
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE

A comprehensive and systematic analysis of Dihydrolipoamide S-acetyltransferase (DLAT) as a novel prognostic biomarker in pan-cancer and glioma
Department of Hematology, West China Hospital, Sichuan University, Chengdu, 610041, China
* Corresponding Authors: JINRONG YANG. Email: ; TING NIU. Email:
# These authors have contributed equally to this work and share the first authorship
Oncology Research 2024, 32(12), 1903-1919. https://doi.org/10.32604/or.2024.048138
Received 28 November 2023; Accepted 21 February 2024; Issue published 13 November 2024
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Background: Dihydrolipoamide S-acetyltransferase (DLAT) is a subunit of the pyruvate dehydrogenase complex (PDC), a rate-limiting enzyme complex, that can participate in either glycolysis or the tricarboxylic acid cycle (TCA). However, the pathogenesis is not fully understood. We aimed to perform a more systematic and comprehensive analysis of DLAT in the occurrence and progression of tumors, and to investigate its function in patients’ prognosis and immunotherapy. Methods: The differential expression, diagnosis, prognosis, genetic and epigenetic alterations, tumor microenvironment, stemness, immune infiltration cells, function enrichment, single-cell analysis, and drug response across cancers were conducted based on multiple computational tools. Additionally, we validated its carcinogenic effect and possible mechanism in glioma cells. Results: We exhibited that DLAT expression was increased in most tumors, especially in glioma, and affected the survival of tumor patients. DLAT was related to RNA modification genes, DNA methylation, immune infiltration, and immune infiltration cells, including CD4+ T cells, CD8+ T cells, Tregs, and cancer-associated fibroblasts. Single-cell analysis displayed that DLAT might regulate cancer by mediating angiogenesis, inflammation, and stemness. Enrichment analysis revealed that DLAT might take part in the cell cycle pathway. Increased expression of DLAT leads tumor cells to be more resistant to many kinds of compounds, including PI3Kβ inhibitors, PKC inhibitors, HSP90 inhibitors, and MEK inhibitors. In addition, glioma cells with DLAT silence inhibited proliferation, migration, and invasion ability, and promoted cell apoptosis. Conclusion: We conducted a comprehensive analysis of DLAT in the occurrence and progression of tumors, and its possible functions and mechanisms. DLAT is a potential diagnostic, prognostic, and immunotherapeutic biomarker for cancer patients.Graphic Abstract
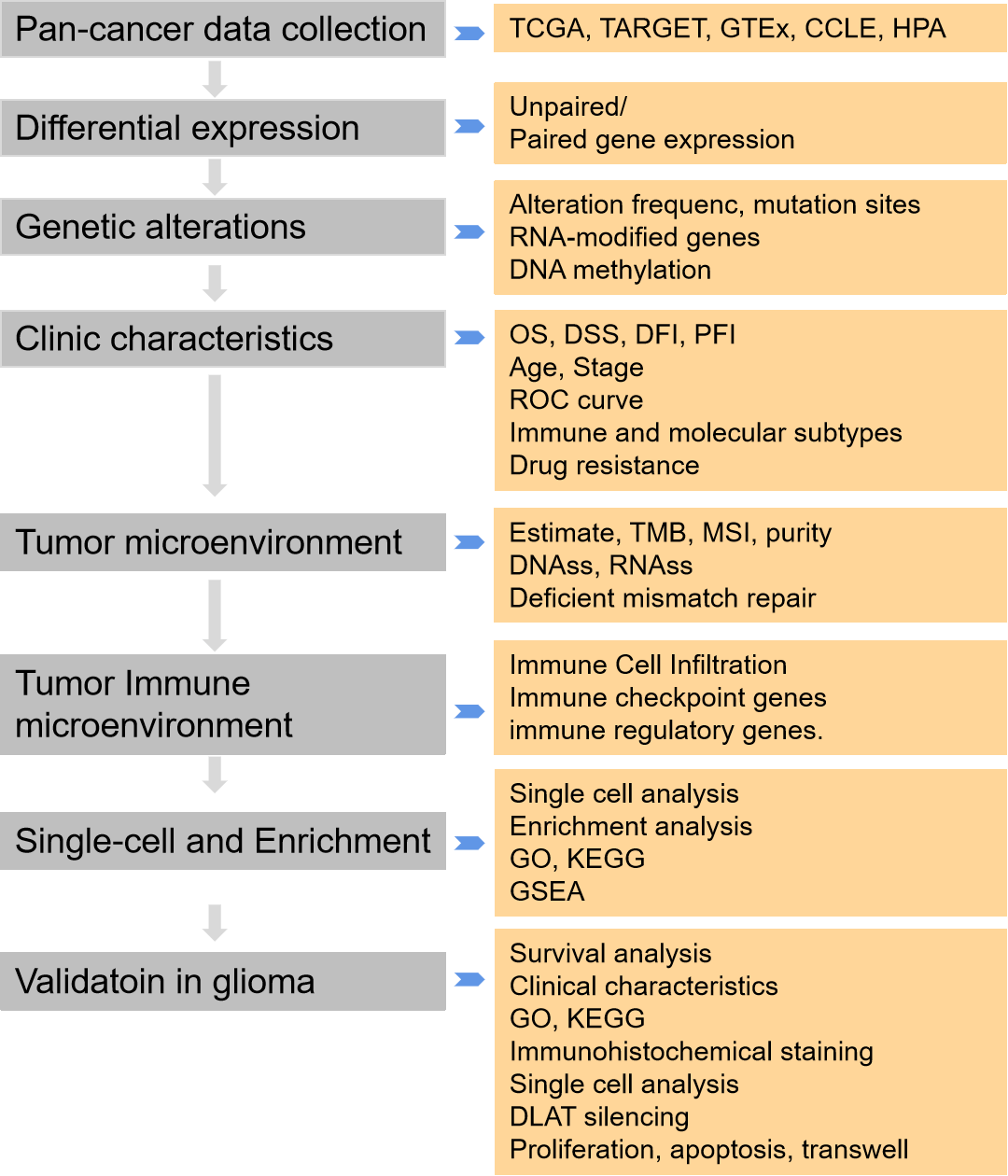
Keywords
Dihydrolipoamide S-acetyltransferase (DLAT); Glioma; Prognostic; Immunological
Supplementary Material
Supplementary Material FileCite This Article
APA Style
ZHOU, H., YU, Z., XU, J., WANG, Z., TAO, Y. et al. (2024). A comprehensive and systematic analysis of Dihydrolipoamide S-acetyltransferase (DLAT) as a novel prognostic biomarker in pan-cancer and glioma. Oncology Research, 32(12), 1903–1919. https://doi.org/10.32604/or.2024.048138
Vancouver Style
ZHOU H, YU Z, XU J, WANG Z, TAO Y, WANG J, et al. A comprehensive and systematic analysis of Dihydrolipoamide S-acetyltransferase (DLAT) as a novel prognostic biomarker in pan-cancer and glioma. Oncol Res. 2024;32(12):1903–1919. https://doi.org/10.32604/or.2024.048138
IEEE Style
H. ZHOU et al., “A comprehensive and systematic analysis of Dihydrolipoamide S-acetyltransferase (DLAT) as a novel prognostic biomarker in pan-cancer and glioma,” Oncol. Res., vol. 32, no. 12, pp. 1903–1919, 2024. https://doi.org/10.32604/or.2024.048138
 Copyright © 2024 The Author(s). Published by Tech Science Press.
Copyright © 2024 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools