| Journal of Renewable Materials |
DOI: 10.32604/jrm.2021.015475
ARTICLE
New Biobased Polyurethane Materials from Modified Vegetable Oil
1Laboratoire de Chimie Appliquée et Environnement–ECOMP—Faculté des Sciences, Université Mohamed Premier, Oujda, 60 000, Maroc
2University of Grenoble Alpes, CNRS, CERMAV, Grenoble, Cedex 9, France
3University of Grenoble Alpes, CNRS, Grenoble INP, Grenoble, F-38000, France
4University of Monastir, Faculty of Sciences of Monastir, Research Unit of Applied Chemistry and Environment, Monastir, 5019, Tunisia
5Higher Institute of Technological Studies (ISET) of Ksar-Hellal, Ksar-Hellal, 5070, Tunisia
*Corresponding Authors: Mohamed Naceur Belgacem. Email: naceur.belgacem@grenoble-inp.fr; Ramzi Khiari. Email: khiari_ramzi2000@yahoo.fr
Received: 21 December 2020; Accepted: 19 January 2021
Abstract: Bio-based polyurethanes (PUs) have been occurred a large attention nowadays. It was found to be an alternative to the petrochemical based materials to the fact of their weak environmental influence, availability, good price and biodegradability. In addition, the nature shows several bio-derived compounds as raw materials for the synthesis of polyols, including the vegetable oils, polyphenol, terpene, and other bio-renewable sources. With the aim to develop a new family of biobased polyurethanes (PUs) via vegetable oils, the elaboration of new Jojoba-based PUs was performed by catalyst-free polycondensation reaction of a synthesized Jojoba diol with various diisocyanates such us toluene diisocyanate (TDI) and isophorone diisocyanate (IPDI). All the compounds were characterized by FTIR and NMR spectroscopies, and their properties were determined by gel permeation chromatography, differential scanning calorimetry and thermogravimetric analysis. The obtained results show renewable vegetable oils-based PUs materials can be preparing using a new environmentally ways and giving various good properties performances.
Keywords: Biopolymers; Jojoba oil; polyurethane; polyol; thiol-ene
Polyurethanes (PUs), are considered as one of the main used synthetic polymer families in the world [1]. At the industrial scale, they were obtained through a polycondensation reaction of polyisocyanate with polyol. These obtained materials as described in literatures, it can be observed in several fields namely: coatings [2], adhesives [3], foams [4]. This large application can be explained due to their good mechanical and thermal properties, including abrasion resistance, high toughness as well as chemical resistance [5]. Currently, PUs materials are prepared from fossil resource mainly from petrol. However, scientific research is oriented to substitute them by abundant renewable raw materials [6]. In fact, the sustainable development takes actually, a valuable place due to the decrease of petroleum resources, greenhouse effect, and new legislations concerning the protection of environment.
Thus, many studies highlighted the possibility of substitution of petroleum based raw materials by agro-resources such as, lignin [7], cellulose [8], starch [9] and vegetable oils (VOs) [10]. These last materials are known to be more useful because of their biodegradability, low price, availability in various varieties forms and low toxicity [11,12]. Indeed, the main compounds in oil seed crops are triglycerides, which are formed by the combination of a glycerol molecule tethered to three fatty acids. Some works evidenced that VOs-based PUs can be considered as materials with promising characteristics thanks to the hydrophobic character of triglyceride [13]. In this context, much efforts have been deployed during the past few decades to elaborate oil-based polymeric polyols from VOs including polyesteramides [14], polyetheramides [15] epoxies [16], and various PU materials [17–19]. It is well known that the fatty acid composition differs for each oil affecting consequently the mechanical, chemical, and thermal properties of the resulting PU materials [20]. For example, linseed [21] and palm oil [22] possesses 6.6 and 1.7 unsaturation per triglyceride, respectively. The use of VOs for the elaboration of PUs materials requires the presence of hydroxyls functions. Nevertheless, except castor oil, VOs present no hydroxyls function. Therefore, it is necessary to functionalize them by hydroxyl moieties at the unsaturation or ester groups.
Among several methods used for the production of polyols from VOs, the most used methods are epoxidation followed by ring-opening of epoxides; hydroformylation succeeded by hydrogenation; ozonolysis and transesterification. Thiol-ene coupling could also be employed to introduce alcohol or other reactive function such us acid or amine [17,22]. Thanks to its high efficiency, rapidity and few steps, thiol-ene coupling reaction are considered as one of the most powerful technique used for obtaining polyols from VOs [23]. Radical Thiol-ene process occurs in three steps. The first one consists of the creation of free radicals adduct by thermal or photochemical initiation followed by the thiyl radical’s formation and finally the addition of these radicals to the unsaturation according to an Anti-Markovnikov process [24]. One of the least studied VOs to produce polymer materials are JO, it is the only known plant to contain a liquid wax in its seeds [25]. Unlike other VOs, jojoba oil (JO) does not contain any triglycerides in its composition [26]. However, this oil is formed by the combination of a fatty acid and fatty alcohol that are both mono unsaturated. JO took much interest for industrial, pharmaceutical and cosmetic fields. In fact, it was used as a lubricant, an ingredient in medicines and dental implants [25,27].
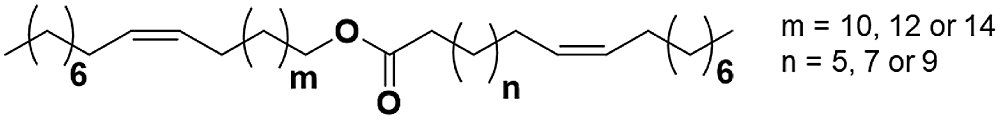
Figure 1: Chemical structure of JO ester
The chemical structure of the JO esters is shown in Fig. 1, and depending on the growing environment of the plant, (n) and (m) are compromise between 5 and 14. The unique structure of JO allows us to obtain a bi functional precursor, unlike the others, that could be used to the synthesis of new linear polymer materials. Therefore, the aim of this paper is to elaborate new PU materials from modified JO. The modification is carried out by thiol-ene coupling with mercaptoethanol, and then the PUs was elaborate by two steps polycondensation reaction with toluene diisocyanate (TDI) or isophorone diisocyanate (IPDI). The obtained materials were characterized by FTIR and 1H NMR spectroscopies and also by several methods such as TGA, DSC and SEC.
Methanol (CH3OH, CAS:67-56-1), dichloromethane (CH2Cl2, CAS:75-09-2), 2,4-Diisocyanato-1-methylbenzene (TDI, CAS:584-84-9), and 5-isocyanato-1-(isocyanatomethyl)-1,3,3-trimethylcyclohexane (IPDI, CAS:4098-71-9) were purchased from Sigma Aldrich. Jojoba diol was synthetized as described in our foregoing work [27].
The functional chemistry of prepared and used samples was studied by the Fourier Transform Infrared Spectroscopy (FTIR) and nuclear magnetic resonance (1H, 13C NMR). The FTIR analysis was carried out using a Perkin-Elmer spectrophotometer (USA) as an instrument. The test was measured at wavelength varied from 4000 to 500 cm−1 and the acquisition conditions of this analysis and resolution are 16 scans and 4 cm-1, respectively. Concerning the NMR spectra analysis, it was performed using a Bruker Avance 400 MHz spectrometer. The test was done at 25°C in deuterated chloroform. The chemical shifts were reported in part per million relatives to tetramethylsilane. Obviously, the spin multiplicity is given by s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet.
The thermal properties were determined by the differential scanning calorimetry (DSC) analysis and the thermogravimetric analyses (TGA). The test of TGA were studied using a TGA Q50 (TA instrument) at a heating rate of 15°C min−1. 15 mg of prepared material was put on a platinum pan and heated 25°C to 750°C under air flow (60 mL min−1). Concerning the DSC method, the test was performed out on a NETZSCH DSC200 calorimeter. The test consists of 10 mg of prepared samples which were sealed in hermetic aluminium pans or on a stainless-steel high-pressure capsule. The thermal behaviours were examined at 10°C min−1 between −150 and 200°C to detect reaction enthalpy, glass transition as well as crystallization/fusion processes. All the reported temperatures are onset values and the test were done at least in duplicate.
The molar mass distribution of the prepared PU was determinate by gel permeation chromatography (GPC) method. The test was established using a Varian ProStar Model 210 equipped with an RI refractive index detector as device. The test consist two PL gel 5 μm were fixed at 70°C with a 0.8 mL min−1 flow rate of DMF with 0.1% of LiBr, calibrated using PMMA standards, several injection volume was typically 20 µL at a concentration of 10 mg.mL−1.
VOs is suitable model substrates for starting the preparation of PU materials. In fact, the unsaturation’s can be easily modified by thiol ene coupling with mercaptoethanol. This direct route allows obtaining only a bifunctional diol from jojoba oil. The characterization of this monomer is done in previous work, [27] where it was provided that jojoba oil could be used as promising reagent to starting for preparing PU materials by a simple way and approach.
1H NMR (δ, 400 MHz, ppm): 0.80 (H1, CH3, t); 1.21 (H2, CH2, l); 1.33 (H3, CH2, m); 1.48 (H4, CH2–CH, m); 1.55 (H5, CH2, m); 2.22 (H6, CH2–C=O, t); 2.52 (H7, CH–S, td); 2.64 (H8, CH2–S, t); 3.62 (H9, CH2–OH, t); 3.98 (H10, CH2–O, t)
13C NMR (δ, 100.6 MHz, ppm): 14.1 (C1); 27.2-35.8 (C2-6); 41.2 (C8); 45.8 (C7’); 60.5 (C9); 174.1 (C11)
The synthesis of prepolymers (Fig. 2) was done in a three necked flask. The reaction was established under a magnetic stirrer, a nitrogen inlet and an addition funnel in order to add the several reagents: (5g, 1eq of Jodiol) and (2.32g, 2eq) of toluene diisocyanate for Pr1, and (2.32g, 2eq) of isophorone diisocyanate for Pr2. The reaction is occurred without catalyst at 80°C during 4 hours in the oil bath. In the case of TDI, THF was used as solvent.
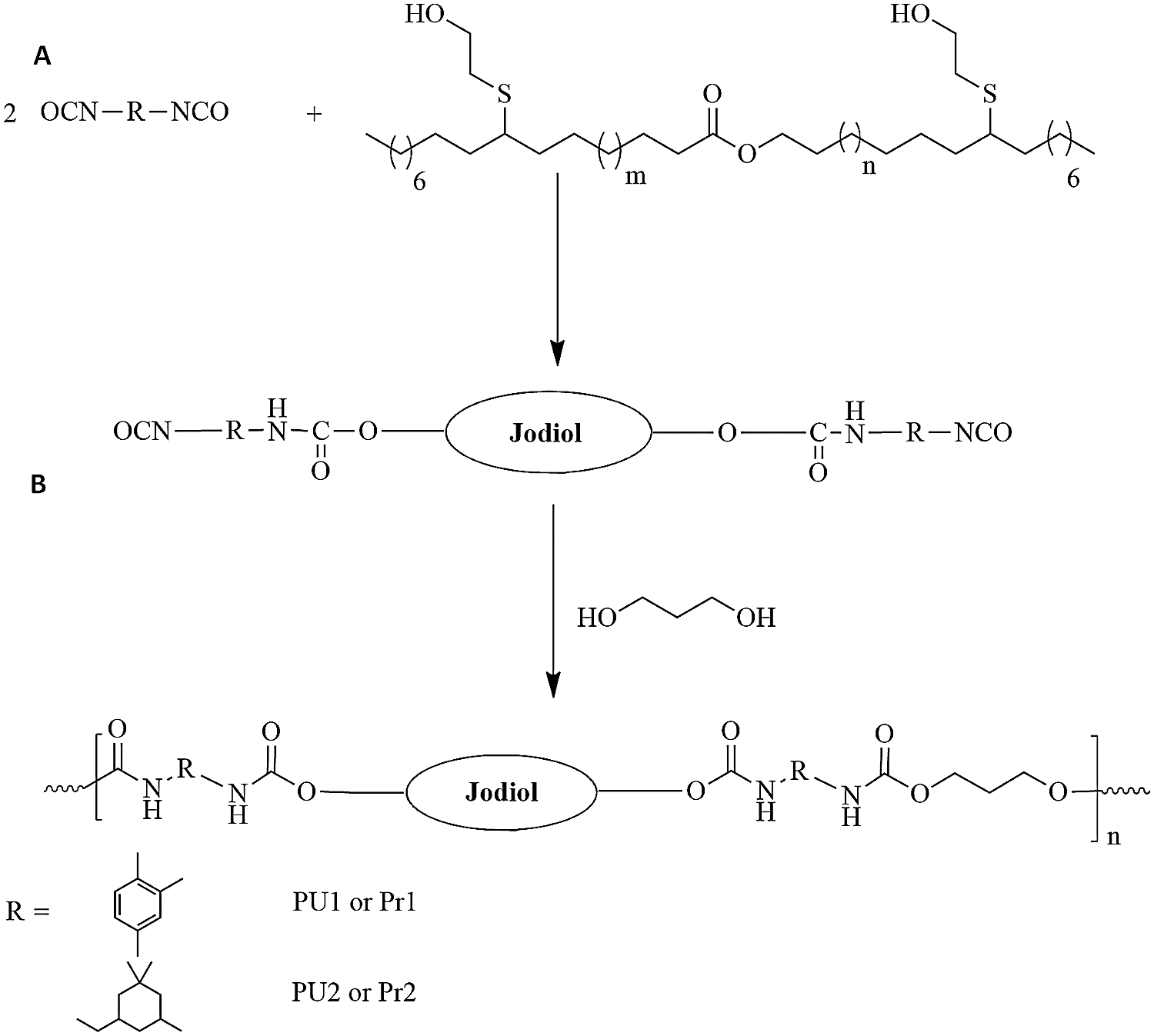
Figure 2: (A) Synthesis of prepolymers and (B) Synthesis of final PU materials
FTIR (δ, cm−1, Fig. 3): 3300 (N–H groups), 2260 (NCO free isocyanate), 2925-2855 (CH aliphatic compounds), 1700–1730 cm−1 (C=O ester and urethane), 1530 (N–H urethane).
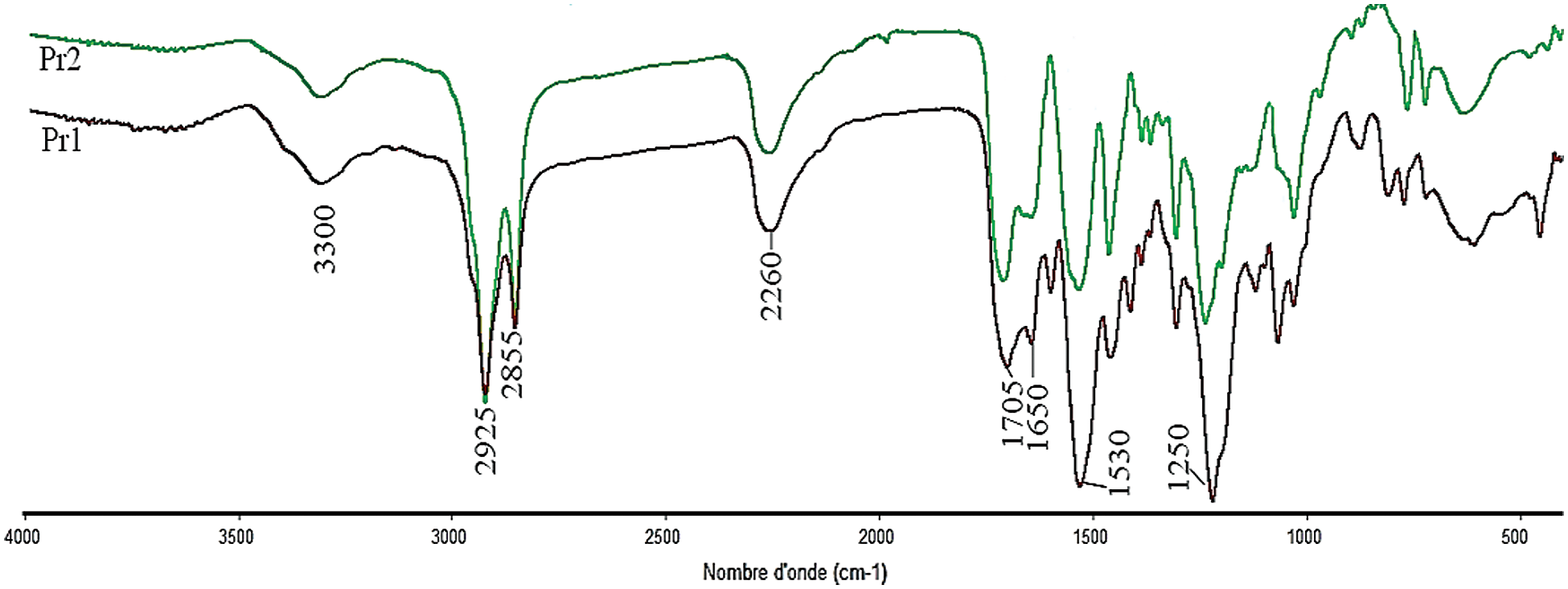
Figure 3: FTIR spectra of Pr1 and Pr2
1H NMR (Fig. 4A, δ, 400 MHz, ppm): δ = 0.85 ppm (6H, CH3–CH2 ,t); δ = 1.1–1.6 ppm (64H, CH2–CH2, m); δ = 2,1 ppm (6H,CH3–ph, d); δ = 2.2 ppm (2H, H5, CH2C=O, t); δ = 2.5−2.6 ppm (2H, H6 H–CS–, m), δ = 2.75 ppm (4H, H7, CH2–S, t), δ = 4 ppm (2H, H10, –CH2 –OCO, t); δ = 4.25 ppm (8H, H9, –CH2–OCO, t); δ = 6.5−7.2 ppm (6H, H10,H11,H12, H–Ph, d).
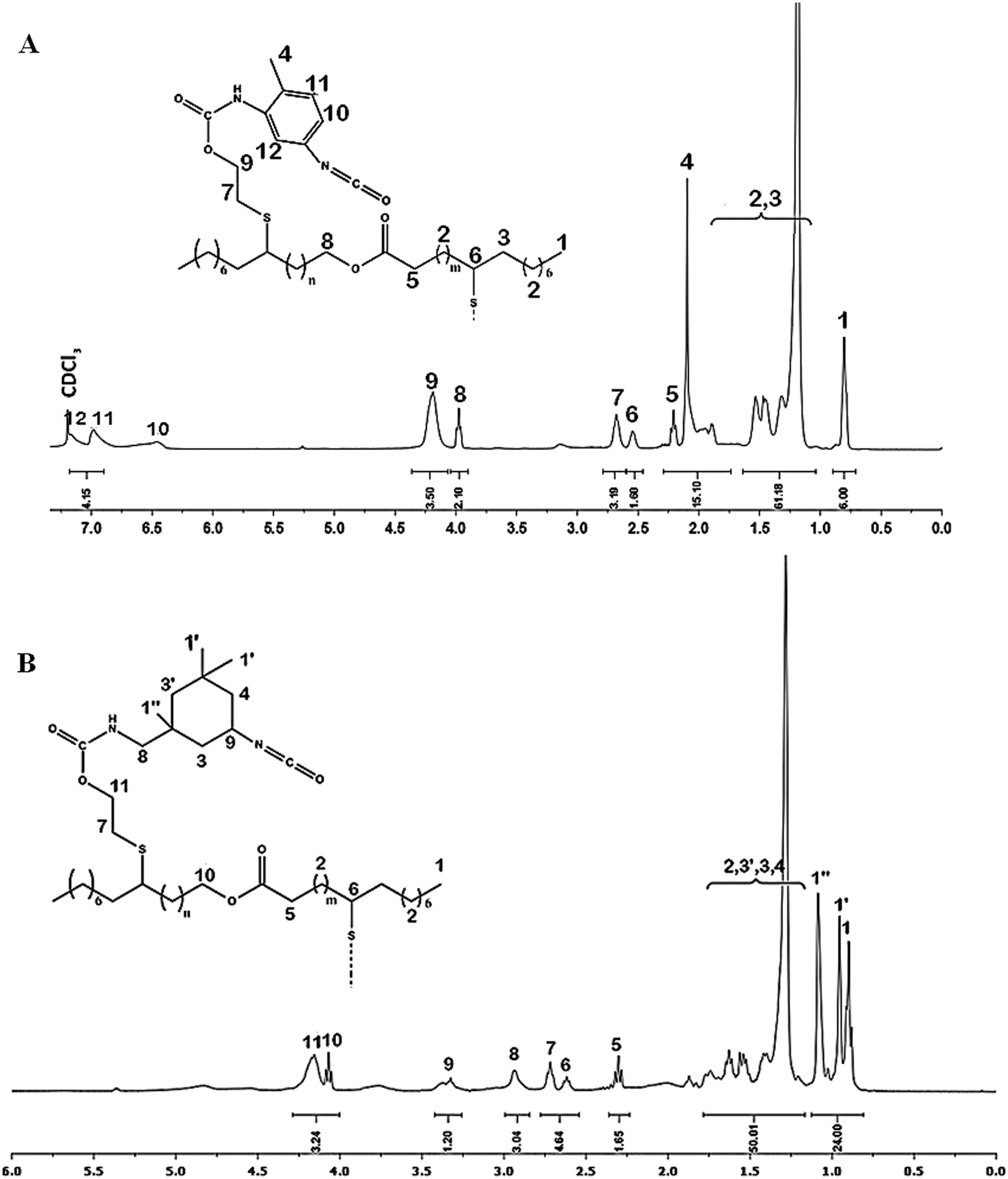
Figure 4: NMR spectrum of (A) Pr1 and (B) Pr2 in CDCl3
1H NMR (Fig. 4B, δ, 400 MHz, ppm): δ = 0.85–1.2 ppm (24H, H1,H1′,H1,CH3–CH2, t); δ = 1.25–1.75 ppm (64H, H2,H3,H3′,H4, CH2–CH2, m); δ = 2.3 ppm (4H, H5, CH2C=O, t); δ = 2.5–2.6 ppm (2H, H6, CH2–HCS–, m), δ = 2.75 ppm (4H, H7 CH2–S–, t), δ = 2.9 ppm (4H, H8, NH–CH2, d), δ = 3.4 ppm (2H, H9, CH-NCO, m), δ = 4 ppm (2H, H10 , -CH2-CH2-OCO, t); δ = 4.25 ppm (8H, H11, -CH2-CH2-OCO, t).
PUs materials were formed by the addition of chain extenders to the liquid prepolymers. 1 eq of 1,3-propanediol were added to Pr1 to obtain PU1, and Pr2 added to get PU2. All the formulations were occurred for 4 h at 80°C.
PU1 and PU2 FTIR (δ, cm−1, Fig. 5): 3300 (N-H groups), 2921-2851 (CH aliphatic), 1700–1730 cm−1 (C=O ester and urethane), 1530 (N-H urethane).
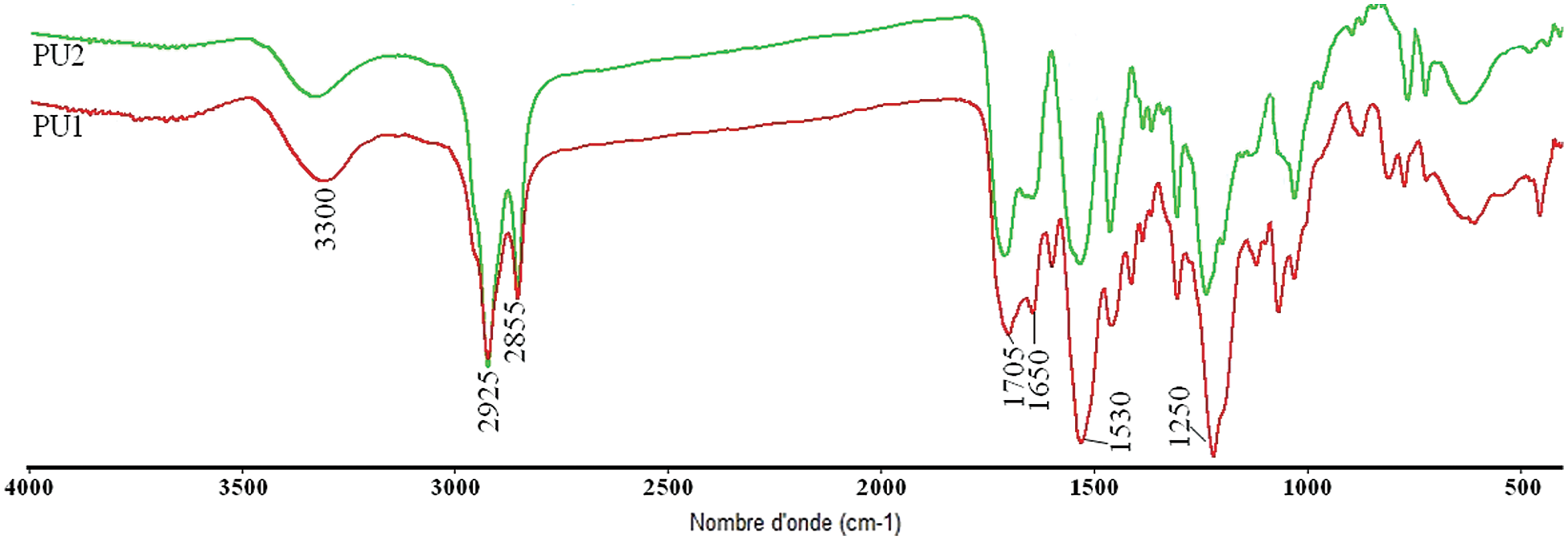
Figure 5: FTIR spectrum of PU1 and PU2
1H NMR (Fig. 6A, δ, 400 MHz, ppm): δ = 0.85 ppm (6H, CH3–CH2 ,t); δ = 1.1–1.6 ppm (64H, CH2–CH2, m); δ = 1.7 - 2 ppm (6H, H4,CH3–ph, d); δ = 2.2 ppm (2H, H5, CH2C=O, t); δ = 2.5–2.6 ppm (2H, H6 H–CS–, m), δ = 2.75 ppm (4H, H7, CH2–S, t), δ = 4 ppm (2H, H8, –CH2 –OCO, t); δ = 4.25 ppm (8H, –CH2–OCO, t); δ = 6.5–7.2 ppm (6H, H10,H11,H12,H–Ph, d).
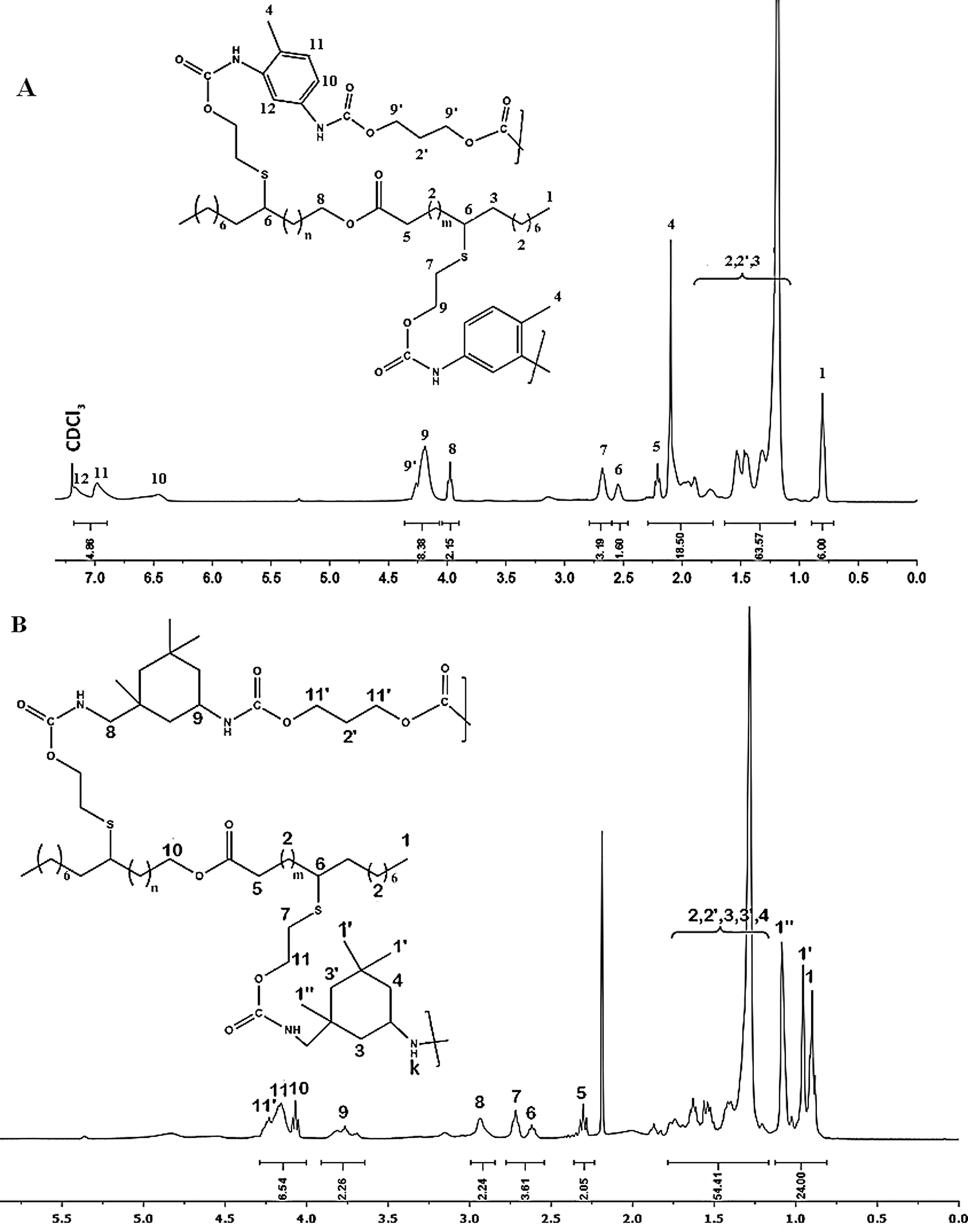
Figure 6: NMR spectrum of (A) PU1 (B) PU2 in CDCl3
1H NMR (Fig. 6B, δ, 400 MHz, ppm): δ = 0.85–1.2 ppm (24H, H1,H1′,H1″,CH3–CH2, t); δ = 1.25–1.75 ppm (64H, H2,H2′,H3,H3′,H4 CH2–CH2, m); δ = 2.3 ppm (2H, H5, CH2C=O, t); δ = 2.5–2.6 ppm (2H, H6, CH2–HCS–, m), δ = 2.75 ppm (4H, H7, CH2–S–, t), δ = 2.9 ppm (4H, H8, NH–CH2, d), δ = 3.75 ppm (8H, H9, NH-CH, m), δ = 4 ppm (2H, H10, –CH2–CH2–OCO, t); δ = 4.1–4.25 ppm (8H, –CH2–CH2–OCO, t).
3.1 Jojoba Diol and Prepolymer Preparation
Jojoba diol (Jodiol) was prepared as reported in previously work [27]. Number of hydroxyl groups was 1.98 hydroxyl functions per molecule [27]. The obtained polyol is used to elaborate new polyurethane material by two-step synthesis: (A) Isocyanate-terminated prepolymer synthesis from excess diisocyanate and jodiol to obtain prepolymer; (B) Reaction of prepolymer with diol to build high molecular weight polyurethane chains. The prepolymer synthesis is displayed at Fig. 2A. It is an isocyanate terminated molecule that was obtained by reaction of an excess of diisocyanate and Jodiol. In this work TDI and IPDI were used to obtain Pr1 and Pr2 respectively. TDI is an aromatic cyclic isocyanate while IPDI is an aliphatic diisocyanate.
The characterisation of theses monomers was occurred by FTIR and NMR 1H. The FTIR spectrum of both prepolymers, presented in Fig. 3, showed the presence of main characteristic band at around 2250 cm−1, which corresponds to the stretching vibration of NCO group and other band at around 1730 cm−1 attributed to the stretching of C=O group. Finally, the presence of N-H stretching vibration around 3300 cm−1 demonstrates that the prepolymerization reaction was successfully occurred.
In addition of protons signals of Jodiol, 1H NMR of Pr1 demonstrate the apparition of the characteristic protons signals H11,12,10 of the aromatic ring between 6.25 and 7.2 ppm and methylene group attached to the ring at 2.1 ppm. 1H NMR of Pr2 demonstrate the apparition of new peaks at 0.8–1.2 and 3.4 ppm attributed to CH3 protons of IPDI and CH proton in α position of the free isocyanate function respectively, as well as CH2 Protons in α position of the urethane group at 2.9 ppm. These spectral findings clearly confirmed the structure of intermediates before the polymerization.
Two different PU materials were synthetized from the Pr1 and Pr2 by the addition of the biobased 1-3 propanediol as chain extender. The synthesis pathways are presented at Fig. 2B. This final step leads to the formation of linear PUs with good flexibility and transparency films. The characterization of the materials was performed by FTIR and NMR.
FTIR spectrum presented in Fig. 5, demonstrate the formation of urethane linkages by observing the desired bands at their respective positions. The disappearance of the characteristic peak of NCO group at 2260 cm−1 for PU 1 and PU 2, while the band at 3200–3500 cm−1 represent the overlapping of NH stretching vibrations, the stretching vibrations of –C=O of urethane and Jodiol appears at 1700–1750 cm−1. N–H deformation vibration of PUs was also observed between 1520 and 1530 cm−1. The 1H NMR study confirmed the formation of the linear polyurethanes.
1H NMR spectrum of PU1 (Fig. 6A), display the apparition of the signals H12, H11 and H10 in the range of 6.4–7.2 ppm and they were attributed to the protons of phenyl groups of TDI, while the signals H9 and H9’ in α position of urethane groups appears at 4.2–4.5 ppm. For the PU2, NMR spectrum (Fig. 6B) show the apparition of the signal H9 at 3.70 ppm referred to the proton of asymmetric carbon of the IPDI cycle, while the methylene protons H1’ and H1” of IPDI appears between 0.9 and 1.15 ppm. The addition of propane diol is demonstrated by the presence of an additional signal at 4.25 ppm attributed to protons in α position of urethane groups. The above results confirm the formation of PUs materials.
3.3 Thermal Characterization and SEC Analysis
First of all, PU materials were characterized by DSC in order to determine their glass transition temperature (Tg). DSC analysis curve in Fig. 7a demonstrates the presence of glass transition at −30°C and −22°C, for PU1 and PU2, respectively. These low Tg are expected and were caused by the aliphatic structure of the macrodiol and propane diol. In addition, these two values are very close, and the small difference is due to the structure of PU1 which has an aromaticity compared to the PU2. No residual exothermic reaction was observed for all PU materials, which implies, that the materials totally formed. In addition, no endothermal peaks were seen in both samples which imply that the materials do not present any change of state until 200°C.
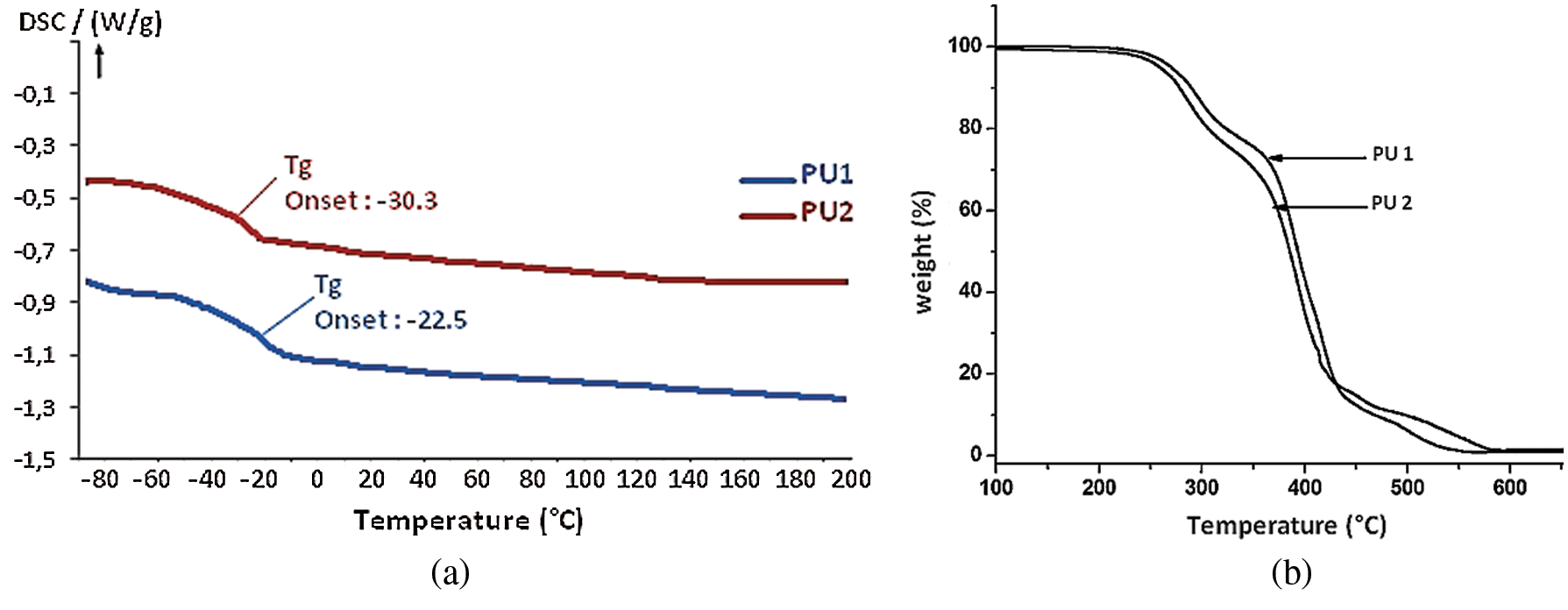
Figure 7: DSC (a) and TGA (b) curves of PU1 and PU2
TGA analyses were carried out for all PUs in order to investigate their stability under air atmosphere (Fig. 7b) with a heating rate of 15°C min−1. It is noticed that the PU1 is slightly stable than the PU2 this is due to the presence of the aromatic ring in the PU1. Both PUs are stable until 250°C and showed three-step degradation just like the PU materials synthetized in our previous work by the use of Methylene bisphenyl isocyanate and hexamethylene diisocyanate. The most thermo-liable section within the structure between 350°C and 450°C is attributed to the aliphatic long chain of jojoba oil which consist the soft segment. These materials exhibited a 5% weight loss between 230 and 250°C, then the mass decreases gradually until 550°C to reach 100% of degradation.
SEC analysis was performed on the PU1 and the PU2 and the result is summarized in the Fig. 8. It is clear that the 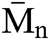 of both materials was pretty much the same, PU1 show a
of both materials was pretty much the same, PU1 show a 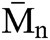 of 17.000 while PU2 show a
of 17.000 while PU2 show a 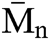 of 15,000 g mol−1 with a dispersity of 1.9. This result is similar to the PUs obtained from methylene diisocyanate and hexamethylene diisocyanate and jojoba diol presented in our previous work [27].
of 15,000 g mol−1 with a dispersity of 1.9. This result is similar to the PUs obtained from methylene diisocyanate and hexamethylene diisocyanate and jojoba diol presented in our previous work [27].
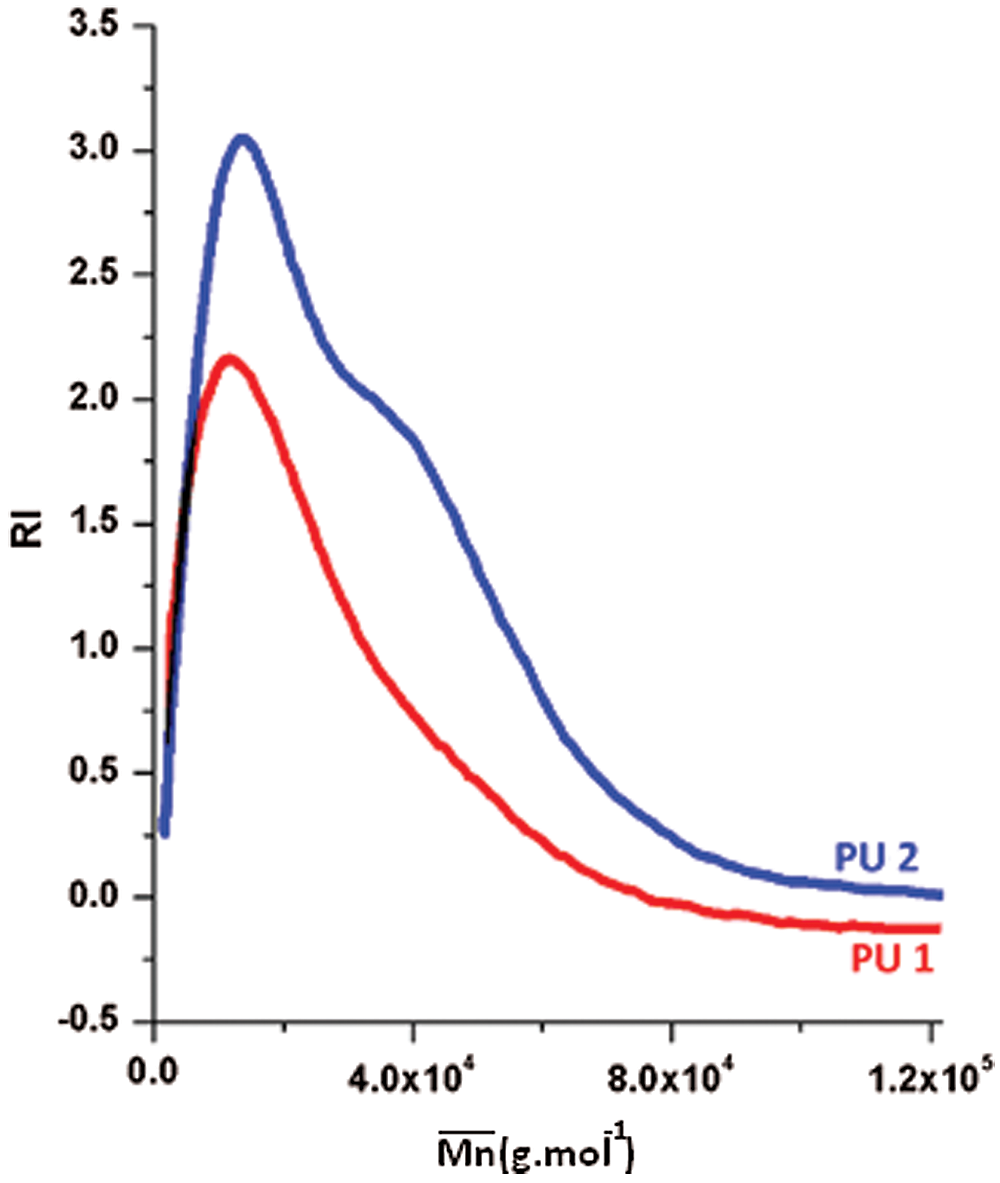
Figure 8: GPC curves of PU1 and PU2
In this work, modified jojoba oil is used to develop new linear PU materials, by converting jojoba oil to its corresponding alcohol. The fully characterised polyurethanes were found to be flexible polymers and exhibited a good thermal stability. It was shown that a desired PU could be prepared by monitoring the nature of the used disocyanate. These partially bio-based polyurethanes represent a good alternative for a partial substitution of petro-based counterpart. In addition, their enhanced solubility allows them to be reinforced with cellulose nanocrystals or cellulose nanofibrils, thus yielding strong nanocomposites from bioresource raw material.
Funding Statement: The authors acknowledge the “CMPTM Project 17TM22” and to the “PHC-UTIQUE CMCU” (18G1132), as well as to the Tunisian Ministry of Higher Education and LabEx Tec 21 for the financial support.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
1. Kamaci, M. (2020). Polyurethane-based hydrogels for controlled drug delivery applications. European Polymer Journal, 123(8), 109444. DOI 10.1016/j.eurpolymj.2019.109444. [Google Scholar] [CrossRef]
2. Wazarkar, K., Sabnis, A. (2018). Development of cardanol-based polyol via click chemistry and crosslinking with melamine formaldehyde resin for coating applications. Journal of Renewable Materials, 6(4), 438–449. DOI 10.7569/JRM.2017.634159. [Google Scholar] [CrossRef]
3. Shen, Y., He, J., Xie, Z., Zhou, X., Fang, C. et al. (2019). Synthesis and characterization of vegetable oil based polyurethanes with tunable thermomechanical performance. Industrial Crops and Products, 140, 111711. DOI 10.1016/j.indcrop.2019.111711. [Google Scholar] [CrossRef]
4. Beverte, I., Shtrauss, V., Kalpinsh, A., Lomanovskis, U., Cabulis, U. et al. (2020). Dielectric permittivity of rigid rapeseed oil polyol polyurethane biofoams and petrochemical foams at low frequencies. Journal of Renewable Materials, 8(9), 1151–1170. DOI 10.32604/jrm.2020.010215. [Google Scholar] [CrossRef]
5. Rane, A. V., Kanny, K., Abitha, V. K., Jadhav, S. Mulge, S. et al. (2017). Applications of polyurethane based composites and nanocomposites. Polyurethane Polymers, 97(6), 559–573. DOI 10.1016/B978-0-12-804065-2.00020-6. [Google Scholar] [CrossRef]
6. Alagi, P., Hong, S. C. (2015). Vegetable oil-based polyols for sustainable polyurethanes. Macromolecular Research, 23(12), 1079–1086. DOI 10.1007/s13233-015-3154-6. [Google Scholar] [CrossRef]
7. de Almeida Machado, J. G., de Carvalho, J. P. R. G., Neves, A. C. C., Lopes, F. P. D., Monteiro, S. N. et al. (2019). Impact energy evaluation of natural castor oil polyurethane matrix composites reinforced with jute fabric. Green Materials Engineering, Springer, Cham, pp. 63–68. DOI 10.1007/978-3-030-10383-5. [Google Scholar] [CrossRef]
8. Tian, D., Wang, F., Yang, Z., Niu, X., Wu, Q. et al. (2019). High-performance polyurethane nanocomposites based on UPy-modified cellulose nanocrystals. Carbohydrate Polymers, 219(6070), 191–200. DOI 10.1016/j.carbpol.2019.05.029. [Google Scholar] [CrossRef]
9. Illy, N., Fache, M., Menard, R., Negrell, C., Caillol, S. et al. (2015). Phosphorylation of bio-based compounds: the state of the art. Polymer Chemistry, 6(35), 6257–6291. DOI 10.1039/C5PY00812C. [Google Scholar] [CrossRef]
10. Gadhave, R. V., Kasbe, P. S., Mahanwar, P. A., Gadekar, P. T. (2019). Synthesis and characterization of lignin-polyurethane based wood adhesive. International Journal of Adhesion and Adhesives, 95(2), 102427. DOI 10.1016/j.ijadhadh.2019.102427. [Google Scholar] [CrossRef]
11. Mokhtari, C., Malek, F., Manseri, A., Caillol, S., Negrell, C. (2019). Reactive jojoba and castor oils-based cyclic carbonates for biobased polyhydroxyurethanes. European Polymer Journal, 113(4), 18–28. DOI 10.1016/j.eurpolymj.2019.01.039. [Google Scholar] [CrossRef]
12. Alam, M., Akram, D., Sharmin, E., Zafar, F., Ahmad, S. (2014). Vegetable oil based eco-friendly coating materials: A review article. Arabian Journal of Chemistry, 7(4), 469–479. DOI 10.1016/j.arabjc.2013.12.023. [Google Scholar] [CrossRef]
13. Patil, C. K., Rajput, S. D., Marathe, R. J., Kulkarni, R. D., Phadnis, H. et al. (2017). Synthesis of bio-based polyurethane coatings from vegetable oil and dicarboxylic acids. Progress in Organic Coatings, 106(2014), 87–95. DOI 10.1016/j.porgcoat.2016.11.024. [Google Scholar] [CrossRef]
14. Nilawar, S., Dasgupta, Q., Madras, G., Chatterjee, K. (2019). Degradable poly(ester amide)s from olive oil for biomedical applications. Emergent Materials, 2(2), 153–168. DOI 10.1007/s42247-019-00032-w. [Google Scholar] [CrossRef]
15. Pawar, M. S., Kadam, A. S., Yemul, O. S. (2015). Development of polyetheramide based corrosion protective polyurethane coating from mahua oil. Progress in Organic Coatings, 89, 143–149. DOI 10.1016/j.porgcoat.2015.08.017. [Google Scholar] [CrossRef]
16. Mustapha, R., Rahmat, A. R., Abdul Majid, R., Mustapha, S. N. H. (2019). Vegetable oil-based epoxy resins and their composites with bio-based hardener: A short review. Polymer-Plastics Technology and Materials, 58(12), 1311–1326. DOI 10.1080/25740881.2018.1563119. [Google Scholar] [CrossRef]
17. Omrani, I., Farhadian, A., Babanejad, N., Shendi, H. K., Ahmadi, A. et al. (2016). Synthesis of novel high primary hydroxyl functionality polyol from sunflower oil using thiol-yne reaction and their application in polyurethane coating. European Polymer Journal, 82, 220–231. DOI 10.1016/j.eurpolymj.2016.07.021. [Google Scholar] [CrossRef]
18. Yakushin, V., Abolins, A., Vilsone, D., Sevastyanova, I. (2019). Polyurethane coatings based on linseed oil phosphate ester polyols with intumescent flame retardants. Fire and Materials, 43(1), 92–100. DOI 10.1002/fam.2672. [Google Scholar] [CrossRef]
19. Zhang, C., Madbouly, S. A., Kessler, M. R. (2015). Biobased polyurethanes prepared from different vegetable oils. ACS Applied Materials & Interfaces, 7(2), 1226–1233. DOI 10.1021/am5071333. [Google Scholar] [CrossRef]
20. Zlatanić, A., Lava, C., Zhang, W., Petrović, Z. S. (2004). Effect of structure on properties of polyols and polyurethanes based on different vegetable oils. Journal of Polymer Science Part B: Polymer Physics, 42(5), 809–819. DOI 10.1002/polb.10737. [Google Scholar] [CrossRef]
21. Mahendran, A. R., Aust, N., Wuzella, G., Kandelbauer, A. (2012). Synthesis and characterization of a bio-based resin from linseed oil. Macromolecular Symposia, 311(1), 18–27. DOI 10.1002/masy.201000134. [Google Scholar] [CrossRef]
22. Ranaweera, C. K., Ionescu, M., Bilic, N., Wan, X., Kahol, P. K. et al. (2017). Biobased polyols using thiol-ene chemistry for rigid polyurethane foams with enhanced flame-retardant properties. Journal of Renewable Materials, 5(1), 1–12. DOI 10.7569/JRM.2017.634105. [Google Scholar] [CrossRef]
23. Zhang, C., Garrison, T. F., Madbouly, S. A., Kessler, M. R. (2017). Recent advances in vegetable oil-based polymers and their composites. Progress in Polymer Science, 71, 91–143. DOI 10.1016/j.progpolymsci.2016.12.009. [Google Scholar] [CrossRef]
24. Khairi, M. M. (2019). Genetics and Breeding of Jojoba Simmondsia chinensis (Link) Schneider. Advances in Plant Breeding Strategies: Industrial and Food Crops, Springer, Cham, pp. 237–276. DOI 10.1007/978-3-030-23265-8. [Google Scholar] [CrossRef]
25. Cappillino, P., Kleiman, R., Botti, C. (2003). Composition of Chilean jojoba seeds. Industrial Crops and Products, 17(3), 177–182. DOI 10.1016/S0926-6690(02)00096-1. [Google Scholar] [CrossRef]
26. Mokhtari, C., Malek, F. (2020). New bio-based polyhydroxyurethane material. Materials Today: Proceedings, 31(2), S12−S15–S15. DOI 10.1016/j.matpr.2020.05.028. [Google Scholar] [CrossRef]
27. Mokhtari, C., Malek, F., Caillol, S., Negrell, C. (2018). Synthesis of bio-based polyurethanes from jojoba oil. European Journal of Lipid Science and Technology, 120(3), 1700414–7. DOI 10.1002/ejlt.201700414. [Google Scholar] [CrossRef]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |