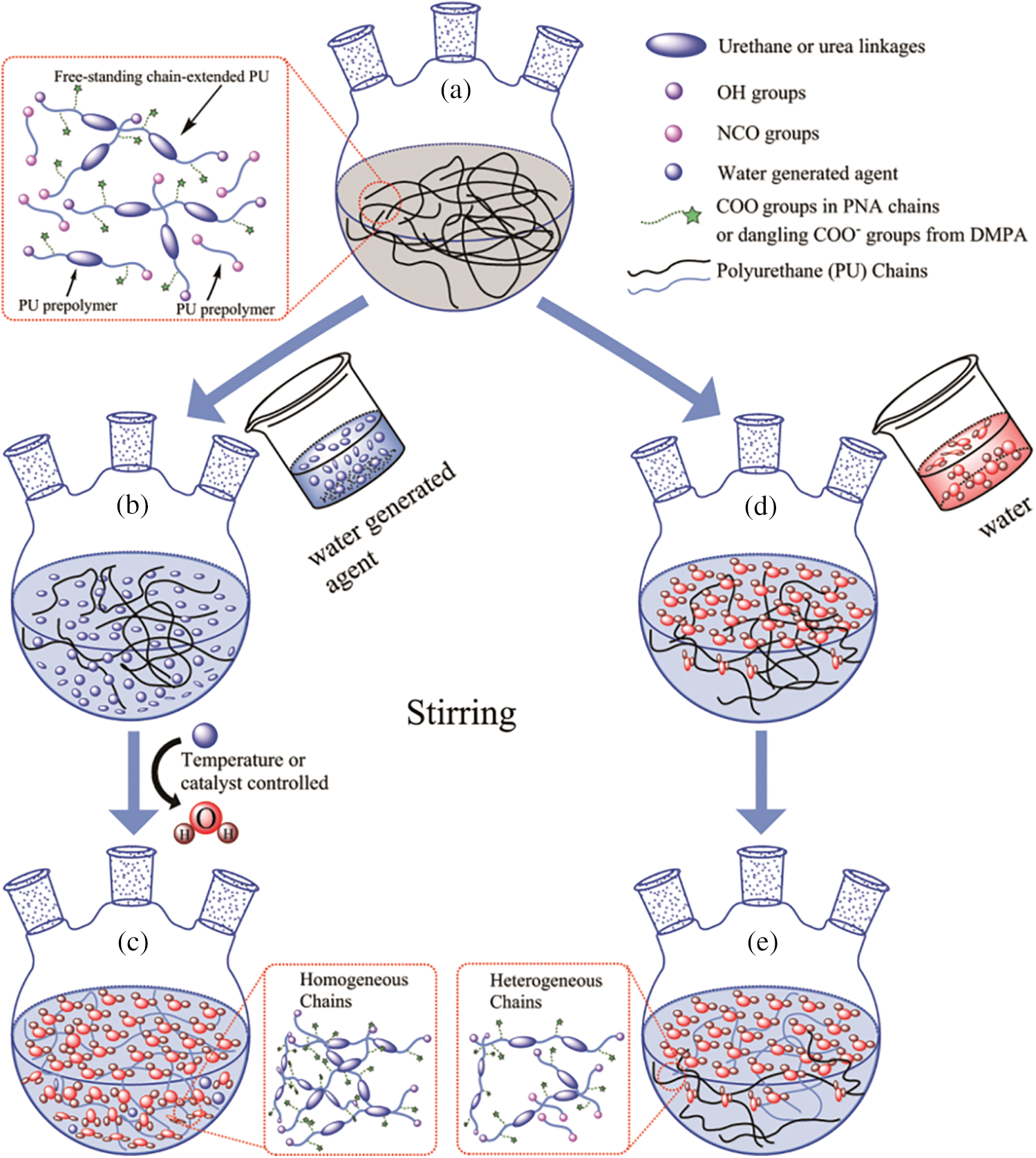
Figure 1: Preparation of WPU dispersions through the in situ WGP and external water addition methods [22]
Isophorone diisocyanate (IPDI, 98 wt% purity, purchased from Jingchun Chemical, Shanghai, China) is used at room temperature. The oligomer glycol is poly (neopentyl glycol adipate) (PNA, molecular weight (Mw) = ca. 2000). The hydrophilic chain extender is dimethylolpropionic acid (DMPA). 1, 4-butanediol (BDO, 99.5 wt% purity, purchased from Fuchen chemical, Tianjin, China.) is used as chain extender. H2O2 (30 wt%, Mw = 34.01), KOH, and dibutyltin dilaurate (DBTDL) are purchased from Qingxi Chemical, Shanghai, China. Acetone (Jingchun Chemical, Shanghai, China) and 1-methyl-2-pyrrolidone (NMP, 99 wt% purity) are used as solvents. Acetone about 4–8 mL was used throughout the traditional process.
2.2 Synthesis of Waterborne Polyurethane Prepolymer
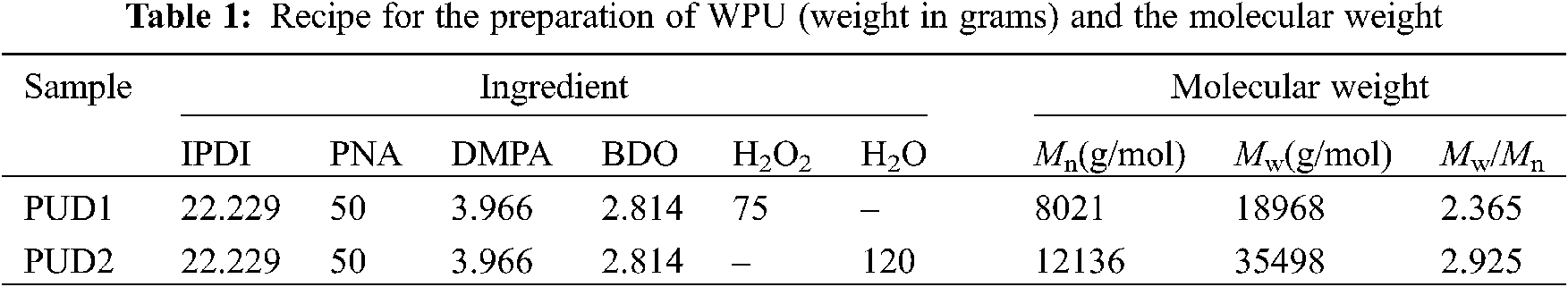
Waterborne polyurethane dispersions (PUD) samples by addition of deionized water or hydrogen peroxide with the hard-/soft-segment molar ratio of 4 and the appropriate NCO/OH ratio of 1.2 were synthesized according to our previous works [8,9]. In this work, PNA was used as oligomer polyol followed by drying in an oven for 2 h at 120°C, and then PNA was mixed with IPDI to produce polyurethane prepolymer. The reaction was carried out at 85°C for 3 h following by the addition of DMPA at 60°C to graft hydrophilic groups onto polyurethane prepolymer. The reaction was continued at 85°C for another 2 h. After that, BDO was added to chain extend the prepolymer for 1 h. After the reaction, WPU prepolymer can be obtained.
2.3 Preparation of WPU by Deionized Water and Hydrogen Peroxide, Respectively
Hydrogen peroxide was employed as water generating agent, and added dropwise to obtain waterborne polyurethane dispersion (PUD1). To control the decomposition of the hydrogen peroxide to produce water, the temperature was adjusted to increase regularly to 50°C by setting the heater. In comparison, deionized water was added in the same way to prepare another waterborne polyurethane dispersion (PUD2). In the preparation of PUD2, to decrease the viscosity of polyurethane prepolymer, acetone and 1-methyl-2-pyrrolidone (NMP) were employed as solvents. The pH values of the polyurethane dispersions were adjusted in the range of 8.5–9.0 by using KOH solution. At last, the sample PUD2 was put into a vacuum drying oven for 1 h to remove solvents with conditions of 50°C and 0.05 MPa.
2.4 Characterization and Property Measurements
Fourier transform infrared spectroscopy (FTIR) was used to identify the structure of the WPU. The infrared spectra of the dried polyurethane films were obtained using a Fourier transform IR spectrophotometer (SHIMADZU FTIR-8400S (CE)) and recorded in the transmission mode at room temperature by averaging 20 scans at a resolution of 16.0 cm−1. The spectra were analyzed in the frequency range of 4000–400 cm−1. Proton nuclear magnetic resonance spectroscopy (1H-NMR) spectra were obtained on a Bruker-400 MHz spectrometer using sodium 2, 2-dimethyl-2-silapentane-5-sulfonate (DSS) as an internal standard, with CDCl3 as solvent. The report of some multiplet peaks of the two samples was as follows: 1H-NMR (400 MHz, CDCl3, δ in ppm) PUD1: 9.38 (s, 1H), 9.26 (s, 1H), 8.81–8.65 (m, 1H), 7.50 (s, 1H), 7.29 (s, 1H), 7.06 (m, 1H), 4.77 (d, J = 2.4 Hz, 1H), 4.19 (s, 1H), 4.14 (s, 1H), 4.10–3.93 (m, 18H), 3.90 (d, J = 11.0 Hz, 1H), 3.77 (d, J = 11.0 Hz, 2H), 3.70 (s, 1H), 3.44–3.36 (m, 1H), 3.27 (s, 1H), 2.94 (s, 1H), 2.87 (d, J = 11.9 Hz, 1H), 2.74 (s, 1H), 2.51 (s, 1H), 2.35 (s, 20H), 2.17 (td, J = 12.4, 6.2 Hz, 5H), 2.03 (d, J = 3.2 Hz, 1H), 1.67 (s, 24H), 1.51 (d, J = 5.3 Hz, 1H), 1.42 (s, 1H), 1.25 (d, J = 7.5 Hz, 1H), 1.18 (s, 1H), 1.07 (s, 8H), 0.98 (dd, J = 11.5, 6.1 Hz, 27H), 0.89 (d, J = 6.7 Hz, 6H), 0.01 (s, 1H).
PUD2: 7.54 (s, 1H), 7.29 (s, 1H), 7.02 (s, 1H), 4.78 (d, J = 3.0 Hz, 1H), 4.20 (s, 1H), 4.09 (s, 1H), 4.07–3.96 (m, 5H), 3.91 (d, J = 11.0 Hz, 1H), 3.78 (d, J = 10.9 Hz, 1H), 3.71 (s, 1H), 3.44–3.36 (m, 1H), 3.28 (s, 1H), 2.94 (s, 1H), 2.87 (s, 1H), 2.75 (s, 1H), 2.52 (s, 1H), 2.39 (d, J = 23.0 Hz, 5H), 2.18 (td, J = 12.7, 6.4 Hz, 1H), 2.05 (dd, J = 15.1, 7.8 Hz, 1H), 1.67 (s, 7H), 1.36 (d, J = 7.4 Hz, 1H), 1.23 (t, J = 7.1 Hz, 1H), 1.15 (s, 1H), 1.05 (d, J = 18.5 Hz, 3H), 0.99 (dd, J = 11.6, 6.0 Hz, 7H), 0.89 (d, J = 6.7 Hz, 2H), 0.17–(−0.15) (m, 1H).
The molecular weights (number-average molecular weights (Mn) and weight-average molecular weights (Mw)) were presented by gel permeation chromatography (GPC) (USA Waters, ALLIANCE) analysis with a DAWN EOS (λ = 690.0 nm) and RI detector (Shodex RI-71). In detection, the eluent was terahydrofuran (THF) with a flow rate of 0.500 mL/min. The particle sizes and the particle size distribution were carried out in Coulter LS230 system (Beckman Coulter, Miami, FL 33196-2500 USA) with laser diffraction and polarized light detectors. Differential scanning calorimetry (DSC) were performed on a NETZSCH DSC machine (200 F3 Maia®) with the setting conditions (temperature range of −100°C–150°C, heating rate of 10°C/min, N2 flow rate of 30 mL/min). The micro-morphology of polyurethane films was performed by the Field Emission Scanning Electron Microscope (FE-SEM) (JSM-6700F, resolution of 133 eV, accelerating voltage of 20 kV) with Energy Dispersive X-ray spectrometers (EDS, Oxford INCA). The EDS detection was repeated for three times. The micro-morphology of polyurethane dispersions was performed by transmission electron microscopy (TEM, accelerating voltage of 300 kV) with a JEM-3010 microscope. The polyurethane dispersion was diluted in deionized water with concentration of 1 wt%. Water contact angle (WCA) was performed with the dropped water volume of 2 μL on an OCA 20 (Dataphysics, Germany). The gel content, swelling capability and water resistance were measured gravimetrically at the relative humidity of 30% and room temperature as follows. Firstly, WPU dispersions were coagulated by CaCl2 aqueous solution (2 wt% CaCl2) at 60°C and then the obtained coagulum was dried in an oven at 60°C for 48 h. Secondly, a 300-mesh stainless steel grid was first weighed (W1) and then placed in a Soxhlet system (temperature of heating of 85°C and circulating cooling of 5°C, respectively) in which tetrahydrofuran (THF) was maintained under reflux for 1 h. The humidified grid was weighed (W2) and dried in an oven at 60°C for 3 h. Thirdly, about 0.5 g of coagulum was placed in the grid and weighed (W3) rapidly, and the samples in grid were extracted with THF under reflux in the Soxhelt for 24 h. The samples were in the form of fragments and the extraction time should be long enough; only in this way could the samples be extracted in the maximum sol fraction. After that, the swollen samples in humidified grid were then weighed (W4) and dried in the 60°C oven for 12 h. Finally, the dried samples and grid were weighed (W5). The gel content, representing the degree of crosslinking of WPU, was defined as
The swelling capability was calculated as
Owing to the rapid evaporation of THF in weighing the humidified grid, the values of gel content and swelling capability have tolerances.
The water resistance values of the samples were measured by immersing the films (5 mm × 5 mm in size) in a beaker with deionized water. The percentage of swelling for a particular film was determined by measuring its weight changes using the equation:
where W0 is the weight of the dried film and W is the weight after water absorption.
A novel anionic waterborne polyurethane dispersion (PUD1) was synthesized via in situ WGP by using ca. 2000 g/mol PNA (PNA-2000) as the soft segment, IPDI as the diisocyanate and H2O2 as the water generating agent (Fig. S1), as shown in Figs. 1a–1c. Firstly, the agent, which can generate water under certain conditions such as catalyst, temperature, time, etc., is added into WPU prepolymer and dispersed uniformly by suitable stir and shear force. Secondly, the conditions are controllable for the added agent to generate water (Fig. 1c). In this way, the uniformly dispersed agent produces water nearly at the same time, creating a homogeneous water environment to emulsify, chain extend and disperse WPU prepolymer to form homogeneous polyurethane dispersion with narrow molecular weight distribution. For comparative purpose, a normal waterborne polyurethane dispersion (PUD2) emulsified and chain extended by direct addition of deionized water was prepared (Figs. 1a–1e). Owing to the high viscosity of WPU prepolymer, the added water is hard to disperse in prepolymer initially, resulting in a heterogeneous reaction between water and WPU prepolymer (Figs. 1d and 1e) and obtaining heterogeneous polyurethane chains. Both of the samples had an equal concentration of hard and soft segment based on the molar ratio of 4 according to previous research [8,9]. The ingredients of the WPU are shown in Table 1.
FTIR spectra present the main peaks of the two PUD samples in the range from 400 to 4000 cm−1. It is reported that hydrogen bonding may induce variations of characteristic peaks in both frequency and peak area, such as a wide and shifting peak in an obvious range. These variations can be presented by FTIR spectroscopy precisely [8,10,19–22]. The typical functional groups are mainly N–H and C=O from urethane groups in polyurethane [19], of which stretching vibrations are in the range of 3200–3400, and 1600–1800 cm−1, respectively. As shown in Fig. 2a, after the peak-fit processing, it is obvious that C=O stretching vibrations of PUD1 show four main peaks at 1643, 1705, 1728, and 1743 cm−1, respectively, which may be ascribed to urea hydrogen bonded ordered carbonyl group, free carbonyl group from 1, 6-adipic acid of PNA, urethane hydrogen bonded disordered carbonyl group, and free urethane carbonyl group. While that of sample PUD2 presents three main peaks at 1650, 1735, and 1743 cm−1, respectively, which may be ascribed to urea hydrogen bonded disordered carbonyl group, urethane hydrogen bonded disordered carbonyl group, and free urethane carbonyl group. The two typical peaks at 1643 and 1728 cm−1 of sample PUD1 are lower than that of PUD2. As illustrated in Fig. S2a, the typical peaks of aliphatic N–H group, amide II (urea stretching C–N group + bending N–H vibration group), and amide I (urethane hydrogen bonded N–H vibration) can be found at 771, 1558 cm−1 for sample PUD1 and 1542 cm−1 for sample PUD2, and 3363 cm−1, respectively. The appearance of N–H peaks suggests the formation of polyurethane for PUD1 and PUD2 samples [21]. Notably, according to Painter [19], three different hydrogen bonding on N–H and C=O stretching vibration bands may be found for polyurethane, such as “free”, “hydrogen bonded disordered”, and “hydrogen bonded ordered”, as shown in Fig. S3. In the case of our research, the N–H region (3200–3400 cm−1) for sample PUD1 is distinctly broader than that of PUD2, indicating that N–H hydrogen bonding strength of PUD1 may be higher than PUD2 [23,24].

Figure 2: (a) FTIR main region for carbonyl group from 1800 to 1600 cm−1 for PUD1 and PUD2; (b) the cartoon of waterborne polyurethane chains (d1) the two kinds of hydrogen bonds in PUD1, (d2) hard-hard hydrogen bonding in PUD2, (d3) cycle-like structure near the interface of hard and soft domains in PUD1
The frequency of carbonyl group decreases, and the frequency of amide II increases a little for PUD1 comparing with PUD2. These should be caused by some kind of strong function in PUD1 molecules, which may be hydrogen bonding [13,25]. Furthermore, as shown in Fig. 2, the peak belonging to the urea hydrogen bonded carbonyl group of sample PUD1 is significantly stronger than that of sample PUD2. The results suggest stronger hydrogen bonding interactions of sample PUD1 synthesized via the new path. Thus, it may facilitate the formation of hard-domains with an increasing hard domains ordering in PUD1 [26].
The aforementioned hydrogen bonding belongs to the hard-hard segment function (shown in Fig. S3). This is the initial significant difference for PUD1 and PUD2. However, according to the previous work [27], it is assumed that there is another kind of hydrogen bonding (shown in Fig. S4) in PUD1, namely the hard-soft segment hydrogen bonding. The structure of the hard-hard segment and hard-soft segment hydrogen bonds in PUD1 and PUD2 are illustrated in Fig. 2b: (d1) and (d2) procedure, respectively. The existence of this hydrogen bonding can induce a significant phase separation formed between the soft and hard domains. As depicted in Figs. S2a and S4, the band assignment for the carbonyl group in 1, 6-adipic acid of PNA-2000 is at about 1732 cm−1. This band can also be seen in PUD1 at about 1705 cm−1 as shown in Fig. 2a. It suggests that the shift of the 1, 6-adipic acid carbonyl group arises from the formed hydrogen bonding between the urethane and 1, 6-adipic acid carbonyl. This hydrogen bonding depicted in Fig. S4, apparently the interaction between hard and soft segments, can also be reflected by the frequencies of ether groups in PNA-2000. The DSC results (Fig. S6) also confirm the phase separation of the two PUDs. The apparent glass transition temperature (Tg) can be observed in the range of −43–(−26°C) and −37–(−27°C) for PUD1 and PUD2, respectively. The different Tg indicate that there may be a certain degree of hard and soft segment mixing, indicating microphase separation in the phase boundary of hard-and soft-segments [26].
As shown in Fig. S2b, two sharp bands at 1242 and 1041 cm−1 can be detected in the PUD2 spectra, ascribing to the symmetric and asymmetric stretching of the ether group in the soft segment. While in PUD1 spectra, the sharp band at 1041 cm−1 disappears and is replaced by a weak band at 1018 cm−1. This shift of the ether group pronounces the interaction of hydrogen bonding formed between N–H in urethane and C=O in the soft segment in PUD1. Meanwhile, the band at 1172 cm−1 in PUD2 spectra should be assigned to the two pendant group of –CH3 in the soft segment, of which frequency can also be seen in PNA spectra in Fig. S2a. Notably, the band of the two pendant group of –CH3 in PUD1 is replaced by a wide and overlapping band, apparently centered near 1110 cm−1, due to the function of the ether group and two pendant group of –CH3. It is suggested that the difference of –CH3 in PUD1 and PUD2 is also caused by the hydrogen bonding described in Fig. S3. In addition, there is a sharp band at 925 cm−1 in PUD1 spectra, while that of PUD2 is weak. This band at 925 cm−1 can be assigned to the stretching of the ether group in cyclic anhydride. It is notable that there is no cyclic anhydride in polyurethane.
Thus, it is speculated that the hydrogen bonding described in Fig. S3 may facilitate the formation of some kind of cycle-like structure near the interface of hard and soft domains, as depicted in the Fig. 2b: (d3) procedure. The impact of the hydrogen bonds on the particle morphology and water swelling property of PUD1 will be described subsequently.
The molecular weights and molecular weight distributions of PUD1 and PUD2 were determined by GPC using THF as the mobile phase and polystyrene as reference. As shown in Table 1 and Fig. S5, the molecular weight distribution of PUD1 is narrower than that of PUD2. This may arise from the insitu generated water, which supplies a homogenous water environment to chain extending the WPU prepolymer. The generated water reacts with the –NCO terminated prepolymer at the same time to produce WPU in homogenous polymer system [13], namely narrower molecular weight distribution than PUD2. The narrow molecular weight distribution of PUD1 corresponds to the assumed homogenous chains illustrated in Fig. 1. Notably, the Mn and Mw of PUD1 are both lower than that of PUD2, indicating that the molecule chain of PUD1 is shorter than that of PUD2. It assumes that the PUD with different molecule chains length may conduct different micro morphology, which can be attested by TEM analysis.
The most powerful method to measure the intensity of urethane, urea, biuret and other groups seems to be 1H-NMR [22], which is able to detect and quantify chemical-shift effects of hydrogen atoms in different constitutional situations. The structure and 1H-NMR results [28] of the samples are shown in Fig. 3. It can be seen that the 1H-NMR spectroscopy of PUD1 and PUD2 are quite similar, indicating that most groups of the molecules chains for both the two samples are similar. Signal at 7.29 ppm ascribes to CDCl3, which was used as a solvent during measurement. The signals from 0.88–1.00 ppm which are complex are attributed to the H protons of IPDI. According to Brame et al. [29], two sharp peaks at 1.67, 4.19 ppm (PUD1) and 1.67, 4.20 ppm (PUD2) are assigned to the methylene protons of –CH2CH2OOCNH–, –CH2CH2OOCNH– in BDO blocks, respectively. Meanwhile, the peak at 1.67 ppm is also ascribed to the methylene protons of –CH2CH2CH2– in PNA blocks. Peaks at 1.25 ppm in PUD1 and 1.23 ppm in PUD2 are assigned to the two pendant group of –CH3 in the PNA chain. As stated from the shifts of methylene protons, the chain extension reaction had completed to synthesize polyurethane. The two extremely weak peaks at 7.06, 7.50 ppm (PUD1) and 7.02, 7.54 ppm (PUD2) may be attributed to the imino group of –CH2NHCOO–, –NHCOOCH2– linked with alicyclic, respectively. Two weak peaks at 9.26 and 9.38 ppm for PUD1 can be observed in the expansion figure in Fig. 3a. These two peaks may be attributed to –NHCONCONH– units. Meanwhile, another weak peak in the range of 8.81–8.65 ppm was reported, which may be attributed to –NHCONH– units. The presence of –NHCONH– and –NHCONCONH– units are considered to be the reason for the side reaction during the synthesis of polyurethane.

Figure 3: (a) 1H-NMR spectrum of the PUD1 and PUD2, (b) 1H-NMR spectrum and monomer structure of the PUD1
Commonly, there are urea and biuret in waterborne polyurethane [8]. However, these characteristic groups of side products in synthesizing polyurethane can only be observed in PUD1. Yet FTIR results showed that the presence of the urea characteristic group of –NHCONH– in both samples. The peak of –NHCONH– for PUD2 may be covered by the strong unimodal of the solvent in 1H-NMR spectrum. As presented in 1H-NMR, FT-IR and the GPC results, It assumes that the mainly monomer structure of the polyurethane based PNA-2000 can be constructed as the inset structure in Fig. 3b.
The particle size, particle size distribution and particle specific surface area, appearance of the molecular chains, and surface of the films were investigated and the results are shown in Fig. 4. As depicted in Figs. 4c and 4f, both dispersions present bimodal profile, similar particle size and particle size distribution. The particle specific surface area of PUD1 and PUD2 are 57.9 and 61.1 m2/mL, respectively. The mean particle size of PUD1 is 65 nm while that of PUD2 is 69 nm. These suggest that the water produced in the reaction through the in situ WAP contributes to the polyurethane chains formation into smaller particles, but the effect is not obvious for the statistical data.
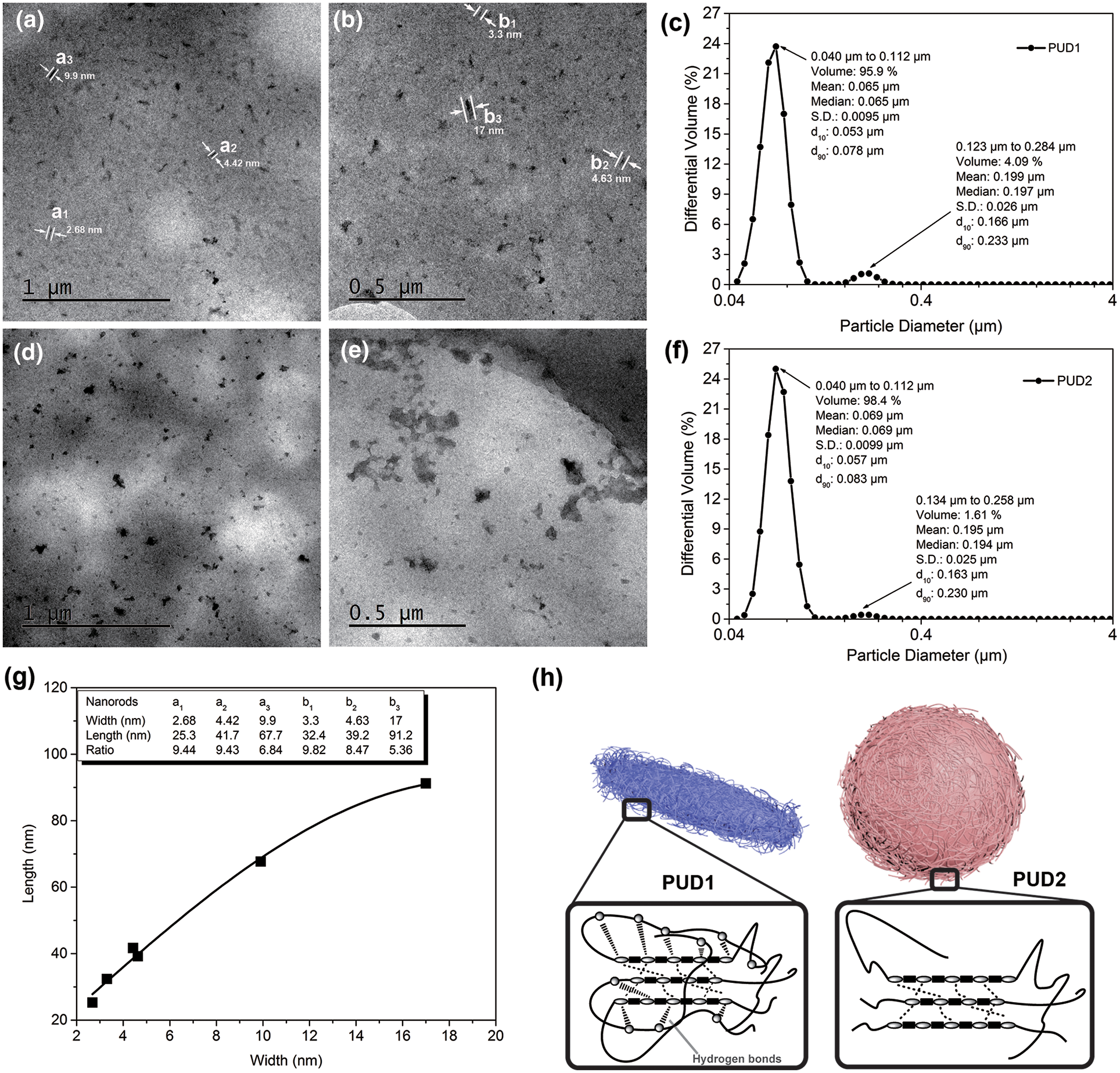
Figure 4: TEM micrographs of polyurethane dispersions diluted by deionized water to the concentration of 1 wt%. PUD1: (a) and (b); PUD2: (d) and (e); particle size and particle size distribution of the (c) PUD1 and (f) PUD2; (g) the relation between length and width values of the selected nanorods in (a) and (b); (h) cartoon for the nanoparticles and aggregation morphology of polyurethane chains for PUD1 and PUD2
Thus, TEM experiment was performed to observe the morphology of these nanoparticles. According to Runt et al. [30], the morphology of polyurethane consisted of both randomly oriented cylinders and domains that appear more or less in the appearance of “spheres”, which are most likely a combination of spherical domains and portions of cylindrical domains. In this research, PNA with the molecular weight of about 2000 g/mol was used to synthesize PUD1 and PUD2. As expected, the PUD2 particles formed analogous sphere shapes [31,32], shown in Fig. 4d, and the size of spherical particles is in the range from 20 to 226.7 nm. Furthermore, Fig. 4e shows that some of the spherical particles aggregate together and form blocks on the membrane. The variation of PUD2 particles shown in Figs. 4d and 4e is essentially in accordance with the kinetic modeling of aggregation and gel formation in quiescent dispersions of polymer colloids postulated by Morbidelli et al. and confirmed by Madbouly et al. [33] and Lattuada et al. [34]. In terms of this model (shown in Fig. S7), the aggregates interconnect with each other by directly experiencing their nearest neighbors to form a network occupying the total volume of the dispersion. This aggregation depicted in Figs. 4d and 4e notably presents the formation process of polyurethane films. What most inspires great interest is the morphology of PUD1, whose particles have a pronounced nanorod shape (Figs. 4a, 4b, and S8) [35] and has been demonstrated by the SEM morphology in the previous work [9]. Since most macromolecular chains are in great length with infinitesimally small cross profile, they curl to form a ball of string morphology. In this way, some macromolecular chains can be observed like a spherical particle via the high-resolution TEM.
In this research, sample PUD2 is in this situation, whose aggregation morphology is depicted as the cartoon of Fig. 4h. However, this situation is no more suitable for sample PUD1 with the morphology of nanorods. The results of TEM show that it is the effect of water product decomposed by hydrogen peroxide in synthesizing WPU that the novel polyurethane dispersions present the nanorod morphology compared with that of external deionized water addition process. It is well known that the higher the degree of hydrogen bonds in polyurethane, the easier it is for ordered structure forms [36].
According to Shin et al. [37], a kind of rod-coil molecule, consisting of hydrophilic and hydrophobic parts, can self-assemble into a variety of architecture in aqueous solution, such as spherical particles, capsules, fibers, tubes, ribbons, etc. Expectedly, the hard/soft segments are exactly the hydrophilic/hydrophobic parts in PUD1 and PUD2. In terms of FTIR results stated above, it is speculated that the most likely explanation for the nanorods in PUD1 involves some orientational chains, which are attributed to the function of hard-soft segment hydrogen bonding in PUD1 (as shown in Fig. 2b: (d3) procedure) and the aggregation of polyurethane chains is depicted in Fig. 4h [38]. This inference of the poly(urethane-urea)s orientational chains can be further demonstrated in DSC and TG analysis in our previous work [27]. As shown in Figs. 4a and 4b, the widths of randomly selected nanorods were marked (a1–a3, and b1–b3) on the images, and the lengths of them were measured to calculate the length-width ratio (listed in Fig. 4g). The discrete points were fitted well by a Gaussian sum, suggesting that the length-width ratio presents basically a Gaussian relation. Meanwhile, the minimum dimension of the polymer nanorod which is found on the images is about 2.48 nm. This value is significant because it occurred only through the change in the method of water addition. It was never thought that such a small nanorod can be obtained through such a facile method. This type of water addition opens a wide field of potential research in WPU synthesis with particles at nanoscale, and the kinetics research of polyurethane prepolymer emulsion reaction can be further studied by water-generating rate in the future work.
Fig. 5 illustrates the surface morphology and hydrophilic property of the prepared PUD1 and PUD2 films. The PUD1 sample shows relative rough surface with chaotic wrinkles in Fig. 5a, and the PUD2 sample presents a typical smooth surface with several oriented wrinkles for polyurethane film [7]. The rough surface leads to different surface property, such as the hydrophilic property. As depicted in the inset diagrams of Figs. 5a and 5b, the average water contact angles of PUD2 film is ca. 30°, while that of PUD1 film is ca. 70°. It indicates that the rough surface of PUD1 may significantly increase the water contact angle of polyurethane. Thus, the novel strategy may supply an effective way to increase the hydrophobicity of waterborne polyurethane by changing the surface structure in preparing polyurethane dispersion.

Figure 5: Surface morphology and property of prepared PUD samples. (a) SEM micrographs of PUD1 show wrinkle and rough surface; (b) SEM micrographs of PUD2 with typical smooth surface for polyurethane film. The inset photographs in (a, b) depict the water contact angle
To further investigate the hydrolytic stability, the swelling behavior of the samples in deionized water and organic solvent (THF) was performed in 7 days or longer. The degradability of both the samples was performed by putting their films through water swelling measurement [39–42]. As depicted in Fig. 6, the weights variation of the films immersed in water with aging time were obtained. The best fitting curves for the discrete weight datas of PUD1 and PUD2 films were calculated by Eq. (3) and fitted using Lorentzian sum. The left and right vertical coordinates in Fig. 6 belong to water swelling of PUD1 and PUD2, respectively. Unlike the rising tendency of WPU films swelling in the previous works [8,43,44], the saturated swollen weights of PUD1 and PUD2 increase firstly, and then decrease largly to achieve a constant value, as shown in Fig. 6.
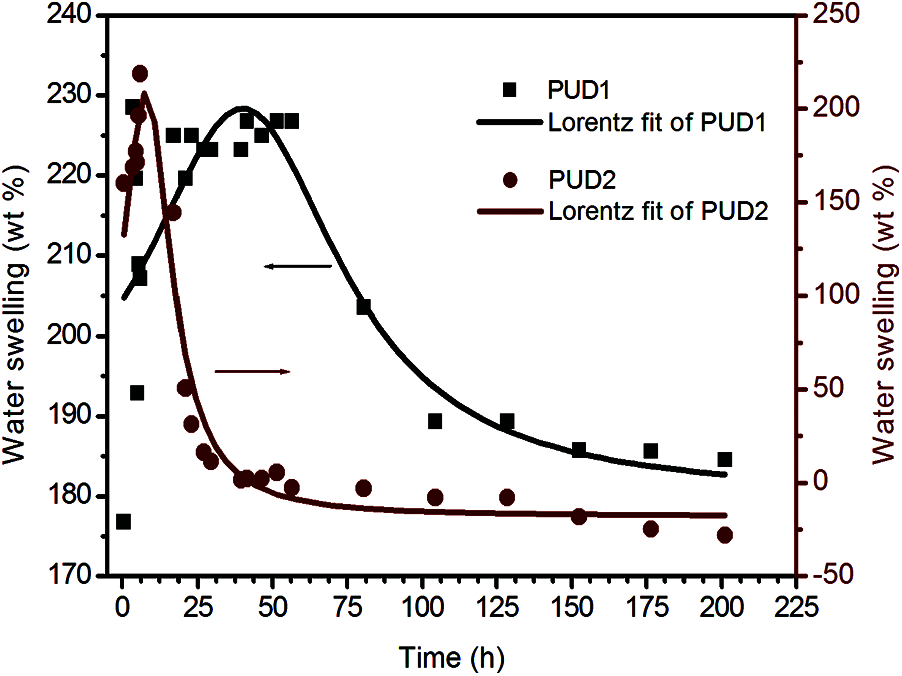
Figure 6: Water swelling of the PUD1 and PUD2 immersed in water with aging time
Thus, the water swelling curves are divided into three stages, namely increasing stage (IS), decreasing stage (DS), and constant stage (CS). The water swelling percent of both PUD1 and PUD2 is high, indicating that they are accessible to water absorption. This may be caused by the presence of large amount of ester bonds in soft segment (PNA-2000). It is well known that the bonds more susceptible to hydrolysis present in poly (ester urethanes) are the ester bonds. The hydrolysis of urethane bonds is one order of magnitude slower relative to ester bonds [45,46]. The hydrolysis process of ester and urethane bonds has been depicted in detail [47]. Hereby, the soft segment with ester bonds will hydrolyse at a greater rate than the hard segment with urethane bonds, signifying that the overall degradation rate of polyurethane with ester and urethane bonds will be governed by the soft segment. In this situation, the samples have a large amount of ester bonds due to the PNA-2000, indicating that the degradation rate of PUD1 and PUD2 is governed by the soft segment, or the interaction of hard segment and soft segment. The percentage of water swelling of the PUD1 is 189.2 ± 0.1 wt%, as shown in Table 2, presenting a good water absorbance property for PUD1 film, indicating that PUD1 has nice water hydrolysis resistance. This may be resulted from the numerous ester groups and strong hydrogen bonding in PUD1. As to PUD2, the water swelling is −28.1 ± 0.1, implying that the film hydrolyses mostly after water immersing, owning an inferior of water swelling resistance.
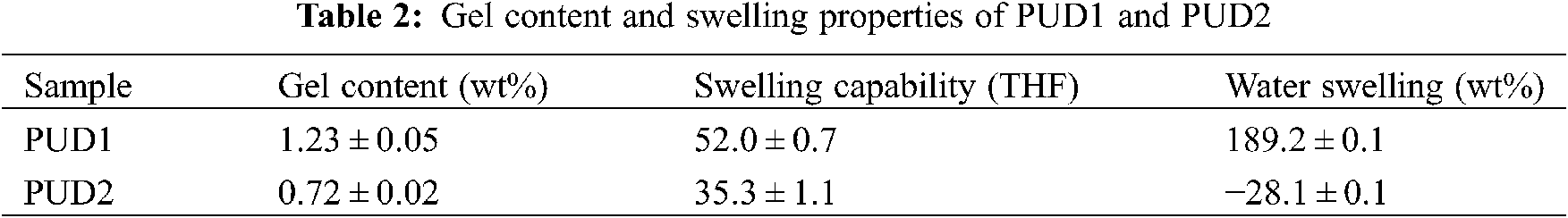
It is noted that at CS after immersing time, the water swelling percentage of PUD1 is nearly the same with the original water swelling before immersing time, while that of PUD2 decreases largely (from 168.6 to −28.1 wt%) after immersing. The decrease of saturated weight of both the samples implies that the polyurethane films are accessible to water via hydrolysis. This result reflects that the polyurethanes in this research are degradable for potential application in biomedical material. It is desirable to characterize the samples′ swelling rate, which can be obtained from the slope of the curves. Comparing the IS and DS of PUD2 with PUD1, the swelling rates of PUD2 are both larger and the degradation times are shorter. It suggests that the PUD2 is easier in the absorption of water and in hydrolysis. Furthermore, for the PUD1, the discrete points are more and the peak is wider than that of PUD2 at the largest water swelling weight, as depicted in Fig. 6. It suggests that PUD1 is more stable than PUD2 at the largest water absorption. It is observed from Fig. 6 that the swelling span (the subtraction of water swelling at the largest and smallest value) of PUD1 is about 65 wt% and that of PUD2 is about 250 wt%, which is approximately 3.8 times as large as that of PUD1. In addition, the weight of PUD2 film after water absorption decreases after immersing in water for about 25 h, while that of PUD1 decreases after 90 h. All the differences of water swelling between PUD1 and PUD2 stated above suggest that water resistance of PUD1 is much better than PUD2.
It is assumed that the differences arise from the interaction of hard and soft segment but not the soft segment for the content of hard and soft segments in PUD1 and PUD2 are the same (as shown in Table 1). According to the FTIR results, the hard segment dissolution in soft segment is more for PUD2, namely, the phase separation is higher for PUD1. Thus, the degree of water absorption and degradation of PUD1 is less affected by soft segment. Furthermore, the hydrogen bonding function in PUD1 is stronger, which retards water absorption and degradation for the film [44]. This indicates that the PUD1 synthesized in a homogeneous water environment presents well water resistance and degradation.
The gel content values, reflecting the degree of crosslinking of WPU, are presented in Table 2. The coagulum of WPU dispersions by 2 wt% CaCl2 aqueous solutions at 60°C is shown in Fig. S9. It can be seen that the morphology of PUD1 coagulum is a 3D network structure, while that of PUD2 shows relative smooth film. It is known that polymer gel is influenced by the interaction of chemical bond, ionic bond, or hydrogen bonding, etc., indicating the polymer network accounting for the whole volume of a fluid [48]. According to Flory theory, partial ordered gel structures can be formed through the interaction of physical binding. It is suggested that the PUD1 gel, shown in Fig. S9 presenting a partial 3D network, is the function of the abundant hydrogen bonds in PUD1 hard and soft domains. That is the reason for the difference of PUD1 and PUD2 gel morphology. The gel content of PUD1 is higher than PUD2. The reason for this is that the gel can be formed in a homogeneous water environment. What should be noteworthy is that the gel fraction in both the PUD1 and PUD2 is low, which may be considered negligible.
This result suggests that both the two samples mainly consist of linear chains without crosslinking. Moreover, the swelling capability value of PUD1 is lower than that of PUD2 in Table 2, indicating that PUD1 presents a slightly lower densely cross-linked network. Because the more densely cross-linked networks, the lesser the networks swell in the solvent, and the smaller is the swelling capability [49]. It also suggests that cross-linked network forms may hardly in a homogeneous water environment for polyurethane.
A facile method, in situ water generating process (in situ WGP) was developed to synthesize WPU dispersion with rod-like nanoparticles and decreasing organic solvent (no acetone). According to the results of FTIR, 1H-NMR, TEM and water swelling, the sample (PUD1) synthesized via this novel process has a larger phase separation and more intense hard and soft segment interaction comparing with the external water addition synthesis process, which is mainly caused by the large amount of hydrogen bonds. The PUD1 had two kinds of hydrogen bonds that are hard-hard segment and hard-soft segment interaction hydrogen bonds. In addition, polyurethane obtained via in situ WGP presents stronger interaction between hard and soft segments than that prepared through external water addition process. It is suggested that the chains of the PUD1 aggregate to form nanorods under the function of strong hydrogen bonds. It was anticipated that polyurethane synthesized from in situ WGP could be potentially applied as degradable material, functional coatings and binders. Meanwhile, owing to the green process and waterborne polymers, the preparation of water-based inks, coatings and even adhesives may be easier. The in situ WGP may pave the way to study the kinetics of the emulsification, chain extension and dispersion process for WPU prepolymer to obtain dispersions in different morphology and performance for wide application.
Funding Statement: The author acknowledges the financial support provided by the National Natural Science Foundation of China [Grant No. 51802259], China Postdoctoral Science Foundation Funded Project [Grant No. 2019M663785], the Opening Project of Shanxi Key Laboratory of Advanced Manufacturing Technology [Grant No. XJZZ202001], the Scientific Research Project of Shaanxi Education Department [Grant No. 20JS108], the Promotion Program for Youth of Shaanxi University science and technology association [Grant No. 20190415], Fund of Key Laboratory of Processing and Quality Evaluation Technology of Green Plastics of China National Light Industry council [Grant No. PQETGP2019003], the Innovation Guidance of Technology Program of Shaanxi Province [Grant No. 2020CGXNG-022], Xi’an Programs for Science and Technology Plan [Grant No. 2020KJRC0090]. The author also acknowledges the previous work [https://www.sciencedirect.com/science/article/pii/S030094401630323X].
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
1. Ludwig, R. (2001). Water: From clusters to the bulk. Angewandte Chemie International Edition, 40(10), 1808–1827. DOI 10.1002/(ISSN)1521-3773. [Google Scholar] [CrossRef]
2. Duer, M., Veis, A. (2013). Water brings order. Nature Materials, 12(12), 1081. DOI 10.1038/nmat3822. [Google Scholar] [CrossRef]
3. Nihonyanagi, S., Yamaguchi, S., Tahara, T. (2010). Water hydrogen bond structure near highly charged interfaces is not like ice. Journal of the American Chemical Society, 132(20), 6867–6869. DOI 10.1021/ja910914g. [Google Scholar] [CrossRef]
4. Chong, S. H., Ham, S. (2014). Interaction with the surrounding water plays a key role in determining the aggregation propensity of proteins. Angewandte Chemie International Edition, 53(15), 3961–3964. DOI 10.1002/anie.201309317. [Google Scholar] [CrossRef]
5. Wang, C., Zhang, J., Wang, H., He, M., Ding, L. et al. (2021). Simultaneously improving the fracture toughness and flame retardancy of soybean oil-based waterborne polyurethane coatings by phosphorus-nitrogen chain extender. Industrial Crops and Products, 163, 113328. DOI 10.1016/j.indcrop.2021.113328. [Google Scholar] [CrossRef]
6. Zhu, A., Wang, H., Zhang, C., Rui, M., Zhou, C. et al. (2017). A facile, solvent-free and scalable method to prepare poly (aniline-co-5-aminosalicylic acid) with enhanced electrochemical activity for corrosion protection. Progress in Organic Coatings, 112, 109–117. DOI 10.1016/j.porgcoat.2017.07.006. [Google Scholar] [CrossRef]
7. Zhang, C., Garrison, T. F., Madbouly, S. A., Kessler, M. R. (2017). Recent advances in vegetable oil-based polymers and their composites. Progress in Polymer Science, 71, 91–143. DOI 10.1016/j.progpolymsci.2016.12.009. [Google Scholar] [CrossRef]
8. Wang, C., Zhang, J., Huang, J., Wang, H., He, M. et al. (2021). Flame retardant modified bio-based waterborne polyurethane dispersions derived from castor oil and soy polyol. European Journal of Lipid Science and Technology, 123(4), 2000248. DOI 10.1002/ejlt.202000248. [Google Scholar] [CrossRef]
9. Zhou, X., Fang, C., Lei, W., Su, J., Li, L. et al. (2017). Thermal and crystalline properties of waterborne polyurethane by in situ water reaction process and their potential application as biomaterial. Progress in Organic Coatings, 104, 1–10. DOI 10.1016/j.porgcoat.2016.12.001. [Google Scholar] [CrossRef]
10. Williams, S. R., Wang, W., Winey, K. I., Long, T. E. (2008). Synthesis and morphology of segmented poly (tetramethyleneoxide)-based polyurethanes containing phosphonium salts. Macromolecules, 41(23), 9072–9079. DOI 10.1021/ma801942f. [Google Scholar] [CrossRef]
11. Kang, S. Y., Ji, Z., Tseng, L. F., Turner, S. A., Villanueva, D. A. et al. (2018). Design and synthesis of waterborne polyurethanes. Advanced Materials, 30(18), 1706237. DOI 10.1002/adma.201706237. [Google Scholar] [CrossRef]
12. Nasrullah, M. J., Bahr, J. A. (2009). Automated parallel polyurethane dispersion synthesis and characterization. Journal of Coatings Technology and Research, 6(1), 1–10. DOI 10.1007/s11998-008-9102-z. [Google Scholar] [CrossRef]
13. Asplund, J. O. B., Bowden, T., Mathisen, T., Hilborn, J. (2006). Variable hard segment length in poly(urethane urea) through excess of diisocyanate and vapor phase addition of water. Macromolecules, 39(13), 4380–4385. DOI 10.1021/ma060361k. [Google Scholar] [CrossRef]
14. Shahrousvand, M., Sadeghi, G. M. M., Shahrousvand, E., Ghollasi, M., Salimi, A. (2017). Superficial physicochemical properties of polyurethane biomaterials as osteogenic regulators in human mesenchymal stem cells fates. Colloids and Surfaces B: Biointerfaces, 156, 292–304. DOI 10.1016/j.colsurfb.2017.04.059. [Google Scholar] [CrossRef]
15. Kim, C. K., Kim, B. K., Jeong, H. M. (1991). Aqueous dispersion of polyurethane ionomers from hexamethylene diisocyanate and trimellitic anhydride. Colloid and Polymer Science, 269, 895–900. DOI 10.1007/BF00657306. [Google Scholar] [CrossRef]
16. Madbouly, S. A., Otaigbe, J. U. (2009). Recent advances in synthesis, characterization and rheological properties of polyurethane and POSS/polyurethane nanocomposites dispersions and films. Progress in Polymer Science, 34(12, 283–1332. DOI 10.1016/j.progpolymsci.2009.08.002. [Google Scholar] [CrossRef]
17. Dieterich, D. (1981). Aqueous emulsions, dispersions and solutions of polyurethanes; synthersis and properties. Progress in Organic Coatings, 9(3), 281–340. DOI 10.1016/0033-0655(81)80002-7. [Google Scholar] [CrossRef]
18. Matyjaszewski, K., Tsarevsky, N. V. (2009). Nanostructured functional materials prepared by atom transfer radical polymerization. Nature Chemistry, 1(4), 276–288. DOI 10.1038/nchem.257. [Google Scholar] [CrossRef]
19. Mattia, J., Painter, P. (2007). A comparison of hydrogen bonding and order in a polyurethane and poly(urethane−urea) and their blends with poly(ethylene glycol). Macromolecules, 40(5), 1546–1554. DOI 10.1021/ma0626362. [Google Scholar] [CrossRef]
20. Yilgor, I., Yilgor, E., Guler, I. G., Ward, T. C., Wilkes, G. L. (2006). FTIR investigation of the influence of diisocyanate symmetry on the morphology development in model segmented polyurethanes. Polymer, 47(11), 4105–4114. DOI 10.1016/j.polymer.2006.02.027. [Google Scholar] [CrossRef]
21. Zhou, X., Song, Y., Wang, D., Fang, C., Xie, L. et al. (2021). Functional nano-fillers in waterborne polyurethane/acrylic composites and the thermal, mechanical and dielectrical properties. Journal of Applied Polymer Science, 138(33), e50822. DOI 10.1002/app.50822. [Google Scholar] [CrossRef]
22. Zhou, X., Hao, Y., He, X., Zhou, D., Xie, L. et al. (2020). Protean morphology of waterborne polyurethane dispersion: An overview of nanoparticles from sphere to irregular elongated shape. Progress in Organic Coatings, 146, 105742. DOI 10.1016/j.porgcoat.2020.105742. [Google Scholar] [CrossRef]
23. Wu, Y., Xu, Y., Wang, D., Zhao, Y., Weng, S. et al. (2004). FT-IR spectroscopic investigation on the interaction between nylon 66 and lithium salts. Journal of Applied Polymer Science, 91(5), 2869–2875. DOI 10.1002/(ISSN)1097-4628. [Google Scholar] [CrossRef]
24. Das, S., Cox, D. F., Wilkes, G. L., Klinedinst, D. B., Yilgor, I. et al. (2007). Effect of symmetry and H-bond strength of hard segments on the structure-property relationships of segmented, nonchain extended polyurethanes and polyureas. Journal of Macromolecular Science Part B–Physics, 46(5), 853–875. DOI 10.1080/00222340701388805. [Google Scholar] [CrossRef]
25. Zhang, J. Y., Beckman, E. J., Piesco, N. P., Agarwal, S. (2000). A new peptide-based urethane polymer: Synthesis, biodegradation, and potential to support cell growth in vitro. Biomaterials, 21(12), 1247–1258. DOI 10.1016/S0142-9612(00)00005-3. [Google Scholar] [CrossRef]
26. Wang, C. B., Cooper, L. (1983). Morphology and properties of segmented polyether polyurethaneureas. Macromolecules, 16(5), 775–786. DOI 10.1021/ma00239a014. [Google Scholar] [CrossRef]
27. Tang, J., Javaid, M. U., Pan, C., Yu, G., Berry, R. M. et al. (2020). Self-healing stimuli-responsive cellulose nanocrystal hydrogels. Carbohydrate Polymers, 229, 115486. DOI 10.1016/j.carbpol.2019.115486. [Google Scholar] [CrossRef]
28. Prabhakar, A., Chattopadhyay, D. K., Jagadeesh, B., Raju, K. V. S. N. (2005). Structure investigations of polypropylene glycol (PPG) and isophorone diisocyanate (IPDI)-based polyurethane prepolymer by 1D and 2D NMR spectroscopy. Journal of Polymer Science Part A: Polymer Chemistry, 43(6), 1196–1209. DOI 10.1002/(ISSN)1099-0518. [Google Scholar] [CrossRef]
29. Zhou, X., Deng, J., Fang, C., Yu, R., Lei, W. et al. (2020). Preparation and characterization of lysozyme@carbon nanotubes/waterborne polyurethane composite and the potential application in printing inks. Progress in Organic Coatings, 142, 105600. DOI 10.1016/j.porgcoat.2020.105600. [Google Scholar] [CrossRef]
30. Garrett, J. T., Lin, J. S., Runt, J. (2002). Influence of preparation conditions on microdomain formation in poly(urethane-urea) block copolymers. Macromolecules, 35(1), 161–168. DOI 10.1021/ma010915d. [Google Scholar] [CrossRef]
31. Lu, Y., Larock, R. C. (2008). Soybean-oil-based waterborne polyurethane dispersions: Effects of polyol functionality and hard segment content on properties. Biomacromolecules, 9(11), 3332–3340. DOI 10.1021/bm801030g. [Google Scholar] [CrossRef]
32. Li, Q. A., Sun, D. C. (2007). Synthesis and characterization of high solid content aqueous polyurethane dispersion. Journal of Applied Polymer Science, 105(5), 2516–2524. DOI 10.1002/(ISSN)1097-4628. [Google Scholar] [CrossRef]
33. Madbouly, S. A., Otaigbe, J. U., Nanda, A. K., Wicks, D. A. (2005). Rheological behavior of aqueous polyurethane dispersions: Effects of solid content, degree of neutralization, chain extension, and temperature. Macromolecules, 38(9), 4014–4023. DOI 10.1021/ma050453u. [Google Scholar] [CrossRef]
34. Lattuada, M., Sandkühler, P., Wu, H., Sefcik, J., Morbidelli, M. (2004). Kinetic modeling of aggregation and gel formation in quiescent dispersion of polymer colloids. Macromolecular Symposia, 206(1), 307–320. DOI 10.1002/(ISSN)1521-3900. [Google Scholar] [CrossRef]
35. Kumar, A. P., Depan, D., Tomer, N. S., Singh, R. P. (2009). Nanoscale particles for polymer degradation and stabilization—Trends and future perspectives. Progress in Polymer Science, 34(6), 479–515. DOI 10.1016/j.progpolymsci.2009.01.002. [Google Scholar] [CrossRef]
36. Zhang, J., Hu, C. P. (2008). Synthesis, characterization and mechanical properties of polyester-based aliphatic polyurethane elastomers containing hyperbranched polyester segments. European Polymer Journal, 4, 3708–3714. DOI 10.1016/j.eurpolymj.2008.08.019. [Google Scholar] [CrossRef]
37. Shin, S., Gihm, S. H., Park, C. R., Kim, S., Park, S. Y. (2013). Water-soluble fluorinated and PEGylated cyanostilbene derivative: An amphiphilic building block forming self-assembled organic nanorods with enhanced fluorescence emission. Chemistry of Materials, 25(16), 3288–3295. DOI 10.1021/cm400988a. [Google Scholar] [CrossRef]
38. Zhou, X., Su, J., Wang, C., Fang, C., He, X. et al. (2020). Design, preparation and measurement of protein/CNTs hybrids: A concise review. Journal of Materials Science & Technology, 46, 74–87. DOI 10.1016/j.jmst.2020.01.008. [Google Scholar] [CrossRef]
39. Liao, W. Z., Lu, Y. J., Fu, J. N., Ning, Z. X., Yang, J. G. et al. (2015). Preparation and characterization of dictyophora indusiata polysaccharide-zinc complex and its augmented antiproliferative activity on human cancer cells. Journal of Agricultural and Food Chemistry, 63(29), 6525–6534. DOI 10.1021/acs.jafc.5b00614. [Google Scholar] [CrossRef]
40. Zhang, C., Madbouly, S. A., Kessler, M. R. (2015). Biobased polyurethanes prepared from different vegetable oils. ACS Applied Materials & Interfaces, 7(2), 1226–1233. DOI 10.1021/am5071333. [Google Scholar] [CrossRef]
41. Zhang, C., Wu, H., Kessler, M. R. (2015). High bio-content polyurethane composites with urethane modified lignin as filler. Polymer, 69(9), 52–57. DOI 10.1016/j.polymer.2015.05.046. [Google Scholar] [CrossRef]
42. Zhang, C., Schneiderman, D. K., Cai, T., Tai, Y. S., Fox, K. et al. (2016). Optically active β-methyl-δ-valerolactone: Biosynthesis and polymerization. ACS Sustainable Chemistry & Engineering, 4(8), 4396–4402. DOI 10.1021/acssuschemeng.6b00992. [Google Scholar] [CrossRef]
43. Loh, X. J., Sng, K. B. C., Li, J. (2008). Synthesis and water-swelling of thermo-responsive poly(ester urethane)s containing poly(3-adipic acidpoly(ethylene glycol) and poly(propylene glycol). Biomaterials, 29(22), 3185–3194. DOI 10.1016/j.biomaterials.2008.04.015. [Google Scholar] [CrossRef]
44. Wen, T. C., Wang, Y. J., Cheng, T. T., Yang, C. H. (1999). The effect of DMPA units on ionic conductivity of PEG–DMPA–IPDI waterborne polyurethane as single-ion electrolytes. Polymer, 40(14), 3979–3988. DOI 10.1016/S0032-3861(98)00625-9. [Google Scholar] [CrossRef]
45. Tatai, L., Moore, T. G., Adhikari, R., Malherbe, F., Jayasekara, R. et al. (2007). Thermoplastic biodegradable polyurethanes: The effect of chain extender structure on properties and in-vitro degradation. Biomaterials, 28(36), 5407–5417. DOI 10.1016/j.biomaterials.2007.08.035. [Google Scholar] [CrossRef]
46. Hiltunen, K., Tuominen, J., Seppälä, J. V. (1998). Hydrolysis of lactic acid-based poly(ester-urethane)s. Polymer International, 47(2), 186–192. DOI 10.1002/(ISSN)1097-0126. [Google Scholar] [CrossRef]
47. Stark, N. M. (2016). Opportunities for cellulose nanomaterials in packaging films: A review and future trends. Journal of Renewable Materials, 4(5), 313–326. DOI 10.7569/JRM.2016.634115. [Google Scholar] [CrossRef]
48. Oliaei, E., Heidari, B. S., Davachi, S. M., Bahrami, M., Davoodi, S. et al. (2016). Warpage and shrinkage optimization of injection-molded plastic spoon parts for biodegradable polymers using taguchi, ANOVA and artificial neural network methods. Journal of Materials Science & Technology, 32(8), 710–720. DOI 10.1016/j.jmst.2016.05.010. [Google Scholar] [CrossRef]
49. Lopez, A., Contraires, E. D., Canetta, E., Creton, C., Keddie, J. L. et al. (2011). Waterborne polyurethane-acrylic hybrid nanoparticles by miniemulsion polymerization: Applications in pressure-pensitive adhesives. Langmuir, 27(7), 3878–3888. DOI 10.1021/la104830u. [Google Scholar] [CrossRef]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |