| Oncologie |
DOI: 10.32604/oncologie.2022.020648
REVIEW
The Effect of Oncogene Proteins of Human Papillomaviruses on Apoptosis Pathways in Prostate Cancer
1Infectious and Tropical Diseases Research Center, Tabriz University of Medical Sciences, Tabriz, Iran
2Immunology Research Center, Tabriz University of Medical Sciences, Tabriz, Iran
3Department of Microbiology, Faculty of Medical Sciences, Islamic Azad University, Kazerun, Iran
4Drug Applied Research Centre, Tabriz University of Medical Sciences, Tabriz, Iran
5Department of Virology, Faculty of Medicine, Iran University of Medical Sciences, Tehran, Iran
6Student Research Committee, Iran University of Medical Sciences, Tehran, Iran
7Department of Virology, Faculty of Medicine, Tabriz University of Medical Sciences, Tabriz, Iran
*Corresponding Author: Hossein Bannazadeh Baghi. Email: hbannazadeh@tbzmed.ac.ir; hb.zadeh@gmail.com
Received: 05 December 2021; Accepted: 28 March 2022
Abstract: The ability of host cells to activate apoptosis is perhaps the most potent weapon for helping cells eliminate viruses. Human papillomaviruses (HPV) activate several pathways, enabling the infected cells to avoid extrinsic and intrinsic apoptosis pathways. The incapacity of prostatic epithelial cells to induce apoptosis leads to the invasive development of prostate cancer. For the pathogenesis of prostate cancer, several risk factors have been reported; for example, some viruses and infectious diseases have been proposed as causative agents for their relation to prostate diseases. According to several studies, high-risk human papillomaviruses cause malignancy by interfering with the apoptotic and inflammatory pathways; these viruses, such as HPV16 and HPV18, block apoptotic pathways and result in prostate cancer. This review is dedicated to presenting a summary of oncogenes (E5, E6, and E7) HR-HPVs’ functions on signaling pathways, inflammation in prostate tumorigenesis, and emphasizing the link between these oncogenes with apoptosis and prostate cancer.
Keywords: Apoptosis; inflammation; oncoproteins; prostate cancer
Prostate cancer (PCa) is considered the fifth important cause of death worldwide and the second-most common cancer diagnosed in men [1]. There is not much data on the progression and precise mechanisms associated with PCa; however, several factors might contribute to men getting PCa, including ethnicity, age of more than 50 years, genetic mutations (inherited and acquired), and infections [2]. Moreover, inflammation and chronic infection cause malignancy in some organs, including the liver, stomach, prostate, cervical, and colon [3]. Also, various etiological factors contribute to prostatic inflammation (prostatitis) induction, such as hormonal variations, infection, dietary factors, and cell injury. The cellular injury resultant may cause a loss of tolerance to prostatic antigens and result in a self-sustaining autoimmune reaction [3,4]. There are approximately 1,400 human pathogens which comprise 220 kinds of viruses; that few of these, referred to oncogenic viruses, are identified to cause malignancy via some strategies. The behavior of the tumor is influenced by virally mediated mechanisms affecting tumor cells and stromal cells in the tumor microenvironment. The virally induced mechanisms include apoptosis suppression, tumor metabolism alternation, anti-tumor immunity alternation, tumor microenvironment inflammation promotion, angiogenesis initiation, cellular proliferation stimulation, enhancing tumor invasion, and metastasis [5]. It is also been observed that HPV infection may impact the occurrence of autoimmunity in people with limited ability and could be related to the inflammation by this virus [6]. It has been indicated that HPV accounts for almost 600,000 cases of cancers (e.g., oropharyngeal, cervix, vulvovaginal, penile, and anal) and genital warts, and recurrent respiratory infection papillomatosis (RRP) of the lungs worldwide. HPV is a sexually transmitted virus that contains more than 200 genotypes, and approximately 40 infect the genital area. It has been shown that the prostate might be a target for HPV infections because of its anatomical location [7]. Even though HPV gene sequences were found in most usual, benign, and malignant prostate tissues [8,9], HPV infection possibly modulates the behavior of PCa cells via affecting apoptosis mechanisms, anoikis, anti-tumor immunity suppression, and resistance mechanisms, which results in metastasis [10]. However, the potential role of HR-HPV infection in PCa development has not yet been established [11]. Apoptosis, known as programmed cell death (PCD), is a tightly regulated process that plays a significant role in adult tissues’ growth and homeostasis; it can also function as an antiviral defense mechanism. It has indicated that the deregulation of cell death has a crucial role in developing many diseases, such as cancer. The oncogenic viruses use various pathways to prevent the apoptotic process, permitting the proliferation of damaged and infected cells. In this way, HPV has developed some strategies to evade elimination by apoptosis that allow the infected cells to escape both intrinsic and extrinsic apoptosis pathways [12]. HPVs of all varieties have the same genetic structure and generate eight proteins, containing six early proteins (E1, E2, E4, E5, E6, and E7) and two late proteins (L1 and L2). Antiapoptotic oncoproteins, including E5, E6, and E7 prevalent in high-risk strains, are expected to play a significant role in malignant transformation [13]. During the viral cycle, the HPV E2 protein is a transcription factor that represses the production of viral oncogenes and stimulates viral DNA replication; this protein is explicitly inactivated, in HPV18-associated cancer cells [14]. Proapoptotic proteins E2 and E7 are also considered; as a result, apoptosis and the regulation of the three oncoproteins E5, E6, and E7 are closely linked [13]. It has shown that prostate tumor cells can obtain intracellular survival pathways and variations in chemokine/cytokines and growth factor signal transduction. These characteristics enable them to elude both apoptotic pathways and, promote androgen-independent aggressive behavior, which is resistant to all types of conventional treatment [15]. The recurrent chronic inflammation of unidentified stimuli in the adult prostate has interested a wide range of research to recognize possible infectious factors that might induce prostatic inflammation [16]. Investigations demonstrate that HPV vaccines are efficient for inhibiting infections and lesions which may cause cancer [17]. Understanding the mechanisms of carcinogenesis is essential for therapy, the design of new alternative drugs and vaccine development. This review is dedicated to discussing the effect of HR-HPVs’ oncogenes (E5, E6, and E7) on signaling pathways and inflammation in prostate tumorigenesis, emphasizing the relationship of these oncogenes with apoptosis and PCa.
Natural selection has created cancer barriers, which prevent cancer progression by affecting the genetic makeup of multicellular organisms. Cell cycle arrest, apoptosis, limits on the total number of future cell divisions, cell adhesion, and asymmetric cell division are the five modifications that can be characterized as barriers to metastatic cancer based on current information. Such barriers are distinguished from constraints, reducing or preventing cancer progression [18,19]. Oncogenic selection refers to selection that promotes the evolution of tumor cells from healthy cells. Also, the oncogenic selection is a primarily destructive process in which cells lose regulatory systems that have been improved through time by natural selection to maintain cells working in the organism’s genetic interest. Natural selection encourages the evolution of anti-oncogenesis features, while oncogenic selection favors their abolition [18,20]. The simultaneous compromise of four cancer barriers enhances oncogenesis by allowing the formation of various populations of infected and dividing cells that are also attracted to the tumor’s periphery [18].
Furthermore, oncogenic occurrences of infectious causes of human cancers that abrogate barriers are important causes of cancer; however, those that loosen constraints aggravate the problem. It is critical to distinguish between necessary and aggravating factors since inhibiting important reasons will prevent cancer [21,22]. One of the reasons for cancer recurrence is the process of barriers inhibition. In this regard, the diversity of genetic mutations due to mutagenic agents exposure such as radiation and carcinogenic chemicals and interruptions in DNA replication in multicellular organisms lead to the transformation of normal cells into malignant cells [18,23]. Proliferation, survival, angiogenesis, and metabolic-related hallmarks are all conveyed to a tumor cell by oncogenic mutations that constitutively activate RAS, a potent regulator of the MAPK and PI3K-AKT-mTOR cascades. Uncontrolled growth and genetic instability are characteristics of mutations that inactivate the tumor suppressor gene p53 (TP53), a DNA damage-induced activator of growth inhibition and death [24].
Since apoptosis is a cancer-prevention mechanism in most cell types, a malignant cell cannot survive death. However, intact apoptotic systems can still allow oncogenesis in particular cell types if the cell has a high threshold for apoptosis initiation [18,25]. Viruses and bacteria may worsen tumors by enhancing the mutation-driven mechanism of tumorigenesis by increasing the participation of mutagenesis molecules after inflammation [26]. Nevertheless, if they undermine cancer barriers, they may operate as fundamental causes. When a virus can replicate its genome in tandem with a host cell, it can improve its viability while avoiding survival and reproduction. Viruses can get this ability by disabling cancer-fighting mechanisms such as cell cycle arrest, apoptosis, telomerase regulation, and cell adhesion [18,19]. Indeed, research into tumor virus proteins has shown exact strategies for removing oncogenesis barriers [21,27]. Tumor viruses interfere with the system by intervening directly with significant biochemical barriers, such as the p53 and pRB proteins, perhaps because the resultant proliferation helps the tumor viruses survive and reproduce [28].
3 Importance of Prostate Cancer
Since the PCa is caused by the accumulation of genetics and epigenetics variations, leading to disabling of inhibitor genes, tumor activated, and oncogenes [29,30]. Many pathologic organisms because of prostate infection induce inflammatory reactions. These agents include vector-borne organisms sexually similar to Neisseria Gonorrhea [31], Chlamydia trachomatis [32], Trichomonas vaginalis [33], Treponema pallidum [34], and bacteria that are transferred via sexual ways, like Propionibacterium acnes [35]. Pathogens have been discovered in the prostate; the prevalence and occurrence vary. For example, Treponema pallidum is an uncommon cause of granulomatous prostatitis and a unique inflammatory pattern in the prostate. There were many various sexually transmitted illnesses (mostly gonorrhea), acute prostate inflammation, and prostate abscesses before the discovery and use of antibiotics, which occurred before 1937 [31]. Moreover, several viruses, including HSV2, HPV, HHV8, EBV [36], CMV [37], and BK viruses, can infect the prostate [38]. Although early detection of PCa, which may be at risk of spreading, can be treated before it spreads, this function diminishes the risk of dying from PCa in some men [39].
4 Prostate Cancer and Human Papillomavirus Infection
HPVs, belonging to the papillomaviridae family, are small non-enveloped double-stranded circular DNA viruses that form a diverse and large virus group, with more HPV being continuously found. They cause a broad range of diseases, such as invasive tumors. HPV might infect the inner lining of tissues or the epithelial lining of the skin and is classified as mucosal or cutaneous type. The cutaneous HPV, as epidermitrophic types, infect the hand-foot skin and mucosal types of HPV infect the coating of the throat, mouth, anogenital epithelium, or respiratory tract. The mucosal HPV types in the α-papillomavirus genus are the main reason for cervical, anal, penile, vulvar, oral, and vaginal cancers. Besides, in the α-papillomavirus genus, benign mucosal HPV types result in benign genital condylomas. The main known HPV genera including alpha-papillomavirus, beta-papillomavirus, gamma-papillomavirus, nu-papillomavirus, and mu-papillomavirus. Also, HPVs may be categorized into low- and high-risk types according to their relationship with precursor lesions and cervical cancer. The HPV 2, 6, 11, 42, 43, and 44 types are considered low risk and high-risk HPV types consist of 16, 18, 31, 33, 34, 35, 39, 45, 51, 52, 56, 58, 59, 66, 68, and 70. A main global health problem worldwide is sexually transmitted infections (STIs). As mentioned above, HPV is a common STI worldwide [4,40], and the earlier is also associated with the etiology of cervical cancer and other anatomical sites [5,7]. The initial assertions of the etiological role of sexually transmitted infections in the progression of PCa date back to the 1950 s [16], and some mechanisms were then offered to clarify this connection. PCa is the second most common malignancy in males; in 2008, there were an estimated 903,500 new cancer cases and 258,400 deaths globally, making it the sixth-leading reason of mortality in men [41]. Except for perhaps obesity [3], smoking [2], and a sedentary lifestyle, no variable risk factors have been recognized for PC, despite wide research efforts [42]. Also, the procedure was studied for bacterial infections such as gonorrhea, resulting in PCa via the prostate inflammation and prostate atrophy phases. For viral infections, especially herpes viruses, the transforming features of viruses are more important [17]. Furthermore, numerous sexually transmitted diseases and infections that last longer increase the risk of PCa development due to a higher risk of prostate involvement [43]. According to a recent meta-analysis, Moghoofei et al. [2] have been reported that there is a significant positive link between HPV (especially genotype 16) infection and the risk of PCa. Moreover, the HPV can influence the biology of PCa and leads to enhanced cancer cells aggressiveness owing to the oncogenic activities of high-risk HPVs and the presence of HPV infection in the specimens of PCa. A few studies have investigated PCa cells’ immortalization through the HPV genes. Firstly, Naghashfar et al. [44] reported the expression of HPV18 E6/E7 genes by a retroviral vector prompted the immortalization of PCa cells. Besides, numerous studies reported that knock outing E6 and E7 genes results in apoptosis or susceptibility of cancer cells [45]. Generally, it could be deduced that the HPV oncogenic genes expression is vital for developing cancer induced by HPV. These researches and also a high incidence of HPV in PCa support the hypothesis of the HPV role in PCa progression [45]. Recently, it has been described that the expression level of HPV E7 oncoprotein was considerably greater in HPV-infected PCa samples comparing the HPV-positive prostate control [46]. This possibly shows the key role of HPV oncoproteins in the PCa development, but more researches are required.
5 Human Papillomaviruses and Replication
Papillomavirus particles share a usual non-enveloped icosahedral structure, and the HPV-DNA genome is divided, generally, into three central regions: Early, Late, and Long Control Region (LCR). The late region of papillomavirus genomes encodes L1 and L2 capsid proteins [19], and also the early part encodes six common open reading frames, including E1, E2, E4, E5, E6, and E7 [18].
Moreover, it was shown that integrin α-6 is the initial candidate of the cellular receptors, and α-6, β-4, and β-1 are mainly involved in HPV binding. The α-6 integrin can invoke a transductive signal pathway to commence replication of DNA in keratinocytes that are very significant for viral replication [30,31]. Additionally, using heparinase, heparan sulfate proteoglycans (HSPGs) on keratinocytes might be eliminated and diminish HPV-11 VLPs binding up to 80%–90%; thus, it indicates that HPVs interact with cell-surface HSPGs [31,32].
HPV uses several pathways for entering the cells; the first pathway is caveolar endocytosis, the second and third pathways are clathrin-mediated endocytosis and clathrin- and caveolin-independent endocytosis. The most common pathway of cell-penetrating is a nonclathrin, noncaveolin pathway which resembles macro-pinocytosis. The low pH encountered in the late endosomal sections is necessary for the exposure of the DNA-virus genome [22,25]. It found 50 to 100 episomal copies per cell in the basal layer of productive warts, indicating that the virus’s early genome replication occurs before being retained in infected cells as a stable multicopy plasmid [34,35]. However, reproduction of vegetative viral DNA happens in the differentiated squamous epithelium cells. The E1 and E2 proteins are the first viral proteins to be expressed that are indispensable to initial amplification [9]. The HPV does not encode any other enzymes involved in the replication to amplify the viral genome and should take the host’s replication system. The E1 and E2 proteins enroll cellular DNA polymerases and further need extra enzymes for the viral genome replication. Generally, differentiating cells cannot support DNA synthesis, given they have suppressed the cell cycle upon departing the basal epithelial layer. However, HPVs can stimulate cellular replication systems to replicate vegetative viral DNA by E6 and E7 oncogenes. These oncogenes are involved in the virus’s life cycle by recovering the cellular environment to allow amplification of the viral genome in differentiated and growth-arrested cells that may usually be inefficient for DNA replication. They accumulate as infectious particles following genome amplification, and capsid proteins such as L1 and L2 are expressed after the genome amplifies. However, the association between genome replication and capsid protein assemblages is not yet fully understood. It is speculated that the infectious virions assembly in the higher epithelial layers needs E2 and the capsid proteins L1 and L2 [34]. In this way, the L1 accumulates inside capsomeres in the cytoplasm before nuclear relocation and is rescued into PML bodies only afterward L2 has combined and displaced the PML component and sp100 [44]. While papillomavirus particles can assemble without L2, the presence of L2 enhances proper packaging [45] and boosts viral infectivity [46]. Finally, virus release demands an effective escape from the cornified cell envelope that the E4 protein might promote. The E4 could interrupt the keratin network and influence the cornified envelope integrity [34]. The genome organization of HPV has been shown in Fig. 1.
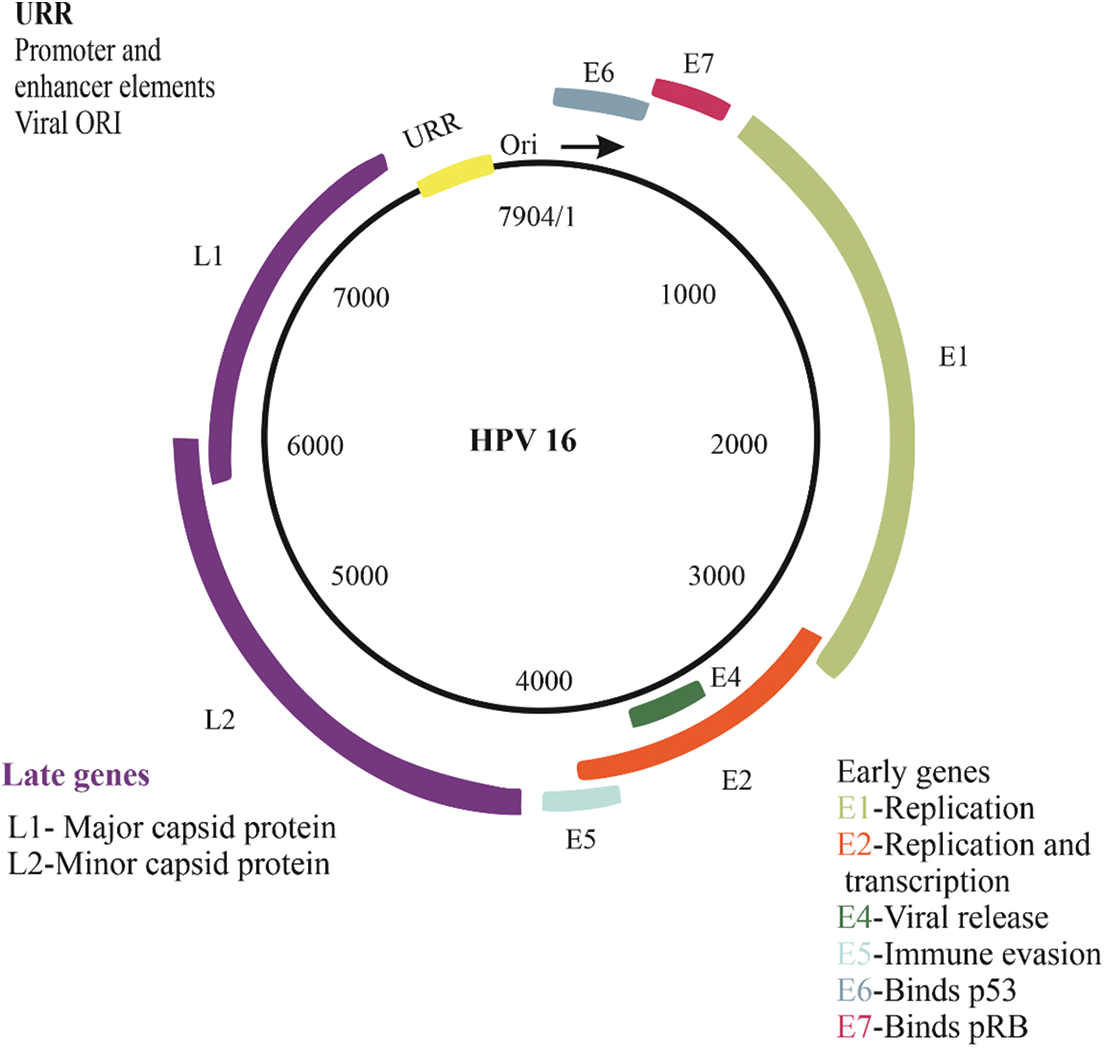
Figure 1: Genome organization of HPV
The HPV genome is divided into two regions: early and late, and the placement of each section changes based on the genome’s position and when genes are expressed during the viral replication cycle. Early region, which includes E2, E1, E4, E5, E6, and E7, is important in viral propagation and transcription control. L1 major capsid proteins and L2 minor capsid proteins, which encode the virus’s structural proteins, make up the late region. The upstream regulatory region (URR), which comprises promoter, enhancer, and viral origin elements, is also in controlling and regulating primary replication transcription. polycistronic transcripts, which utilize the major polyadenylation signal, are generated in this transcription area.
HPV oncoproteins (E5, E6, and E7), as the main drivers of HPV carcinogenesis, are involved in all cancer hallmarks [47,48]. HPV-mediated cancers are highly dependent on the expression of oncogenes (E6 and E7) for survival and continued propagation [49]. The mechanisms of hypermethylation and HPV viral genome integration induce the unbalanced expression of the E5, E6, and E7 oncoproteins that are critical factors for the carcinogenic cascade in human papillomavirus-mediated cancers [6,47]. For instance, interactions of E6, which are related to the malignant progression of human papillomavirus-infected cells with various host cell proteins, deregulates vital cellular functions and develops numerous cancer hallmarks, leading to the proliferation and survival of genomically defective cancer cells. The HPVs are frequently integrated with premalignant lesions and various oropharyngeal and anogenital cancers; however, this is not part of the virus’s life cycle. Integration is a dead-end for a virus because it can no longer form a circular genome that might be assembled and transferred to another host. To present, all HPV integration events that have been thoroughly explored have been roughly linked to HPV oncogenesis [50]. The HPV integration process might be found in premalignant lesions; nevertheless, the cells with integrated HPV enhanced as cells develop into invasive cancer [51]. Generally, integration leads to dysregulated expression of E6 and E7 oncogenes, which stimulates cell proliferation, abolishes cell cycle checkpoints, and results in progressive genetic instability [52]. Human papillomavirus-associated cancers might include either extrachromosomal or integrated viral DNA or both [53].
7 Apoptosis and Resistance to Apoptosis in Prostate Cancer
All cancer cells, regardless of the type or region, have cancer hallmarks; that includes angiogenesis and apoptosis evasion [54,55]. One of the significant functions of apoptosis is the inhibition of cancer. Usually, it is the inherent pathway prevented in cancer, but there is a wide range of means to impede apoptosis. Where apoptosis is lost, this allows tumor cells to survive longer and gives additional time for the accumulating mutations that can enhance invasiveness, the progression of tumor cells, stimulation of angiogenesis, interfering with differentiation, deregulation of cell proliferation [56]. Therefore, the apoptosis resistance provided using E6 and E7 oncoproteins might be thought of as a factor contributing to the progression of human cancer. A variety of genes contribute to apoptosis modulation, and among these, the members of the B-cell lymphoma 2 (Bcl-2) family are vigorously involved in cancer pathogenesis. The Bcl-2 expression, as an anti-apoptotic agent, was found to be significantly increased after transfection of cells with E6 and E7. Abnormal Bcl-2 activation aligned with the finding of a significant enhancement in the resistance to apoptosis of cells transfected with E6 and E7 oncoproteins [57]. Tumor cells are resistant to chemotherapeutic drugs by several mechanisms, including defective apoptotic signaling, downregulated pro-apoptotic signals, and upregulated anti-apoptotic signals [58]. Androgen independent prostate tumor cells are highly resistant to therapeutic factors, which activate apoptosis through the caspase cascade [59].
8 Modulation of Apoptotic Pathways by HPV Encoded Proteins
Apoptosis is an essential mechanism via which the host could eliminate infectious cells, and also the ability to eschew apoptosis might elevate virus survival [60]. Papillomavirus oncoproteins have been revealed to interfere in apoptotic pathways; for instance, some proteins (e.g., E5 and E6) might protect host cells from apoptosis, whereas E6 has pro-apoptotic attributes, and also E7 might act as an anti-apoptotic or pro-apoptotic factor, based on the cell sort. Overexpression of prominent oncogenes, such as E6 and E7, in cell alteration primarily affects in resistance of changed cells to apoptosis. The capability to eschew apoptosis is the main sign of carcinogenesis [61]. The evasion of apoptosis is one of cancer’s essential characteristics, promoting tumor growth and progression of metastases [61,62]. Several studies have revealed that HPVs infection can affect the pathogenesis of PCa. The papillomavirus has been shown to interfere with apoptosis via its oncogenic E5, E6, and E7 proteins, resulting in decreased apoptosis, cell cycle arrest, and cancer in cells [63–66]. It is worth noting that the mechanism of apoptosis involves extrinsic and intrinsic or mitochondria pathways (Table 1).
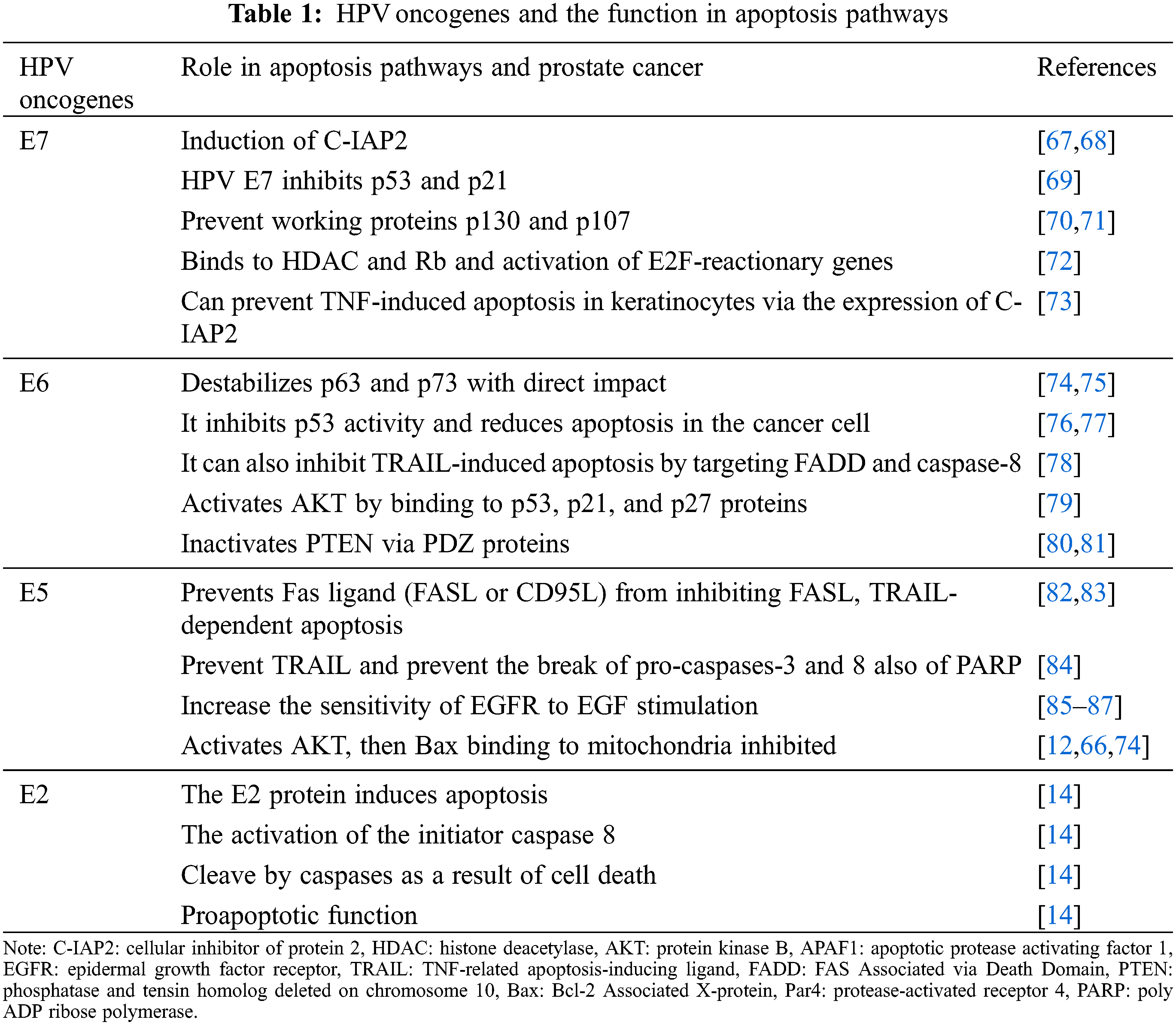
The extrinsic apoptotic pathway is activated when receptor trimerization occurs, and adaptor molecules and pro-caspase-8 are recruited to the death-induced signaling complex (DISC). Caspase-8 activation causes downstream executioner caspases-3 and -7 to activate, resulting in cell death/apoptosis [88]. These receptors can also promote cell proliferation, survival, and death. Studies have been shown that HPV oncogenes can inhibit apoptosis induced by interference with apoptotic pathway molecules [89] (Fig. 2). Early genes E5, E6, and E7 systematize are the major infectious agent converting proteins in HPV. These oncoproteins are efficient for transformation and immortalization in primary human keratinocytes in vitro [52–54]. Moreover, the E2 protein induces apoptosis; in this way, this apoptotic activity is mediated by the amino-terminal domain. The activation of the initiator caspase-8, part of the extrinsic apoptosis pathway, is required for the apoptotic pathway induced by E2, so caspases cleave E2, leading to cell death. The HPV18 E2 protein may inhibit viral oncogenes’ proliferative functions, making E2 inactivation essential for carcinogenic progression [14]. On the other hand, E7 proteins from HPV16 and HPV18 persuade resistance to tumor necrosis factor instead of proliferative result on primary human keratinocytes induction of cellular inhibitor of protein 2 (c-IAP2) has been detected in cells expressing HPV16 E6 and E7 [57]. TNFR interacts with Fas Associated via Death Domain (FADD) and TNFR1-associated death domain protein (TRADD) by its second death in the intracellular region, and this binding eventually activates caspases [90,91]. By binding with complex family proteins (pRB, p130, and p107) and starting E2F-dependent transcription, HPV-E7 can interrupt the G1-S checkpoint and allow the cell to continue into the S phase. The mentioned proteins (pRB, p130, and p107) and their interactions with the E2F transcription factor family members play critical roles in the cell cycle and apoptosis regulation; also, the E7 oncoprotein prevents proteins p130 and p107 function [70,71]. The p130 and p107 adjust E2F Transcription Factor 4 (E2F4) and E2F5, which are critical in cell cycle adjustment [74]. Besides, the E5 alone can result in cancer, augmenting the carcinogenic impression of E6 and E7 [92]. A study proposed that E5 might action a central role in the primary stage of tumorigenesis [93], and inhibits the extrinsic apoptotic pathway by downregulating the Fas receptor and disrupting the development of the DISC [55]. HPV16 E5 can also block Fas-L and TNF-related apoptosis-inducing ligand (TRAIL)-induced apoptosis by downregulating Fas receptor expression and preventing both recruitment of FADD and forming the DISC [83,94]. Kabsch and Alonso, in their study, demonstrated that E5 prevents Fas-mediated apoptosis by reducing the Fas receptor. Besides, E5 prevents TRAIL signaling by participating in the TRAIL DISC formation, thereby preventing the break of pro-caspases-3, pro-caspases-8, and PARP [84]. Epidermal growth factor receptor (EGFR) is stimulated using several ligands, including EGF and TGFα. The EGF/EGFR can increase cell proliferation and reduce apoptotic cells via AKT, MAPK, and COX-2 proteins [95]. Furthermore, HPV16 E5 promotes EGFR recycling to the cell surface and increases EGFR phosphorylation levels while necessitating EGF binding [96]. It has shown that E5 has a key role in enhanced sensitivity of EGFR to EGF stimulation [85–87]. E6 is another important oncogene of the papillomaviruses that is expressed at high levels, and also it can reduce apoptosis by altering several pathways. Following this fact, the findings indicate that reduction in this gene induces apoptosis in papillomavirus cancer cells [65]. E6 has been shown to prevent apoptosis triggered by TNF, FAS, and TRAIL by accelerating the degradation of pro-apoptotic proteins like FADD and/or procaspase-8, or by interacting with proteins that constitute the DISC [78] (Fig. 2). In early stage, decreased FAS expression or mutations in this gene have been observed in PCa patients.It has also been shown that in PCa malignancy, the loss of TRAILR1 and 2 and the defects in these pathways may be due to the influence of papillomavirus oncogenes [79]. This is followed by the fact that PCU and DU145 PCa cells are sensitive to apoptosis via TRAIL and defects in this pathway lead to further proliferation of cancerous cells, suggesting that the papillomavirus may decrease apoptosis in PCa [90].
Figure 2: E5, E6, E7 HPV function in the external pathway
The external pathway begins with private pathways and leads to cell death by inactivating effective caspases. Personal pathways such as the mediator receptor, Fas-L binding to the Fas-L receptor, and downstream pathways inhibit apoptosis. TRail route; Oncogenic HPV genes include E5 and E6, which bind to FADD and inhibit apoptosis. Also, E6 HPV binds to the TNFR pathway, leading to its non-binding to tumor necrosis factor (TNF) and TRADD, thereby inactivating caspase-8 and inhibiting apoptosis.
External stimuli (ultraviolet radiation, oxidative stress, hunger, etc.) trigger the intrinsic apoptotic pathway, which results in pores in the mitochondrial membrane and the release of mitochondrial inner membrane proteins (cytochrome c, SMAC) into the cytosol. Released cytochrome c and pro-caspase-9 create the apoptosome, which activates caspase-9, which subsequently activates downstream executioner caspases-3 and -7, causing apoptosis [88]. Studies have shown that E5, by direct connection, activates AKT, then Bax binding to mitochondria inhibited; as a result, cytochrome C is released and suppresses apoptosis [14,66,74] (Fig. 3). E5 also inhibits apoptosis by directly binding to the p53 tumor suppressor and prevents its function; on the other hand, by binding to Bak, Myc can also inhibit apoptosis via E6AP [12,66,74] (Fig. 3). p53 as a tumor suppressor can inhibit cell propagation through apoptosis, so mutations in the p53 gene have been described in many human tumors, with p53 enhancing or decreasing the expression of its target genes through transcription-dependent function. It has been reported that phosphorylated p53 is activated and can induce apoptosis through increased expression of apoptotic promoter genes such as Bax and Puma [88,97]. Many studies described the ability of HPV-16 E5 to communicate with the EGFR signaling pathway [98–100]. It has indicated that E5 activating EGFR can control cell proliferation, gene transcription, metastasis, and angiogenesis through the PI3 K-Art pathway [101] (Fig. 3).
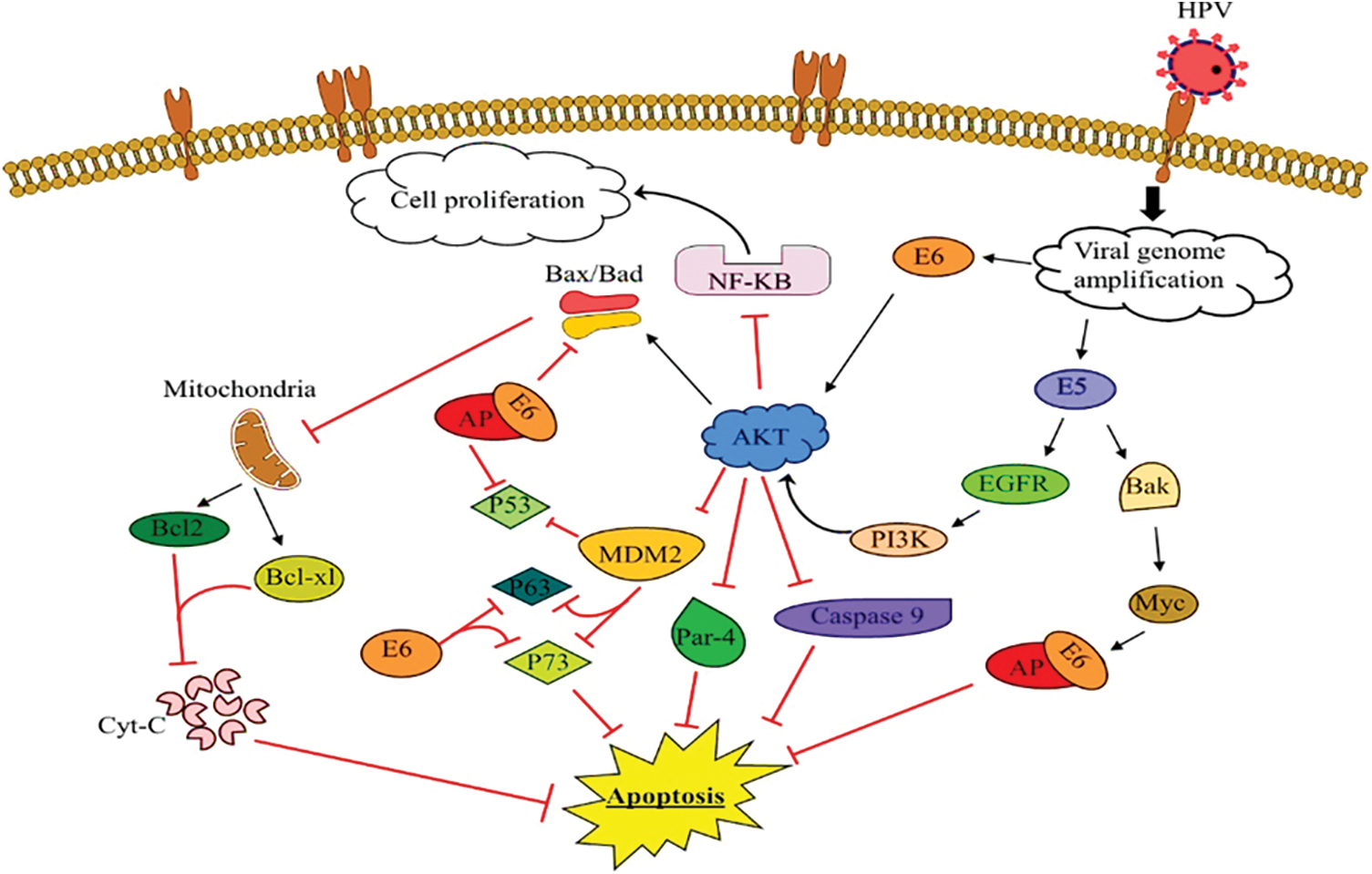
Figure 3: The function of E5, E6 of HPV in the intrinsic pathway in prostate cancer
Binding of E5 HPV oncoproteins to EGFR followed by increased PI3 K activity and AKT production and activation, which resulted in inhibition of increased NF-KB and cell proliferation and inhibition of caspase-9 and Par-4, and finally, they inhibit apoptosis. Also, binding E6 HPV directly to AKT, enhancing AKT activity, inhibiting Bak and Bax activity, and E6 binding through E6AP to Bak led to inhibition of mitochondrial binding and increased Bcl-2 and Bcl-xl, and failure to release cytochrome c and ultimately inhibit apoptosis. On the other hand, inhibition of MDM2 binding to p53 by increasing AKT, also binding E6 to p53, p63, p73 and inhibiting apoptosis.
Also, HPV-16 E5 can protect cells from UV-induced apoptotic cell death by activating the MAP kinase and PI3 K-Art pathways [102]. Lastly, the E5 protein may prevent hydrogen peroxide-mediated apoptosis via activating ubiquitin–proteasome-induced degradation of Bax as a pro-apoptotic protein [103]. Thus, the E5 protein could inhibit HPV-infected cells from reacting to apoptotic stimulation [104]. On the other hand, E6 inhibits the intrinsic apoptotic pathway by inactivating p53, Bax, and Bak, which prevents MOMP and the release of cytochrome C [74], and also by directly binding to p63 [75] (Fig. 3). Smeets et al. have been shown that E6 prevents p53 activity and reduces apoptosis in tumor cells [105]. It has been illustrated that E6/E6AP may exclusively target cellular pathways of p53 that are intended to stimulate the transcription of downstream pro-apoptotic proteins such as Bax, Fas, Puma, Apaf-1, and PIG, which are all part of the intrinsic apoptotic cascade [67]. Bak breakdown by E6 prevents the release of apoptosis-inducing factor (AIF) from the mitochondria into the nucleus, preventing UV-induced cell death [106]. E6 also activates AKT by binding to p53, p21, and p27 proteins and reducing their expression; AKT activation prevents Bad, Bax, and caspase-9 activity, and deletion of p21 destroys p53’s ability to stop the G1 cell cycle [107,108]. Findings indicate that AKT signaling is involved in TRAIL resistance in the PCa cell. Also in studies showing that Bcl-2 pathways, induced via the activation of AKT and NF-KB pathways might control the sensitivity of TRAIL to PCa cells [74,90]. The AKT has wide downstream aims that control cell duplication, cell growth, angiogenesis, and cell survival [109,110]. Many studies have manifested that E6 may activate the apoptosis pathway via several mechanisms. For example, the E6 inactivates PTEN via PDZ proteins, increasing pAKT and enhancing cell duplication [80–81,111]. There is a p53 mutation in many cancers, and in PCa, the frequency of mutation in p53 is variable [112]; it seems in androgen-independent tumors, the p53 mutation is abundant [113]. Overexpression of p53 blocks androgen signal; however, decreased expression of p53 and increased carcinogenicity of p53 are vital for the progression of PCa [114]. Denmeade et al. [115] have shown that the growth of androgen-dependent PCa is dependent on the balance between androgen p53, and cutting the balance may be involved in the development of PCa. In humans, there is also a reverse association between the expression of Par4 and Bcl-2 in the benign prostatic tumor epithelium and primary and metastatic PCa [116]. In living cells, AKT kinase activated by E6 has been exposed to phosphorylate Par4 in human prostate tumor cells, leading to par4 silencing and ultimately inhibiting apoptosis. In the end, it can be interpreted that in PCa, various anti-apoptotic pathways such as Bcl-2 are unregulated and p53, Par4 genes are inhibited, and AKT, NF-KB proliferative pathways are also activated [79,117,118]. Moreover, E7 binds to HDAC (histone deacetylase) and Rb and abate their repression of E2F, leading to the activation of E2F-reactionary genes. Besides, E7 as an anti-apoptotic factor, causes the cell to enter the S phase that cellular replication factors are activated [72]. Yuan et al. [73] recently, offered that by the expression of c-IAP2, E7 can prevent tumor necrosis factor (TNF)-induced apoptosis in keratinocytes. Another study found that E7 overexpression in fibroblasts inhibited the release of caspase-8, which postponed Fas and TNF-induced apoptosis [119]. On the other hand, E7, has been reported to play a significant role in interferon-alpha-mediated apoptosis in mouse lymphoma cells and has a pro-apoptotic impact [120]. In genital keratinocytes, overexpression of E7 promotes spontaneous cell death and increases the cells’ susceptibility to TNF-induced apoptosis [121]. The intrinsic pathway is controlled using the Bcl-2 protein family [122]. It has been shown that anti-apoptotic Bcl-2 proteins are overexpressed in cancer cells, expressed in 70% of androgen-independent tumors, and reduce the sensitivity of chemotherapy-induced apoptosis cancer cells [123,124]. Besides, it has been reported that p53 can act as an apoptosis suppressor by inhibiting the expression of Bcl-2 protein [125]. Presumably, as the high-risk E6 oncoprotein can inhibit p53, it directly promotes tumor progression by eliminating this protein’s inhibitory impact on the Bcl-2 [76]. On the other hand, HPV E5 protein prevents apoptosis via decreasing activity of Bax protein, a pro-apoptotic protein, which can act as a helper for E6/E7 oncoproteins to inhibit apoptosis [123]. A negative correlation between Bcl-2 and p53 has been observed in several cancer cells, but some studies have indicated that the expression of both proteins was increased in HPV-induced cancer cells. A study was conducted by Grace and et al. considerably positive association between the Bcl-2/p53 expression and HPV-mediated squamous cell carcinoma. They suggested that HPV through the deregulation of apoptosis pathways can cause cervical lesions [126]. The theory hypothesized that high-risk HPV E6 oncoprotein causes non-functional Bcl-2 and p53 proteins to co-express in cervical cancer by removing p53’s inhibitory impact on Bcl-2 [127]. Wang et al. examined the relationship between p53 and Bcl-2 expression in HPV-positive and HPV-negative breast cancer cases. It was shown that in the HPV positive tumors compared with HPV negative tumors, the p53 and Bcl-2 expression was substantially increased in HPV positive tumors [128]. Another study showed that the activity of Bcl-2 and p53 decreased and increased, respectively, in PCa samples infected with HPV. As well as, they have been reported that there had been a statistically meaningful negative and positive relationship between the measure of E6/E7 expression with that of p53 and Bcl-2, respectively. Therefore, HPV infection possibly assists in tumor development by promoting apoptosis resistance in PCa cells [46]. Survivin is the smallest member of the inhibitor of apoptosis protein (IAP) family which plays a key role in the determination of cell survival via preventing apoptosis, and the survivin expression was enhanced in many cancer types [129]. For example, in PCa, the expression level of survivin was regularly upregulated from low to high grade PCa [130]. Negative regulation of survivin expression by p53 made the hypothesis that by inhibiting p53, HPV-E6 protein leads to overexpression of survivin and subsequently, contributes to the immortalization of cancer cells [131]. Burberry et al. found the expression of endogenous survivin mRNA was increased following the transduction of E6 and E7 into human embryonic fibroblast cells [131]. In one study, Muzio et al. investigated the survivin expression in the oral premalignant lesions and oral cancer infected with HPV. They noticed that the expression level of survivin was considerably high in HPV-positive samples comparing HPV-negative samples. It can be concluded that HPV directly or indirectly influences expression of survivin [132]. Furthermore, Fatemipour et al. reported that the amount of survivin in HPV-infected PCa samples was considerably higher than in HPV-negative PCa samples. E6, E7, and survivin were also shown to have a substantial favorable connection.
Proteins E5, E6, and E7 of HPV are contributed to the apoptotic pathway and play a crucial role in the resistance to apoptotic cell death. The virus’ capability to escape apoptosis and prevent the extinction of infected cells leads to tumor growth and metastasis, resulting in cancer. HPV infection may induce chronic prostatic inflammation caused by sexually transmitted infections (STIs), which may increase PCa risk. HPV is known as the original etiological factor in cervical cancer studies. Targeting E5, E6, and E7 effectively inhibits apoptosis inflammation and prevents precancerous intraepithelial lesions and their progression to cancer. A thorough understanding of the biology of carcinogenic viruses and host defense mechanisms can facilitate therapeutic approaches. These findings suggest that targeting HPV oncoproteins could be a wise strategy for treating HPV-related cancers. Given that the mechanisms involved in oncogenic virus infection are better known, up-to-date and significant insights into the molecular mechanisms of apoptosis are on the way.
Acknowledgement: This project was supported by the Infectious and Tropical Diseases Research Center (IR.TBZMED.REC.1397.1046), Tabriz University of Medical Sciences, Tabriz, Iran.
Ethics Approval and Informed Consent Statement: Not applicable.
Author Contributions: Robabeh Faghani Baladehi: Literature search, manuscript preparation, and manuscript review. Ahad Bazmani, Mohammad Yousef Memar, Javid Sadri Nahand, and Abolfazl Jafari-Sales: Edit and review the manuscript. Parisa Shiri Aghbash: Figure design, edit the manuscript. Hossein Bannazadeh Baghi: Design the study, corresponding author.
Funding Statement: The authors received no specific funding for this study.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
1. Rawla, P. (2019). Epidemiology of prostate cancer. World Journal of Oncology, 10(2), 63. DOI 10.14740/wjon1191. [Google Scholar] [CrossRef]
2. Moghoofei, M., Keshavarz, M., Ghorbani, S., Babaei, F., Nahand, J. S. et al. (2019). Association between human papillomavirus infection and prostate cancer: A global systematic review and meta-analysis. Asia-Pacific Journal of Clinical Oncology, 15(5), 59–67. DOI 10.1111/ajco.13124. [Google Scholar] [CrossRef]
3. de Marzo, A. M., Platz, E. A., Sutcliffe, S., Xu, J., Grönberg, H. et al. (2007). Inflammation in prostate carcinogenesis. Nature Reviews Cancer, 7(4), 256–269. DOI 10.1038/nrc2090. [Google Scholar] [CrossRef]
4. Sfanos, K. S., de Marzo, A. M. (2012). Prostate cancer and inflammation: The evidence. Histopathology, 60(1), 199–215. DOI 10.1111/j.1365-2559.2011.04033.x. [Google Scholar] [CrossRef]
5. Blaylock, R. L. (2019). Viruses and tumor cell microenvironment: A brief summary. Surgical Neurology International, 10, 1–9. DOI 10.25259/SNI_351_2019. [Google Scholar] [CrossRef]
6. Hemmat, N., Mokhtarzadeh, A., Aghazadeh, M., Jadidi-Niaragh, F., Baradaran, B. et al. (2020). Role of microRNAs in epidermal growth factor receptor signaling pathway in cervical cancer. Molecular Biology Reports, 47(6), 4553–4568. DOI 10.1007/s11033-020-05494-4. [Google Scholar] [CrossRef]
7. Aydin, M., Bozkurt, A., Cikman, A., Gulhan, B., Karabakan, M. et al. (2017). Lack of evidence of HPV etiology of prostate cancer following radical surgery and higher frequency of the Arg/Pro genotype in turkish men with prostate cancer. International Brazilian Journal of Urology, 43(1), 36–46. DOI 10.1590/s1677-5538.ibju.2015.0429. [Google Scholar] [CrossRef]
8. Singh, N., Hussain, S., Kakkar, N., Singh, S. K., Sobti, R. C. et al. (2015). Implication of high risk human papillomavirus HR-HPV infection in prostate cancer in Indian population–A pioneering case-control analysis. Scientific Reports, 5(1), 1–4. DOI 10.1038/srep07822. [Google Scholar] [CrossRef]
9. Yang, L., Xie, S., Feng, X., Chen, Y., Zheng, T. et al. (2015). Worldwide prevalence of human papillomavirus and relative risk of prostate cancer: A meta-analysis. Scientific Reports, 5(1), 1–10. DOI 10.1038/srep14667. [Google Scholar] [CrossRef]
10. Mahmoudi, S., Jafari-Sales, A., Nasiri, R., Baghi, H. B. (2020). Prostate cancer and human papillomavirus infection: A recent literature review. Reviews in Medical Microbiology. DOI 10.1097/MRM.0000000000000261. [Google Scholar] [CrossRef]
11. Pascale, M., Pracella, D., Barbazza, R., Marongiu, B., Roggero, E. et al. (2013). Is human papillomavirus associated with prostate cancer survival? Disease Markers, 35(6), 607–613. DOI 10.1155/2013/735843. [Google Scholar] [CrossRef]
12. Yuan, C. H., Filippova, M., Duerksen-Hughes, P. (2012). Modulation of apoptotic pathways by human papillomaviruses (HPVMechanisms and implications for therapy. Viruses, 4(12), 3831–3850. DOI 10.3390/v4123831. [Google Scholar] [CrossRef]
13. Jiang, P., Yue, Y. (2014). Human papillomavirus oncoproteins and apoptosis. Experimental and Therapeutic Medicine, 7(1), 3–7. DOI 10.3892/etm.2013.1374. [Google Scholar] [CrossRef]
14. Demeret, C., Garcia-Carranca, A., Thierry, F. (2003). Transcription-independent triggering of the extrinsic pathway of apoptosis by human papillomavirus 18 E2 protein. Oncogene, 22(2), 168–175. DOI 10.1038/sj.onc.1206108. [Google Scholar] [CrossRef]
15. McKenzie, S., Kyprianou, N. (2006). Apoptosis evasion: The role of survival pathways in prostate cancer progression and therapeutic resistance. Journal of Cellular Biochemistry, 97(1), 18–32. DOI 10.1002/(ISSN)1097-4644. [Google Scholar] [CrossRef]
16. Sfanos, K. S., Isaacs, W. B., de Marzo, A. M. (2013). Infections and inflammation in prostate cancer. American Journal of Clinical and Experimental Urology, 1(1), 3. [Google Scholar]
17. Hemmat, N., Bannazadeh Baghi, H. (2019). Association of human papillomavirus infection and inflammation in cervical cancer. Pathogens and Disease, 77(5), ftz048. DOI 10.1093/femspd/ftz048. [Google Scholar] [CrossRef]
18. Ewald, P. W., Swain Ewald, H. A. (2013). Toward a general evolutionary theory of oncogenesis. Evolutionary Applications, 6(1), 70–81. DOI 10.1111/eva.12023. [Google Scholar] [CrossRef]
19. Ewald, P. W., Swain Ewald, H. A. (2012). Infection, mutation, and cancer evolution. Journal of Molecular Medicine, 90(5), 535–541. DOI 10.1007/s00109-012-0891-2. [Google Scholar] [CrossRef]
20. Casás-Selves, M., DeGregori, J. (2011). How cancer shapes evolution and how evolution shapes cancer. Evolution Education and Outreach, 4, 624–634. [Google Scholar]
21. Ewald, P. W., Ewald, H. A. S. (2014). Joint infectious causation of human cancers. Advances in Parasitology, 84, 1–26. DOI 10.1016/B978-0-12-800099-1.00001-6. [Google Scholar] [CrossRef]
22. Dalton-Griffin, L., Kellam, P. (2009). Infectious causes of cancer and their detection. Journal of Biology, 8(7), 1–5. DOI 10.1186/jbiol168. [Google Scholar] [CrossRef]
23. Darido, C., Georgy, S., Jane, S. (2016). The role of barrier genes in epidermal malignancy. Oncogene, 35(44), 5705–5712. DOI 10.1038/onc.2016.84. [Google Scholar] [CrossRef]
24. Mesri, E. A., Feitelson, M. A., Munger, K. (2014). Human viral oncogenesis: A cancer hallmarks analysis. Cell Host & Microbe, 15(3), 266–282. DOI 10.1016/j.chom.2014.02.011. [Google Scholar] [CrossRef]
25. Savage, P., Stebbing, J., Bower, M., Crook, T. (2009). Why does cytotoxic chemotherapy cure only some cancers? Nature Clinical Practice Oncology, 6(1), 43–52. DOI 10.1038/ncponc1260. [Google Scholar] [CrossRef]
26. Moss, S. F., Blaser, M. J. (2005). Mechanisms of disease: Inflammation and the origins of cancer. Nature Clinical Practice Oncology, 2, 90–97. DOI 10.1038/ncponc0081. [Google Scholar] [CrossRef]
27. Pandey, N. V. (2020). DNA viruses and cancer: Insights from evolutionary biology. VirusDisease, 31(1), 1–9. DOI 10.1007/s13337-019-00563-0. [Google Scholar] [CrossRef]
28. Swain Ewald, H. A., Ewald, P. W. (2020). Integrating the microbiome into the barrier theory of cancer. Evolutionary Applications, 13(7), 1701–1707. DOI 10.1111/eva.13066. [Google Scholar] [CrossRef]
29. Gonzalgo, M. L., Isaacs, W. B. (2003). Molecular pathways to prostate cancer. The Journal of Urology, 170(6), 2444–2452. DOI 10.1097/01.ju.0000085381.20139.b6. [Google Scholar] [CrossRef]
30. Shand, R. L., Gelmann, E. P. (2006). Molecular biology of prostate-cancer pathogenesis. Current Opinion in Urology, 16(3), 123–131. DOI 10.1097/01.mou.0000193384.39351.64. [Google Scholar] [CrossRef]
31. Pelouze, P. S. (1941). Gonorrhea in the male and female: A book for practitioners. WB Saunders Compnay. [Google Scholar]
32. Poletti, F., Medici, M., Alinovi, A., Menozzi, M. G., Sacchini, P. et al. (1985). Isolation of chlamydia trachomatis from the prostatic cells in patients affected by nonacute abacterial prostatitis. The Journal of Urology, 134(4), 691–692. DOI 10.1016/S0022-5347(17)47387-3. [Google Scholar] [CrossRef]
33. Gardner, J. W., Culberson, D., Bennett, B. (1986). Trichomonas vaginalis in the prostate gland. Archives of Pathology & Laboratory Medicine, 110(5), 430–432. [Google Scholar]
34. Thomson, L. (1920). Syphilis of the prostate. American Journal of Syphilis, 4, 323–341. [Google Scholar]
35. Cohen, R. J., Shannon, B. A., McNEAL, J. E., Shannon, T., Garrett, K. L. (2005). Propionibacterium acnes associated with inflammation in radical prostatectomy specimens: A possible link to cancer evolution? The Journal of Urology, 173(6), 1969–1974. DOI 10.1097/01.ju.0000158161.15277.78. [Google Scholar] [CrossRef]
36. Spînu, D., Bratu, O., Aungurenci, A., Marcu, D., Ursaciuc, C. et al. (2015). Epstein barr virus and cytomegalovirus in prostate–A controversial subject. Modern Medicine, 22(3), 259–263. [Google Scholar]
37. Sutcliffe, S., Till, C., Gaydos, C. A., Jenkins, F. J., Goodman, P. J. et al. (2012). Prospective study of cytomegalovirus serostatus and prostate cancer risk in the prostate cancer prevention trial. Cancer Causes & Control, 23(9), 1511–1518. DOI 10.1007/s10552-012-0028-5. [Google Scholar] [CrossRef]
38. Gorish, B. M. T., Ournasseir, M. E. H., Shammat, I. M. (2019). A correlation study of BK polyoma virus infection and prostate cancer among Sudanese patients-immunofluorescence and molecular based case-control study. Infectious Agents and Cancer, 14(1), 25. DOI 10.1186/s13027-019-0244-7. [Google Scholar] [CrossRef]
39. Mulhem, E. (2014). What are the benefits and harms of screening for prostate cancer? Cochrane Clinical Answers. [Google Scholar]
40. Tavakolian, S., Goudarzi, H., Eslami, G., Dayyani, F., Kazeminezhad, B. et al. (2020). Prevalence of human papilloma virus and Epstein–Barr virus in tumorous and adjacent tissues of colorectal cancer in Iran. Gene Reports, 20, 100774. DOI 10.1016/j.genrep.2020.100774. [Google Scholar] [CrossRef]
41. Rawla, P., Sunkara, T., Gaduputi, V. (2019). Epidemiology of pancreatic cancer: Global trends, etiology and risk factors. World Journal of Oncology, 10(1), 10–27. DOI 10.14740/wjon1166. [Google Scholar] [CrossRef]
42. Fatemipour, M., Nahand, J. S., Azar, M. E. F., Baghi, H. B., Taghizadieh, M. et al. (2021). Human papillomavirus and prostate cancer: The role of viral expressed proteins in the inhibition of anoikis and induction of metastasis. Microbial Pathogenesis, 152, 104576. DOI 10.1016/j.micpath.2020.104576. [Google Scholar] [CrossRef]
43. Caini, S., Gandini, S., Dudas, M., Bremer, V., Severi, E. et al. (2014). Sexually transmitted infections and prostate cancer risk: A systematic review and meta-analysis. Cancer Epidemiology, 38(4), 329–338. DOI 10.1016/j.canep.2014.06.002. [Google Scholar] [CrossRef]
44. Naghashfar, Z., DiPaolo, J., Woodworth, C., Passaniti, A. (1996). Immortalization of human adult prostatic adenocarcinoma cells by human papilloma virus HPV16 and—18 DNA. Cancer Letters, 100(1–2), 47–54. DOI 10.1016/0304-3835(95)04071-4. [Google Scholar] [CrossRef]
45. Pal, A., Kundu, R. (2020). Human papillomavirus E6 and E7: The cervical cancer hallmarks and targets for therapy. Frontiers in Microbiology, 10, 3116. DOI 10.3389/fmicb.2019.03116. [Google Scholar] [CrossRef]
46. Nahand, J. S., Esghaei, M., Monavari, S. H., Moghoofei, M., Kiani, S. J. et al. (2020). The assessment of a possible link between HPV-mediated inflammation, apoptosis, and angiogenesis in prostate cancer. International Immunopharmacology, 88, 106913. DOI 10.1016/j.intimp.2020.106913. [Google Scholar] [CrossRef]
47. Estêvão, D., Costa, N. R., da Costa, R. M. G., Medeiros, R. (2019). Hallmarks of HPV carcinogenesis: The role of E6, E7 and E5 oncoproteins in cellular malignancy. Biochimica et Biophysica Acta (BBA)-Gene Regulatory Mechanisms, 1862(2), 153–162. DOI 10.1016/j.bbagrm.2019.01.001. [Google Scholar] [CrossRef]
48. Aghbash, P. S., Hemmat, N., Nahand, J. S., Shamekh, A., Memar, M. Y. et al. (2021). The role of Th17 cells in viral infections. International Immunopharmacology, 91, 107331. DOI 10.1016/j.intimp.2020.107331. [Google Scholar] [CrossRef]
49. Goodwin, E. C., Yang, E., Lee, C. J., Lee, H. W., DiMaio, D. et al. (2000). Rapid induction of senescence in human cervical carcinoma cells. Proceedings of the National Academy of Sciences, 97(20), 10978–10983. DOI 10.1073/pnas.97.20.10978. [Google Scholar] [CrossRef]
50. McBride, A. A., Warburton, A. (2017). The role of integration in oncogenic progression of HPV-associated cancers. PLoS Pathogens, 13(4), e1006211. DOI 10.1371/journal.ppat.1006211. [Google Scholar] [CrossRef]
51. Shukla, S., Mahata, S., Shishodia, G., Pande, S., Verma, G. et al. (2014). Physical state & copy number of high risk human papillomavirus type 16 DNA in progression of cervical cancer. The Indian Journal of Medical Research, 139(4), 531–543. [Google Scholar]
52. Jeon, S., Allen-Hoffmann, B. L., Lambert, P. F. (1995). Integration of human papillomavirus type 16 into the human genome correlates with a selective growth advantage of cells. Journal of Virology, 69(5), 2989–2997. DOI 10.1128/jvi.69.5.2989-2997.1995. [Google Scholar] [CrossRef]
53. Kristiansen, E., Jenkins, A., Holm, R. (1994). Coexistence of episomal and integrated HPV16 DNA in squamous cell carcinoma of the cervix. Journal of Clinical Pathology, 47(3), 253–256. DOI 10.1136/jcp.47.3.253. [Google Scholar] [CrossRef]
54. Arbiser, J. L., Bonner, M. Y., Gilbert, L. C. (2017). Targeting the duality of cancer. npj Precision Oncology, 1, 1–7. DOI 10.1038/s41698-017-0026-x. [Google Scholar] [CrossRef]
55. Xu, W., Jing, L., Wang, Q., Lin, C. C., Chen, X. et al. (2015). Bax-PGAM5L-drp1 complex is required for intrinsic apoptosis execution. Oncotarget, 6(30), 30017. DOI 10.18632/oncotarget.5013. [Google Scholar] [CrossRef]
56. Pfeffer, C. M., Singh, A. T. (2018). Apoptosis: A target for anticancer therapy. International Journal of Molecular Sciences, 19(2), 448. DOI 10.3390/ijms19020448. [Google Scholar] [CrossRef]
57. Du, J., Chen, G., Vlantis, A. C., Chan, P., Tsang, R. et al. (2004). Resistance to apoptosis of HPV 16-infected laryngeal cancer cells is associated with decreased Bak and increased Bcl-2 expression. Cancer Letters, 205(1), 81–88. DOI 10.1016/j.canlet.2003.09.035. [Google Scholar] [CrossRef]
58. Chen, L., Zeng, Y., Zhou, S. F. (2018). Role of apoptosis in cancer resistance to chemotherapy. In: Current understanding of apoptosis-programmed cell death. [Google Scholar]
59. Howell, S. (2000). Resistance to apoptosis in prostate cancer cells. Molecular Urology, 4(3), 225–229. [Google Scholar]
60. Gandarillas, A., Goldsmith, L., Gschmeissner, S., Leigh, I., Watt, F. (1999). Evidence that apoptosis and terminal differentiation of epidermal keratinocytes are distinct processes. Experimental Dermatology, 8(1), 71–79. DOI 10.1111/j.1600-0625.1999.tb00350.x. [Google Scholar] [CrossRef]
61. Hanahan, D., Weinberg, R. A. (2000). The hallmarks of cancer. Cell, 100(1), 57–70. DOI 10.1016/S0092-8674(00)81683-9. [Google Scholar] [CrossRef]
62. Olechowska-Jarząb, A., Ptak-Belowska, A., Brzozowski, T. (2016). Therapeutic importance of apoptosis pathways in pancreatic cancer. Folia Medica Cracoviensia, 56(161–70. [Google Scholar]
63. Singh, N., Josefsson, A., Hussain, S., Hugosson, J. (2018). Human papillomavirus (HPV) infection as an emerging risk factor in prostate cancer. Journal of Global Oncology, 4(Supplement 2). [Google Scholar]
64. Divya, C. S., Pillai, M. R. (2006). Antitumor action of curcumin in human papillomavirus associated cells involves downregulation of viral oncogenes, prevention of NFkB and AP-1 translocation, and modulation of apoptosis. Molecular Carcinogenesis, 45(5), 320–332. DOI 10.1002/(ISSN)1098-2744. [Google Scholar] [CrossRef]
65. Butz, K., Denk, C., Ullmann, A., Scheffner, M., Hoppe-Seyler, F. (2000). Induction of apoptosis in human papillomaviruspositive cancer cells by peptide aptamers targeting the viral E6 oncoprotein. Proceedings of the National Academy of Sciences, 97(12), 6693–6697. DOI 10.1073/pnas.110538897. [Google Scholar] [CrossRef]
66. Garnett, T., Duerksen-Hughes, P. (2006). Modulation of apoptosis by human papillomavirus (HPV) oncoproteins. Archives of Virology, 151(12), 2321–2335. DOI 10.1007/s00705-006-0821-0. [Google Scholar] [CrossRef]
67. Vats, A., Trejo-Cerro, O., Thomas, M., Banks, L. (2021). Human papillomavirus E6 and E7: What remains? Tumour Virus Research, 11, 200213. DOI 10.1016/j.tvr.2021.200213. [Google Scholar] [CrossRef]
68. Miao, X., Deng, Z., Wang, S., Weng, H., Zhang, X. et al. (2021). IAP-1 promoted cisplatin resistance in nasopharyngeal carcinoma via inhibition of caspase-3-mediated apoptosis. American Journal of Cancer Research, 11(3), 640–667. [Google Scholar]
69. Westrich, J. A., Warren, C. J., Klausner, M. J., Guo, K., Liu, C. W. et al. (2018). Human papillomavirus 16 E7 stabilizes APOBEC3A protein by inhibiting cullin 2-dependent protein degradation. Journal of Virology, 92(7), DOI 10.1128/JVI.01318-17. [Google Scholar] [CrossRef]
70. Eldakhakhny, S. (2018). Crosstalk between high-risk human papillomavirus E7 and P63 in cervical cancer. (Doctoral Dissertation). University of Manchester. [Google Scholar]
71. Onci, D., Coppola, V., Musumeci, M., Addario, A., Giuffrida, R. et al. (2008). The miR-15a–miR-16-1 cluster controls prostate cancer by targeting multiple oncogenic activities. Nature Medicine, 14(11), 1271–1277. DOI 10.1038/nm.1880. [Google Scholar] [CrossRef]
72. Longworth, M. S., Laimins, L. A. (2004). The binding of histone deacetylases and the integrity of zinc finger-like motifs of the E7 protein are essential for the life cycle of human papillomavirus type 31. Journal of Virology, 78(7), 3533–3541. DOI 10.1128/JVI.78.7.3533-3541.2004. [Google Scholar] [CrossRef]
73. Yuan, H., Fu, F., Zhuo, J., Wang, W., Nishitani, J. et al. (2005). Human papillomavirus type 16 E6 and E7 oncoproteins upregulate c-IAP2 gene expression and confer resistance to apoptosis. Oncogene, 24(32), 5069–5078. DOI 10.1038/sj.onc.1208691. [Google Scholar] [CrossRef]
74. Chen, J. (2015). Signaling pathways in HPV-associated cancers and therapeutic implications. Reviews in Medical Virology, 25, 24–53. DOI 10.1002/rmv.1823. [Google Scholar] [CrossRef]
75. di Como, C. J., Urist, M. J., Babayan, I., Drobnjak, M., Hedvat, C. V. et al. (2002). P63 expression profiles in human normal and tumor tissues. Clinical Cancer Research, 8(2), 494–501. [Google Scholar]
76. Liang, X. H., Mungal, S., Ayscue, A., Meissner, J. D., Wodnicki, P. et al. (1995). Bcl-2 protooncogene expression in cervical carcinoma cell lines containing inactive p53. Journal of Cellular Biochemistry, 57(3), 509–521. DOI 10.1002/(ISSN)1097-4644. [Google Scholar] [CrossRef]
77. Zhu, J., Kamara, S., Wang, Q., Guo, Y., Li, Q. et al. (2021). Novel affibody molecules targeting the HPV16 E6 oncoprotein inhibited the proliferation of cervical cancer cells. Frontiers in Cell and Developmental Biology, 9, 1150. DOI 10.3389/fcell.2021.677867. [Google Scholar] [CrossRef]
78. Martina, R., Jan, B., Michal, M. (2021). Cell death in head and neck cancer pathogenesis and treatment. Cell Death and Disease, 12(2), 1–17. [Google Scholar]
79. Castilla, C., Congregado, B. N., Chinchón, D., Torrubia, F. J., Japón, M. A. et al. (2006). Bcl-xL is overexpressed in hormone-resistant prostate cancer and promotes survival of LNCaP cells via interaction with proapoptotic Bak. Endocrinology, 147(10), 4960–4967. DOI 10.1210/en.2006-0502. [Google Scholar] [CrossRef]
80. Adey, N. B., Huang, L., Ormonde, P. A., Baumgard, M. L., Pero, R. et al. (2000). Threonine phosphorylation of the MMAC1/PTEN PDZ binding domain both inhibits and stimulates PDZ binding. Cancer Research, 60(1), 35–37. [Google Scholar]
81. Contreras-Paredes, A., de la Cruz-Hernández, E., Martínez-Ramírez, I., Dueñas-González, A., Lizano, M. (2009). E6 variants of human papillomavirus 18 differentially modulate the protein kinase B/phosphatidylinositol 3-kinase (akt/PI3 K) signaling pathway. Virology, 383(1), 78–85. DOI 10.1016/j.virol.2008.09.040. [Google Scholar] [CrossRef]
82. Whang, S. N., Filippova, M., Duerksen-Hughes, P. (2015). Recent progress in therapeutic treatments and screening strategies for the prevention and treatment of HPV-associated head and neck cancer. Viruses, 7(9), 5040–5065. DOI 10.3390/v7092860. [Google Scholar] [CrossRef]
83. Duerksen-Hughes, P., Yuan, C. H., Filippova, M., Krstenansky, J. (2018). Compositions for preventing cancers associated with human papilloma viruses. United States patent application US 15/744,711. EP16825052.0. [Google Scholar]
84. Absch, K., Alonso, A. (2002). The human papillomavirus type 16 E5 protein impairs TRAIL-and FasL-mediated apoptosis in haCaT cells by different mechanisms. Journal of Virology, 76(23), 12162–12172. DOI 10.1128/JVI.76.23.12162-12172.2002. [Google Scholar] [CrossRef]
85. Pim, D., Collins, M., Banks, L. (1992). Human papillomavirus type 16 E5 gene stimulates the transforming activity of the epidermal growth factor receptor. Oncogene, 7(1), 27–32. [Google Scholar]
86. Crusius, K., Auvinen, E., Steuer, B., Gaissert, H., Alonso, A. (1998). The human papillomavirus type 16 E5-protein modulates ligand-dependent activation of the EGF receptor family in the human epithelial cell line haCaT. Experimental Cell Research, 241(1), 76–83. DOI 10.1006/excr.1998.4024. [Google Scholar] [CrossRef]
87. Martin, P., Vass, W. C., Schiller, J. T., Lowy, D. R., Velu, T. J. (1989). The bovine papillomavirus E5 transforming protein can stimulate the transforming activity of EGF and CSF-1 receptors. Cell, 59(1), 21–32. DOI 10.1016/0092-8674(89)90866-0. [Google Scholar] [CrossRef]
88. Basukala, O., Banks, L. (2021). The not-so-good, the bad and the ugly: HPV E5, E6 and E7 oncoproteins in the orchestration of carcinogenesis. Viruses, 13(10), 1892. DOI 10.3390/v13101892. [Google Scholar] [CrossRef]
89. Tan, S., Hougardy, B. M., Meersma, G. J., Schaap, B., de Vries, E. G. et al. (2012). Human papilloma virus 16 E6 RNA interference enhances cisplatin and death receptor-mediated apoptosis in human cervical carcinoma cells. Molecular Pharmacology, 81(5), 701–709. DOI 10.1124/mol.111.076539. [Google Scholar] [CrossRef]
90. Lorenzo, P. I., Arnoldussen, Y. J., Saatcioglu, F. (2007). Molecular mechanisms of apoptosis in prostate cancer. Critical Reviews™ in Oncogenesis, 13(1), 1–38. DOI 10.1615/CritRevOncog.v13.i1.10. [Google Scholar] [CrossRef]
91. Yuan, C., Filippova, M., Krstenansky, J., Duerksen-Hughes, P. (2016). Flavonol and imidazole derivatives block HPV16 E6 activities and reactivate apoptotic pathways in HPV+ cells. Cell Death & Disease, 7(1), 2060. DOI 10.1038/cddis.2015.391. [Google Scholar] [CrossRef]
92. Krawczyk, E., Suprynowicz, F. A., Liu, X., Dai, Y., Hartmann, D. P. et al. (2008). Koilocytosis: A cooperative interaction between the human papillomavirus E5 and E6 oncoproteins. The American Journal of Pathology, 173(3), 682–688. DOI 10.2353/ajpath.2008.080280. [Google Scholar] [CrossRef]
93. Stoler, M. H., Rhodes, C. R., Whitbeck, A., Wolinsky, S. M., Chow, L. T. et al. (1992). Human papillomavirus type 16 and 18 gene expression in cervical neoplasias. Human Pathology, 23(2), 117–128. DOI 10.1016/0046-8177(92)90232-R. [Google Scholar] [CrossRef]
94. Scarth, J. A., Patterson, M. R., Morgan, E. L., Macdonald, A. (2021). The human papillomavirus oncoproteins: A review of the host pathways targeted on the road to transformation. The Journal of General Virology, 102(3), 1–28. DOI 10.1099/jgv.0.001540. [Google Scholar] [CrossRef]
95. Chen, J., Elfiky, A., Han, M., Chen, C., Saif, M. W. (2014). The role of Src in colon cancer and its therapeutic implications. Clinical Colorectal Cancer, 13(1), 5–13. DOI 10.1016/j.clcc.2013.10.003. [Google Scholar] [CrossRef]
96. Medda, A., Duca, D., Chiocca, S. (2021). Human papillomavirus and cellular pathways: Hits and targets. Pathogens, 10(3), 262. DOI 10.3390/pathogens10030262. [Google Scholar] [CrossRef]
97. Raver-Shapira, N., Marciano, E., Meiri, E., Spector, Y., Rosenfeld, N. et al. (2007). Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Molecular Cell, 26(5), 731–743. DOI 10.1016/j.molcel.2007.05.017. [Google Scholar] [CrossRef]
98. Straight, S. W., Hinkle, P. M., Jewers, R. J., McCance, D. (1993). The E5 oncoprotein of human papillomavirus type 16 transforms fibroblasts and effects the downregulation of the epidermal growth factor receptor in keratinocytes. Journal of Virology, 67(8), 4521–4532. DOI 10.1128/jvi.67.8.4521-4532.1993. [Google Scholar] [CrossRef]
99. Zhang, B., Srirangam, A., Potter, D. A., Roman, A. (2005). HPV16 E5 protein disrupts the c-Cbl–EGFR interaction and EGFR ubiquitination in human foreskin keratinocytes. Oncogene, 24(15), 2585–2588. DOI 10.1038/sj.onc.1208453. [Google Scholar] [CrossRef]
100. Suprynowicz, F. A., Krawczyk, E., Hebert, J. D., Sudarshan, S. R., Simic, V. et al. (2010). The human papillomavirus type 16 E5 oncoprotein inhibits epidermal growth factor trafficking independently of endosome acidification. Journal of Virology, 84(20), 10619–10629. DOI 10.1128/JVI.00831-10. [Google Scholar] [CrossRef]
101. Dannenberg, A. J., Lippman, S. M., Mann, J. R., Subbaramaiah, K., DuBois, R. N. (2005). Cyclooxygenase-2 and epidermal growth factor receptor: Pharmacologic targets for chemoprevention. Journal of Clinical Oncology, 23(2), 254–266. DOI 10.1200/JCO.2005.09.112. [Google Scholar] [CrossRef]
102. Zhang, B., Spandau, D. F., Roman, A. (2002). E5 protein of human papillomavirus type 16 protects human foreskin keratinocytes from UV B-irradiation-induced apoptosis. Journal of Virology, 76(1), 220–231. DOI 10.1128/JVI.76.1.220-231.2002. [Google Scholar] [CrossRef]
103. Oh, J. M., Kim, S. H., Cho, E. A., Song, Y. S., Kim, W. H. et al. (2010). Human papillomavirus type 16 E5 protein inhibits hydrogen peroxide-induced apoptosis by stimulating ubiquitin–proteasome-mediated degradation of Bax in human cervical cancer cells. Carcinogenesis, 31(3), 402–410. DOI 10.1093/carcin/bgp318. [Google Scholar] [CrossRef]
104. Tetsu, O., McCormick, F. (1999). Β-Catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature, 398(6726), 422–426. DOI 10.1038/18884. [Google Scholar] [CrossRef]
105. Smeets, S. J., van der Plas, M., Schaaij-Visser, T. B., van Veen, E. A., van Meerloo, J. et al. (2011). Immortalization of oral keratinocytes by functional inactivation of the p53 and pRb pathways. International Journal of Cancer, 128(7), 1596–1605. DOI 10.1002/ijc.25474. [Google Scholar] [CrossRef]
106. Gheit, T. (2019). Mucosal and cutaneous human papillomavirus infections and cancer biology. Frontiers in Oncology, 9, 355. DOI 10.3389/fonc.2019.00355. [Google Scholar] [CrossRef]
107. McDonnell, T. J., Troncoso, P., Brisbay, S. M., Logothetis, C., Chung, L. W. et al. (1992). Expression of the protooncogene Bcl-2 in the prostate and its association with emergence of androgen-independent prostate cancer. Cancer Research, 52(24), 6940–6944. [Google Scholar]
108. McDonnel, T. J., Navone, N. M., Troncoso, P., Pisters, L. L., Conti, C. et al. (1997). Expression of BCL-2 oncoprotein and p53 protein accumulation in bone marrow metastases of androgen independent prostate cancer. The Journal of Urology, 157(2), 569–574. DOI 10.1016/S0022-5347(01)65204-2. [Google Scholar] [CrossRef]
109. Rodon, J., Dienstmann, R., Serra, V., Tabernero, J. (2013). Development of PI3 K inhibitors: Lessons learned from early clinical trials. Nature Reviews Clinical Oncology, 10(3), 143–153. DOI 10.1038/nrclinonc.2013.10. [Google Scholar] [CrossRef]
110. Chen, J., Zhao, K. N., Li, R., Shao, R., Chen, C. (2014). Activation of PI3 K/Akt/mTOR pathway and dual inhibitors of PI3 K and mTOR in endometrial cancer. Current Medicinal Chemistry, 21(26), 3070–3080. DOI 10.2174/0929867321666140414095605. [Google Scholar] [CrossRef]
111. Tolkacheva, T., Boddapati, M., Sanfiz, A., Tsuchida, K., Kimmelman, A. C. et al. (2001). Regulation of PTEN binding to MAGI-2 by two putative phosphorylation sites at threonine 382 and 383. Cancer Research, 61(13), 4985–4989. [Google Scholar]
112. Navone, N. M., Troncoso, P., Pisters, L. L., Goodrow, T. L., Palmer, J. L. et al. (1993). p53 protein accumulation and gene mutation in the progression of human prostate carcinoma. Journal of the National Cancer Institute, 85(20), 1657–1669. DOI 10.1093/jnci/85.20.1657. [Google Scholar] [CrossRef]
113. Ruijter, E., van de Kaa, C., Miller, G., Ruiter, D., Debruyne, F. et al. (1999). Molecular genetics and epidemiology of prostate carcinoma. Endocrine Reviews, 20(1), 22–45. DOI 10.1210/edrv.20.1.0356. [Google Scholar] [CrossRef]
114. Nantermet, P. V., Xu, J., Yu, Y., Hodor, P., Holder, D. et al. (2004). Identification of genetic pathways activated by the androgen receptor during the induction of proliferation in the ventral prostate gland. Journal of Biological Chemistry, 279(2), 1310–1322. DOI 10.1074/jbc.M310206200. [Google Scholar] [CrossRef]
115. Denmeade, S. R., Isaacs, J. T. (2002). A history of prostate cancer treatment. Nature Reviews Cancer, 2(5), 389–396. DOI 10.1038/nrc801. [Google Scholar] [CrossRef]
116. Qiu, G., Ahmed, M., Sells, S. F., Mohiuddin, M., Weinstein, M. H. et al. (1999). Mutually exclusive expression patterns of Bcl-2 and Par-4 in human prostate tumors consistent with down-regulation of Bcl-2 by Par-4. Oncogene, 18(3), 623. DOI 10.1038/sj.onc.1202344. [Google Scholar] [CrossRef]
117. Howie, H. L., Koop, J. I., Weese, J., Robinson, K., Wipf, G. et al. (2011). Beta-HPV 5 and 8 E6 promote p300 degradation by blocking AKT/p300 association. PLoS Pathogens, 7(8), e1002211. DOI 10.1371/journal.ppat.1002211. [Google Scholar] [CrossRef]
118. Wu, H. H., Wu, J. Y., Cheng, Y. W., Chen, C. Y., Lee, M. C. et al. (2010). cIAP2 upregulated by E6 oncoprotein via epidermal growth factor receptor/phosphatidylinositol 3-kinase/AKT pathway confers resistance to cisplatin in human papillomavirus 16/18–infected lung cancer. Clinical Cancer Research, 16(21), 5200–5210. DOI 10.1158/1078-0432.CCR-10-0020. [Google Scholar] [CrossRef]
119. Thompson, D. A., Zacny, V., Belinsky, G. S., Classon, M., Jones, D. L. et al. (2001). The HPV E7 oncoprotein inhibits tumor necrosis factor α-mediated apoptosis in normal human fibroblasts. Oncogene, 20(28), 3629–3640. DOI 10.1038/sj.onc.1204483. [Google Scholar] [CrossRef]
120. Thyrell, L., Sangfelt, O., Zhivotovsky, B., Pokrovskaja, K., Wang, Y. et al. (2005). The HPV-16 E7 oncogene sensitizes malignant cells to IFN-α-induced apoptosis. Journal of Interferon & Cytokine Research, 25(2), 63–72. DOI 10.1089/jir.2005.25.63. [Google Scholar] [CrossRef]
121. StoÈppler, H., StoÈppler, M. C., Johnson, E., Simbulan-Rosenthal, C. M., Smulson, M. E. et al. (1998). The E7 protein of human papillomavirus type 16 sensitizes primary human keratinocytes to apoptosis. Oncogene, 17(10), 1207–1214. DOI 10.1038/sj.onc.1202053. [Google Scholar] [CrossRef]
122. Adams, J. M., Cory, S. (2007). Bcl-2-regulated apoptosis: Mechanism and therapeutic potential. Current Opinion in Immunology, 19(5), 488–496. DOI 10.1016/j.coi.2007.05.004. [Google Scholar] [CrossRef]
123. Alibek, K., Irving, S., Sautbayeva, Z., Kakpenova, A., Bekmurzayeva, A. et al. (2014). Disruption of Bcl-2 and Bcl-xL by viral proteins as a possible cause of cancer. Infectious Agents and Cancer, 9(1), 44. DOI 10.1186/1750-9378-9-44. [Google Scholar] [CrossRef]
124. Maji, S., Panda, S., Samal, S. K., Shriwas, O., Rath, R. et al. (2018). Bcl-2 antiapoptotic family proteins and chemoresistance in cancer. Advances in Cancer Research, 1379, 37–75. DOI 10.1016/bs.acr.2017.11.001. [Google Scholar] [CrossRef]
125. Chen, D., Zheng, X., Kang, D., Yan, B., Liu, X. et al. (2012). Apoptosis and expression of the Bcl-2 family of proteins and P53 in human pancreatic ductal adenocarcinoma. Medical Principles and Practice, 21(1), 68–73. DOI 10.1159/000332423. [Google Scholar] [CrossRef]
126. Grace, V. B., Shalini, J. V., Devaraj, S. N., Devaraj, H. (2003). Co-overexpression of p53 and Bcl-2 proteins in HPV-induced squamous cell carcinoma of the uterine cervix. Gynecologic Oncology, 91(1), 51–58. DOI 10.1016/S0090-8258(03)00439-6. [Google Scholar] [CrossRef]
127. Sidransky, D., Hollstein, M. (1996). Clinical implications of the p53 gene. Annual Review of Medicine, 47(1), 285–301. DOI 10.1146/annurev.med.47.1.285. [Google Scholar] [CrossRef]
128. Wang, Y. W., Zhang, K., Zhao, S., Lv, Y., Zhu, J. et al. (2017). HPV status and its correlation with BCL2, p21, p53, Rb, and survivin expression in breast cancer in a Chinese population. BioMed Research International, 2017,1–8. DOI 10.1155/2017/6315392. [Google Scholar] [CrossRef]
129. Li, D., Hu, C., Li, H. (2018). Survivin as a novel target protein for reducing the proliferation of cancer cells. Biomedical Reports, 8(5), 399–406. DOI 10.3892/br. [Google Scholar] [CrossRef]
130. Shariat, S. F., Lotan, Y., Saboorian, H., Khoddami, S. M., Roehrborn, C. G. et al. (2004). Survivin expression is associated with features of biologically aggressive prostate carcinoma. Cancer, 100(4), 751–757. DOI 10.1002/(ISSN)1097-0142. [Google Scholar] [CrossRef]
131. Borbely, A. A., Murvai, M., Kónya, J., Beck, Z., Gergely, L. et al. (2006). Effects of human papillomavirus type 16 oncoproteins on survivin gene expression. Journal of General Virology, 87(2), 287–294. DOI 10.1099/vir.0.81067-0. [Google Scholar] [CrossRef]
132. Muzio, L. L., Campisi, G., Giovannelli, L., Ammatuna, P., Greco, I. et al. (2004). HPV DNA and survivin expression in epithelial oral carcinogenesis: A relationship? Oral Oncology, 40(7), 736–741. DOI 10.1016/j.oraloncology.2003.11.011. [Google Scholar] [CrossRef]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |