International Journal of
Experimental Botany
| Phyton- International Journal of Experimental Botany |
DOI: 10.32604/phyton.2021.014396
ARTICLE
Genome-Wide GRAS Gene Family Analysis Reveals the Classification, Expression Profiles in Melon (Cucumis melo L.)
1Jiangsu Changshu National Agricultural Science and Technology Park, Suzhou, 215500, China
2College of Horticulture, Nanjing Agricultural University, Nanjing, 210095, China
3Changshu Agricultural Science and Technology Development Co., Ltd., Suzhou, 215500, China
*Corresponding Author: Chuntao Qian. Email: ctqian@126.com
Received: 23 September 2020; Accepted: 24 January 2021
Abstract: Melon (Cucumis melo), belonging to the Cucurbitaceae family, is a globally important economic crop. GRAS (GAI, RGA, SCR) genes, which are a type of transcription factor, play a critical role in plant growth and development, including processes such as radial root patterning, light signalling, abiotic/biotic stress, axillary shoot meristem formation, and phytohormone (gibberellin) signal transduction. In this study, the GRAS family in melon was analysed comprehensively with respect to chromosomal location, motif prediction, gene structure, and expression pattern. A total of 37 GRAS genes were first identified in melon, after which a phylogenetic tree was built with the GRAS genes of three model species (Arabidopsis, rice, and sacred lotus) and were divided into nine groups based on the findings of previous studies. Motif and gene structure analysis showed typical conserved domains in all melon GRAS and similar structures in the same subfamilies. The expression analysis of GRAS genes done using RNA-seq data, showed that these genes were differentially expressed in different melon leaves under powdery mildew stress. Furthermore, the real-time quantitative PCR for GRAS genes revealed gene expression corresponding to powdery mildew stress. Our results provide useful information for a better understanding of GRAS genes and provide the foundation for additional functional exploration of the melon GRAS gene family in the powdery mildew stress response.
Keywords: Genome-wide identification; GRAS; melon; stress response
Melon (Cucumis melo L.) is a eudicot diploid species (2n = 24) from the Cucurbitaceae family, which includes other well-known crops such as watermelon, cucumber, and pumpkin. The Cucurbitaceae family is of great economic significance, second only to the Solanaceae family, and among them, melon is one of the most important crops. Melons are planted in multiple climatic environments, and approximately 32 million tons of melons were produced worldwide in 2017 (Food and Agriculture Organization, http://faostat.fao.org). The melon genome was originally sequenced in 2012 [1], and the sequences were improved in 2018 [1,2] along with the development of genetic and genomic resources [3] that greatly advanced molecular-level research focusing on the genomic characterization of important agronomic traits related to abiotic or biotic stress [4–6], fruit ripening [7–9], and sex determination [10].
Melon is susceptible to powdery mildew disease (PM) during the later stage of development. PM is a kind of fungal disease of melon caused by Golovinomyces cichoracearum or Podosphaera xanthii (Px), which is widespread and can cause serious yield losses in melon, and PM has severely hindered the development of melon industry [11–14].
Transcription factors play a significant role in plant development and stress response, and GRAS is a group of transcription factor genes that are widespread in plants. Many studies have shown that GRAS genes respond to plant development and biotic or abiotic stress; these studies have been carried out in model species such as Arabidopsis thaliana [15], Oryza sativa [16], Solanum lycopersicum [17], Nelumbo nucifera [18], and Nicotiana tabacum [19].
The acronym GRAS originated from the characteristic letters of the first known three members, GAI (gibberellic acid insensitive) [20], RGA (repressor of GA1–3 mutant) [21], and SCR (scarecrow) [22]. Normally, GRAS genes contain one or two structural domains, with a protein length of 400–700 amino acid residues [23]. In previous studies, GRAS family members were further divided into 8–13 branches based on phylogenetic relationships and on common features in different species [15–19,24]. Because of their highly homologous gene structures, GRAS genes from the same branches may have similar functions [17], as has been supported by similar gene expression patterns observed in Arabidopsis, rice, and Populus [24]. Since then, many GRAS genes have been identified in plants, including 34 from A. thaliana [16], 60 from O. sativa [24], 38 from N. nucifera [18], 53 from S. lycopersicum [15], 21 from N. tabacum [19] and 106 from Populus tremula [24].
Sequencing and annotation of the melon genome provide an opportunity to identify all the melon GRAS genes [1,2]. In this study, 37 GRAS genes were identified from the melon genome, and analyses of their structure, phylogeny, chromosomal distribution, conserved motifs, and stimulation in response to powdery mildew were performed. The results provide insights into the evolution of the melon GRAS genes and their functions in resisting powdery mildew (PM). Identification and characterization of these GRAS genes may provide opportunities to improve the PM resistance of melon.
All melon protein sequences were downloaded from Melonomics (https://www.melonomics.net/), which is the melon genomic database. The Arabidopsis thaliana GRAS protein sequences were downloaded from TAIR (https://www.arabidopsis.org), Oryza sativa GRAS protein sequences were from the Rice Genome Annotation Project (http://rice.plantbiology.msu.edu/downloads_gad.shtml), and N. nucifera GRAS protein sequences were from the lotus genome database (http://lotus-db.wbgcas.cn/).
2.2 Identification of the GRAS Gene Family in Melon
To identify the GRAS gene in the melon genome (downloaded from https://www.melonomics.net/, genome version:V3.6.1), the HMM profile for GRAS genes was first constructed using 132 protein sequences from O. sativa, A. thaliana, and N. tetragona, and it was used to scan out candidate GRAS genes in the melon genome using HMMER 3.0 [25]. Meanwhile, all melon protein sequences were searched against 132 downloaded GRAS proteins to identify candidate GRAS genes using blastp with the e-value set to 1e–10 [26]. Finally, candidate GRAS genes acquired from two identification methods were combined and used to identify GRAS genes using pfamscan [27,28]. Consequently, genes including the PF03514.13 pfam domain were finally recognized as GRAS genes in melon. The subcellular locations of these GRAS genes were then predicted by ProtComp 9.0 (http://linux1.softberry.com/berry.phtml?topic=protcomppl&group=programs&subgroup=proloc) using the default parameters.
2.3 Chromosomal Location and Phylogeny Analysis of the GRAS Gene Family in Melon
The physical positions of the CmGRAS genes in the chromosomes were located using in-house scripts and drawn using MG2C v2 (http://mg2c.iask.in/mg2c_v2.1/). ClustalW2 [29] was used to perform multiple sequence alignments, and MEGA 7 [30] was used to construct an MP phylogenetic tree based on the amino acid sequences of melon GRAS genes and 132 protein sequences from O. sativa, A. thaliana, and N. tetragona with 500 bootstrap replicates.
2.4 Motif Analysis and Gene Structure Visualization
MEME [31] was used to analyse the conserved motifs in the melon GRAS genes, with the largest number of motifs to seek in GRAS sequences set to 10. Gene structures were visualized using GSDS 2.0 [32].
2.5 Analysis of CmGRAS Gene Expression in Response to Powdery Mildew
The transcriptome data of melon in resistant and susceptible leaves after pathogen inoculation were downloaded from NCBI (SRA identifier: SRP095589) to observe the expression patterns in response to powdery mildew [33]. Raw data were quality controlled by fastp [34] to remove reads containing adapter, reads containing poly-N, and low-quality reads. The clean reads were mapped to the melon reference genome using Tophat [35], and FPKM was calculated using Cufflinks [36] using the default parameters. CmGRA expression profile heatmaps were drawn using the R package pheatmap [37].
2.6 Plant Materials and RT-qPCR Experiment
We selected two plant varieties as experimental materials. They were the powdery mildew (1) disease-resistant (i.e., Elizabeth) and (2) susceptible (i.e., Zhaojun 1) varieties. Muskmelon seeds with full grains and uniform size (26.07 ± 0.82 g/1,000 grain weight for Elizabeth, and 17.22 ± 0.61 g/1,000 grain weight for Zhaojun 1) were selected and then soaked in water at 30°C for 24 h to accelerate germination. The young plants were sowed in a 72-hole burrow plate filled with the following matrix = peat: vermiculite: perlite = 3 V:1 V:1 V). When the seedlings grew to 2 leaves and 1 heart, plants of the two varieties with the same growth conditions were selected to wash their root. Then the plants were transplanted into a water culture plastic box (50 cm × 35 cm × 15 cm). Plants were planted in each box and cultured in Hoagland nutrient solution. The culture conditions were as follows: day temperature 28°C/night temperature 18°C, light 16 h/dark 8 h; relative humidity: 75 ± 5%. The nutrient solution was changed every 2 days. Approximately 100 PM x strains were collected from the farm using the single sporangiophore transfer method. When the seedlings grew to 3 leaves and 1 heart, the strains were inoculated onto seedlings with two or three unfolded leaves. The leafs of muskmelon seedlings were taken for quantitative analysis at 0 h, 24 h, 72 h and 168 h, respectively.
The Primer3 software (http://bioinfo.ut.ee/primer3/) was used to design the RT-qPCR primers. RT-qPCR analysis was used to analyze the GmGRASs. Using the actin gene as an internal control, the standard RT-qPCR with SYBR Premix Ex Taq II (TaKaRa) was repeated at least three times on a CFX96 Real Time System (BioRad). The relative expression profiles were analyzed through the 2−ΔΔCT method with the samples in 0 h point used as controls, then the relative mRNA expression information was obtained [38]. Student’s t-test was used for significance analysis. The Y samples were used as controls.
3.1 Identification of GRAS Family Genes and Chromosomal Locations in Melon
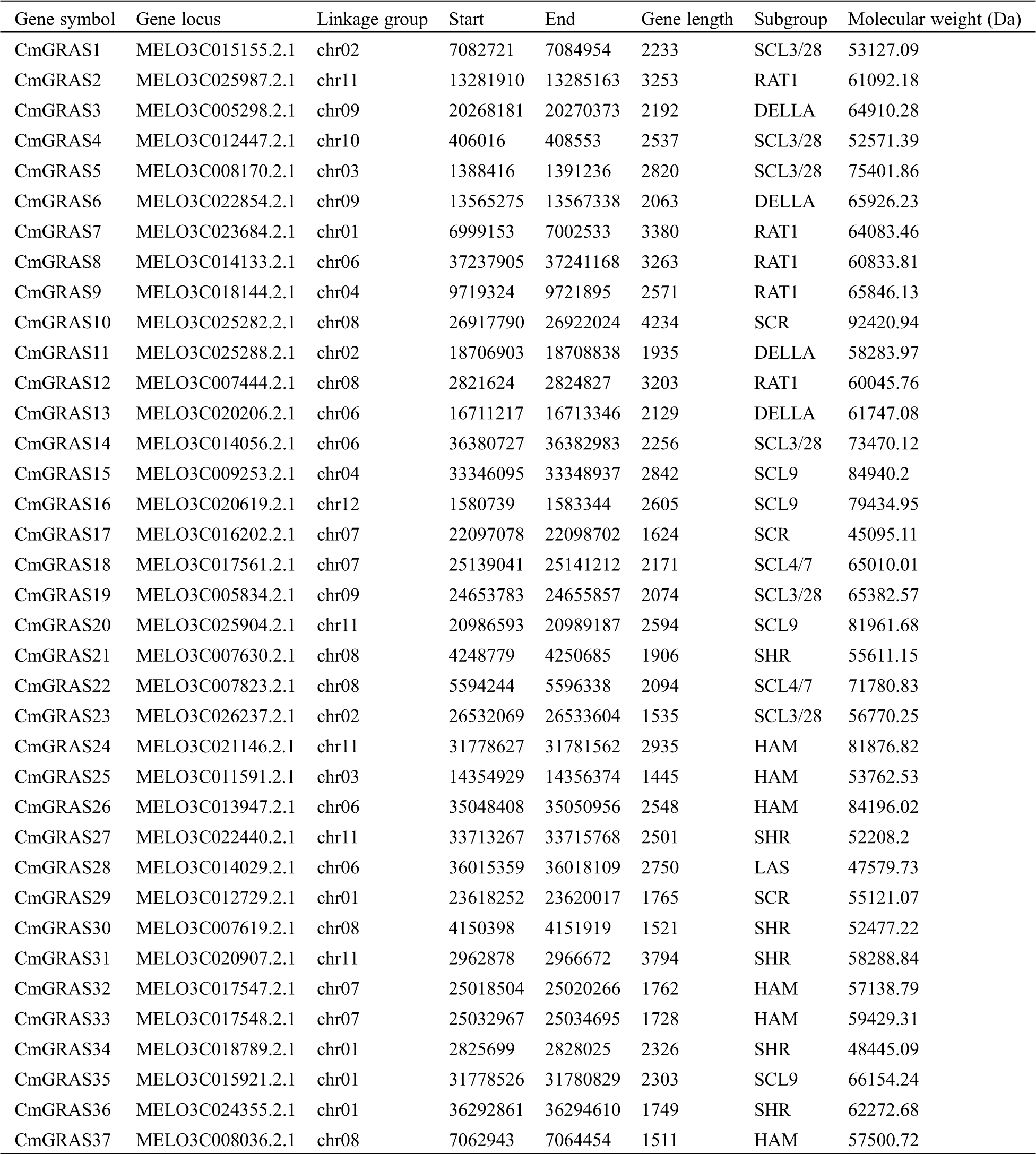
In total, 132 GRAS protein sequences from A. thaliana, O. sativa, and N. nucifera were collected to build a hidden Markov model (HMM), and the search against the melon genome was performed on the model using HMMER 3.0 [25]. Meanwhile, to improve the sensitivity of identifying GRAS homologs, the blastp program included in blast+ 2.29 [26] with an e-value set to 1e–5 was used to find GRAS homologs against 132 collected GRAS proteins in the melon genome. In total, 158 candidate GRAS genes were identified. These candidate melon GRAS proteins were scanned against the Pfam A database using pfamscan, with an e-value of 1e–5, to filter out genes without the GRAS domain PF03514.13 [27,28]. Finally, only 37 melon proteins were identified as GRAS and were labelled as CmGRAS1, CmGRAS2, and so forth (Tab. 1). The length of 37 melon GRAS genes ranged from 1445 to 4234 bp, with an average length of approximately 2382 bp. Of the 37 CmGRAS genes, all were mapped to 12 melon chromosomes (Fig. 1); chromosome 08 contained the greatest number of CmGRAS genes (6%, 16.2%), whereas chromosome 10 and chromosome 12 contained only one CmGRAS gene. The subcellular locations of these GRAS genes were predicted as follows: cytoplasmic (13.51%), extracellular (10.81%), and nuclear (75.68%).
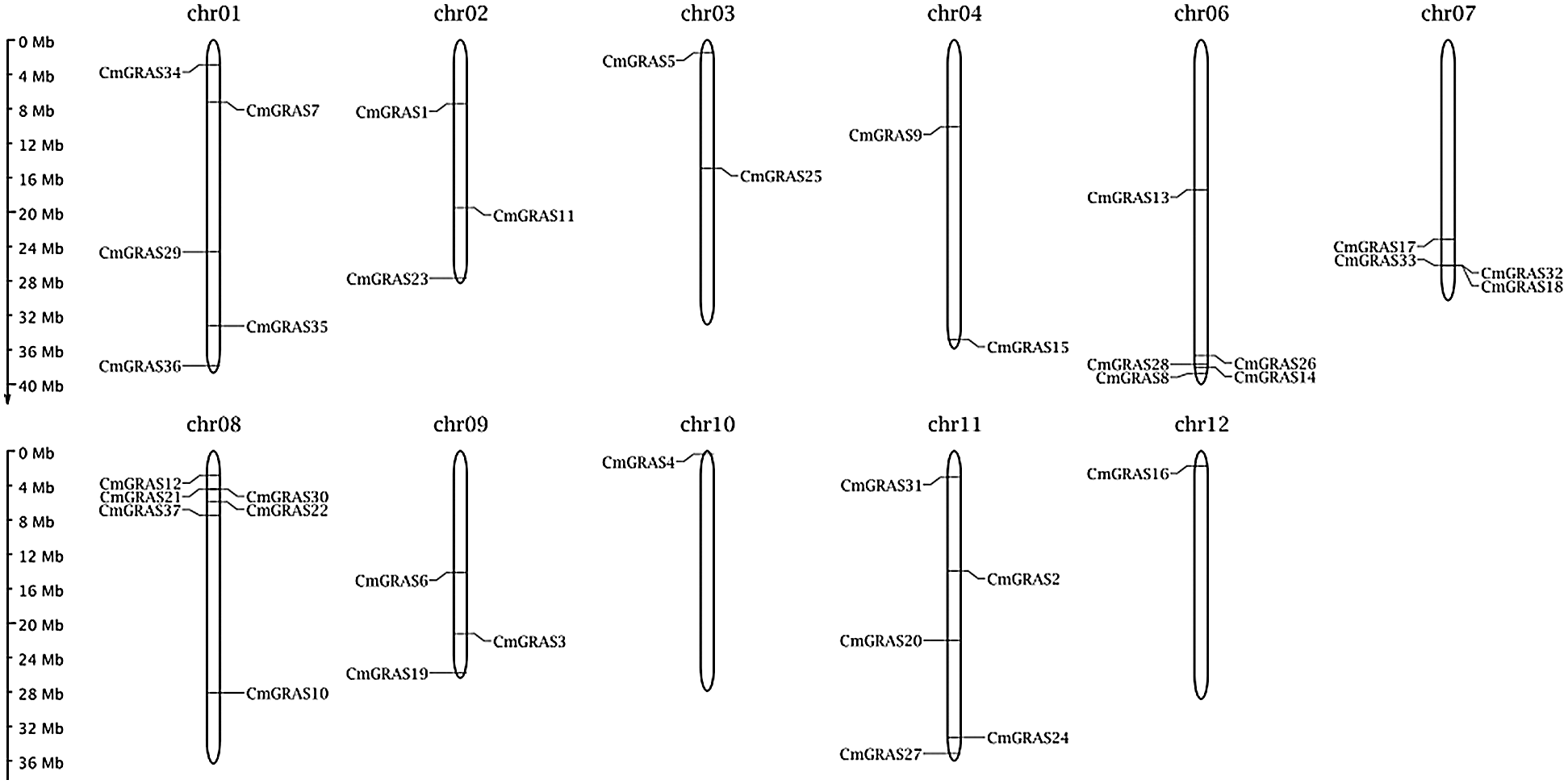
Figure 1: Distribution of CmGRAS genes within the melon chromosomes. Vertical bars represent the chromosomes within the melon genome. The chromosome number is indicated at the top of each chromosome. The scale on the left is in millions of bases (Mb) and indicates the physical length of each linkage group. The positions of each CmGRAS gene are represented by black lines
3.2 Classification and Phylogenetic Analysis of the CmGRAS Genes
Based on multiple alignments of melon with Arabidopsis, rice, and lotus GRAS proteins, a phylogenetic tree was constructed using the maximum parsimony (MP) method, with a bootstrap value of 500. The number of GRAS genes in melon was higher than that in Arabidopsis (34), but less than in rice (60) and in the sacred lotus (38) [18,24]. In previous studies, GRAS genes were divided into nine branches in plants, and the phylogenetic tree generated in this study supported the formal classification. As shown in Fig. 2, among 37 CmGRAS genes, 2, 1, 3, 5, 4, 6, 4, 6, and 6 were found in the SCL4/7, LAS, SCR, SCL 3/28, DELLA, HAM, SCL9, PAT1, and SHR branches, respectively. GRAS genes have been reported to be involved in the development, regulation, and signal transduction [16,20]. In the melon genome, most GRAS genes (31/37) were clustered in six branches, particularly the DELLA and PAT1 branches, whose members in Arabidopsis have been found to be related to signal transduction in the cytoplasm and to the negative regulation of GA signalling [20,21].
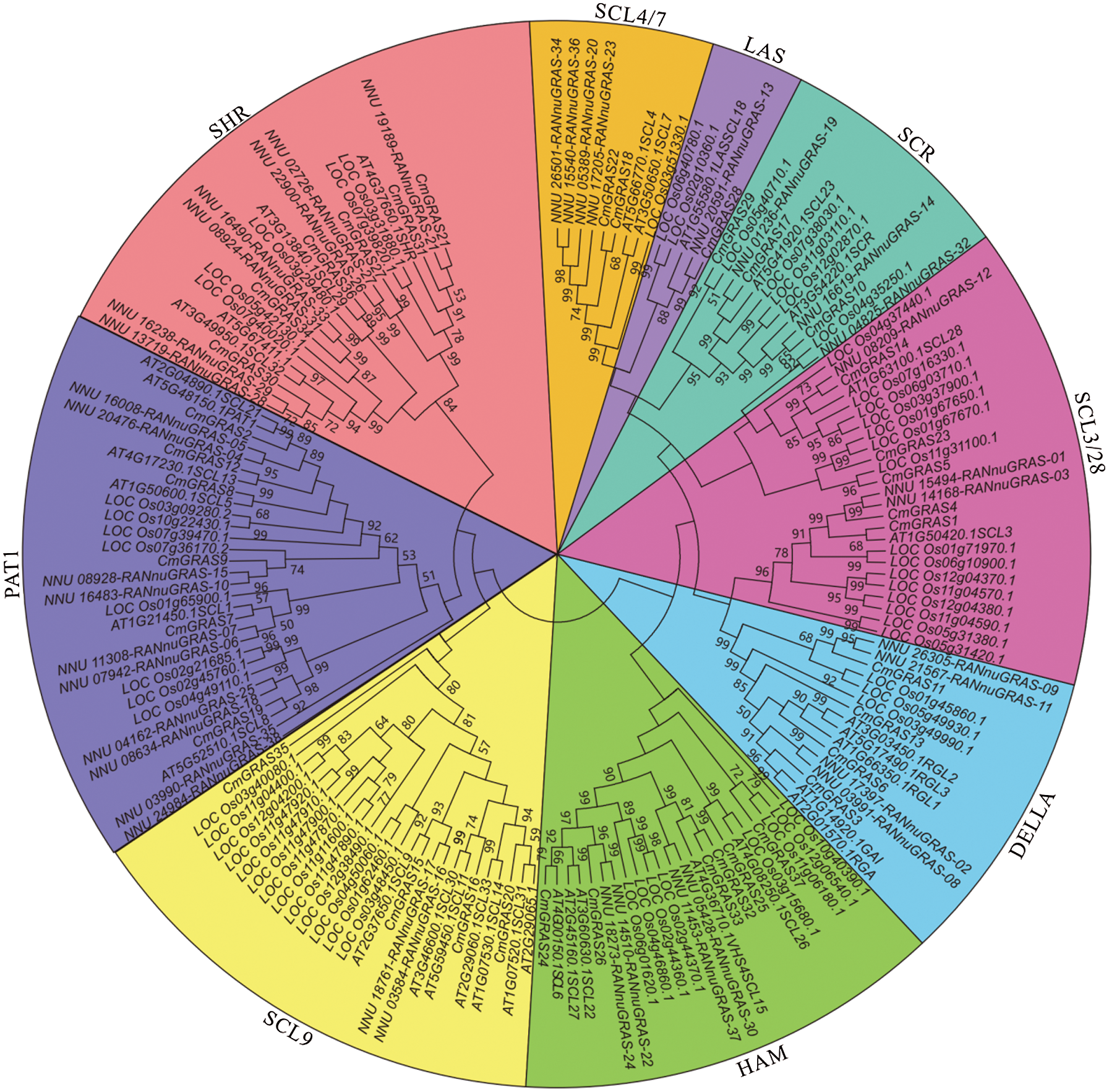
Figure 2: Phylogenetic analysis of the GRAS proteins in melon and 3 other plants. Multiple sequence alignments of GRAS amino acid sequences were performed using ClustalX, and the phylogenetic tree was constructed with MEGA 7 using the maximum parsimony (MP) method and 500 bootstrap replicates. The tree was divided into nine phylogenetic subgroups: SCL4/7, LAS, SCR, SCL 3/28, DELLA, HAM, SCL9, PAT1, and SHR branches. Bootstrap values were set to ≥50%
3.3 Conserved Motifs and Structure of the CmGRAS Genes
Using GRAS phylogenetic relationship data (Fig. 3a), we identified structural features of the melon GRAS, including conserved motifs and the locations of UTRs, exons, and introns. Using the multiple em for motif elicitation (MEME), we identified 10 conserved motifs in the melon GRAS (Fig. 3c). Of the 37 GRAS genes, 14 contained all 10 motifs, and all melon GRAS genes had more than 7 motifs. The gene structures (Fig. 3b) revealed that only eight CmGRAS genes had more than one exon (8/37). In addition, 59.4% (22/37) of CmGRAS genes had no introns, which is less than those of tomato (77.4%), sacred lotus (73.7%), and Arabidopsis (67.6%), but is higher than those of rice (55%) and Populus (54.7%), supporting the notion that most GRAS genes in plants are intronless [15,18,24].
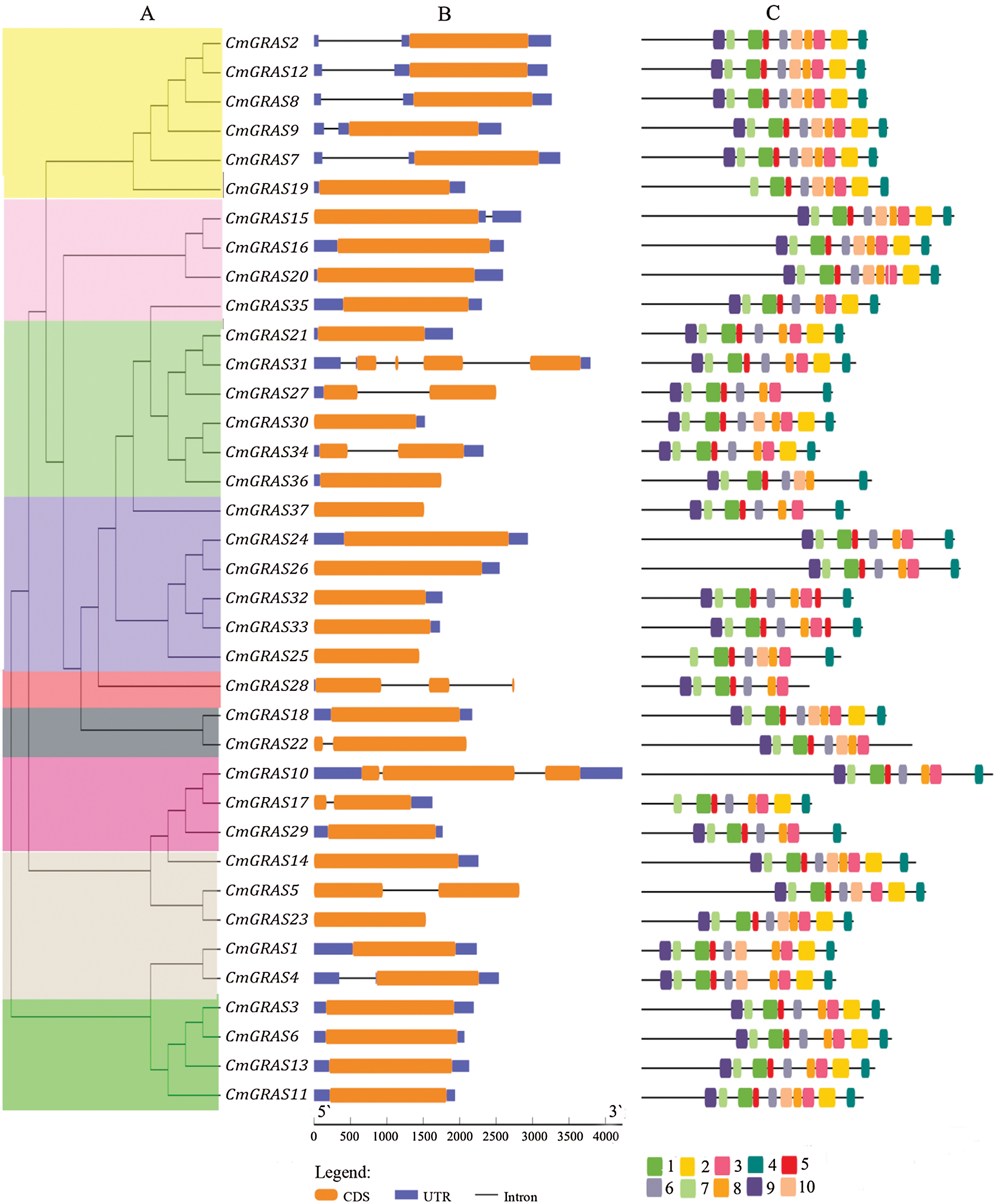
Figure 3: Structure of CmGRAS genes. (A) phylogenetic tree, with coloured regions representing the SCL4/7, LAS, SCR, SCL 3/28, DELLA, HAM, SCL9, PAT1, and SHR branches, as shown in Fig. 2. (B) gene structure, with the brown rectangle representing exon, blue rectangles representing UTR, and blue lines corresponding to introns; protein lengths are shown at the bottom. (C) motif logo, with the amino acid composition of each motif
3.4 Cis-element Analysis of the Promoter Regions of the CmGRAS Genes
Analysis of the 2 kb upstream regions (from the translation start site) of CmGRAS genes revealed the presence of various regulatory elements that are associated with development and abiotic/biotic stress signalling. As shown in Fig. 4, the regulatory elements involved in the regulation of growth and development processes, such as the Dof (DNA binding with one finger) binding site, appear to be enriched in CmGRASs. Stress-responsive cis-regulatory elements identified in this study included basic helix-loop-helix (bHLH) binding site, basic region/leucine zipper motif (bZIP) binding site, and WRKY binding site. Other regulatory elements were also identified, such as the C2H2 zinc finger transcription factor (involved in the occurrence of plant leaves and the regulation of floral organs) binding site and lateral organ boundaries domain (LBD, involved in the development and formation of plant lateral organs) binding sites. In particular, the promoters of 37 CmGRASs contained MBS elements ranging from 1 to 14 copies (Tab. S1), indicating the importance of MYB transcription factors in regulating CmGRASs.

Figure 4: The cis-elements identified in more than ten CmGRAS genes
3.5 Gene Ontology Analysis of GRAS Family Genes
To better understand the functions of CmGRAS family genes, gene ontology (GO) analysis was performed. As shown in Tab. S2, 35 CmGRASs proteins were annotated in terms of molecular function. In total, 31 CmGRASs were annotated as DNA-binding transcription factor activity. 35 CmGRASs were annotated in the category of biological process. The annotated CmGRASs were involved in a variety of biological processes. All CmGRASs took part in regulation of transcription, DNA-templated. In addition, six CmGRASs could participate in response to gibberellin. This is consistent with the study that GRAS family are major players in gibberellin (GA) signalling [16]. Based on the biological process analysis, the main functions of CmGRASs were to bind regulation of transcription, DNA-templated and participate in some other biological processes such as leaf development and regulation of nitrogen utilization. In addition, 35 CmGRASs were annotated in a cellular component. The results showed that CmGRASs were located on the cell nucleus, which was related to their function of transcription factor activity.
GO enrichment analysis was performed to investigate the function of CmGRASs. As shown in Fig. S1 and Tab. S3, 37 GO terms showedenrichment in CmGRASs. Transcription, DNA-templated and regulation of transcription, DNA-templated are the top 2 enriched GO terms, which was related to their function of transcription factors. In addition, some other biological processes, such as response to xenobiotic stimulus and positive regulation of signaling receptor activity were also enriched. It indicated that CmGRASs play an important role in plant stress resistance and biological regulation.
3.6 Expression Patterns of CmGRAS in Response to Powdery Mildew
In this study, we analysed the expression levels of GRAS genes in two types of melon leaves (PM-resistant type MR-1 and susceptible type Topmark), with pathogen inoculation using the published RNA-seq data [33]. Generally, a similar expression pattern was observed in the same branch of GRAS genes; for instance, most members of the DELLA and PAT1 branches exhibited a higher expression level than the GRAS genes of other branches, whereas the SHR subfamily showed a notably low expression level in all samples (Fig. 5).
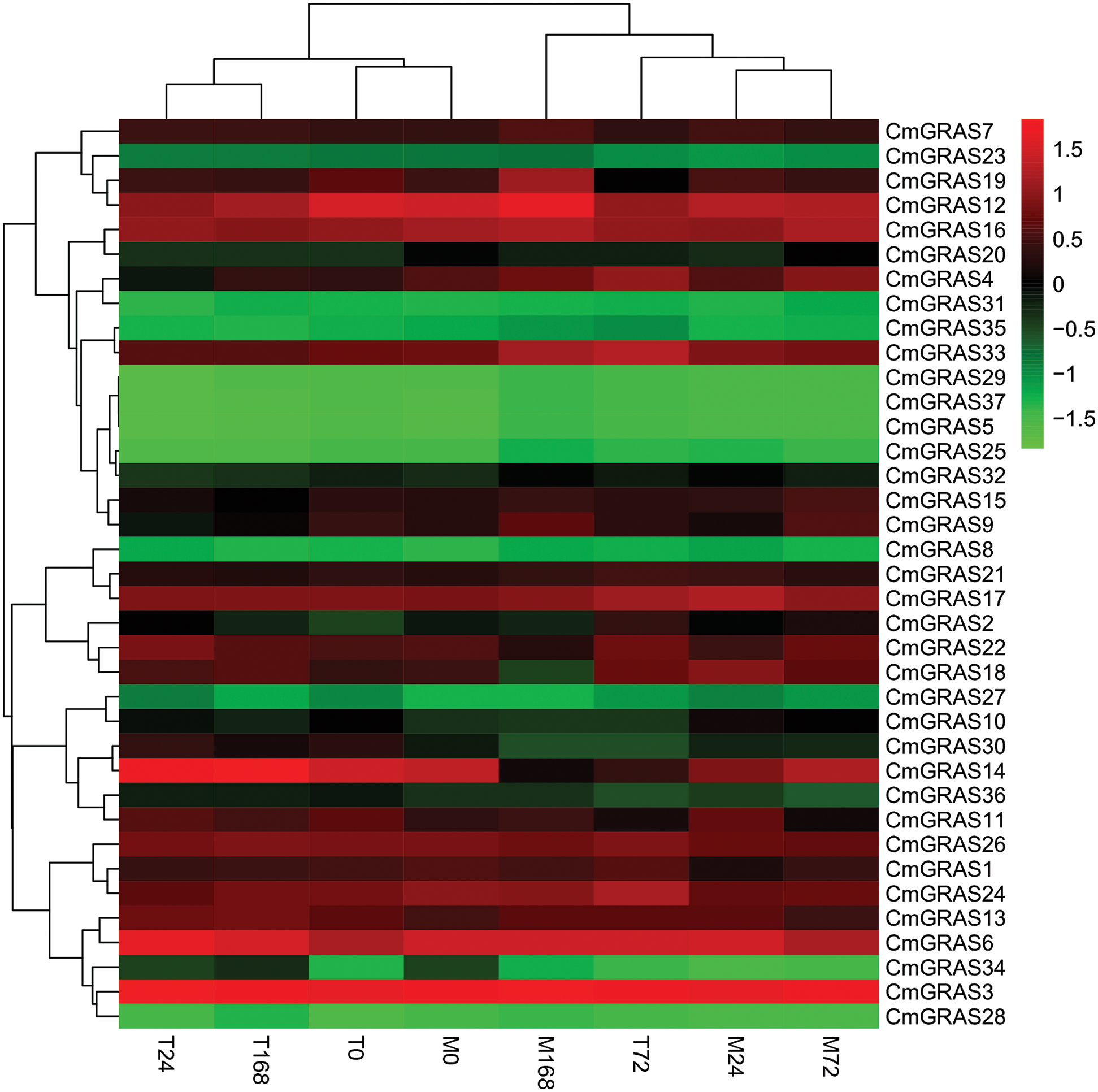
Figure 5: Expression profile analysis of CmGRAS genes in two melon types (PM-resistant and PM-susceptible) inoculated with Podosphaera xanthii (Px) at different time points. T represents the susceptible cultivar “TopMark” and M represents the resistant cultivar “MR-1”; the numbers 0, 24, 72 and 168 represent the time (in h) of Px infection. Transcriptome data (fragments per kilobase million; FPKM) were used to measure the expression levels of CmGRAS genes. The coloured scale used for the different expression levels is shown
However, few PM-resistance genes have been identified in melon. Recent studies have demonstrated that GRAS genes are involved in the responses to the interactions with fungi in plants such as Lotus japonicus and Petunia hybrid [39–41]. There is compelling evidence that GRAS are plant transcription factors that regulate resistance to PM in melon. Interestingly, GRAS genes were expressed differentially at different time points after inoculation. For example, members of the SCL3/28 subfamily, such as CmGRAS14, and members of the SCL4/7 subfamily, such as CmGRAS18, showed higher expression in the susceptible cultivar than in the resistant type at 168 h post-inoculation (Fig. 5), indicating that they may play a critical role in the response to PM.
To further confirm the expression of CmGRASs in other powdery mildew resistant and sensitive materials, ten CmGRASs were further analyzed using qRT-PCR (Fig. 6). As shown in Fig. 6, there has differential expression genes between powdery mildew resistant and powdery mildew sensitive varieties in different time points. It indicates that CmGRASs has a stress response under pathogen invasion. By GO enrichment analysis, the CmGRASs were enrichment in the GO term of response to xenobiotic stimulus. These results indicated that CmGRAS family is an important gene resource for the improvement of powdery mildew resistant in Cucumis melo L.
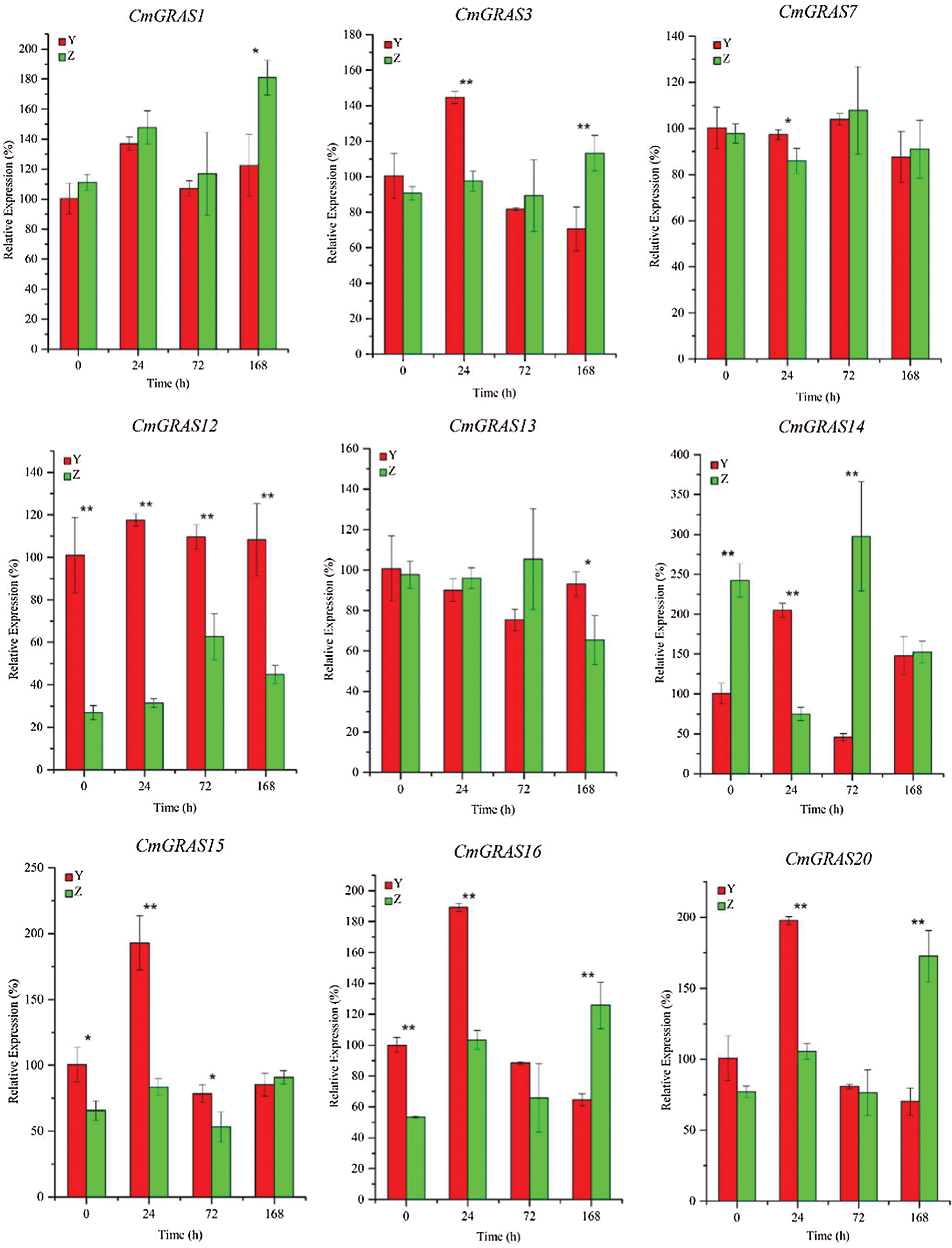
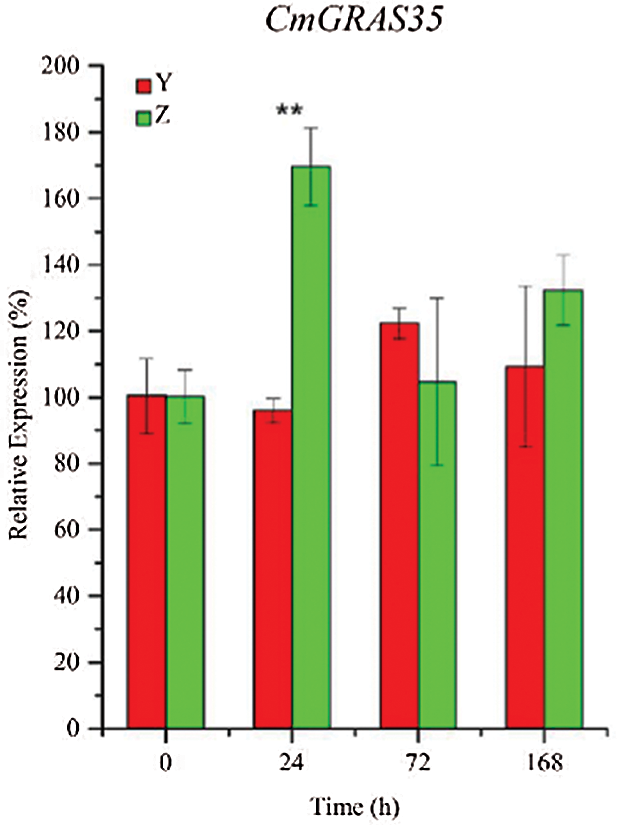
Figure 6: Relative expression profiles of CmGRASs at different time points. Error bars represent means ± SD (n = 3). Three independent experiments were performed for each sample. The significant levels were labeled with asterisk “*” indicated p value < 0.05, and “**” indicated p value < 0.01
In this study, a total of 37 melon GRAS genes were identified, and we focused on the GRAS genes involved in the response to powdery mildew infection. Additionally, we investigated the characteristics of melon GRAS genes, such as physical distribution, classification, and gene structure. In these different stages of RNA-seq data, the differential expression patterns of CmGRAS genes showed that these genes play different roles in melon powdery mildew responses. Additionally, CmGRAS gene expression analysis revealed that some were markedly upregulated or downregulated in response to powdery mildew. In recent decades, many studies have focused on the functions of GRAS genes, and various investigations have revealed that GRAS genes are related to plant growth, development, and stress response in Solanum lycopersicum, Gossypium hirsutum, Medicago truncatula, and Vitis vinifera [15]. Our results also revealed significant differences in the powdery mildew stress-induced GRAS expression, indicating the involvement of these GRAS genes in powdery mildew stress tolerance in melon. Accordingly, our study establishes a structural and functional framework for investigating melon GRAS proteins. Although the melon genome has already been sequenced, identification of melon GRAS genes and investigation of their functions, such as powdery mildew response, are still in their early stages. Our results will facilitate further studies on the functions of GRAS genes important in the abiotic stress response and the development of molecular breeding programs that can be used to enhance abiotic stress tolerance in melon.
We identified 37 GRAS gene family genes in melon genome for the first time, and revealed their structure and evolutionary characteristics. In addition, we revealed the response pattern of this gene family when melon was infected by powdery mildew. Our results provide useful information for a better understanding of GRAS genes and provide the foundation for additional functional exploration of the melon GRAS gene family in powdery mildew stress response.
Acknowledgement: Yanfei Bi and Bin Wei performed the data analyses and wrote the manuscript. Ying Meng and Zhongzhao Li contributed to analysis and manuscript preparation. Zhenghui Tang and Feng Yin contributed to the conception of the study. Chuntao Qian designed the experiment and revised the manuscript.
Funding Statement: This work was supported by the Science and Technology Project of Suzhou (SNG2017083) and the Science and Technology Project of Changshu (CN201701).
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
1. Garcia-Mas, J., Benjak, A., Sanseverino, W., Bourgeois, M., Mir, G. et al. (2012). The genome of melon (Cucumis melo L.). Proceedings of the National Academy of Sciences, 109(29), 11872–11877. DOI 10.1073/pnas.1205415109. [Google Scholar] [CrossRef]
2. Ruggieri, V., Alexiou, K. G., Morata, J., Argyris, J., Pujol, M. et al. (2018). An improved assembly and annotation of the melon (Cucumis melo L.) reference genome. Scientific Reports, 8(1), 1–9. DOI 10.1038/s41598-018-26416-2. [Google Scholar] [CrossRef]
3. Ezura, H., Fukino, N. (2009). Research tools for functional genomics in melon (Cucumis melo L.Current Status and Prospects. Plant Biotechnology, 26(4), 359–368. DOI 10.5511/plantbiotechnology.26.359. [Google Scholar] [CrossRef]
4. Jiao, Z., Sun, J., Wang, C., Dong, Y., Xiao, S. et al. (2018). Genome-wide characterization, evolutionary analysis of WRKY genes in cucurbitaceae species and assessment of its roles in resisting to powdery mildew disease. PLoS One, 13(12), e0199851. DOI 10.1371/journal.pone.0199851. [Google Scholar] [CrossRef]
5. Joobeur, T., King, J. J., Nolin, S. J., Thomas, C. E., Dean, R. A. (2004). The fusarium wilt resistance locus Fom-2 of melon contains a single resistance gene with complex features. The Plant Journal, 39(3), 283–297. DOI 10.1111/j.1365-313X.2004.02134.x. [Google Scholar] [CrossRef]
6. Natarajan, S., Kim, H. T., Thamilarasan, S. K., Veerappan, K., Park, J. I. et al. (2016). Whole genome re-sequencing and characterization of powdery mildew disease-associated allelic variation in melon. PLoS One, 11(6), e0157524. DOI 10.1371/journal.pone.0157524. [Google Scholar] [CrossRef]
7. Pech, J. C., Bouzayen, M., Latché, A. (2008). Climacteric fruit ripening: Ethylene-dependent and independent regulation of ripening pathways in melon fruit. Plant Science, 175(1–2), 114–120. DOI 10.1016/j.plantsci.2008.01.003. [Google Scholar] [CrossRef]
8. Ríos, P., Argyris, J., Vegas, J., Leida, C., Kenigswald, M. et al. (2017). ETHQV 6.3 is involved in melon climacteric fruit ripening and is encoded by a NAC domain transcription factor. The Plant Journal, 91(4), 671–683. DOI 10.1111/tpj.13596. [Google Scholar] [CrossRef]
9. Pereira, L., Santo Domingo, M., Ruggieri, V., Argyris, J., Phillips, M. A. et al. (2020). Genetic dissection of climacteric fruit ripening in a melon population segregating for ripening behavior. Horticulture Research, 7(1), 1–18. DOI 10.1038/s41467-020-16924-z. [Google Scholar] [CrossRef]
10. Ge, C., Zhao, W., Nie, L., Niu, S., Fang, S. et al. (2020). Transcriptome profiling reveals the occurrence mechanism of bisexual flowers in melon (Cucumis melo L.). Plant Science, 301, 110694. DOI 10.1016/j.plantsci.2020.110694. [Google Scholar] [CrossRef]
11. Gao, C., Sun, J., Dong, Y., Wang, C., Xiao, S. et al. (2020). Comparative transcriptome analysis uncovers regulatory roles of long non-coding RNAs involved in resistance to powdery mildew in melon. BMC Genomics, 21(1), 125. DOI 10.1186/s12864-020-6546-8. [Google Scholar] [CrossRef]
12. Hong, Y. J., Hossain, M. R., Kim, H. T., Park, J. I., Nou, I. S. (2018). Identification of two new races of podosphaera xanthii causing powdery mildew in melon in South Korea. The Plant Pathology Journal, 34(3), 182. DOI 10.5423/PPJ.OA.12.2017.0261. [Google Scholar] [CrossRef]
13. Polonio, Á., Pineda, M., Bautista, R., Martínez-Cruz, J., Pérez-Bueno, M. L. et al. (2019). RNA-Seq analysis and fluorescence imaging of melon powdery mildew disease reveal an orchestrated reprogramming of host physiology. Scientific Reports, 9(1), 1–16. DOI 10.1038/s41598-019-44443-5. [Google Scholar] [CrossRef]
14. Jones, J. G., Korir, R. C., Walter, T. L., Everts, K. L. (2020). Reducing chlorothalonil Use in fungicide spray programs for powdery mildew, anthracnose, and gummy stem blight in melons. Plant Disease, 104(12), 3213–3220. DOI 10.1094/PDIS-04-20-0712-RE. [Google Scholar] [CrossRef]
15. Huang, W., Xian, Z., Kang, X., Tang, N., Li, Z. (2015). Genome-wide identification, phylogeny and expression analysis of GRAS gene family in tomato. BMC Plant Biology, 15(1), 1–18. DOI 10.1186/s12870-015-0590-6. [Google Scholar] [CrossRef]
16. Pysh, L. D., Wysocka-Diller, J. W., Camilleri, C., Bouchez, D., Benfey, P. N. (1999). The GRAS gene family in arabidopsis: Sequence characterization and basic expression analysis of the SCARECROW-like genes. The Plant Journal, 18(1), 111–119. DOI 10.1046/j.1365-313X.1999.00431.x. [Google Scholar] [CrossRef]
17. Tian, C., Wan, P., Sun, S., Li, J., Chen, M. (2004). Genome-wide analysis of the GRAS gene family in rice and arabidopsis. Plant Molecular Biology, 54(4), 519–532. DOI 10.1023/B:PLAN.0000038256.89809.57. [Google Scholar] [CrossRef]
18. Wang, Y., Shi, S., Zhou, Y., Zhou, Y., Yang, J. et al. (2016). Genome-wide identification and characterization of GRAS transcription factors in sacred lotus (Nelumbo nucifera). PeerJ, 4, e2388. DOI 10.7717/peerj.2388. [Google Scholar] [CrossRef]
19. Chen, Y. Q., Tai, S. S., Wang, D. W., Ding, A. M., Sun, T. T. et al. (2015). Homology-based analysis of the GRAS gene family in tobacco. Genetics and Molecular Research, 14(4), 15188–15200. DOI 10.4238/2015.November.25.7. [Google Scholar] [CrossRef]
20. Peng, J., Carol, P., Richards, D. E., King, K. E., Cowling, R. J. et al. (1997). The arabidopsis GAI gene defines a signaling pathway that negatively regulates gibberellin responses. Genes & Development, 11(23), 3194–3205. DOI 10.1101/gad.11.23.3194. [Google Scholar] [CrossRef]
21. Silverstone, A. L., Ciampaglio, C. N., Sun, T. P. (1998). The arabidopsis RGA gene encodes a transcriptional regulator repressing the gibberellin signal transduction pathway. The Plant Cell, 10(2), 155–169. DOI 10.1105/tpc.10.2.155. [Google Scholar] [CrossRef]
22. Di Laurenzio, L., Wysocka-Diller, J., Malamy, J. E., Pysh, L., Helariutta, Y. et al. (1996). The SCARECROW gene regulates an asymmetric cell division that is essential for generating the radial organization of the arabidopsis root. Cell, 86(3), 423–433. DOI 10.1016/S0092-8674(00)80115-4. [Google Scholar] [CrossRef]
23. Bolle, C. (2004). The role of GRAS proteins in plant signal transduction and development. Planta, 218(5), 683–692. DOI 10.1007/s00425-004-1203-z. [Google Scholar] [CrossRef]
24. Liu, X., Widmer, A. (2014). Genome-wide comparative analysis of the GRAS gene family in populus. Arabidopsis and Rice. Plant Molecular Biology Reporter, 32(6), 1129–1145. DOI 10.1007/s11105-014-0721-5. [Google Scholar] [CrossRef]
25. Mistry, J., Finn, R. D., Eddy, S. R., Bateman, A., Punta, M. (2013). Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Research, 41(12), e121–e121. DOI 10.1093/nar/gkt263. [Google Scholar] [CrossRef]
26. Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J. et al. (2009). BLAST+: Architecture and applications. BMC Bioinformatics, 10(1), 1–9. DOI 10.1186/1471-2105-10-421. [Google Scholar] [CrossRef]
27. Finn, R. D., Bateman, A., Clements, J., Coggill, P., Eberhardt, R. Y. et al. (2014). Pfam: The protein families database. Nucleic Acids Research, 42(D1), D222–D230. DOI 10.1093/nar/gkt1223. [Google Scholar] [CrossRef]
28. Li, W., Cowley, A., Uludag, M., Gur, T., McWilliam, H. et al. (2015). The EMBL-eBI bioinformatics web and programmatic tools framework. Nucleic Acids Research, 43(W1), W580–W584. DOI 10.1093/nar/gkv279. [Google Scholar] [CrossRef]
29. Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A. et al. (2007). Clustal W and clustal X version 2.0. Bioinformatics, 23(21), 2947–2948. DOI 10.1093/bioinformatics/btm404. [Google Scholar] [CrossRef]
30. Kumar, S., Stecher, G., Tamura, K. (2016). MEGA7: Molecular evolutionary genetics analysis version 7. 0 for bigger datasets brief communication. Molecular Biology and Evolution, 33(7),1870–1874. DOI 10.1093/molbev/msw054. [Google Scholar] [CrossRef]
31. Bailey, T. L., Boden, M., Buske, F. A., Frith, M., Grant, C. E. et al. (2009). MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Research, 37(2), W202–W208. DOI 10.1093/nar/gkp335. [Google Scholar] [CrossRef]
32. Hu, B., Jin, J., Guo, A. Y., Zhang, H., Luo, J. et al. (2015). GSDS 2.0: An upgraded gene feature visualization server. Bioinformatics, 31(8), 1296–1297. DOI 10.1093/bioinformatics/btu817. [Google Scholar] [CrossRef]
33. Zhu, Q., Gao, P., Wan, Y., Cui, H., Fan, C., Liu, S. et al. (2018). Comparative transcriptome profiling of genes and pathways related to resistance against powdery mildew in two contrasting melon genotypes. Scientia Horticulturae, 227, 169–180. DOI 10.1016/j.scienta.2017.09.033. [Google Scholar] [CrossRef]
34. Chen, S., Zhou, Y., Chen, Y., Gu, J. (2018). Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics, 34(17), i884–i890. DOI 10.1093/bioinformatics/bty560. [Google Scholar] [CrossRef]
35. Kim, D., Pertea, G., Trapnell, C., Pimentel, H., Kelley, R. et al. (2013). Tophat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biology, 14(4), 1–13. DOI 10.1186/gb-2013-14-4-r36. [Google Scholar] [CrossRef]
36. Trapnell, C., Williams, B. A., Pertea, G., Mortazavi, A., Kwan, G. et al. (2010). Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature Biotechnology, 28(5), 511–515. DOI 10.1038/nbt.1621. [Google Scholar] [CrossRef]
37. Kolde, R., Kolde, M. R. (2015). Package ‘pheatmap’. R Package, 1(7), 790. https://mran.microsoft.com/snapshot/2017-09-01/web/packages/pheatmap/pheatmap.pdf [Google Scholar]
38. Guilfoyle, T., Hagen, G., Ulmasov, T., Murfett, J. (1998). How does auxin turn on genes?. Plant Physiology, 118(2), 341–347. DOI 10.1104/pp.118.2.341. [Google Scholar] [CrossRef]
39. Xue, L., Cui, H., Buer, B., Vijayakumar, V., Delaux, P. M. et al. (2015). Network of GRAS transcription factors involved in the control of arbuscule development in lotus japonicus. Plant Physiology, 167(3), 854–871. DOI 10.1104/pp.114.255430. [Google Scholar] [CrossRef]
40. Rich, M. K., Courty, P. E., Roux, C., Reinhardt, D. (2017). Role of the GRAS transcription factor ATA/RAM1 in the transcriptional reprogramming of arbuscular mycorrhiza in petunia hybrida. BMC Genomics, 18(1), 1–14. DOI 10.1186/s12864-017-3988-8. [Google Scholar] [CrossRef]
41. Hartmann, R. M., Schaepe, S., Nübel, D., Petersen, A. C., Bertolini, M. et al. (2019). Insights into the complex role of GRAs transcription factors in the arbuscular mycorrhiza symbiosis. Scientific Reports, 9(1), 1–15. DOI 10.1038/s41598-019-40214-4. [Google Scholar] [CrossRef]
Figure S1: The GO enrichment analysis of CmGRASs.
Table S1: Regulatory elements in the 2 kb upstream regions of the CmGRAS genes.
Table S2: The GO annotation of all CmGRASs.
Table S3: The enrichment score, gene counts and qvalue of GO enrichment analysis.
Table S4: Primer sequence.
Table S5: GenBank accession number.
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |