| Journal of Renewable Materials |
DOI: 10.32604/jrm.2021.014476
ARTICLE
Synthesis and Characterization of Thermoplastic Poly(Ester Amide)s Elastomer (TPEaE) Obtained from Recycled PET
Institute of Organic and Polymeric Materials, Research and Development Center of Smart Textile Technology, National Taipei University of Technology, Taipei, 10608, Taiwan
*Corresponding Authors: Chin-Wen Chen. Email: cwchen@ntut.edu.tw; Syang-Peng Rwei. Email: f10714@ntut.edu.tw
Received: 30 September 2020; Accepted: 09 November 2020
Abstract: A series of thermoplastic polyester elastomer (TPEE) and thermoplastic poly(ester amide)s elastomer (TPEaE) copolymers were obtained by depolymerizing PET (polyethylene terephthalate) by which the waste PET can be efficiently recovered and recycled into value-added products from a practical and economical point of view. The structure of TPEE and TPEaE was identified using nuclear magnetic resonance (NMR) and Fourier transform infrared spectroscopy (FT-IR). Differential scanning calorimetry (DSC) data showed that the melting temperature (Tm) decreased with the amide content increased. The glass transition temperature (Tg) was increased as introducing the amide group, and the formation of amide-ester and amide-amide hydrogen bonds increased the intermolecular chain force. The intrinsic viscosity (η) showed the tendency of increment from TPEE (0.53 dL g−1) to TPEaE-5% (0.72 dL g−1) due to the reinforcement of hydrogen bond and chain entanglement.
Keywords: Recycle polyethylene terephthalate (r-PET); depolymerization; thermalplastic poly(ester amide)s copolymer (TPEaE)
Poly(ethylene terephthalate) (PET) is widely used in engineering, textile fiber, and beverage bottle because of its low-cost, low energy requirement, excellent mechanical properties, and chemical resistance [1,2]. Thus, the excessive productions of PET result in a large amount of PET waste [3,4]. Recycling PET (r-PET) is one of the critical tasks for reducing environmental pollution and managing wastes. Therefore, there is a growing interest in the development of new materials based on recycled PET by mechanical or chemical recycling from waste [5–8].
PET mechanical recycling is a relatively simple and low investment, which provides an efficient method to resolve plastic pollution. Several studies have been conducted to investigate the effect of the chain extender or plastic resin on r-PET [9–11]. For instance, Mohammadreza et al. studied the r-PET blends with different content of chain extender and poly(butylene terephthalate) (PBT) to modify their thermal and mechanical properties [10]. However, the complexity and contamination of PET waste make mechanical recycling extremely challenging [12]. Moreover, mechanical recycling contains two types of degradation: degradation caused by reprocessing (thermal-mechanical degradation) and degradation during lifetime [13], limiting its feasibility, and led to weak properties and low-grade performance of PET.
On the contrary, chemical recycling provides an alternative recovery approach by depolymerizing the PET into well-defined monomer, oligomer, or other chemical substances as precursors to value-added productions for industrial and commercial applications [5,6,14,15]. Recently, Leng et al. depolymerize the r-PET to obtain PET derived additives, which can be combined with scrap tires to modify asphalt binders [16]. Scremin et al. successfully prepare the urethane adhesives based on PET’s chemical recycling [17]. Several methods to depolymerize PET for chemical recycling, such as alcoholysis, hydrolysis, ammonolysis, aminolysis, and hydrogenation [7]. In all solvolytic reactions, alcoholysis is the most common way to chemical recycling PET scrap without colorants or dyes nowadays [18,19]. The alcoholysis of PET in the presence of ethylene glycol (EG) leads to monomers (bis(2-hydroxyethyl) terephthalate, EG, and other PET glycosylate, etc. [20]. These compounds are utilized as reactants for the precursors in the synthesis of polyester derivative, e.g., thermoplastic polyester elastomer (TPEE). The synthesis of TPEE with terephthalate group is based on the polymerization of hard segment (PET, (poly(trimethylene terephthalate), and PBT) and soft segment (poly(ethylene glycol) and poly(tetramethylene glycol) (PTMG)). The most common commercial TPEE material is Hytrel (Dupont), which is composed of the hard segment of PBT and the soft segment of PTMG (PBT-block-PTMG) [21]. Moreover, the presence of PET as a hard segment (PET-block-PTMG) exhibit similar properties and have the same application as Hytrel. The phase separation and crystallization process of PET segments in the amorphous region of PTMG have been reported [22]. Recently, Paszkiewicz et al. incorporate cyclohexanedimethanol (CHDM) into PET-block-PTMG to investigate thermal and mechanical properties [23]. Thus, the modification with an amide bond to increase intermolecular forces and improve the thermal and mechanical properties of PET-block-PTMG has been investigated.
It is well-known that polyamides with hydrogen bonds possess excellent physical and thermal properties due to the interchain force [24]. Many research groups incorporate the amide group into polyester via chemical synthesis to form poly(ester amide) (PEA) copolymer [25–28]. The PEA, combine with the favorable properties of both polyesters and polyamides, has attracted a class of promising products. For instance, Gao et al. conjugate the amide bond into PET backbone to increase water and gas barrier properties thanks to the mutual attraction of intermolecular hydrogen force [29]. Winnacker et al. mention that PEA increases medical applications due to excellent mechanical properties and biodegradability [30]. All these reports reveal that introducing the amide bond into polyester to form PEA analogs may be an effective way to modify polyester’s thermal and mechanical behavior.
In this study, the introduction of 1,6-hexanediamine (HMDA) into PET-block-PTMG was developed to form thermoplastic poly(ester amide) elastomer (TPEaE) via an environmentally friendly procedure. The hard segment of TPEaE was prepared by alcoholysis of r-PET to obtain the low average polymerization number oligomers. The soft segment (PTMG) was added to gain chain flexibility. Then, the HMDA with an amino group forming hydrogen bonding was regarded as a functional group to increase intermolecular attractions. The structures and compositions were investigated using 1H NMR and FT-IR spectroscopy. The mechanical properties, crystallinity, and thermal properties were identified using the tensile test, XRD, DSC, and DMA.
Recycled poly(ethylene terephthalate) (r-PET, Hong-Sheng Green Wealth Co., Ltd, ChangHua, Taiwan). Ethylene glycol (EG, commercial-grade) was purchased from Emperor Chemical Co., Ltd. (Taipei, Taiwan). Poly(tetramethylene ether)glycol (PTMG, Mn~1000 g mol−1), 1,6-hexanediamine (HMDA, 98% purity), titanium(IV) butoxide (TBT, 97%), and trifluoroacetic acid-d (d-TFA, 99.5%) were purchased from Aldrich (Westport Center. Louis, United States). Phenol (97%) was purchased from the Aencore chemical (Whitehorse Road, Surrey Hills, Australia). 1,1,2,2-Tetrechloroethane (97%) was purchased from Showa Chemical Industry Co., Ltd. (Shimo-Meguro Meguro-Ku, Tokyo, Japan). CHINOX 1010 (98%) was purchased from Double Bonding Partnership (Taipei, Taiwan).
2.1.2 Synthesis of TPEE and TPEaE Copolymer
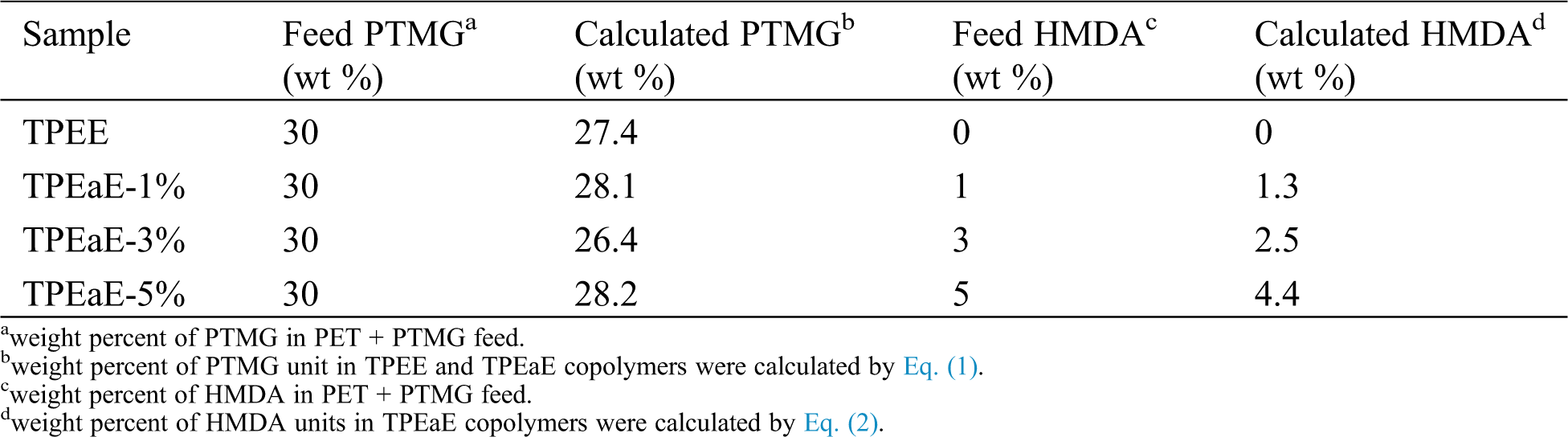
All the synthesizing process are shown in Scheme 1. The r-PET chip (420 g) and EG (20g, 0.322 mol) were charged into the stainless steel 2 L reactor to obtain PET oligomer (Scheme 1a). The reactor with the chemicals was heated to 275°C in an atmosphere of nitrogen for 2 h for depolymerization reaction. Then, the PTMG (180 g, 0.18 mol) and a different weight ratio of HMDA (0, 1, 3, and 5 wt%) were added into the reactor with 400 ppm of TBT as the catalyst and 1000 ppm of CHINOX 1010 as antioxidants for transesterification and amidation (Scheme 1b). After 1 h reaction time, the pressure was then carefully reduced (P < 30 mbar) for 30 min and then further reduce (P < 1 mbar) for 1.5–2 h to removed by-products for polycondensation. The stirring torque was recorded, and the reaction was finished when it reached approximately 1.2 times the reference value. The composition of the different samples was listed in Tab. 1. It was denoted as TPEE and TPEaE-n% (n = 1, 3, and 5), where TPEE stands for 0 wt% of HMDA in the copolymer, and 1, 3, and 5 stand for the weight ratio of HMDA in the copolymer.
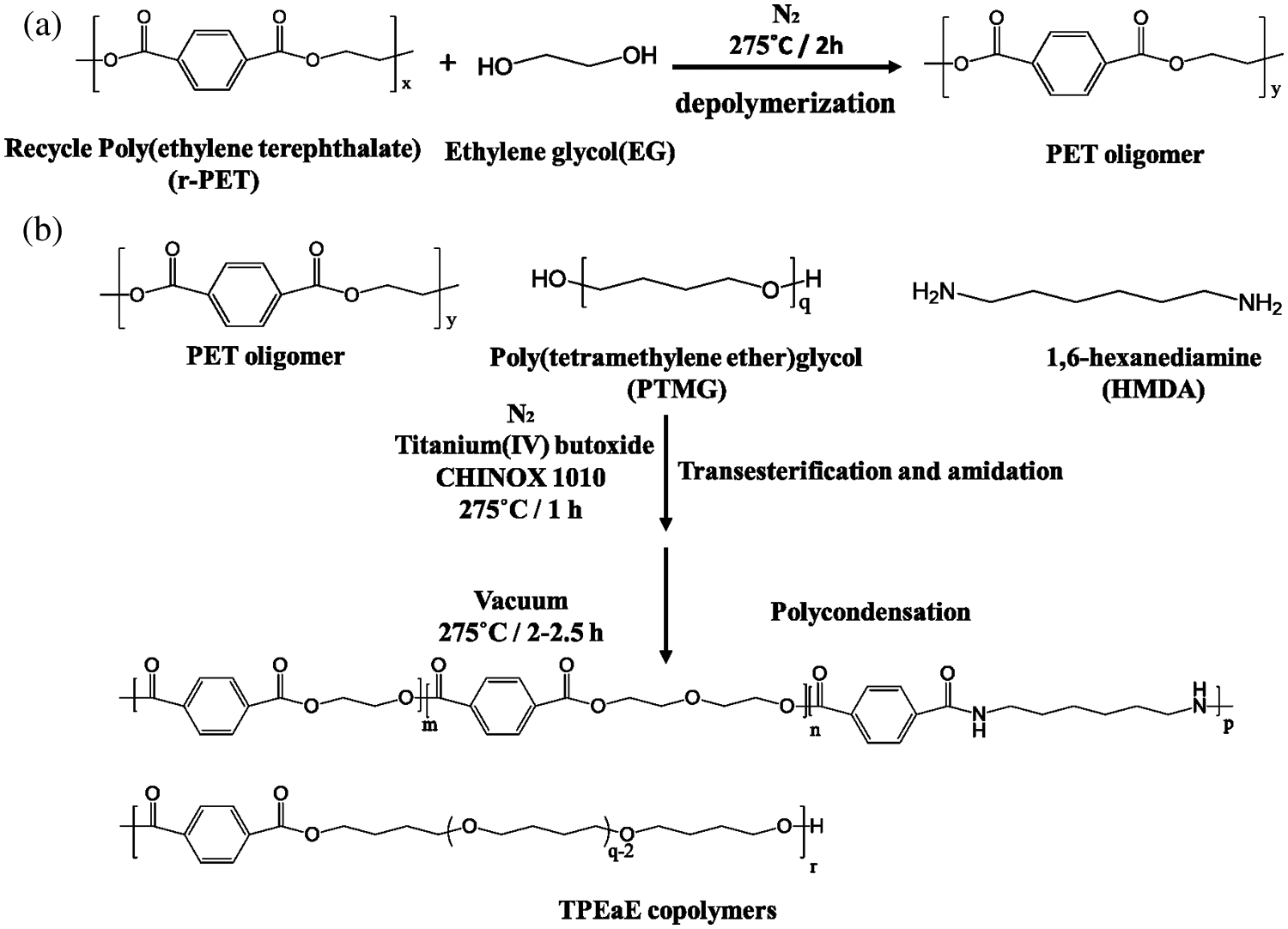
Scheme 1: (a) depolymerization of PET, (b) synthesis of TPEE, and TPEaE-n% copolymers
Table 1: Molecular characterization of TPEE and TPEaE-n% copolymers
2.2.1 Nuclear Magnetic Resonance (1H NMR) Spectroscopic Analysis
1H (300 MHz, Billerica, Massachusetts, US) NMR spectrometer was recorded on a Bruker. Each sample (5–10 mg) was dissolved in 1 mL of d-TFA, and the measurement was carried out with 16 scans at room temperature.
2.2.2 Infrared Spectroscopic Analysis
Fourier transform infrared (FT-IR) spectroscopy was recorded with a PerkinElmer spectrum One (Cinnaminson, New Jersey, USA) in attenuated total reflection (ATR) mode. The sample was analyzed at a resolution of 4 cm−1 with 16 scans over the wavenumber range of 650–4000 cm−1.
2.2.3 Differential Scanning Calorimetry
Thermal analysis of polymers was conducted using a PerkinElmer DSC 800 (Waltham, MA, USA). The sample (3–6 mg) was performed from room temperature to 300°C to erase thermal history and cooling scans with a heating rate of –10 °C min−1, than second heating scans with a heating rate of 10°C min−1 in the range 0 to 300°C. All measurements were carried out in a nitrogen atmosphere using aluminum pans.
2.2.4 Dynamic Mechanical Analysis
The viscoelastic properties of polymers were analyzed using a ThechMax DMS 6100 (Tokyo, Japan). The testing was made in compression mode at 200 mN with heating from –100 to 150°C at a heating rate of 10°C min−1 and a fixed frequency of 1 Hz.
2.2.5 Thermogravimetric Analysis
Thermal gravimetric analysis was carried out with 5–10 mg samples using STA 7200 HITACHI (Tokyo, Japan). The testing was performed from 50 to 600°C range at a heating rate of 10°C min−1 in a nitrogen atmosphere that was flowing at a rate of 50 ml min−1.
The powder of the copolymers was prepared, and then the XRD pattern of the sample was recorded over 2θ angles from 10° to 35° using a Malvern Panalytical X’Pert3 powder diffractometer (Malvern, UK) with CuKα radiation at a scanning speed of 0.2° min−1.
Tensile tests were conducted using a Cometech (Taichung, Taiwan) QC-505M2F by ASTM638 type IV standard at a strain rate of 100 mm min−1. Dumb-bell shaped specimens with 3 × 6 mm2 cross-section were prepared using a HAAKE MiniJet injection molding machine (Thermo scientific). The sample was loading of powders within the cylinder at 250°C for 2 min. Then the pressure of 280 bar was applied to inject the sample into the mold. The as-obtained sample was drawn out from the mold until cooled to room temperature. The average value of all the data was obtained on five specimens.
Intrinsic viscosity (η) was determined using an Ubbelohde viscometer at 25°C in a phenol/1,1,2,2-tetrachloroethane (60/40, w/w) with a concentration of 0.1 dL g−1. To ensure that samples were dissolved entirely, each sample was maintained 80°C and cooled to room temperature. The following empirical equation was applied:
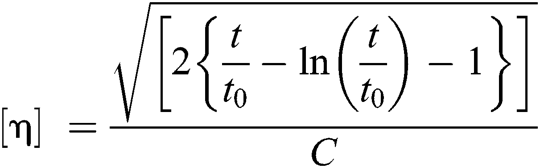
where C was the concentration of solution; t0 is the pure solvent’s flow time, and t is the solution’s flow time.
3.1 Chemical Structure of Copolymers
The chemical structure of PET oligomer and TPEE and TPEaE-n% copolymers were characterized using 1H NMR and FT-IR. The value of the intrinsic viscosity of PET oligomer was 0.12 dL g−1. The result of 1H NMR and FT-IR for PET oligomer was presented in Fig. S1 and Fig. S2. The 1H NMR spectra of TPEE and TPEaE-n% (n = 1, 3, and 5) were shown in Fig. 1, and the chemical shifts were summarized in Tab. 2. Moreover, the composition in the polymer can be calculated from 1H NMR analysis, according to Eqs. (1) and (2). Where Ic, Ik, Ii, Ie, and Ib were the abbreviations of the integral intensities of chemical shift c, k, i, e, and b, respectively, and their molecular weight were 192 g mole−1 (–C8H4O2–OCH2CH2O–), 72 g mole−1 (–OCH2CH2CH2CH2–), 88 g mole−1 (–OCH2CH2CH2CH2O, 236 g mole−1 (–C8H4O2–OCH2CH2OCH2CH2O–), 244 g mole−1 (–C8H4O2–NHCH2CH2CH2CH2CH2CH2NH–), and the results were shown in Tab. 1.
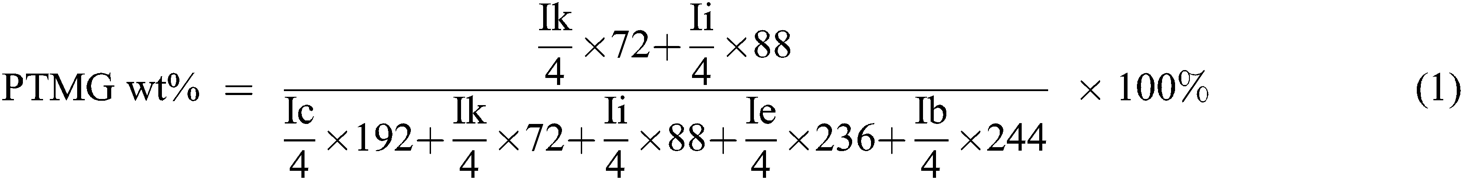
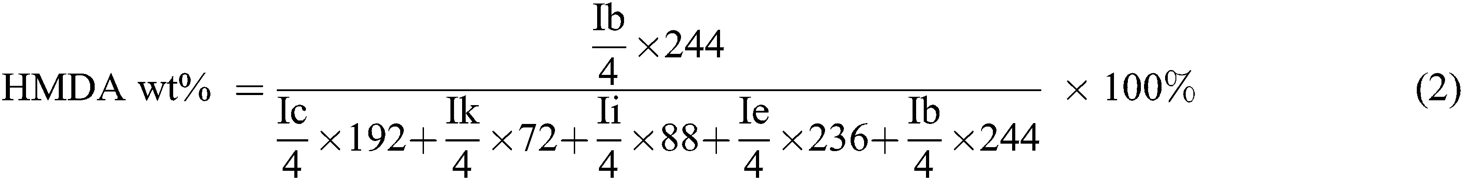
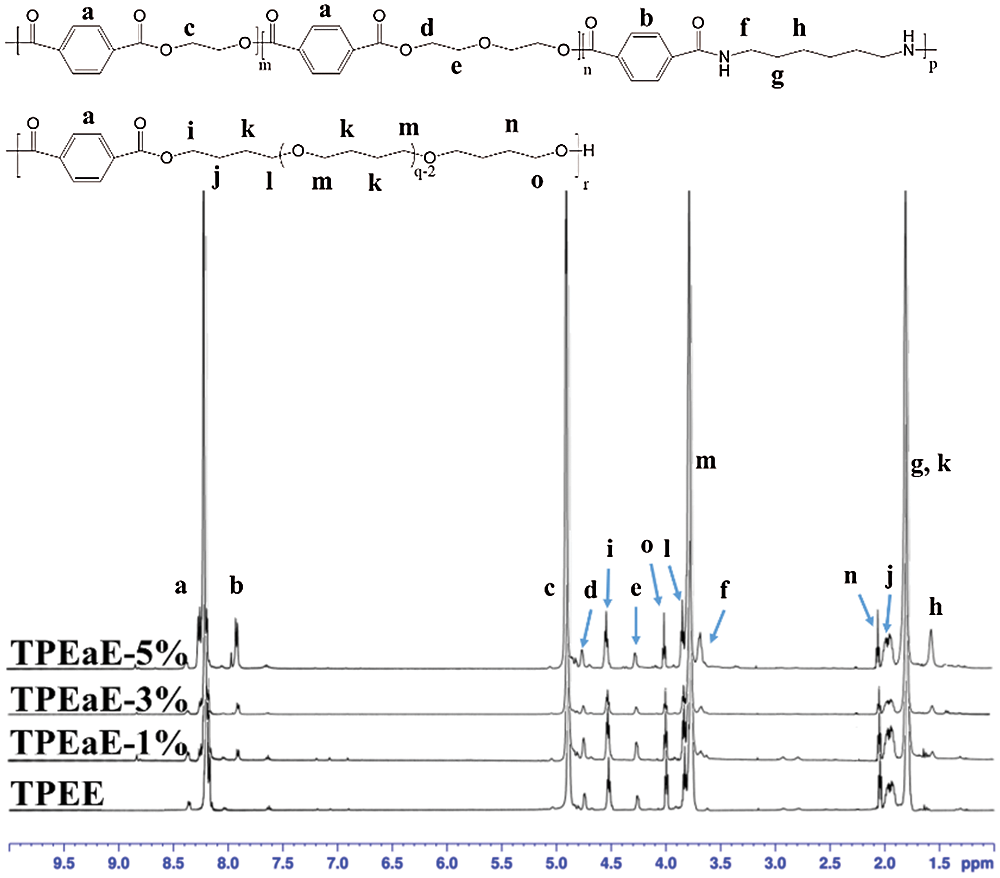
Figure 1: 1H NMR spectra of TPEE and TPEaE-n% copolymers
Table 2: the chemical shifts of TPEE and TPEaE-n% copolymers determined in 1H NMR
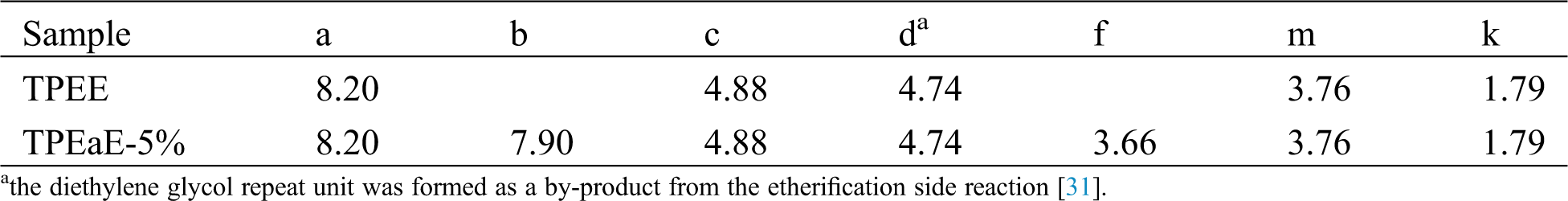
Fig. 2 showed the FTIR spectra of TPEE and TPEaE-n% copolymers. The characteristic peaks of the benzene ring, such as C=C, were observed at 1578 cm−1. The strong absorption around 1715 cm−1 was attributed to the C=O of the ester group. The bond peaks around 1095 cm−1 and 1017 cm−1 belong to H-C-H and C-O of the ester group. The C-N and N-H absorption of amide group bonds were found at 1579 cm−1.
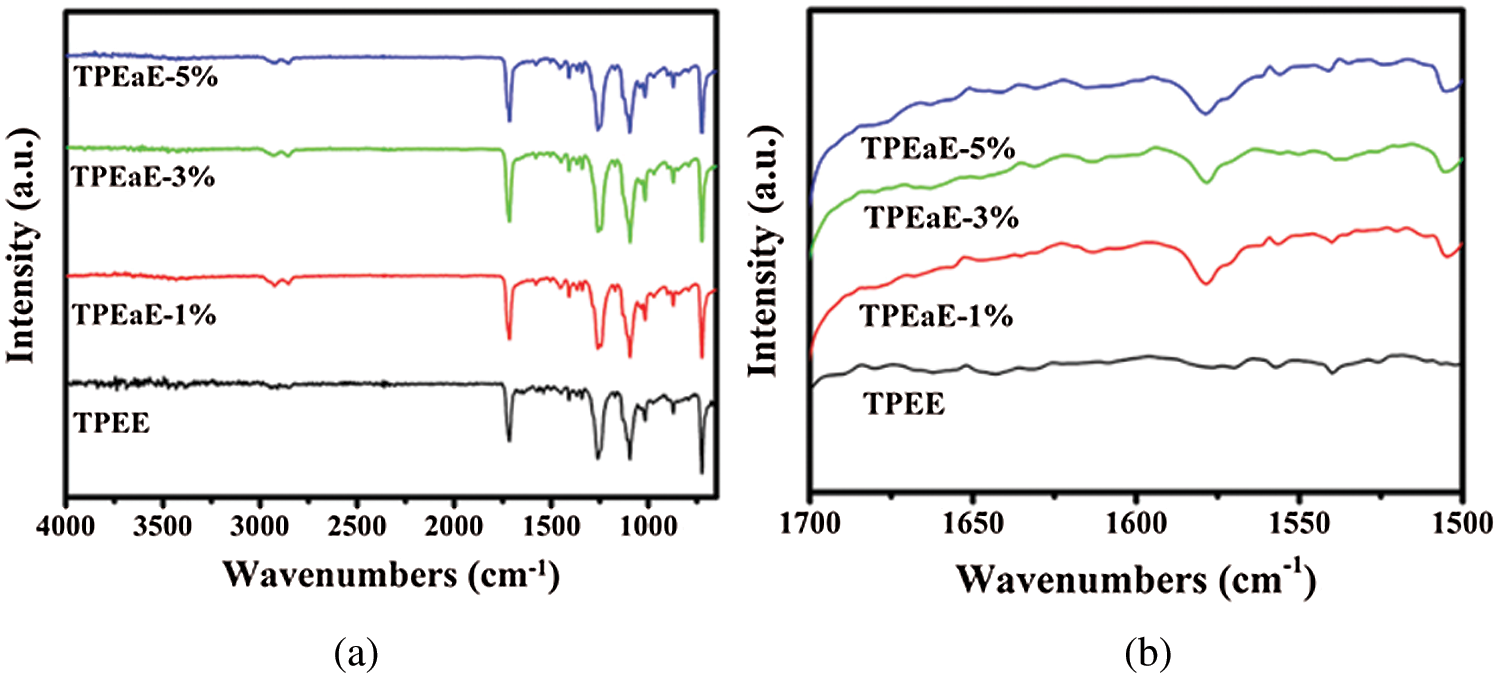
Figure 2: (a) FT-IR spectra, and (b) details of FT-IR spectra in the region 1700 to 1500 cm−1 for TPEE and TPEaE-n% copolymers
3.2 Thermal Property of Copolymers
The TPEE and TPEaE-n% copolymers’ thermal behaviors were measured using DSC, and the results were shown in Tab. 3 and Fig. 3. As shown in Fig. 3, the melting temperature and fusion enthalpies of poly(ester amide)s were obtained from the first heating scans to erase thermal history and cooling scans with a heating rate of −10°C min−1, than second heating scans with a heating rate of 10°C min−1 in the range 0 to 300°C. Typically, the Tm values of PEA would be increased when the increment of uniform diamide content, suggesting the replacement of esters by amide residue, would lead to hydrogen bonding formation to increase Tm [32,33]. However, the Tm values decreased as the ratio of HMDA increased because of the diamide segment of uneven length for neat TPEE, destroying the regularity of chain order [34,35]. Besides, Gao et al. indicate the amide-bond units of the PEA structure facilitate its crystallization process during the cooling cycle due to the formation of crystal nuclei from amide units [29]. Thus, the crystallization temperature (Tc) shows the tendency of decline with the introduction of the HMDA in our study. The reason was inferred that even though the amide-bond might expedite the crystallization process, the diamide segment’s varying lengths disturb the crystallization zone (hard segment), leading to a decrease in the Tc value. The DSC peak becomes broader and less accurate with increasing the amide content, suggesting that the ester amide segment’s higher content possessed a wide variety of lamellar sizes and composition [34,35]. The viscoelastic mechanical properties of synthesized copolymers were using DMA. The Tg indicated the temperature at which the chain segments undergo co-ordinated molecular motions [36]. As seen in Fig. 4 and Tab. 3, the values of Tg was found to increase with increasing HMDA content from TPEE (−2.26°C) to TPEaE-5% (21.59°C) because the amide group could form a hydrogen bond, resulting in increasing intermolecular interaction and decreasing mobility of polymer chain [29,37]. Hibbs et al. revealed that adding the bis-ester diamide monomers into PET structure leads to an increase in Tg. The tendency to increment Tg was ascribed to hydrogen bonding or the increasing rigidity of the polymer backbone, which suppresses the polymer chain [38]. Gao et al. also observed incorporating 4 mol % content amide-bond unit to PET; Tg value was located in 8.1°C higher than that of virgin PET [29]. Our study also manifested that the high content amide-bond unit’s chain movement was more complicated than the TPEE.
Table 3: Thermal properties of TPEE and TPEaE-n% copolymers
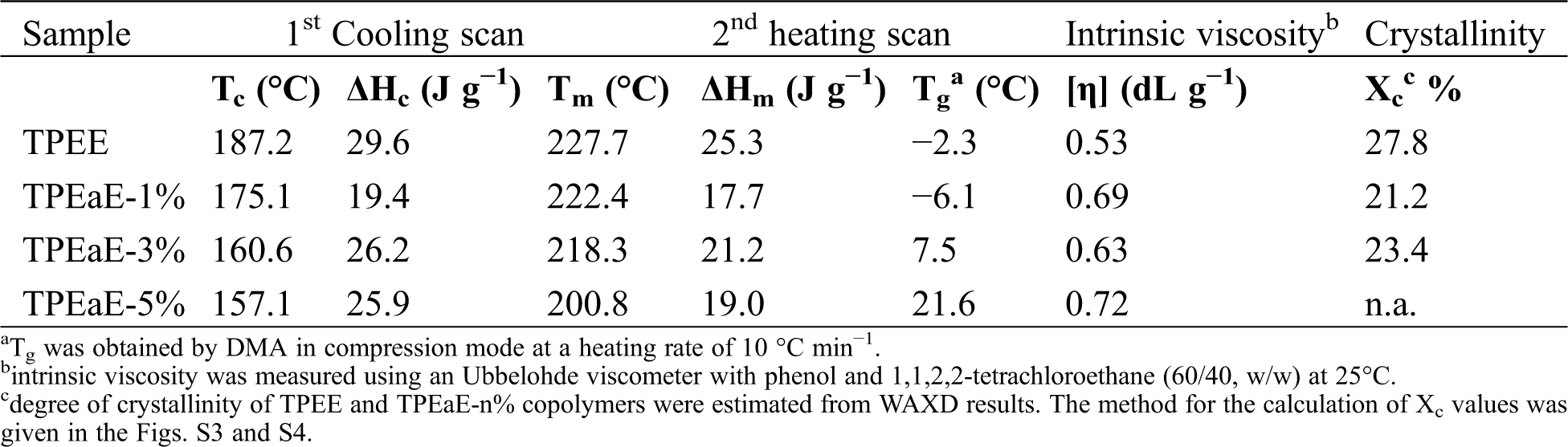
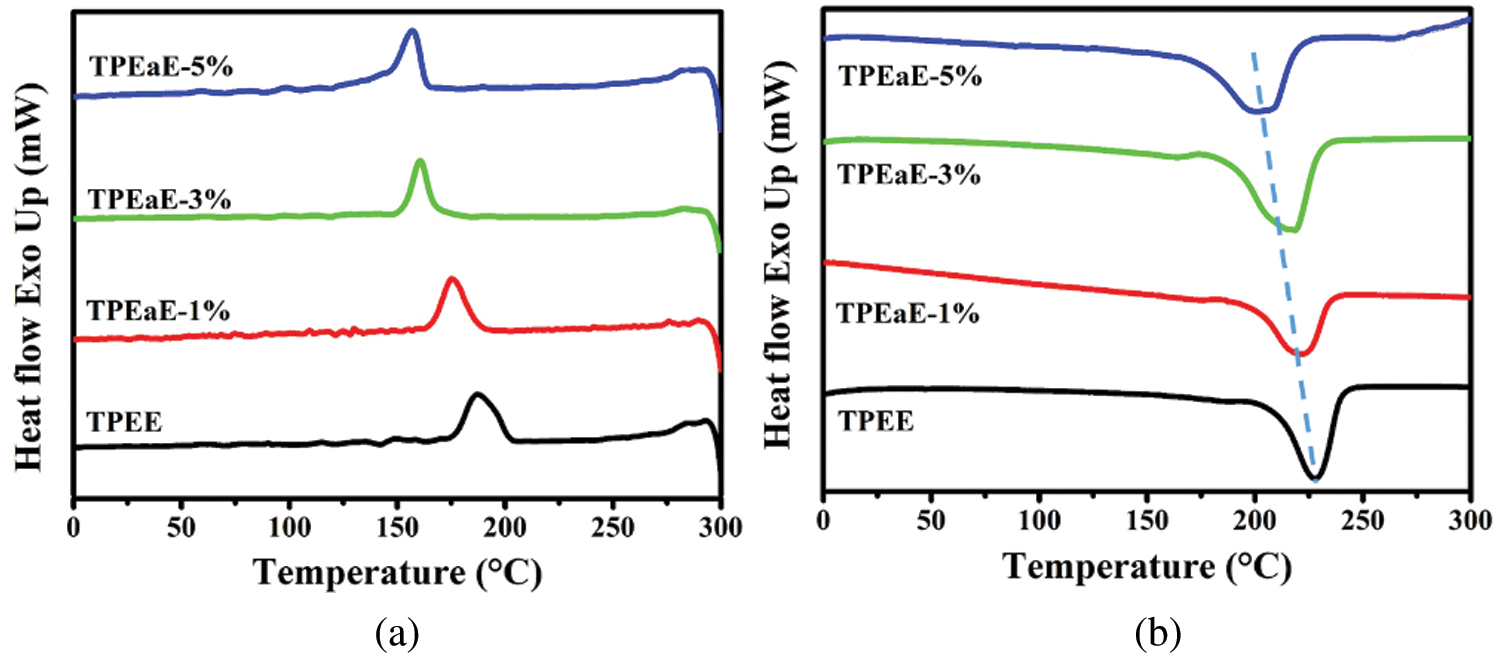
Figure 3: (a) DSC cooling trace from 300 to 0°C, and (b) second heating trace from 0 to 300°C for TPEE and TPEaE-n% copolymers
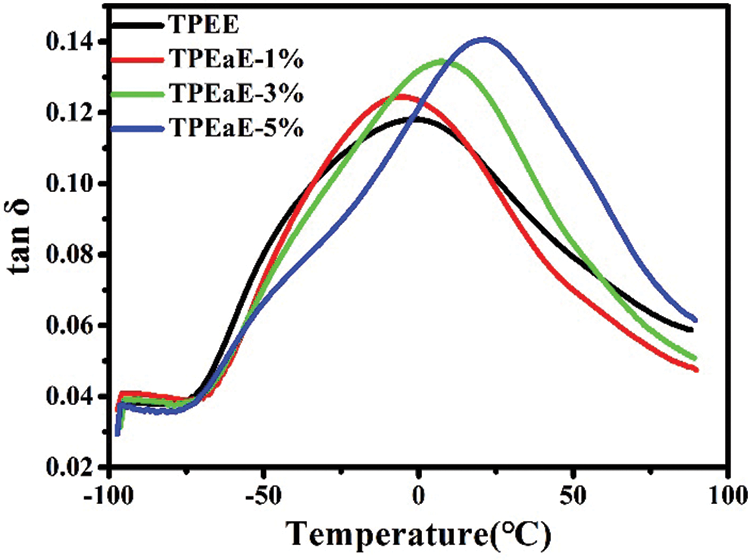
Figure 4: Tan δ of TPEE and TPEaE-n% copolymers
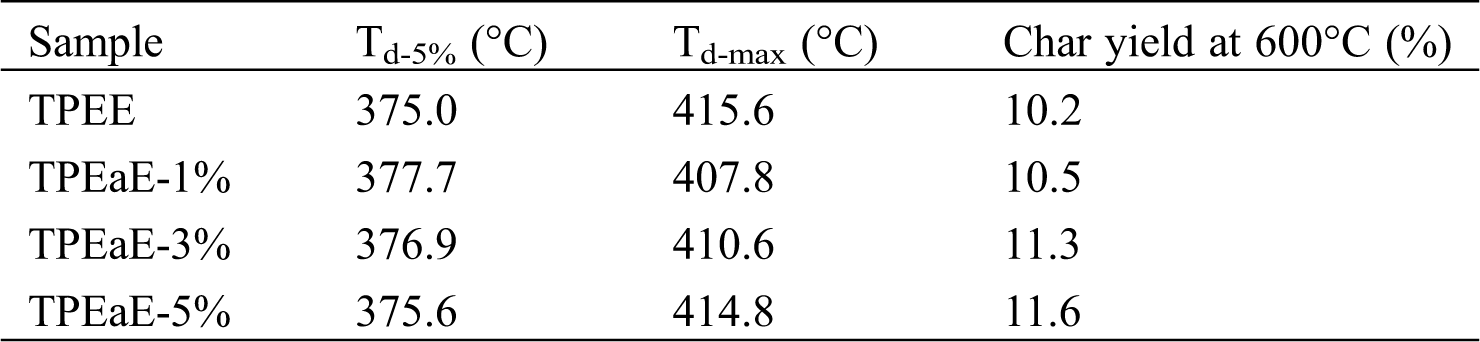
The thermal decomposition of TPEE and TPEaE-n% copolymers at a weight loss of 5 % (Td-5%), the maximum rate of decomposition (Td-max), and the char yield at 600°C are presented in Fig. 5 and Tab. 4. Td-5% showed the tendency to increase slightly and then decreased as the amide content increasing. Thus, the Td-max showed the opposite trend of decreasing and then increased. Different decompositions could be inferred that the number of methylene groups of the HMDA has little influence on thermal decomposition [39,40]. Thus, the amide bond plays a positive role in thermal properties simultaneously [29].
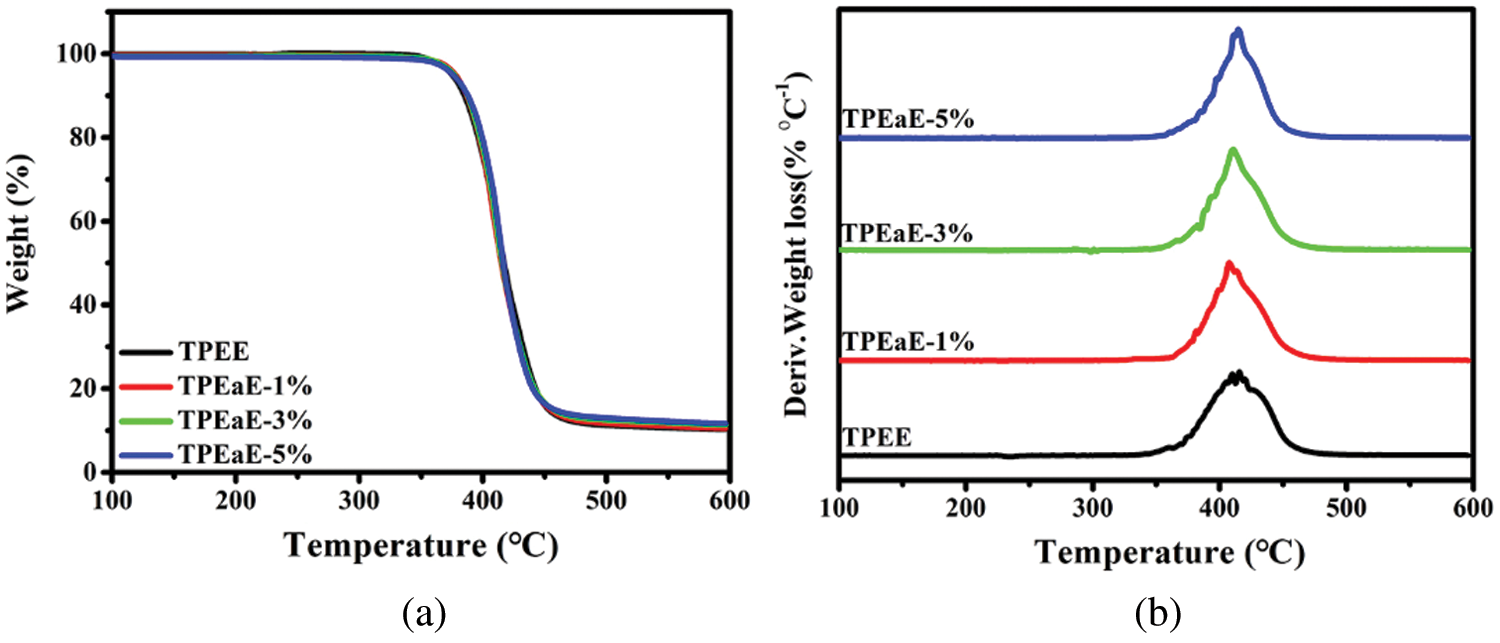
Figure 5: (a) TGA, and (b) derivative weight loss curves of TPEE and TPEaE-n% copolymers
Table 4: Thermal degradation properties of TPEE and TPEaE-n% copolymers
The crystallization structures of synthesized polymers were investigated using WAXD analysis (λ = 0.154 nm), and the WAXD curves were presented in Fig. 6. The main diffraction peaks were observed at the 2θ values of 16.4°, 17.9°, 21.3°, 22.5°, 26.3°, and 32.9°, consistent with the crystalline reflection of PET [41,42]. Moreover, the copolymers’ crystallinity was calculated via WAXD (Tab. 3), and showed a similar melting enthalpy trend through DSC measurement (Tab. 3). Two independent and competing effects of the diamine structure on the crystallization of copolymers were expected: (1) the HMDA formed hydrogen bonding inducing crystallization ability [29,43], (2) the HMDA has the uneven length for neat TPEE structure, restricting the chain order and limiting crystallinity [34,35,43].
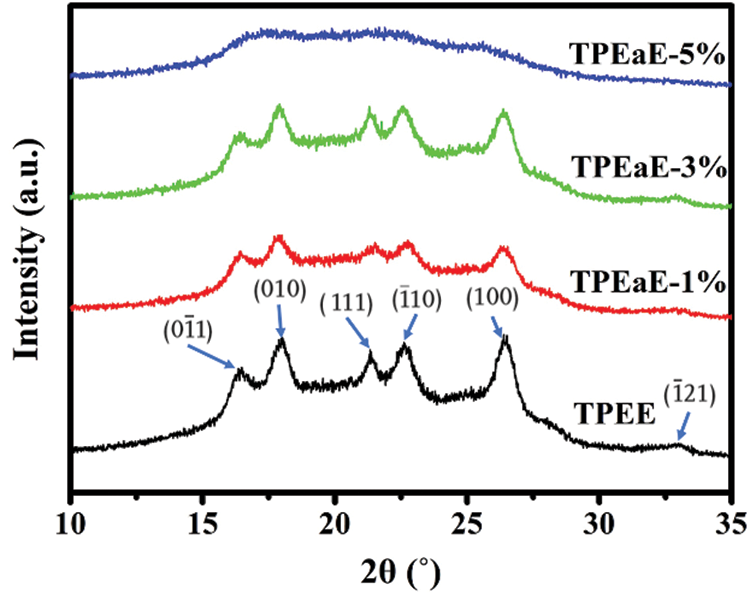
Figure 6: X-ray patterns of TPEE and TPEaE-n% copolymers
3.4 Mechanical Properties of Copolymers
The tensile testing of TPEE and TPEaE-n% copolymers were conducted, and the results were tabulated in Tab. 5 and Fig. 7. The tensile strength and elongation at break were 24.1 to 16.2 MPa, and 563.9 to 314.1%. The tensile strength reminded stable; thus, the elongation at break decreased fiercely as increasing the amide content. Meanwhile, Young’s modulus showed the tendency of increment with the increasing content of the amide-bond unit. With an addition of 5 wt% amide-bond unit in TPEE, Young’s modulus was 1.61 times than that of virgin TPEE (62.6 vs. 38.9 MPa). Such decrement of elongation at break and growth of Young’s modulus has combined the influence of inter-chain force caused by the incorporation of amide-bond units [29].
Table 5: Mechanical properties of TPEE and TPEaE-n% copolymers
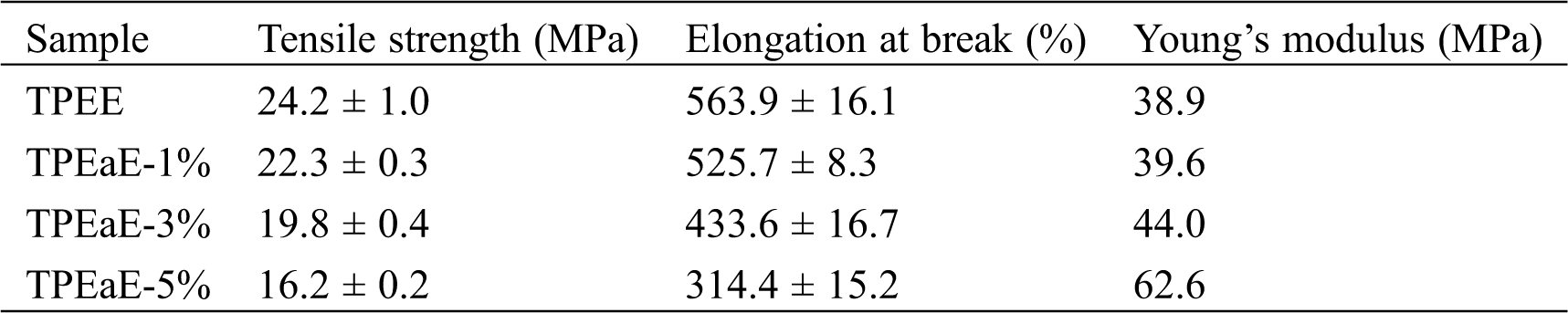
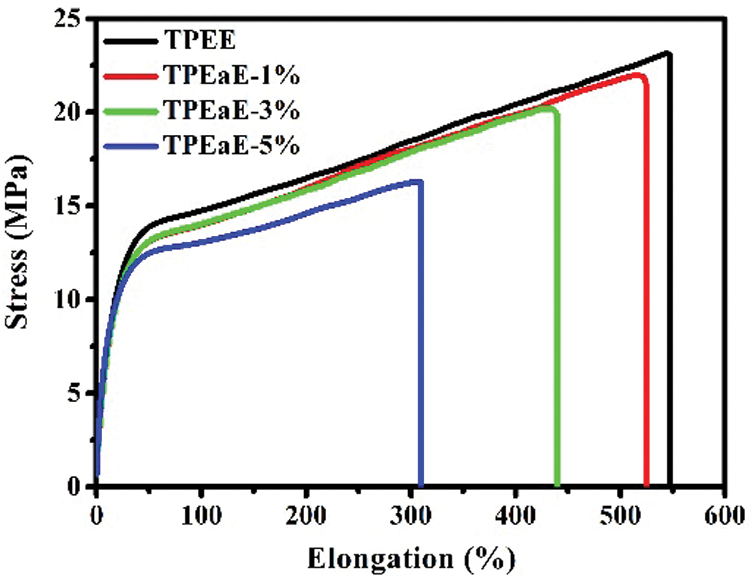
Figure 7: Tensile strength curves of TPEE and TPEaE-n% copolymers
The mechanical and chemical recycling process always included thermal, physical, and hydrolytic degradation, which reduced its molecular weight, intrinsic viscosity and resulted in poor mechanical properties. For mechanical recycling, chain extension was an efficient way to improve recycling properties during extrusion processing [44,45]. The chain extender reacted with the PET end group to rejoin the polymer chain, leading to an increase in polymer molar mass and improved properties. For chemical recycling, crosslinker could link polymer chain by covalent bonding to enhance molar mass and entanglement [46,47]. In our study, as increasing amide content, the intrinsic viscosity (Tab. 1) increased from 0.53 dL g−1 in TPEE to 0.72 dL g−1 in TPEaE-5%. This is probably due to the influence of a reversible physical cross-linked effect by hydrogen bonding. The reversible physical cross-linked reinforced the interaction force and polar entangling between the polymer chain, increasing entanglement and molecular weight [29,48].
A series of TPEE and TPEaE-n % copolymers were synthesized via an environmentally friendly procedure. The precursor of TPEE and TPEaE-n % was obtained by alcoholysis of r-PET, which means the waste PET materials were successfully recycled into value-added products. The structure of each sample was investigated using 1H NMR and FT-IR analyses. As increasing amide content, Tm values showed a decreasing tendency due to the diamide segment of uneven length for neat TPEE, destroying the chain regularity. Tg values were tunable from –2.3 to 21.6°C by controlling the existence of HMDA. Young’s modulus of TPEaE-5% has 1.61 times higher than TPEE because the amide group forms amide-ester or amide-amide hydrogen bonding, leading to increasing intermolecular chain interactions. Moreover, it is worth mentioning that the reversible physical cross-linked hydrogen bonding reinforced the polymer chain's interaction force and entanglement to increase the intrinsic viscosity. In conclusion, this study provides a new approach to manage the r-PET from low-value waste to value-add products.
Acknowledgement: The authors gratefully thank Dr. Sheng-Yen Shen and Prof. Jiang-Jen Lin at National Taiwan University for their valuable discussion and technical support.
Funding Statement: This research was funded by the Ministry of Science and Technology of Taiwan, Grant No. MOST 109-2634-F-027-001.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
1. Benvenuta Tapia, J. J., Tenorio-López, J. A., Martínez-Estrada, A., Guerrero-Sánchez, C. (2019). Application of RAFT-synthesized reactive tri-block copolymers for the recycling of post-consumer R-PET by melt processing. Materials Chemistry and Physics, 229, 474–481. DOI 10.1016/j.matchemphys.2019.02.074. [Google Scholar] [CrossRef]
2. Mouzakis, D. E., Papke, N., Wu, J. S., Karger-Kocsis, J. (2002). Fracture toughness assessment of poly(ethylene terephthalate) blends with glycidyl methacrylate modified polyolefin elastomer using essential work of fracture method. Journal of Applied Polymer Science, 79, 842–852. DOI 10.1002/1097-4628(20010131)79:5<842::AID-APP90>3.0.CO;2-1. [Google Scholar] [CrossRef]
3. Sinha, V., Patel, M. R., Patel, J. V. (2010). Pet waste management by chemical recycling: A review. Journal of Polymers and the Environment, 18(1), 8–25. DOI 10.1007/s10924-008-0106-7. [Google Scholar] [CrossRef]
4. Alfahdawi, I. H., Osman, S. A., Hamid, R., AL-Hadithi, A. I. (2019). Influence of PET wastes on the environment and high strength concrete properties exposed to high temperatures. Construction and Building Materials, 225, 358–370. DOI 10.1016/j.conbuildmat.2019.07.214. [Google Scholar] [CrossRef]
5. Carta, D., Cao, G., D’Angeli, C. (2003). Chemical recycling of poly(ethylene terephthalate) (pet) by hydrolysis and glycolysis. Environmental Science and Pollution Research, 10(6), 390–394. DOI 10.1065/espr2001.12.104.8. [Google Scholar] [CrossRef]
6. Shukla, S. R., Harad, A. M., Jawale, L. S. (2009). Chemical recycling of PET waste into hydrophobic textile dyestuffs. Polymer Degradation and Stability, 94(4), 604–609. DOI 10.1016/j.polymdegradstab.2009.01.007. [Google Scholar] [CrossRef]
7. Ragaert, K., Delva, L., Van Geem, K. (2017). Mechanical and chemical recycling of solid plastic waste. Waste Management, 69, 24–58. DOI 10.1016/j.wasman.2017.07.044. [Google Scholar] [CrossRef]
8. Kathalewar, M., Dhopatkar, N., Pacharane, B., Sabnis, A. Raut, P. et al. (2013). Chemical recycling of PET using neopentyl glycol: reaction kinetics and preparation of polyurethane coatings. Progress in Organic Coatings, 76(1), 147–156. DOI 10.1016/j.porgcoat.2012.08.023. [Google Scholar] [CrossRef]
9. Leal Filho, W., Ellams, D., Han, S., Tyler, D. Boiten, V. J. et al. (2019). A review of the socio-economic advantages of textile recycling. Journal of Cleaner Production, 218, 10–20. DOI 10.1016/j.jclepro.2019.01.210. [Google Scholar] [CrossRef]
10. Nofar, M., Oğuz, H. (2019). Development of PBT/recycled-PET blends and the influence of using chain extender. Journal of Polymers and the Environment, 27(7), 1404–1417. DOI 10.1007/s10924-019-01435-w. [Google Scholar] [CrossRef]
11. Awaja, F., Daver, F., Kosior, E. (2004). Recycled poly(ethylene terephthalate) chain extension by a reactive extrusion process. Polymer Engineering and Science, 44(8), 1579–1587. DOI 10.1002/pen.20155. [Google Scholar] [CrossRef]
12. Park, S. H., Kim, S. H. (2014). Poly (ethylene terephthalate) recycling for high value added textiles. Fashion and Textiles, 1(1), 269. DOI 10.1186/s40691-014-0001-x. [Google Scholar] [CrossRef]
13. Mantia, F. P. L. (1996). Recycling of PVC and mixed plastic waste, pp. 63–76. Ontario: ChemTec Publishing. [Google Scholar]
14. Raheem, A. B., Noor, Z. Z., Hassan, A., Abd Hamid, M. K. Samsudin, S. A. et al. (2019). Current developments in chemical recycling of post-consumer polyethylene terephthalate wastes for new materials production: A review. Journal of Cleaner Production, 225, 1052–1064. DOI 10.1016/j.jclepro.2019.04.019. [Google Scholar] [CrossRef]
15. Merkel, D. R., Kuang, W., Malhotra, D., Petrossian, G. Zhong, L. et al. (2020). Waste PET chemical processing to terephthalic amides and their effect on asphalt performance. ACS Sustainable Chemistry & Engineering, 8(14), 5615–5625. DOI 10.1021/acssuschemeng.0c00036. [Google Scholar] [CrossRef]
16. Leng, Z., Padhan, R. K., Sreeram, A. (2018). Production of a sustainable paving material through chemical recycling of waste PET into crumb rubber modified asphalt. Journal of Cleaner Production, 180, 682–688. DOI 10.1016/j.jclepro.2018.01.171. [Google Scholar] [CrossRef]
17. Scremin, D. M., Miyazaki, D. Y., Lunelli, C. E., Silva, S. A. Zawadzki, S. F. et al. (2019). PET recycling by alcoholysis using a new heterogeneous catalyst: study and its use in polyurethane adhesives preparation. Macromolecular Symposia, 383(1), 1800027. DOI 10.1002/masy.201800027. [Google Scholar] [CrossRef]
18. Oku, A. (1997). Alkali decomposition of poly(ethylene terephthalate) with sodium hydroxide in nonaqueous ethylene glycol: a study on recycling of terephthalic acid and ethylene glycol. Journal of Applied Polymer Science, 63, 595–601. DOI 10.1002/(SICI)1097-4628(19970131)63:5<595::AID-APP7>3.0.CO;2-P. [Google Scholar] [CrossRef]
19. Campanelli, J. R., Kamal, M. R., Cooper, D. G. (1994). Kinetics of glycolysis of poly(ethylene terephthalate) melts. Journal of Applied Polymer Science, 54(11), 1731–1740. DOI 10.1002/app.1994.070541115. [Google Scholar] [CrossRef]
20. Malik, N., Kumar, P., Shrivastava, S., Ghosh, S. B. (2017). An overview on PET waste recycling for application in packaging. International Journal of Plastics Technology, 21(1), 1–24. DOI 10.1007/s12588-016-9164-1. [Google Scholar] [CrossRef]
21. Litvinov, V. M., Bertmer, M., Gasper, L., Demco, D. E. Blümich, B. et al. (2003). Phase composition of block copoly(ether ester) thermoplastic elastomers studied by solid-state NMR techniques. Macromolecules, 36(20), 7598–7606. DOI 10.1021/ma030314h. [Google Scholar] [CrossRef]
22. Li, G., Jiang, J., Jin, J., Yang, S. Wu, C. et al. (2006). Crystallization behavior of modified polyester with varied macromolecular architecture. Journal of Macromolecular Science, Part B, 45(4), 639–652. DOI 10.1080/00222340600770277. [Google Scholar] [CrossRef]
23. Paszkiewicz, S., Szymczyk, A., Pawlikowska, D., Irska, I. Taraghi, I. et al. (2017). Synthesis and characterization of poly(ethylene terephthalate-co-1,4-cyclohexanedimethylene terephtlatate)-block-poly(tetramethylene oxide) copolymers. RSC Advances, 7(66), 41745–41754. DOI 10.1039/C7RA07172H. [Google Scholar] [CrossRef]
24. Ortmann, P., Lemke, T. A., Mecking, S. (2015). Long-spaced polyamides: elucidating the gap between polyethylene crystallinity and hydrogen bonding. Macromolecules, 48(5), 1463–1472. DOI 10.1021/acs.macromol.5b00060. [Google Scholar] [CrossRef]
25. Lynch, J. G., Jaycox, G. D. (2014). Stimuli-responsive polymers. 10. Photo-regulation of optical rotations in azobenzene modified poly(ester-amide)s containing highly structured, atropisomeric backbone geometries. Polymer, 55(16), 3564–3572. DOI 10.1016/j.polymer.2014.06.065. [Google Scholar] [CrossRef]
26. Aharoni, S. M. (1988). Hydrogen-bonded highly regular strictly alternating aliphatic-aromatic liquid-crystalline poly(ester amides). Macromolecules, 21(7), 1941–1961. DOI 10.1021/ma00185a011. [Google Scholar] [CrossRef]
27. Ho, R. M., Chi, C. W., Tsai, C. C., Lin, J. J. (2002). Glass transition and exclusion model in crystallization of polyether–polyester block copolymers with amide linkages. Polymer, 43(4), 1365–1373. DOI 10.1016/S0032-3861(01)00680-2. [Google Scholar] [CrossRef]
28. Abbes, M., Salhi, S., Lefevre, L., Delaite, C. Abid, S. et al. (2014). Poly(ester-amide)s derived from adipic acid, 1,4-butanediol and β-alanine: synthesis and characterization. Journal of Macromolecular Science, Part A, 52(1), 56–63. DOI 10.1080/10601325.2014.976752. [Google Scholar] [CrossRef]
29. Gao, H., Bai, Y., Liu, H., He, J. (2019). Mechanical and gas barrier properties of structurally enhanced poly(ethylene terephthalate) by introducing 1,6-hexylenediamine unit. Industrial & Engineering Chemistry Research, 58(47), 21872–21880. DOI 10.1021/acs.iecr.9b04953. [Google Scholar] [CrossRef]
30. Winnacker, M., Rieger, B. (2016). Poly(ester amide)s: Recent insights into synthesis, stability and biomedical applications. Polymer Chemistry, 7(46), 7039–7046. DOI 10.1039/C6PY01783E. [Google Scholar] [CrossRef]
31. Wu, J., Xie, H., Wu, L., Li, B. G., Dubois, P. et al. (2016). DBU-catalyzed biobased poly(ethylene 2,5-furandicarboxylate) polyester with rapid melt crystallization: synthesis, crystallization kinetics and melting behavior. RSC Advances, 6(103), 101578–101586. DOI 10.1039/C6RA21135F.
32. van Bennekom, A. C. M., Gaymans, R. J. (1997). Amide modified polybutylene terephthalate: Structure and properties. Polymer, 38(3), 657–665. DOI 10.1016/S0032-3861(96)00553-8. [Google Scholar] [CrossRef]
33. van Bennekom, A. C. M., Willemsen, P. A. A. T., Gaymans, R. J. (1996). Amide-modified poly(butylene terepthalatethermal stability. Polymer, 37(24), 5447–5459. DOI 10.1016/S0032-3861(96)00355-2. [Google Scholar] [CrossRef]
34. Lips, P. A. M., Broos, R., van Heeringen, M. J. M., Dijkstra, P. J., Feijen, J. et al. (2005). Synthesis and characterization of poly(ester amide)s containing crystallizable amide segments. Polymer, 46(19), 7823–7833. DOI 10.1016/j.polymer.2005.07.013. [Google Scholar] [CrossRef]
35. Bouma, K. (2000). Polyesteramides based on PET and nylon 2,T Part 3. Properties. Polymer, 41, 3965–3974. [Google Scholar]
36. Scheirs., J., Long, T. E. (2004). Modern polyester: Chemistry and technology of polyesters and copolyester, pp. 31–115. Hoboken, NJ: Wiley Publishing. [Google Scholar]
37. Hao, Y., Chen, M., Zhao, J., Zhang, Z. Yang, W. et al. (2013). Synthesis and properties of polyesteramides having short nylon-610 segments in the main chains through polycondensation and chain extension. Industrial & Engineering Chemistry Research, 52(19), 6410–6421. DOI 10.1021/ie302879t. [Google Scholar] [CrossRef]
38. Hibbs, M. R., Holtzclaw, J., Collard, D. M., Liu, R. Y. F. Hiltner, A. et al. (2004). Poly(ethylene terephthalate) modified with aromatic bisester diamides: Thermal and oxygen barrier properties. Journal of Polymer Science Part A: Polymer Chemistry, 42(7), 1668–1681. DOI 10.1002/pola.11075. [Google Scholar] [CrossRef]
39. Xie, H., Wu, L., Li, B. G., Dubois, P. (2018). Biobased poly(ethylene-co-hexamethylene 2,5-furandicarboxylate) (PEHF) copolyesters with superior tensile properties. Industrial & Engineering Chemistry Research, 57(39), 13094–13102. DOI 10.1021/acs.iecr.8b03204. [Google Scholar] [CrossRef]
40. Terzopoulou, Z., Tsanaktsis, V., Nerantzaki, M., Papageorgiou, G. Z. Bikiaris, D. N. et al. (2016). Decomposition mechanism of polyesters based on 2,5-furandicarboxylic acid and aliphatic diols with medium and long chain methylene groups. Polymer Degradation and Stability, 132, 127–136. DOI 10.1016/j.polymdegradstab.2016.03.006. [Google Scholar] [CrossRef]
41. Wang, D., Luo, F., Shen, Z., Wu, X. Qi, Y. et al. (2017). A study on the crystallization behavior and mechanical properties of poly(ethylene terephthalate) induced by chemical degradation nucleation. RSC Advances, 7(59), 37139–37147. DOI 10.1039/C7RA06823A. [Google Scholar] [CrossRef]
42. Yuan, R., Fan, S., Sun, Y., Wu, D. Wang, X. et al. (2019). Enhanced mechanical properties of PET-based thermoplastic elastomers: Brittle-ductile transition via micro cross-linking technology. Macromolecular Chemistry and Physics, 220(15), 1900206. DOI 10.1002/macp.201900206. [Google Scholar] [CrossRef]
43. Lips, P. A. M., Broos, R., van Heeringen, M. J. M., Dijkstra, P. J. Feijen, J. et al. (2005). Incorporation of different crystallizable amide blocks in segmented poly(ester amide)s. Polymer, 46(19), 7834–7842. DOI 10.1016/j.polymer.2005.07.009. [Google Scholar] [CrossRef]
44. Raffa, P., Coltelli, M. B., Savi, S., Bianchi, S. Castelvetro, V. et al. (2012). Chain extension and branching of poly(ethylene terephthalate) (PET) with di- and multifunctional epoxy or isocyanate additives: An experimental and modelling study. Reactive and Functional Polymers, 72(1), 50–60. DOI 10.1016/j.reactfunctpolym.2011.10.007. [Google Scholar] [CrossRef]
45. Makkam, S., Harnnarongchai, W. (2014). Rheological and mechanical properties of recycled PET modified by reactive extrusion. Energy Procedia, 56, 547–553. DOI 10.1016/j.egypro.2014.07.191. [Google Scholar] [CrossRef]
46. Shah, R. V., Borude, V. S., Shukla, S. R. (2013). Recycling of PET waste using 3-amino-1-propanol by conventional or microwave irradiation and synthesis of bis-oxazin there from. Journal of Applied Polymer Science, 127(1), 323–328. DOI 10.1002/app.37900. [Google Scholar] [CrossRef]
47. Jamdar, V., Kathalewar, M., Dubey, K. A., Sabnis, A. (2017). Recycling of PET wastes using Electron beam radiations and preparation of polyurethane coatings using recycled material. Progress in Organic Coatings, 107, 54–63. DOI 10.1016/j.porgcoat.2017.02.007. [Google Scholar] [CrossRef]
48. Ma, X., Guo, X., Gu, L. (2007). Rheological behavior in blends of PET with ionomeric polyester. European Polymer Journal, 43(8), 3613–3620. DOI 10.1016/j.eurpolymj.2007.05.041. [Google Scholar] [CrossRef]
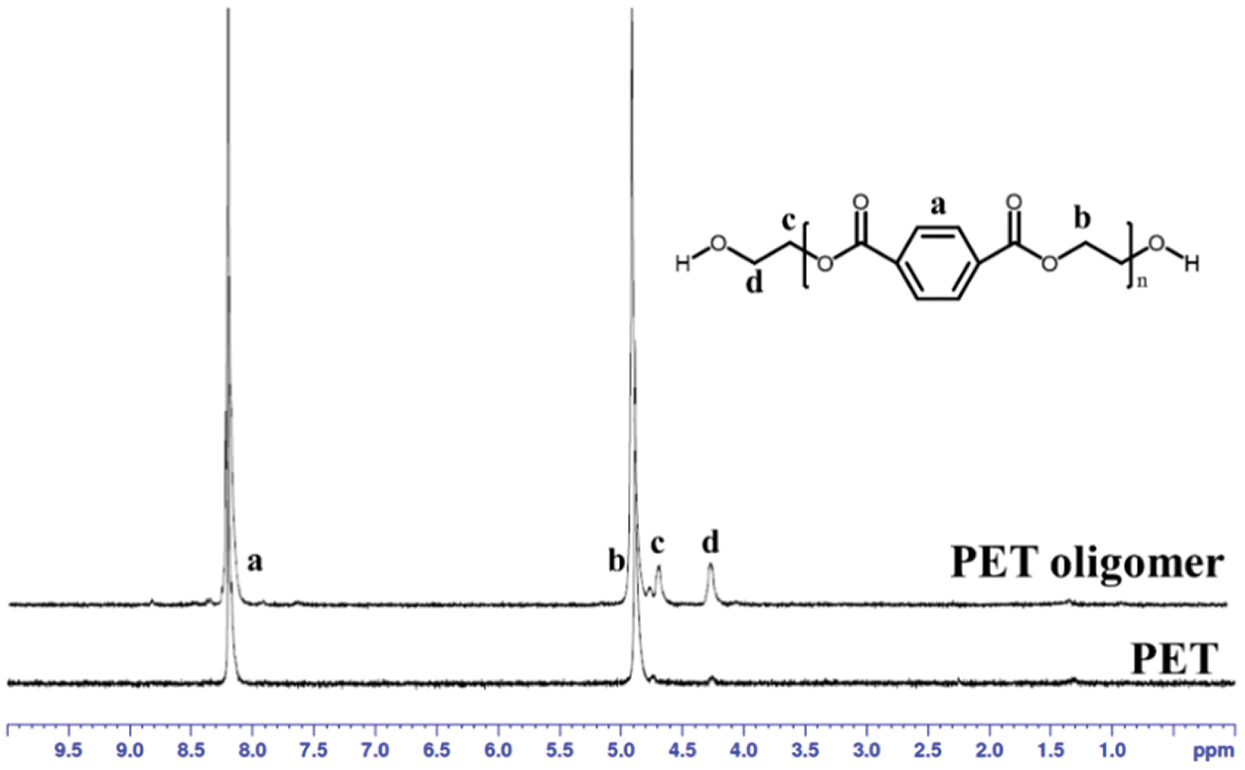
Figure S1: 1H NMR spectra of PET and PET oligomer
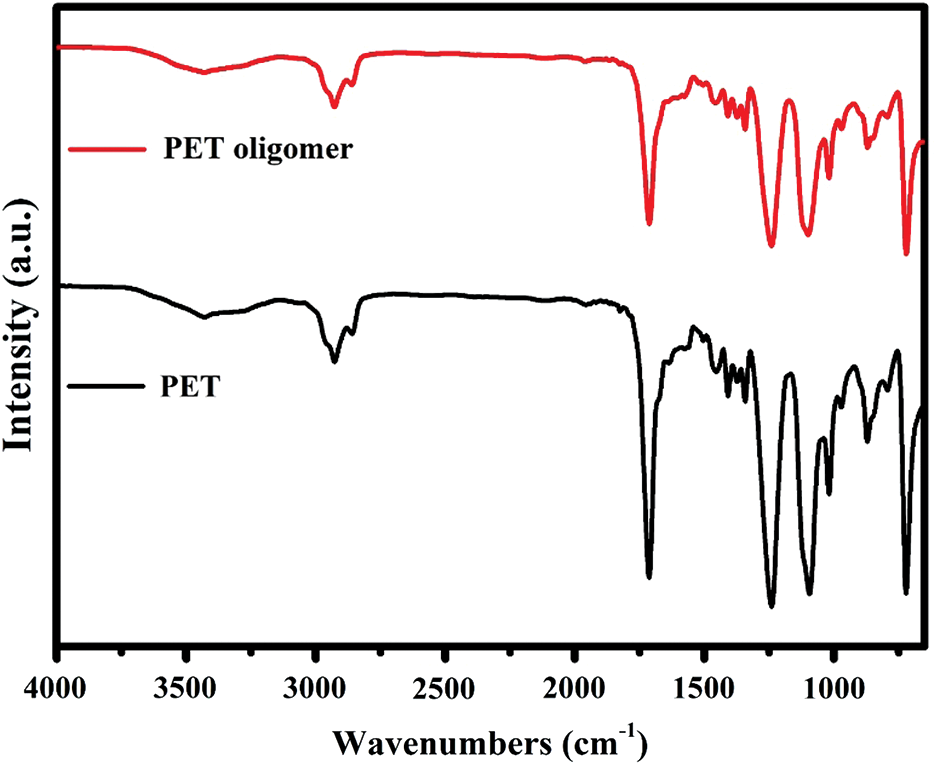
Figure S2: FT-IR spectra of PET and PET oligomer
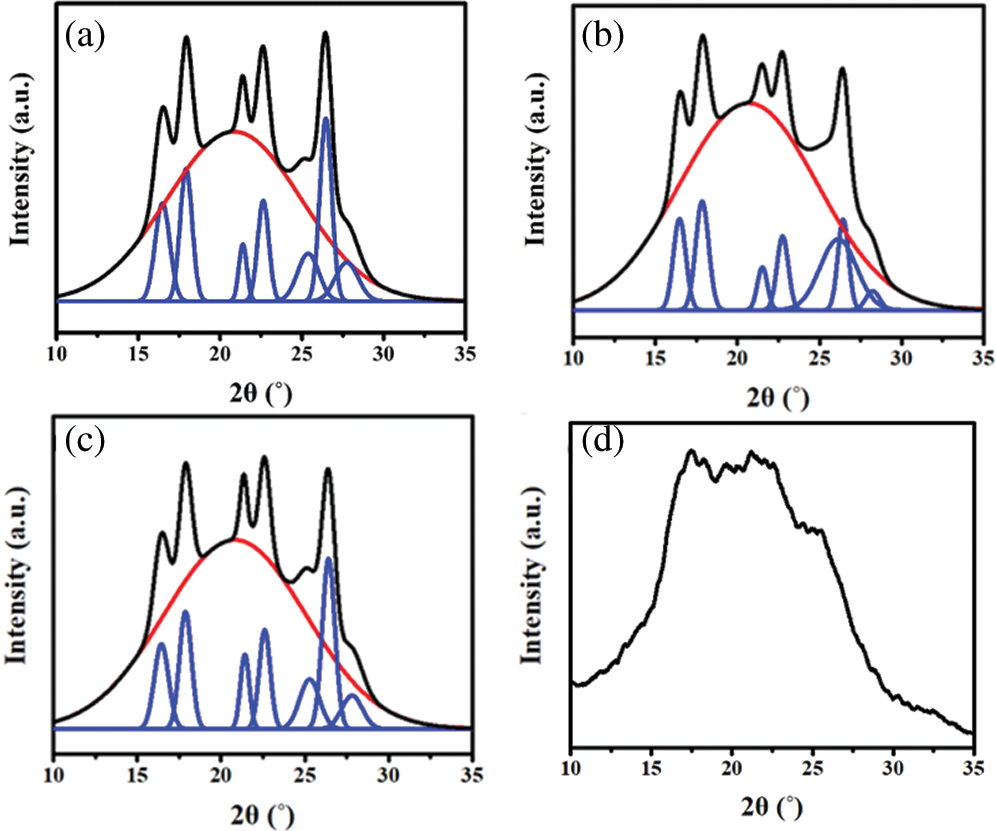
Figure S3: WAXD fitting results of (a) TPEE, (b) TPEaE-1%, (c) TPEaE-3%, and (d) TPEaE-5%
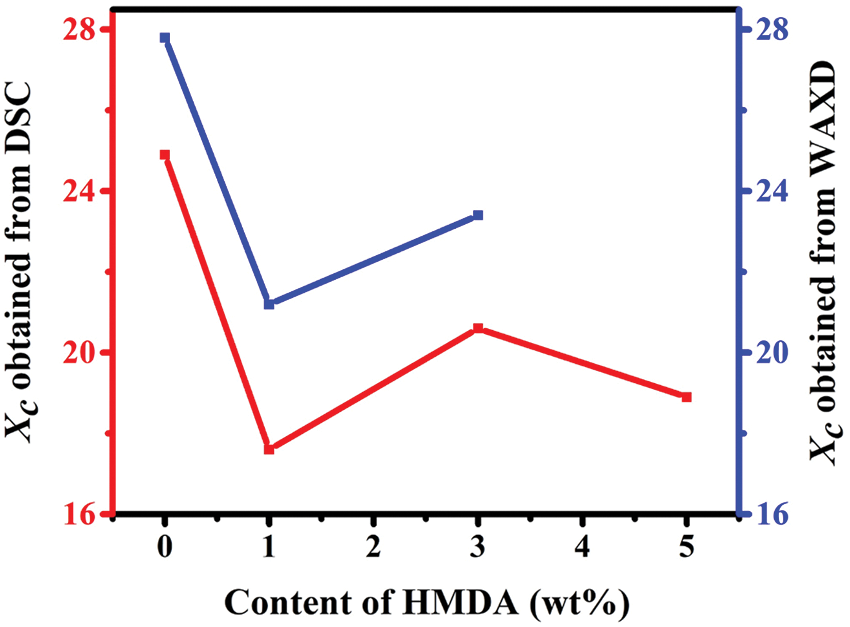
Figure S4: Degree of crystallinity of TPEE and TPEaE-n% copolymers with different content of HMDA evaluated from the DSC data and WAXD data
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |