| Oncologie |
DOI: 10.32604/Oncologie.2021.015929
ARTICLE
Tolerance and Efficacy of Regorafenib according to UGT Pharmacogenetical Status in the Treatment of Metastatic Refractory Colorectal Cancer
1HIA Brest, Rue Colonel Fonferrier, Gastroentérologie, Brest, 29200, France
2CHU Morvan, 2 Av Foch, Oncologie Digestive, Brest, 29200, France
3ICO Paul Papin, Pharmacologie Toxicologie Pharmacogénétique, 15 Rue André Bocquel, Angers, 49055, France
*Corresponding Author: Pierre-Guillaume Poureau. Email: pierre-guillaume.poureau@chu-brest.fr
Received: 24 January 2021; Accepted: 21 April 2021
Abstract: Introduction: Regorafenib is a multi tyrosin-kinase inhibitor prescribed in metastatic refractory colorectal cancer treatment. Its toxicity is significant but inconstant. The metabolism of regorafenib includes oxydation via cytochrome P3A4, then glucuroconjugation. A pharmacogenetical approach of mutational status of Uridine-Diphospho-Glucuronosyltransfersase (UDP-glucuronosyl-transferase, UGT) could be a strategy to optimise the use of regorafenib. Patients and Method: This is a restrospective, unicentric study. All adult patients treated with regorafenib for a metastatic colorectal cancer in our center between 2013 and 2017 were analysed. UGT1A1 polypmorphism was previously researched in the laboratory after written informed consent. Results: Thirty-five patients received regorafenib during the study period. A TA repetition in UGT1A1 gene was present for 16 patients including two Gilbert syndrome. There were no more adverse events on patients with a heterozygous TA repetition or with a Gilbert syndrome compared with patients without UGT polymorphism, whatever the dosage at initiation of regorafenib. Adverse events grade 2 or superior, and grade 3 or superior tended to be more noticeable when the starting dose was 120 or 160 mg per day compared to 80 mg per day, but not statistically significant. No difference was observed on progression-free survival neither depending on UGT status nor depending on initating dose of regorafenib. Conclusion: This is a preliminary study evaluating safety of regorafenib according to UGT1A1 polymorphism. Larger and prospective studies are needed to evaluate dose-escalation strategy in patients with variable activity of glucuroconidation, especially Gilbert syndrome, or with abnormalities in other UGT enzymes such as UGT1A9.
Keywords: Regorafenib; UGT polymorphism; metastatic colorectal cancer
Colorectal cancer is the third most common cancer in the world in incidence and the second in mortality [1]. Several randomised placebo-controlled trials [2,3] have shown a survival benefit in patients with metastatic colorectal cancer treated with regorafenib who progressed on standard therapies. Regorafenib is now allowed from third line setting in this indication. However, the toxicity of this molecule is significant and constitutes a limiting factor, but quite unpredictable.
Anticancer therapies are more and more personalised, according to molecular signature, and/or to toxicity profile. The aim of this personalised-medicine strategy is to optimise treatment by preventing toxicities. One of the ways to understand inter-individual variability is to research germinal polymorphism by pharmacogenetic analysis, such as the Uridine-Diphospho-Glucuronosyltransfersase (UDP-glucuronosyltransferase, UGT).
Dose-adjustment of irinotecan according to UGT1A1*28(rs8175347) status has already been demonstrated and is completely absorbed into our clinical practice [4]. Regorafenib is a multi tyrosin-kinase inhibitor that blocks the activity of several protein kinases involved in tumor pathogenesis (such as vascular endothelial growth factor receptors 1, 2 and 3; angiopoietin 1 receptor; stem-cell growth factor receptor; RET and BRAF; beta-type platelet-derived growth factor receptor; and fibroblast growth factor receptor). This molecule is mainly metabolised in the liver by oxydation via cytochrome P3A4, but also by glucuroconjugation. Exploiting the same pharmacogenetical approach of UGT polymorphism status could be a strategy to optimise regorafenib uses, as well as the use of irinotecan. This strategy was suggested by experts [5] in previous articles, but to our knowledge it has never been tested so far.
The main objective of this study was to determine whether a polymorphism in the UGT gene was associated with a lower tolerance of regorafenib. The secondary objectives were to determine whether a polymorphism in the UGT gene was associated with decreased efficacy of regorafenib, and whether dose-escalation strategy was better tolerated.
This is a retrospective, unicentric study performed in Brest, France. Analysed patients were all adults treated with regorafenib for a metastatic colorectal cancer in our center between 2013 and 2017 in third line or more. Age, gender, prior anticancer treatments, performance status at treatment initiation, mutational status of RAS and RAF, starting dose of regorafenib and adjustments, duration of treatment, adverse events and progression-free survival data were collected. All patients provided written informed consent as required by Good Clinical Practices guidelines and ethics commitee.
UGT1A1 polymorphism (UGT1A1*28(rs8175347)) was previously researched by the laboratory « Eurofins-Biomnis ». The method used for determination of UGT1A1 polymorphism has been previously described [6], and consisted of the Pyrosequencing technology, a real-time sequencing method, to detect the UGT 1A1 TATA box polymorphisms and mutations in the coding regions. Results were given as Wild type (*1/*1), heterozygote with presence of one allele UGT1A1*28 (*1/*28), or homozygote mutation corresponding to Gilbert syndrome (*28/*28).
Statistical analyses were performed with Excel© software v.2003 and in BiostaTGV website. Discrete variables were expressed in percentage of the studied population and compared with the Fisher’s exact test. Non discrete variables were expressed as mean and compared with the Student’s test. Survival curves were created using the Kaplan-Meier method and compared with the Log-rank test. A p value ≤ 0.05 was considered statistically significant.
Thirty-five patients received regorafenib during the study period. Main characteristics are described in Tab. 1. A polymorphism in UGT gene was present for 16 (46%) patients, including two Gilbert syndrome. There were no significant differences between the two groups UGT heterozygous or homozygous (i.e., UGT-*28*28), except for starting dose. When the subgroup of patients treated with regorafenib started at 80 milligram per day (mg/d) and 120 mg/d (dose-escalation) were grouped together, there was no longer any difference in the distribution between the two groups of *28 and Wild Type UGT (OR = 3.97, IC95% [0.69; 29.9], p = 0.13). Most frequent adverse events are reported in Tab. 2 and were mainly asthenia (46%), hand-foot syndrome (37%), diarrhoea (29%), arterial high blood pressure (26%) and anorexia (14%).
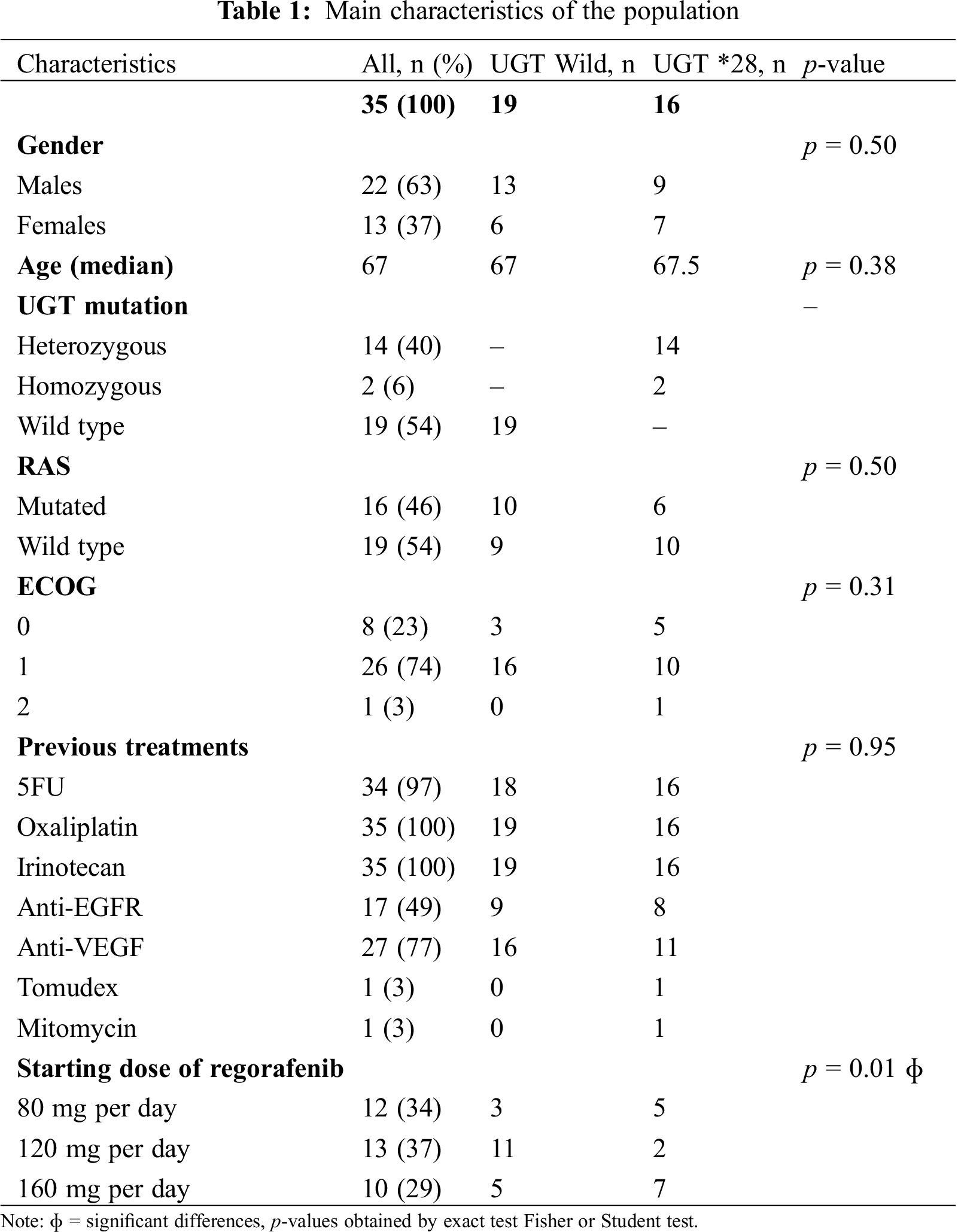
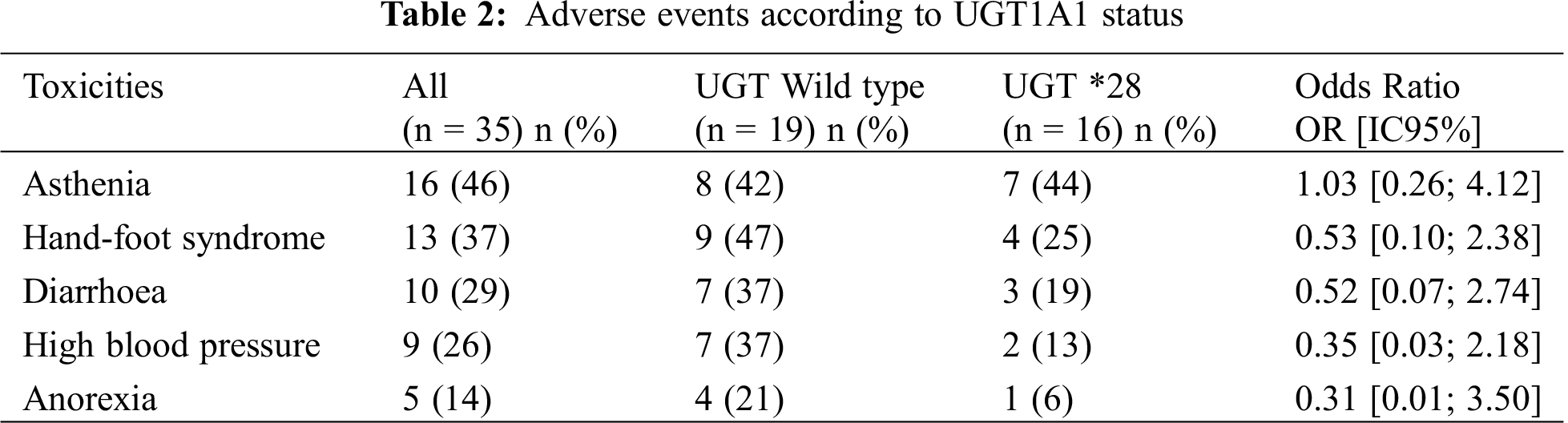
There were no more adverse events in patients with a heterozygote status or with a Gilbert syndrome compared with patients without UGT polymorphism (Tab. 2), whatever the starting dose of regorafenib.
Two patients were Gilbert syndrome. The first one received 160 mg per day from the outset and the treatment was discontinued because of deterioration in general condition. The second one started the treatment at 80 mg per day, then 120 mg per day without ever reaching 160 mg per day, with a clinical benefit for 84 days.
Adverse events ≥ grade 2 and ≥ grade 3 tended to be more frequent when the starting dose was 120 or 160 mg per day (78% and 52%, respectively) compared to 80 mg per day (67% and 17%, respectively), but this result was not statistically significant, whatever the subsequent strategy of dose escalation or de-escalation (Tab. 3).
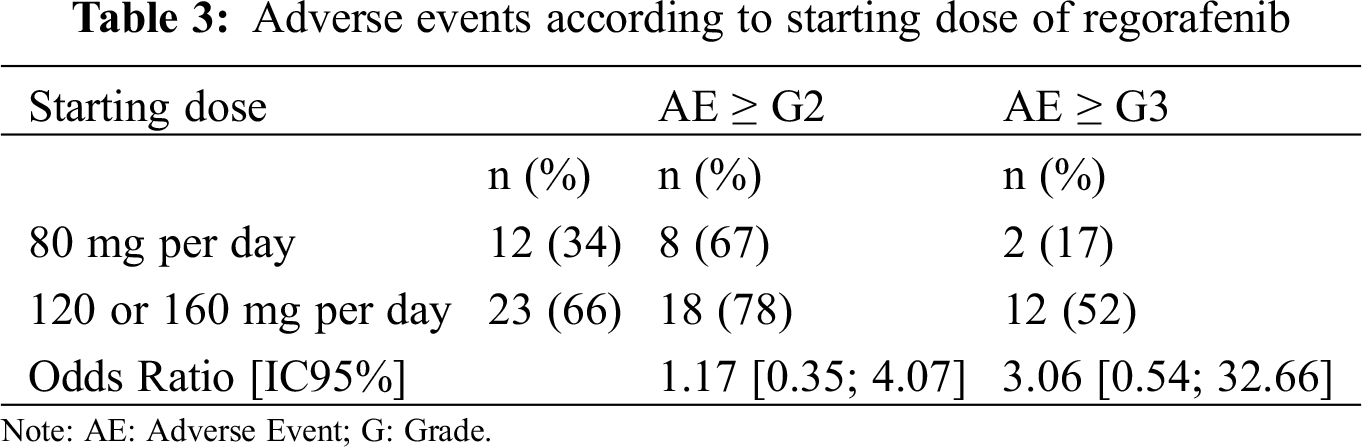
Considering the population with a dose-escalation strategy (i.e., starting dosage at 80 mg or 120 mg per day), more patients received an increasing dose of regorafenib when they were UGT Wild type compared with patients with *28 allele (56% vs. 33%). Similarly, 67% of patients with UGT polymorphism have never been able to tolerate an augmentation, whereas only 25% of the patients without TA repetition have not been able to tolerate an augmentation. These differences were marked in absolute value but not statistically significant (OR = 0.6, IC95% [0.08; 3.31], p = 0.6 and OR = 2.5, IC95% [0.47; 16.17], p = 0.27, respectively).
The median progression-free survival (PFS) of the whole cohort was 90 days. No difference was observed on PFS, neither depending on UGT status (Wild type compared to heterozygote or Gilbert syndrome) nor depending on starting dose (80 mg per day compare to 120 or 160 mg per day) (p = 0.98). PFS curves are presented in Fig. 1.
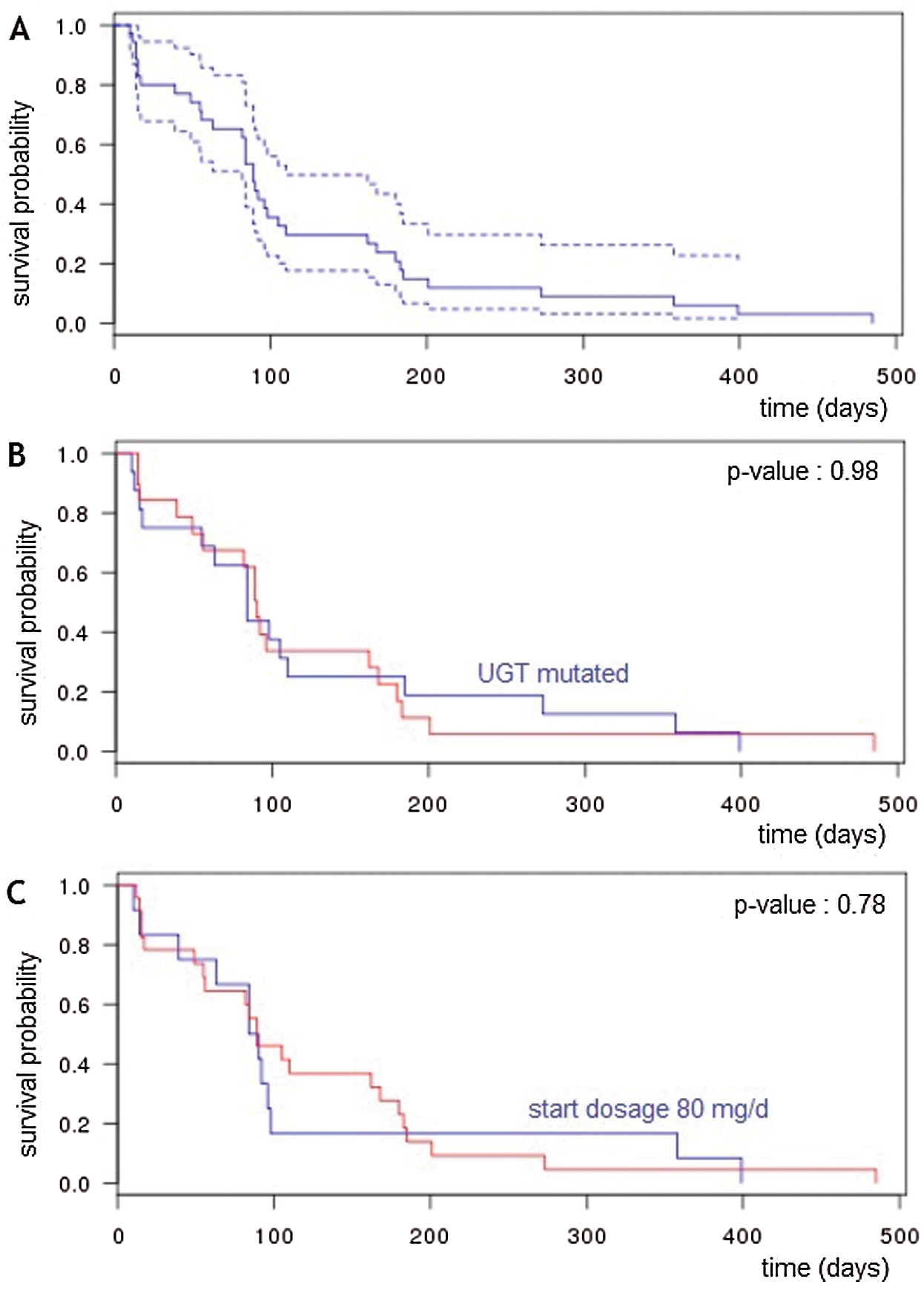
Figure 1: Progression-free survival (A) Progression-free survival for the whole population. (B) Progression-free survival for patient UGT Wild type or not. (C) Progression-free survival for patients with starting dose at 80 mg/d or 120–160 mg/d
To our knowledge, this is the first cohort assessing safety of regorafenib according to mutational status of UGT1A1.
Of the thirty-five patients of the cohort, a TA repetition in UGT1A1 gene was present for 16. There were no more adverse events on patients with a heterozygous TA repetition or with a Gilbert syndrome compared with patients without UGT-polymorphism, whatever the initiating dose of regorafenib. Adverse events grade ≥ 2 and ≥ grade 3 tended to be more noticeable when the starting dose was 120 or 160 mg per day as opposed to 80 mg per day. Most patients with UGT polymorphism have never been able to tolerate a dose-escalation compared with patients without UGT polymorphism. No difference was observed on progression-free survival neither depending on UGT status nor depending on starting dose.
Following oral administration, regorafenib reaches peak plasma level at a median time of four hours. The main circulating metabolites of regorafenib are M-2 (N-oxide) and M-5 (N-oxide/N-desmethyl). Both of them have similar in vitro activity as regorafenib. The elimination half-lives for regorafenib, M-2 metabolite and M-5 metabolite are 28 h, 25 h and 51 h, respectively. Approximately, 71% is excreted in faeces and 19% in urine, mostly as glucoronides. [7] Regorafenib is metabolised by cytochrome P3A4 and glucoronidation using UGT enzymes (Fig. 2).
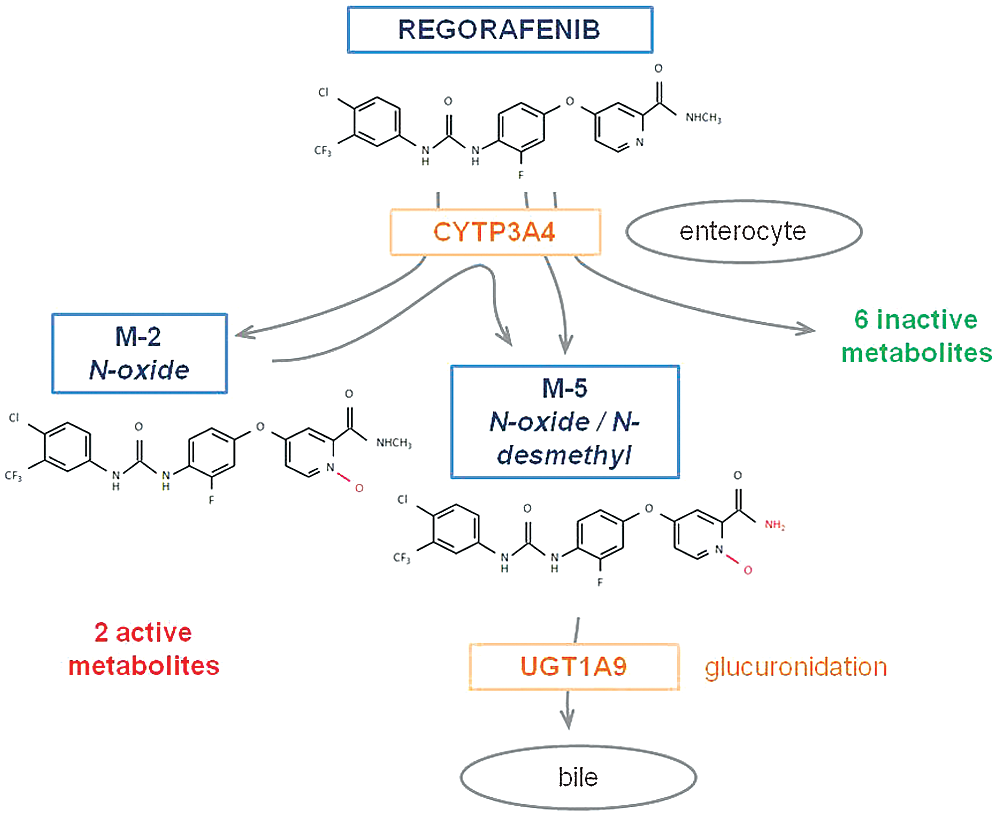
Figure 2: Metabolism of regorafenib
UDP-glucuronosyltransferases (UGTs) are a multigenic family of enzymes responsible for the glucuronidation reaction. In humans, up to 22 UGT isoforms have been identified. Among these families, UGT1A, 2A and 2B are active in biotransformation. The gene coding for the UGT 1A family is located on chromosome 2. There are 12 isoforms for UGT1A (UGT1A1 to UGT1A12).
The importance of UGT-mediated metabolism and its consequences for toxicity has already been demonstrated for irinotecan. Irinotecan is converted to its active form, 7-ethyl-10-hydroxycamptothecin (SN-38). SN-38 is then further metabolised into an inactive metabolite, SN-38G, by the UGT enzyme in the liver. Glucuronidation of SN-38 to SN-38G is a decisive step in the metabolism and detoxification of irinotecan. Genetic polymorphisms of the UGT1A1 gene can alter the glucuronidation of SN-38 and as such have a major influence on the pharmacokinetics and toxicity of irinotecan [8].
The research of higher risk of toxicity with irinotecan is only based on the UGT1A1. That is why we only looked in this study at UGT1A1. This point could be a part of explanation for the lack of difference in regorafenib tolerance. Specific alteration of UGT1A9 should therefore be studied. A polymorphism in UGT1A9 promoter region gene has been reported for 3 patients who experienced acute hepatitis under regorafenib treatment. Moreover, it appears that both of them already experienced significant toxicity with irinotecan-based chemotherapy [9]. However, it is known that the alteration of genes coding for UGT1A1 is frequently associated with other polymorphisms of the superfamily of UGT. Similarly, a heterozygote has a heterogeneous phenotypic expression. It would be necessary to study more people in the specific population of Gilbert syndrome. Indeed, the report of our two patients is noteworthy. One patient very quickly had a poor tolerance at full dose, and the other patient benefitted from dose escalation.
The strategy of dose-escalation was not decided upstream according to UGT1A1 status but was at the physician’s discretion. Moreover, the studied population was a small cohort. There are therefore biases and a lack of statistical power that may explain the absence of difference in the frequency of side effects according to UGT status. However, it is noted that the dose-escalation strategy enables toxicity to be limited without impairing efficacy, which has been confirmed by large cohorts (ReDOS [10] and REARRANGE [11] trials). Especially this dose escalation is more restrained in patients with UGT polymorphism, suggesting that their tolerance is more critical. The RESET trial [12] showed that serum concentrations of regorafenib for patients whose dose was escalated to 160 mg/day were significantly lower than those of the patients whose dose was not escalated. Moreover, concentration of active metabolites correlated with the maximum grade of regorafenib-related adverse events. A recent publication showed that M-5 concentration was associated with the severity of hypertension or rash [13]. These results suggest that dose escalation is limited by higher concentrations of regorafenib and its metabolites, responsible for greater toxicity. These higher concentrations are partly related to the UGT enzyme due to its metabolism. Dose-escalation appeared therefore to be a better strategy and should be prefered especially in patients with alteration of UGT gene, although the low number of patients has not permitted us to demonstrate it.
This is a preliminary study evaluating the safety of regorafenib according to UGT1A1 polymorphism. The possibility of increasing the dosage appears to be more limited in the case of UGT mutation leading to lower level of safety of regorafenib for these patients. Larger prospective studies are needed to evaluate dose-escalation strategy in patients with variable activity of glucuronidation, especially Gilbert syndrome, or with abnormalities in other UGT enzymes such as UGT1A9.
Acknowledgement: We thanks our English friends Esme and Stephen for the proofreading.
Funding Statement: The authors received no specific funding for this study.
Conflicts of Interest: Poureau Pierre-Guillaume: Investigator for MSD, Servier, Atrazeneca, BeiGene studies. Invitation to congress from Servier. Fees from Sanofi, Bayer for participation to boards; Dhamelincourt Estelle: No conflict of interest with this work; Nguyen Jessica: No conflict of interest with this work; Babey Hélène: No conflict of interest with this work; Renaud Emmanuelle: No conflict of interest with this work; Geier Margaux: Invitation to congress from MSD. Fees from BMS, AstraZeneca for participation to boards; Boisdron-Celle Michèle: Biologist at ICO. No conflict of interest with this work; Metges Jean-Philippe: Investigator for MSD, Servier, Astrazeneca, BeiGene, Roche studies. Invitation to congress from MSD. Fees from Sanofi, Bayer, Roche for participation to boards. Editor in chief of Journal Oncologie (ISSN: 1292-3818 Print, ISSN: 1765-2839 Online).
1. Ferlay, J., Steliarova-Foucher, E., Lortet-Tieulent, J., Rosso, S., Coebergh, J. W. W. et al. (2013). Cancer incidence and mortality patterns in Europe: Estimates for 40 countries in 2012. European Journal of Cancer, 49(6), 1374–1403. DOI 10.1016/j.ejca.2012.12.027. [Google Scholar] [CrossRef]
2. Grothey, A., van Cutsem, E., Sobrero, A., Siena, S., Falcone, A. et al. (2013). Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECTAn international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet, 381(9863), 303–312. DOI 10.1016/S0140-6736(12)61900-X. [Google Scholar] [CrossRef]
3. Li, J., Qin, S., Xu, R., Yau, T. C. C., Ma, B. et al. (2015). Regorafenib plus best supportive care versus placebo plus best supportive care in Asian patients with previously treated metastatic colorectal cancer (CONCURA randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncology, 16(6), 619–629. DOI 10.1016/S1470-2045(15)70156-7. [Google Scholar] [CrossRef]
4. Boisdron-Celle, M., Metges, J. P., Capitain, O., Adenis, A., Raoul, J. L. et al. (2017). A multicenter phase II study of personalized FOLFIRI-cetuximab for safe dose intensification. Seminars in Oncology, 44(1), 24–33. DOI 10.1053/j.seminoncol.2017.02.007. [Google Scholar] [CrossRef]
5. Tlemsani, C., Huillard, O., Arrondeau, J., Boudou-Rouquette, P., Cessot, A. et al. (2015). Effect of glucuronidation on transport and tissue accumulation of tyrosine kinase inhibitors: consequences for the clinical management of sorafenib and regorafenib. Expert Opinion on Drug Metabolism & Toxicology, 11(5), 785–794. DOI 10.1517/17425255.2015.1030392. [Google Scholar] [CrossRef]
6. Rouits, E., Boisdron-Celle, M., Dumont, A., Guerin, O., Morel, A. et al. (2004). Relevance of different UGT1A1 polymorphisms in irinotecan-induced toxicity. Clinical Cancer Research, 10(15), 5151–5159. DOI 10.1158/1078-0432.CCR-03-0548. [Google Scholar] [CrossRef]
7. Mross, K., Frost, A., Steinbild, S., Hedbom, S., Büchert, M. et al. (2012). A phase I dose-escalation study of regorafenib (BAY 73-4506an inhibitor of oncogenic, angiogenic, and stromal kinases, in patients with advanced solid tumors. Clinical Cancer Research, 18(9), 2658–2667. DOI 10.1158/1078-0432.CCR-11-1900. [Google Scholar] [CrossRef]
8. Iyer, L., King, C. D., Whitington, P. F., Green, M. D., Roy, S. K. et al. (1998). Genetic predisposition to the metabolism of irinotecan (CPT-11). Role of uridine diphosphate glucuronosyltransferase isoform 1A1 in the glucuronidation of its active metabolite (SN-38) in human liver microsomes. Journal of Clinical Investigation, 101(4), 847–854. DOI 10.1172/JCI915. [Google Scholar] [CrossRef]
9. Sacré, A., Lanthier, N., Dano, H., Aydin, S., Leggenhager, D. et al. (2016). Regorafenib induced severe toxic hepatitis: Characterization and discussion. Liver International, 36(11), 1590–1594. DOI 10.1111/liv.13217. [Google Scholar] [CrossRef]
10. Bekaii-Saab, T. S., Ou, F. S., Ahn, D. H., Boland, P., Ciombor, K. K. et al. (2019). Regorafenib dose-optimisation in patients with refractory metastatic colorectal cancer (ReDOSA randomised, multicentre, open-label, phase 2 study. The Lancet Oncology, 20(8), 1070–1082. DOI 10.1016/S1470-2045(19)30272-4. [Google Scholar] [CrossRef]
11. Argiles, G., Mulet Margalef, N., Valladers Ayerbes, M., Vietez de Prado, J., Gravalos, C. et al. (2019). Results of REARRANGE trial: A randomized phase 2 study comparing different dosing approaches for regorafenib (REG) during the first cycle of treatment in patients (pts) with metastatic colorectal cancer (mCRC). Annals of Oncology, 30(4), iv137–iv151. DOI 10.1093/annonc/mdz153. [Google Scholar] [CrossRef]
12. Suzuki, T., Sukawa, Y., Imamura, C. K., Masuishi, T., Satake, H. et al. (2020). A phase II study of regorafenib with a lower starting dose in patients with metastatic colorectal cancer: Exposure-Toxicity analysis of unbound regorafenib and its active metabolites (RESET Trial). Clinical Colorectal Cancer, 19(1), 13–21. DOI 10.1016/j.clcc.2019.10.004. [Google Scholar] [CrossRef]
13. Kobayashi, K., Sugiyama, E., Shinozaki, A., Wakatsuki, T., Tajima, M. et al. (2021). Associations among plasma concentrations of regorafenib and its metabolites, adverse events, and ABCG2 polymorphisms in patients with metastatic colorectal cancers. Cancer Chemotherapy and Pharmacology, Online ahead of print. [Google Scholar]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |