| Phyton-International Journal of Experimental Botany |
DOI: 10.32604/phyton.2022.021922
ARTICLE
Predicted Functional Shifts Due to Type of Soil Microbiome and Watering of Two Wild Plants in Western Region of Saudi Arabia
1Department of Biochemistry, Faculty of Science, King AbdulAziz University, Jeddah, 21589, Saudi Arabia
2Biological Sciences Department, College of Science & Arts, King Abdulaziz University, Rabigh, 21911, Saudi Arabia
3Department of Biochemistry, College of Science, University of Jeddah, Jeddah, Saudi Arabia
4Department of Chemistry Al-Leith University College, Umm Al Qura University, Makkah, Saudi Arabia
5Department of Biology, College of Sciences, Princess Nourah bint Abdulrahman University, P.O. Box 84428, Riyadh, 11671, Saudi Arabia
6Department of Biology, Faculty of Applied Science, Umm Al-Qura University, Makkah, Saudi Arabia
7Department of Biology, College of Science, University of Jeddah, Jeddah, Saudi Arabia
*Corresponding Author: Rewaa S. Jalal. Email: rsjalal@uj.edu.sa
Received: 13 February 2022; Accepted: 23 March 2022
Abstract: The present study aimed to predict differential enrichment of pathways and compounds in the rhizosphere microbiomes of the two wild plants (Abutilon fruticosum and Nitrosalsola vermiculata) and to predict functional shifts in microbiomes due to water. Amplicon sequencing of 16S rRNA region V3–V4 was done and gene-based microbial compositions were enrolled in PICRUSt to predict enriched pathways and compounds. The results indicated that “ABC transporters” and “Quorum sensing” pathways are among the highest enriched pathways in rhizosphere microbiomes of the two wild plants compared with those of the bulk soil microbiomes. The highest enriched compounds in soil microbiomes of the two wild plants included five proteins and three enzymes participating in one or more KEGG pathways. Six of these eight compounds showed higher predicted enrichment in rhizosphere soil microbiomes, while only one, namely phosphate transport system substrate-binding protein, showed higher enrichment in the surrounding bulk soil microbiomes. In terms of differentially enriched compounds due to watering, only the dual-specific aspartyl-tRNA (Asn)/glutamyl-tRNA (Gln) amidotransferase subunit A showed higher enrichment in rhizosphere soil of the two wild plants after 24 h of watering. Two of the highly enriched compounds namely branched-chain amino acid transport system ATP-binding protein and branched-chain amino acid transport system substrate-binding protein, are encoded by genes stimulated by the plant’s GABA that participates in conferring biotic and abiotic stresses in plants and improves the plant’s growth performance. The 3-Oxoacyl-[ACP] reductase, a member of the short-chain alcohol dehydrogenase/reductase (SDR) superfamily, participates in fatty acids elongation cycles and contributes to plant-microbe symbiotic relationships, while enoyl-CoA hydratase has a reverse action as it participates in “Fatty acid degradation” pathway. The methyl-accepting chemotaxis protein is an environmental signal that sense “Bacterial chemotaxis” pathway to help establishing symbiosis with plant roots by recruiting/colonizing of microbial partners (symbionts) to plant rhizosphere. This information justifies the high enrichment of compounds in plant rhizosphere. The dual-specific aspartyl-tRNA (Asn)/glutamyl-tRNA (Gln) amidotransferase subunit A contributes to the plant ability to respond to watering as it participates in attaching the correct amino acid during translation to its cognate tRNA species, while hydrolyzing incorrectly attached amino acid. These two actions reduce the influence of oxidative stress in generating misfolded proteins and in reducing fidelity of translation.
Keywords: Amplicon sequencing; PICRUSt; KEGG; GABA; symbionts; wild plants
Abutilon fruticosum and Nitrosalsola vermiculata are two non-toxic drought tolerant perennial wild species, native to the western coast of Saudi Arabia, that belong to the families of Malvaceae and Amaranthaceae, respectively [1]. Malvaceae is one of the largest families of angiosperm that harbors wild and economic potentials as it is used in the treatment of several human diseases [2,3]. While plants belong to Amaranthaceae are commonly used as a forage crop for livestock due to their high protein content [4] as well as are well adapted to drought [5].
Soil microorganisms naturally interact together through several mechanisms including microbial co-existence or co-aggregation that helps cohabiting microbes to form biofilms and resist antibiotics and promote sustainability, especially when water is not available [6,7]. However, microbes in the plant root rhizosphere are further interact with the plant through several symbiotic and non-symbiotic relationships, of which is the complex communication network namely quorum sensing (QS) that mediates microbe-microbe as well as microbe-host interactions [8]. During microbe-host interaction, bacteria are intertwined with root rhizosphere by various types of root exudates, specific for certain microbial taxa to promote bacterial growth under drought stress condition [9]. In return, some of these taxa act as plant growth promoting bacteria (PGPB) utilizing several mechanisms [10]. These mechanisms include facilitation of resource acquisition through nitrogen fixation, phosphate solubilization and sequestering iron [11,12]. PGPB also modulate levels of phytohormones like cytokinins, gibberellins, indoleacetic acid and ethylene [11,12]. Accordingly, the study of rhizosphere microbes and their enriched metabolic functions can help in deciphering new mechanisms that can be future candidates for plants that have economic value to improve their ability to tolerate drought stress [13,14]. Examples of these candidate microbes include members of phylum Actinobacteria and other gram-positive taxa [13–15] as it is suggested that gram-positive bacteria have higher ability to tolerate drought stress than gram-negative bacteria due to the differential thickness of cell wall Potts Naylor [16,17]. Bacterial response to drought and rewetting stress is based on types of bacteria and their interaction with the plant host(s) as well as the chemical structure of surrounding soil [18]. Nonetheless, drought and rewetting was considered as a sort of abiotic stress response that can effectively alter architecture of soil and its existing microbes [18].
Amplicon 16S rRNA sequencing is used to address structure and complexity of microbial communities in a given environmental niche. Several bioinformatics tools have been developed to predict microbial pathways, compounds and functional genes from amplicon sequencing data based on mapping 16S rRNA genes of taxa whose genome sequences are not available to homologous taxa with fully sequenced genomes. These tools include PICRUSt, PICRUSt2, Tax4Fun, and FaproTax [19–22] and the target environments include soil microbiomes [23]. Functional prediction studies scope the light on the possible enriched pathways and compounds in an environmental niche without the need to make whole metagenomic sequencing.
The present study aims to use PICRUSt (Phylogenetic Investigation of Communities by Reconstruction of Unobserved States) in predicting differential enrichment of metabolic pathways in the rhizosphere microbiomes of the two wild plants Abutilon fruticosum and Nitrosalsola vermiculata and functional shifts in these microbiomes due to water.
2.1 Watering Experiment and Soil Collection
Watering experiment was conducted in the western Mecca region of Saudi Arabia north of Jeddah City, where A. fruticosum and N. vermiculata perennial plants grow natively in the wild (N21°36.738′ E040°06.913′ & N21°56.607′ E039°01.742′, respectively) [1]. We have selected this area where the two wild plants usually grow side-by-side. According to the information in the link of National Center for Meteorology, Saudi Arabia, this area has received no rainfall for >3 months prior watering experiment. The latter conditions are prerequisites in selecting the area for the watering experiment as previously described [24,25]. Plots (~1 m2 each) of single-grown similar-sized plants of the two wild plants were watered (25 liters dH2O/plot) only once and rhizosphere and surrounding bulk soil samples were collected in three replicates in the morning after 0, 24 and 48 h of watering. Average morning temperature across the three days was 27.4°C. Soil samples were collected in liquid nitrogen and stored at −20°C as previously described [26,27].
2.2 DNA Extraction, Amplicon Sequencing and Functional Analysis of Soil Microbiomes
DNAs were extracted from bulk and rhizosphere soil microbiota at the three time points using CTAB/SDS method and concentration was adjusted to 1 ng/μL prior shipment to Novogene Co., Ltd., Singapore for deep amplicon sequencing of 16S rRNA region V3–V4. Raw read datasets were generated for microbiome samples of surrounding bulk (S) and rhizosphere (R) soils of A. fruticosum and N. vermiculata that were collected in three replicates at the three watering time points, e.g., 0 (S11–S13 or group A for bulk soil & R11–R13 or group D for rhizosphere soil), 24 (S21–S23 or group B for bulk soil & R21–R23 or group E for rhizosphere soil) and 48 h (S31–S33 or group C for bulk soil & R31–R33 or group F for rhizosphere soil).
Recovered raw data were merged using FLASH V1.2.7 [28] and high-quality clean tags were obtained using QIIME software V1.7.0 [29] as previously described [30]. While, chimeric sequences were removed using UCHIME algorithm [31] and effective tags were obtained using Uparse software V7.0.1090 [32]. The data matrices were used in measuring principal coordinate analysis (PCoA) to ensure incorporation of ecological distances. PCoA is displayed by WGCNA package, stat packages and ggplot2 package in R software V2.15.3. While, QIIME software V1.7.0 [33] in Mothur method was performed against SSUrRNA dataset of SILVA database to annotate taxa and detect their abundance following standard protocols [34]. Then, gene-based microbial compositions were enrolled in PICRUSt algorithm V1.0.0 to make inferences from KEGG (Kyoto Encyclopedia of Genes and Genomes) annotated database with pathways and metabolites [19]. The analysis involved functional predictions at KEGG levels II and III and subsequent enriched compounds. Predicted pathways and compounds whose abundance in one soil type was ≥1.5-fold that of the other soil type were considered highly enriched.
2.3 Validation of Soil Enriched Compounds and Real Time PCR
Soils of the three replicates of each of the six groups, e.g., A, B, C, D, E and F, were gathered for the two wild plants. Then, total RNAs were isolated using RNA PowerSoil® Total RNA isolation kit (Mo Bio, cat. No. 12866-25) following manufacturer instructions. DNA contamination was removed using RQ1 RNase-free DNase (Promega, Madison, WI, USA). Primers to amplify partial-length fragment (323 bp) of gatA gene (K02433) of Bacillus subtilis str. 168 (GenBank or Embl acc. No. AL009126.3: 729038-730495 nt) encoding dual-specific aspartyl-tRNA(Asn)/glutaminyl-tRNA (Gln) amidotransferase subunit A (EC: 6.3.5.7 & 6.3.5.6, GenBank acc. No. CAB12488.1) (e.g., FW: 5′ GATGGCCGCAGCGAACATGG 3′, & RV: 5′ CTAAACGGCACTTCGCCCGC 3′, amplicon size = 323 bp) as well as its 16S rRNA (AB042061) partial house-keeping gene fragment (e.g., FW: 5′ TCGCGGTTTCGCTGCCCTTT 3′, & RV: 5′ AAGTCCCGCAACGAGCGCAA 3′, amplicon size = 177 bp) were designed using Netprimer software (http://www.premierbiosoft.com/netprimer/index.html) using standard criteria. Expression level of the metatranscript gatA was detected by real time PCR using Agilent Mx3000P qPCR System (Agilent technology, USA). Maxima™ SYBR Green/ROX real time PCR was done as described [35]. Similar amounts of RNAs were used for the six group samples and calculations were made to detect the expression level of gatA gene in individual samples relative to that of the house-keeping gene, e.g., 16S rRNA.
Amplicon 16S rRNA sequencing was performed for rhizosphere microbiomes and their surrounding bulk soil microbiomes of the two wild plants (A. fruticosum and N. vermiculata). The amount of water used (25 L dH2O/m2 plot) was enough to keep the soil moist during the experiment. Nomenclature of microbiome groups of the two wild plants was based on soil type (e.g., S for bulk soils & R for rhizosphere soils) regardless of watering time points. Further, grouping style ABCDEF refers to the interaction between the two soil types, e.g., S (ABC subgrouping style) & R (DEF subgrouping style), and the three watering time points, e.g., 0 (A & D groups, respectively), 24 (B & E groups, respectively) and 48 h (C & F groups, respectively). The average sequence length per read across the raw data of the two wild plants is ~412 bp. The total number of OTUs referring to different microbial taxonomic ranks (phylum, class, order, family, genus & species) across microbiomes of the two wild plants is 4371. Across highly abundant taxa of the two wild plants, phylum Cyanobacteria was significantly higher in bulk soil, while phyla Proteobacteria, Firmicutes, Chloroflexi and Actinobacteria were significantly higher in rhizosphere soil (Fig. S1). Phylum bacteroidetes was only significantly higher in rhizosphere soil of A. fruticosum. At the genus level, unidentified genus of Cyanobacteria was significantly higher in bulk soil of the two wild plants, while Bacillus, Ammoniphilus, Sphingomonas and Microvirga were significantly higher in rhizosphere soil. These results indicate that bulk soil has a wealth of new microbes of the economically important phylum Cyanobacteria that remains to be deciphered.
The results of principle coordinate in Figs. 1 and 2 indicate complete separation of bulk (subgrouping style ABC) and rhizosphere (subgrouping style DEF) soil microbiomes of the two wild plants, as bulk soil microbiomes are located in the negative position of PCoA 1 (or PC1) for the two wild plants, while rhizosphere microbiomes are located in the positive position of PC1 (Figs. 1 & 2). Bulk soil microbiome group A of A. fruticosum and rhizosphere microbiome groups D and F of N. vermiculata are located in the positive position of PC2, while the other groups for the two wild plants are located in the negative position of PC2. Interestingly, groups E and F of rhizosphere microbiomes collected at 0 and 48 h watering time points, respectively, showed tendency to merge for the microbiomes of the two wild plants indicating that rhizosphere microbiomes collected at 24 h watering time point for the two wild plants respond differently to watering in terms of predicted function compared with those of the other two time points (Figs. 1 & 2).
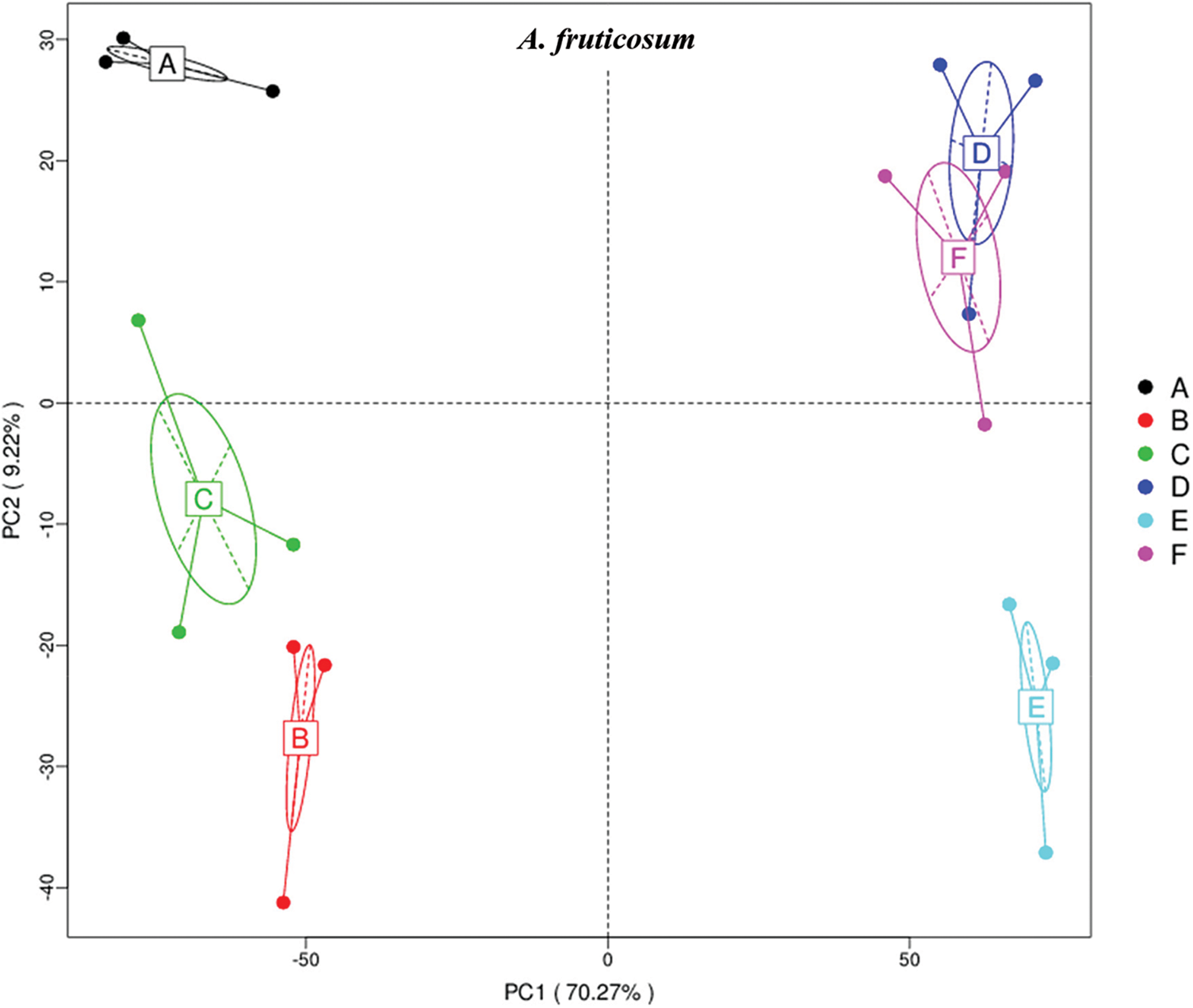
Figure 1: Principle coordinate analysis (PCoA) based on compound enrichment of microbiomes collected from surrounding bulk (subgrouping style ABC) and rhizosphere (subgrouping style DEF) soils of Abutilon fruticosum after 0 (groups A & D, respectively), 24 (groups B & E, respectively) and 48 h (groups C & F, respectively) of watering. A colored dot refers to a given sample in one group, and similar colored dots refer to samples of the same group. X-axis is the first principal coordinate and Y-axis is the second. Number in brackets represents contributions of PCoAs to differences among samples
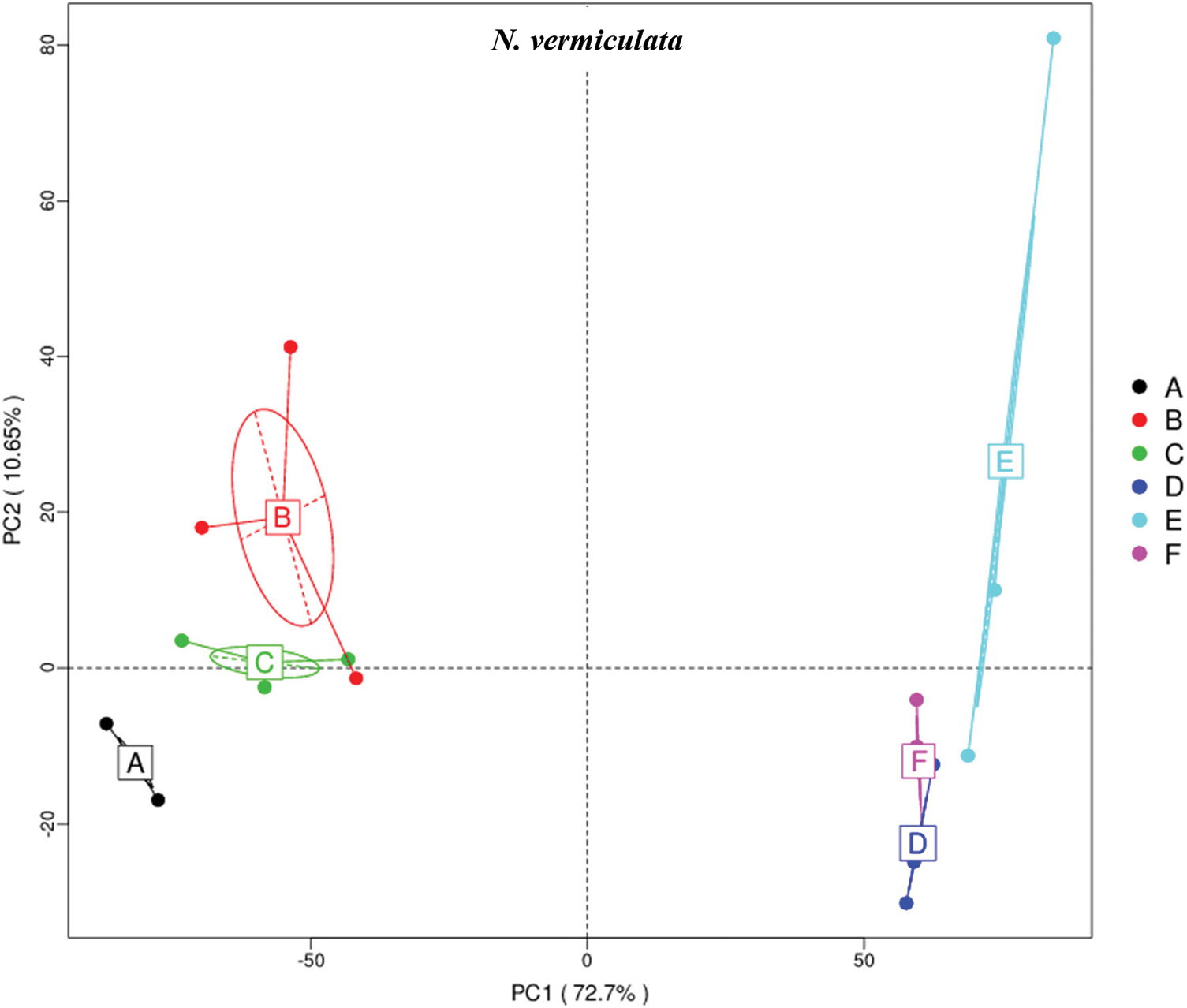
Figure 2: Principle coordinate analysis (PCoA) based on compound enrichment of microbiomes collected from surrounding bulk (subgrouping style ABC) and rhizosphere (subgrouping style DEF) soils of Nitrosalsola vermiculata after 0 (groups A & D, respectively), 24 (groups B & E, respectively) and 48 h (groups C & F, respectively) of watering. A colored dot refers to a given sample in one group, and similar colored dots refer to samples of the same group. X-axis is the first principal coordinate and Y-axis is the second. Number in brackets represents contributions of PCoAs to differences among samples
Enriched KEGG pathways at levels I and II referring to the predicted number of genes across soil type and time after watering were common in microbiomes of the two wild plants (Figs. S2 & S3). The results indicate that predicted level-I KEGG pathways with the largest number of genes in microbiomes of the two wild plants include “Environmental information processing”, “Genetic information processing” and “Metabolism”. Of which, level-II pathways “Membrane transport” derived from level-I pathway “Environmental information processing”, “Replication and repair” derived from level-I pathway “Genetic information processing” as well as “Energy metabolism”, “Carbohydrate metabolism” and “Amino acid metabolism” derived from level-I pathway “Metabolism” were the most enriched pathways in terms of number of genes across the two types of soil microbiomes and watering time points.
Results in Figs. S4 and S5 refer to the top 10 enriched level-III KEGG pathways across microbiome types and time after watering of A. fruticosum and N. vermiculata, respectively. These predicted pathways are “Transporters”, “ATP-binding cassette (or ABC) transporters and Quorum sensing”, “General function prediction only”, “DNA repair and recombination proteins”, “Two component system”, “Purine metabolism”, “Ribosomes”, “Peptidases”, “Oxidative phosphorylation” and “Secretion system”.
Figs. S6 and S7 refer to enriched pathways at the individual level, while Figs. 3 and 4 refer to enriched pathways at the ABCDEF grouping style level for microbiomes of the two wild plants A. fruticosum and N. vermiculata, respectively. The data of level-III predicted pathways at the individual level almost show complete separation of bulk soil and rhizosphere microbiomes, which align with the data at the ABCDEF grouping style level. Figs. 3 and 4 indicate the top 35 enriched pathways of which one of them refers to a pathway with unknown function. The results indicate that the predicted pathways “Transporters” and “ABC transporters and Quorum sensing” were highly enriched in rhizosphere microbiomes of the two wild plants compared with those of the bulk soil microbiomes. The other level-III KEGG predicted pathways, except for “General function prediction only” and “Two component system”, showed opposite results (Figs. 3 & 4). Enrichment of the latter two predicted pathways at the ABCDEF grouping style level differed between soil microbiomes of the two wild plants.
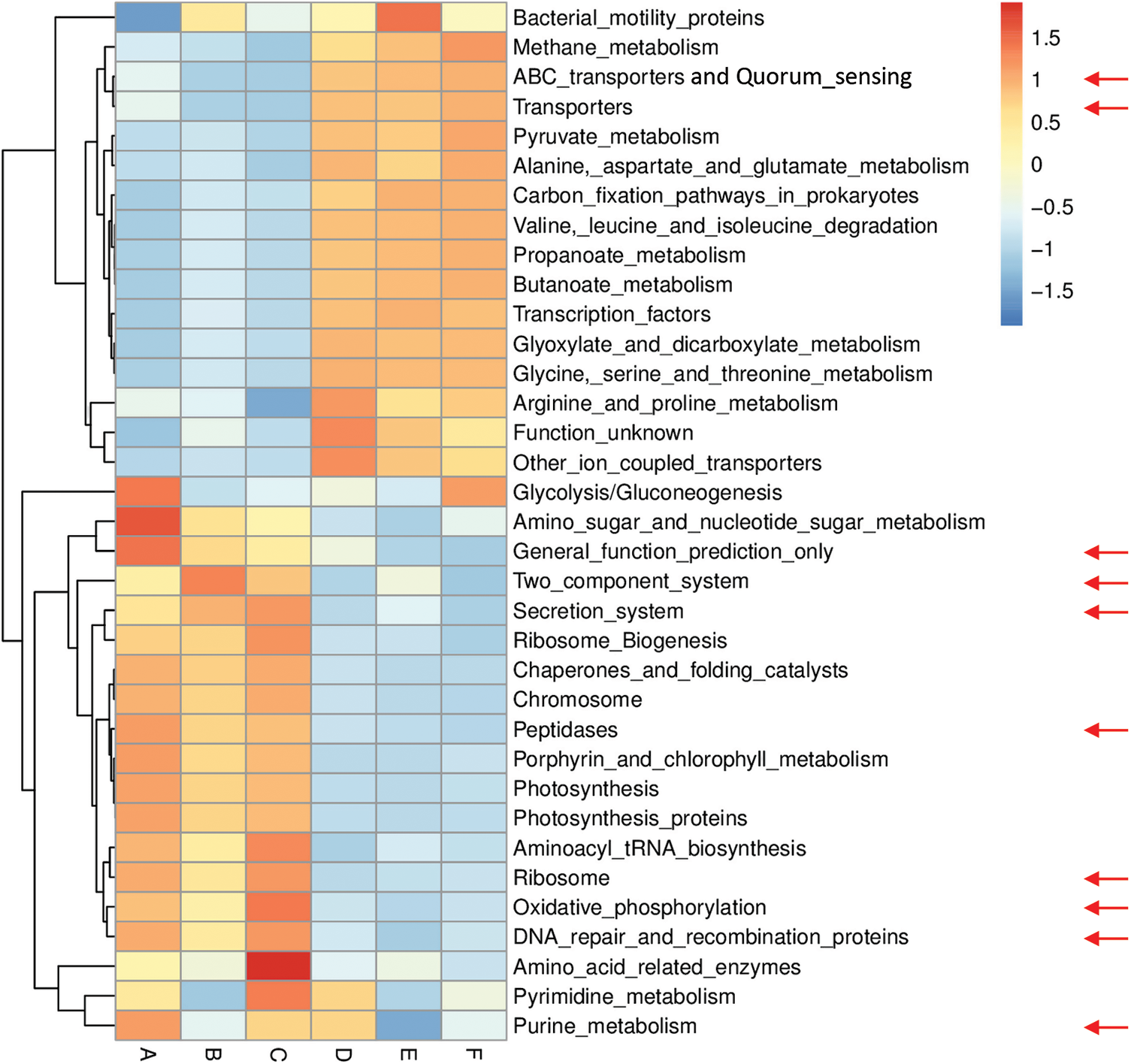
Figure 3: Heat map referring to the top 35 KEGG level-III pathways in terms of enrichment within grouping style ABCDEF microbiomes collected from surrounding bulk (subgrouping style ABC) and rhizosphere (subgrouping style DEF) soils of A. fruticosum after 0 (groups A & D, respectively), 24 (groups B & E, respectively) and 48 h (groups C & F, respectively) of watering. Red arrows refer to the top 10 highly enriched pathways in soil microbiomes (see Fig. S4)
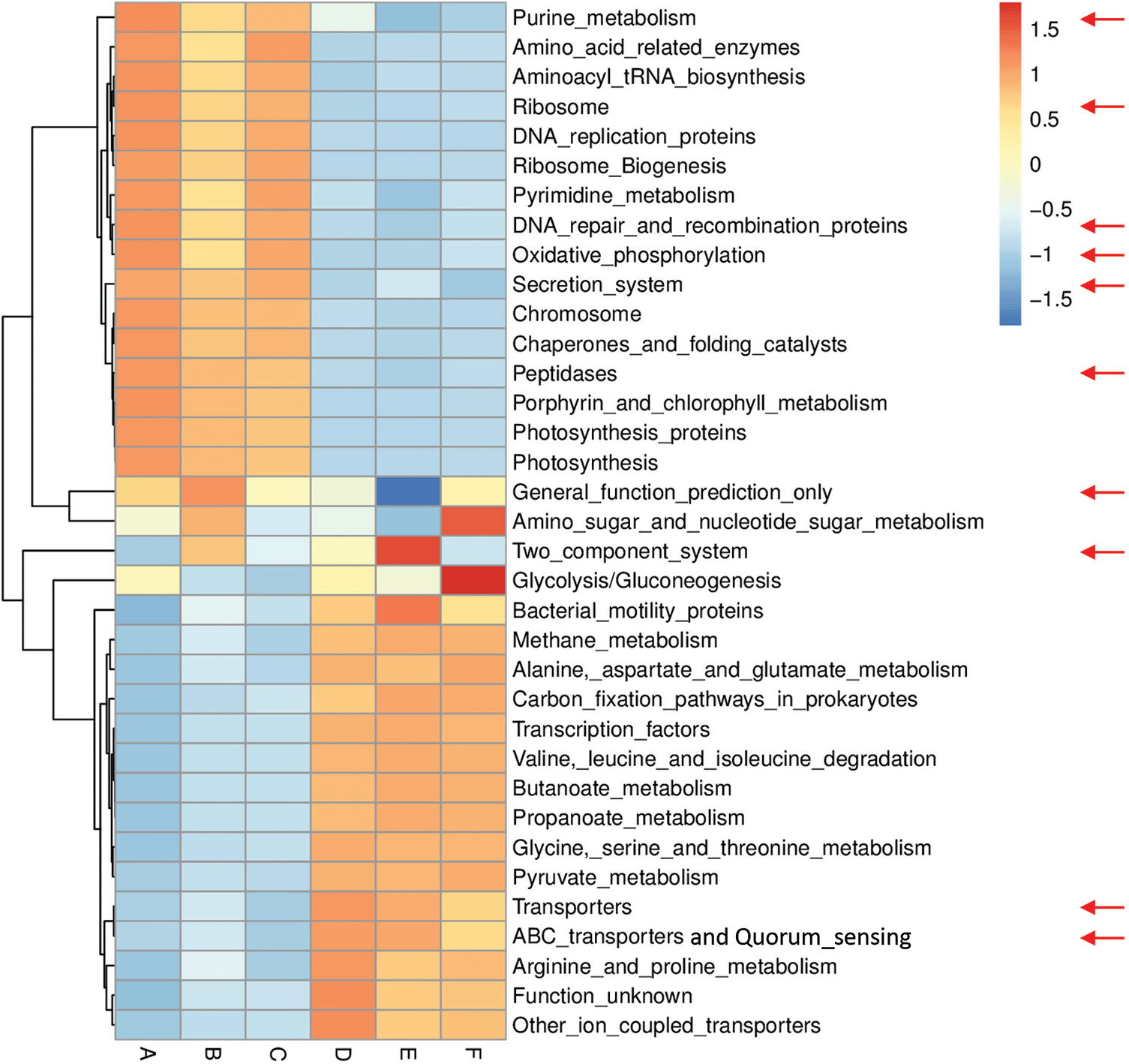
Figure 4: Heat map referring to the top 35 KEGG level-III pathways in terms of enrichment within grouping style ABCDEF microbiomes collected from surrounding bulk (subgrouping style ABC) and rhizosphere (subgrouping style DEF) soils of N. vermiculata after 0 (groups A & D, respectively), 24 (groups B & E, respectively) and 48 h (groups C & F, respectively) of watering. Red arrows refer to the top 10 highly enriched pathways in soil microbiomes (see Fig. S5)
Figs. S8 and S9 refer to level-III KEGG predicted pathways with known function (34) that were differentially enriched in bulk soil vs. rhizosphere microbiomes for the two wild plants A. fruticosum and N. vermiculata, respectively. Across the two wild plants, four predicted pathways, e.g., “Butanoate metabolism”, “Propanoate metabolism”, “ABC transporters and Quorum sensing” and “Valine, leucine and isoleucine degradation”, were highly enriched in rhizosphere soil microbiomes, while three predicted pathways, e.g., “Photosynthesis proteins”, “Porphyrin and chlorophyll metabolism” and “Photosynthesis”, in bulk soil microbiomes (Figs. S8 & S9, respectively). Among the top 10 most enriched level-III KEGG predicted pathways, enrichment of the “ABC transporters and Quorum sensing” pathways in rhizosphere soil microbiomes across the two wild plants was ≥1.5-fold that of bulk soil microbiomes (Figs. S8 & S9). In other words, “ABC transporters and Quorum sensing” predicted pathways in microbiomes of the two wild plants were less enriched in bulk soil microbiomes across time after watering compared with those in rhizosphere soil.
The top 35 enriched compounds in soil microbiomes of the two wild plants A. fruticosum and N. vermiculata in terms of soil type and watering time point at individual level are shown in Figs. S10 and S11, respectively, while those predicted at the ABCDEF grouping style level are shown in Figs. 5 and 6, respectively. The data of compound enrichment at the individual level almost completely separated in bulk soil and rhizosphere microbiomes and align with the data at the ABCDEF grouping style level. KEGG ontology (KO) hierarchy and description of the enriched compounds at grouping style level are shown in Table S1. Only, 27 enriched compounds are common in microbiomes of the two wild plants A. fruticosum and N. vermiculata (Figs. S12 & S13, respectively). Fourteen compounds in rhizosphere soil microbiome of A. fruticosum showed ≥1.5-fold predicted enrichment, while eight compounds in bulk soil microbiome (Fig. S12). Additionally, 11 compounds in rhizosphere soil microbiome of N. vermiculata showed ≥1.5-fold predicted enrichment, while five compounds in bulk soil microbiome (Fig. S13).
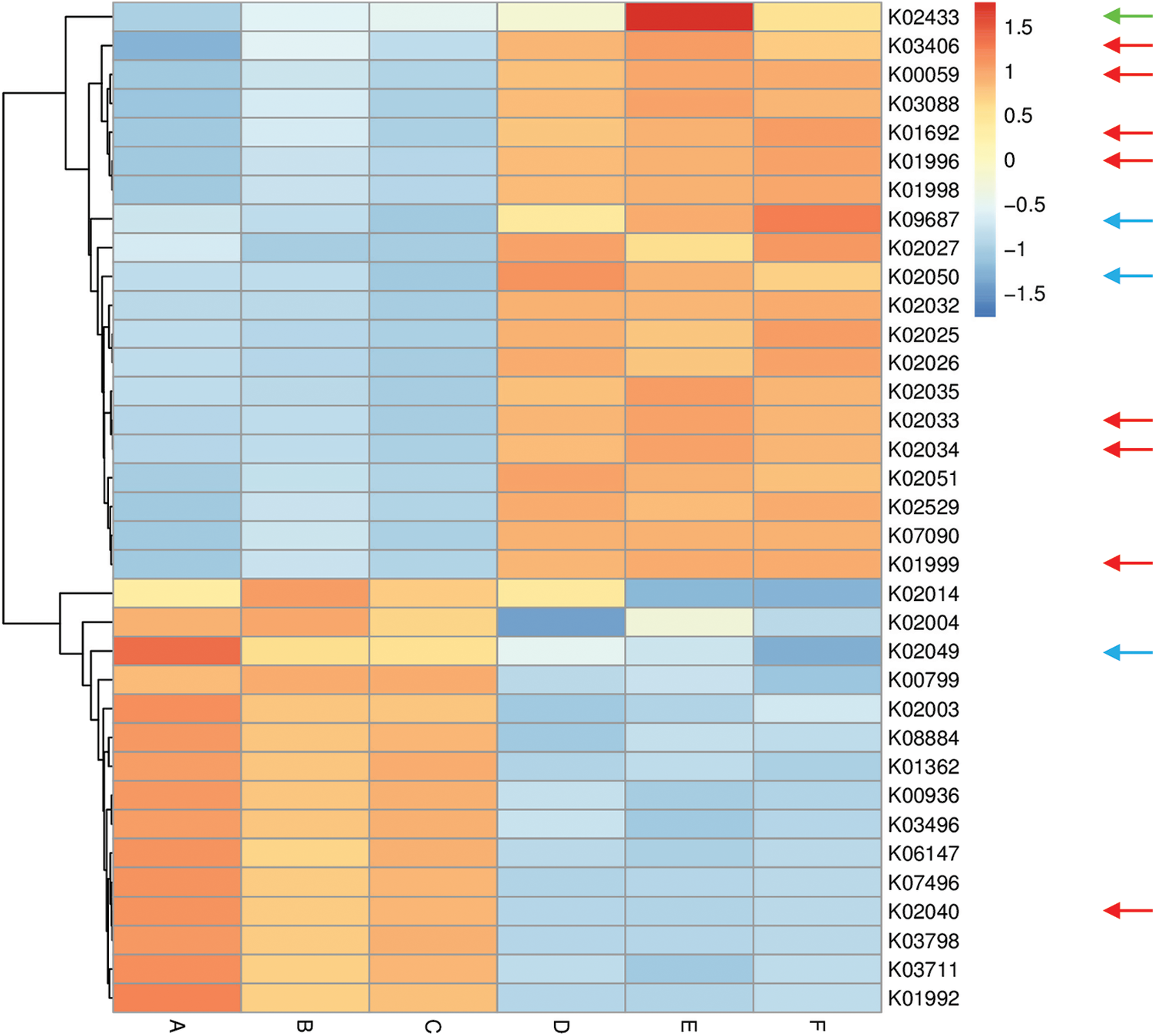
Figure 5: Heat map referring to the top 35 enriched compounds in KEGG pathways in terms of enrichment within grouping style ABCDEF microbiomes collected from surrounding bulk (subgrouping style ABC) and rhizosphere (subgrouping style DEF) soils of A. fruticosum after 0 (groups A & D, respectively), 24 (groups B & E, respectively) and 48 h (groups C & F, respectively) of watering. Numbers after the letter K refer to KO hierarchy of different enriched compounds across microbiomes of the two wild plants. Description of compounds shared in microbiomes of the two wild plants is shown in Table S1. Red arrows refer to compounds (8) that participate in one or more KEGG pathways in microbiomes of the two wild plants (see Table S2) with ≥1.5-fold enrichment in one soil type than the other (see Figs. S8 & S9). Blue arrows refer to compounds (3) with differential enrichment due to watering (see Table S3). Green arrow refers to a compound that participates in one or more KEGG pathways in microbiomes of the two wild plants with 1.5-fold enrichment due to watering (see Table S3 & Fig. 7)
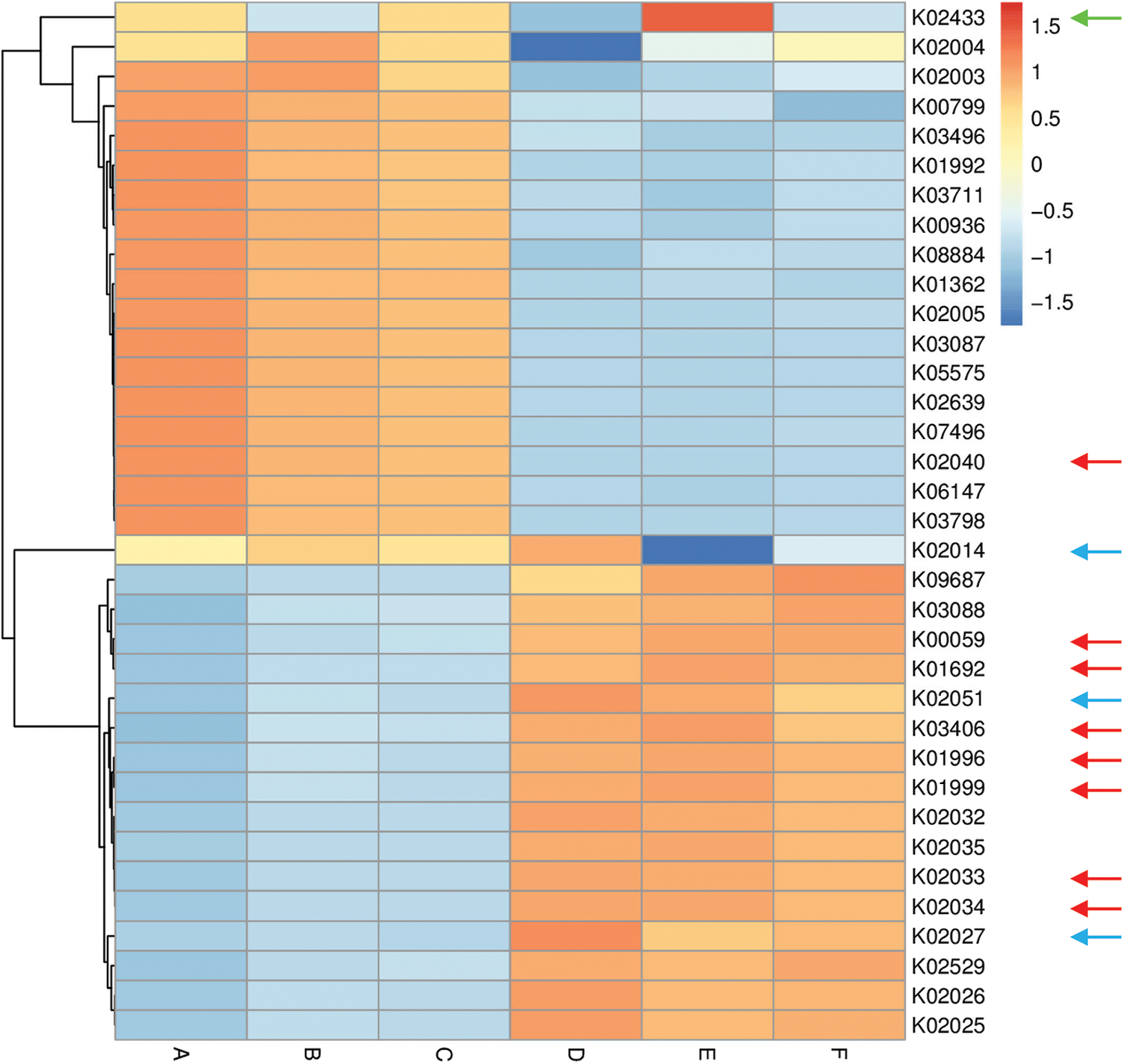
Figure 6: Heat map referring to the top 35 enriched compounds in KEGG pathways in terms of enrichment within grouping style ABCDEF microbiomes collected from surrounding bulk (subgrouping style ABC) and rhizosphere (subgrouping style DEF) soils of N. vermiculata after 0 (groups A & D, respectively), 24 (groups B & E, respectively) and 48 h (groups C & F, respectively) of watering. Numbers after the letter K refer to KO hierarchy of different enriched compounds across microbiomes of the two wild plants. Description of compounds shared in microbiomes of the two wild plants is shown in Table S1. Red arrows refer to compounds (8) participating in one or more KEGG pathways in microbiomes of the two wild plants (see Table S2) with ≥1.5-fold enrichment in one soil type than the other (see Figs. S8 & S9). Blue arrows refer to compounds (3) with differential enrichment due to watering (see Table S3). Green arrow refers to a compound that participates in one or more KEGG pathways in microbiomes of the two wild plants with 1.5-fold enrichment due to watering (see Table S3 & Fig. 7)
Among enriched compounds that showed ≥1.5-fold predicted enrichment in one soil type than the other, only eight participate in one or more KEGG pathways in microbiomes of the two wild plants (Table S2). The two KO terms K02033 and K02034 refer to one metabolic compound namely peptide/nickel transport system permease protein. Differential enrichment of these seven predicted compounds across the two soil microbiomes of A. fruticosum and N. vermiculata is described in Figs. S14 and S15, respectively. These seven compounds were analyzed further for their predicted differential enrichment in microbiomes of the two wild plants due to soil type. Six of them showed ≥1.5-fold predicted enrichment in rhizosphere soil microbiomes of the two wild plants, while only one showed higher predicted enrichment in the surrounding bulk soil microbiomes. The latter compound is phosphate transport system substrate-binding protein (K02040) (Table S2).
In terms of differentially enriched compounds in the soil microbiomes of the two wild plants due to watering, five predicted compounds were detected (Table S3). Only one of them (e.g., K02433), namely dual-specific aspartyl-tRNA(Asn)/glutamyl-tRNA (Gln) amidotransferase subunit A (EC: 6.3.5.6 & EC: 6.3.6.7), is shared in the microbiomes of the two wild plants with 1.5-fold predicted enrichment and participates in two KEGG pathways namely “Aminoacyl-tRNA biosynthesis” and “Metabolic pathways” (Table S2). Differential enrichment pattern of this compound for grouping style ABCDEF across the two wild plants is shown in Fig. 7. While another predicted compound, e.g., sulfonate/nitrate/taurine transport system ATP-binding protein, is shared in microbiomes of the two wild plants but has different KO terms (K02049 & K02050 for A. fruticosum, while K02051 for N. vermiculata) and participates in no KEGG pathway. Thus, the compound with K02433 term was analyzed further for its differentially predicted enrichment in microbiomes of the two wild plants due to watering. This compound was highly enriched in the rhizosphere microbiomes of the two wild plants 24 h after watering when compared with the predicted enrichment level in the surrounding bulk soil microbiomes of the two wild plants (Fig. 7).

Figure 7: Enrichment pattern of aspartyl-tRNA(Asn)/glutamyl-tRNA (Gln) amidotransferase subunit A across different groups of soil microbiomes collected from surrounding bulk (subgrouping style ABC) and rhizosphere (subgrouping style DEF) soils of the two wild plants A. fruticosum and N. vermiculata after 0 (groups A & D, respectively), 24 (groups B & E, respectively) and 48 h (groups C & F, respectively) of watering (see Table S3)
To validate functional prediction data at the enriched compounds level, actual response to watering of the gene encoding K02433 enzyme, e.g., gatA gene, of genus Bacillus in the two soil types of the two wild plants was determined in metatranscriptomes via qPCR (Fig. S16). Sequence of this gene was retrieved from the NCBI for Bacillus subtilis subsp. subtilis str. 168 (acc. No. AL009126.3). The results of qPCR aligned with those of differential enrichment of the dual-specific aspartyl-tRNA (Asn)/glutamyl-tRNA (Gln) amidotransferase subunit A (K02433) due to watering, as expression of gatA gene was higher in rhizosphere soil metatranscriptomes of the two wild plants after 24 h of watering as compared with that after 0 and 24 h of watering (Figure S16).
Among the top 10 enriched level-III KEGG pathways in soil microbiomes of the two wild plants (Figs. S4 & S5), “ABC transporters and Quorum sensing” pathways showed ≥1.5-fold higher enrichment in rhizosphere soil microbiomes than that of bulk soil microbiomes. Four compounds, out of the eight highly enriched compounds in one soil type, participate in pathway(s) “ABC transporters” and/or “Quorum sensing”. The two pathways were twined in the present study as they crosstalk and two compounds in the present study interconnect the two pathways. These two compounds are branched-chain amino acid transport system ATP-binding protein (K01996) and branched-chain amino acid transport system substrate-binding protein (K01999) (Table S2). These compounds were highly enriched in rhizosphere soil microbiomes of the two wild plants (Figs. S14 & S15). Peptide/nickel transport system permease protein (K02033 & K02034) was also highly enriched in rhizosphere soil microbiomes of the two wild plants (Figs. S14 & S15) but participates in “Quorum sensing” pathway, only (Table S2). The fourth compound, namely phosphate transport system substrate-binding protein (K02040), was highly enriched in bulk soil microbiomes of the two wild plants (Figs. S14 & S15) and participates in “ABC transporters” pathway, only (Table S2).
Three other compounds were also highly enriched in rhizosphere soil microbiomes of the two wild plants and participate in pathways other than “ABC transporters” or “Quorum sensing”. These enriched compounds are 3-oxoacyl-[acyl-carrier protein] reductase (K00059), enoyl-CoA hydratase (K01692) and methyl-accepting chemotaxis protein (K03406). These compounds participate in a large number of pathways (Table S2). In addition, aspartyl-tRNA (Asn)/glutamyl-tRNA (Gln) amidotransferase subunit A (K02433), was proven to be the only compound in rhizosphere soil microbiomes of the two wild plants that was highly enriched 24 h after watering (Fig. 7). This compound participates in two pathways, e.g., “Aminoacyl-tRNA biosynthesis” and “Metabolic pathways” (Table S2).
As indicated earlier, three compounds were highly enriched in rhizospheres soil microbiomes of the two wild plants and participate in “Quorum sensing” pathway. Quorum sensing (QS) is a system of natural stimuli and responses in relation to the density of bacterial population. QS system regulates bacterial gene expression especially determinants of pathogen virulence, microbial biofilm formation and antibiotic resistance [36]. Inhibition of QS requires screening of specific molecules of various chemical natures. Recent studies have proven the anti-QS properties of natural herbal medicinal substances [37]. Two, out of these three highly enriched compounds namely branched-chain amino acid transport system ATP-binding protein (K01996) and branched-chain amino acid transport system substrate-binding protein (K01999), are encoded by livF and livK genes, respectively. These two genes are stimulated by the release of plant’s GABA (γ aminobutyric acid) in order to regulate transamination of GABA to succinic semialdehyde (SSA) (Fig. 8). GABA is a non-protein amino acid found in many prokaryotic, ex., bacteria, and eukaryotic, ex., plant, organisms [38]. GABA participates in conferring biotic stress in plants as it acts in promoting degradation of the signal N-(3-oxooctanoyl) homoserine lactone (OC8-HSL) in Agrobacterium tumefaciens, a quorum-sensing signal (or ‘‘quormone’’), via the action of OC8-HSL lactonase (or ATTM). This action limits the progression of QS-dependent infection in plant, hence, reduces virulence of this and possibly other soil pathogens [36,39]. Thus, high enrichment of the compounds K01996 and K01999 is required in the soil rhizosphere to modulate QS and protect the two wild plants from pathogenic bacteria via the transamination of GABA to SSA and downstream reactions (Fig. 8). After transamination, SSA is oxidized to succinate to feed into the TCA cycle, the main source of energy and important part of aerobic respiration [40]. Interestingly, genus Agrobacterium does not exist in either soil type of A. fruticosum or N. vermiculata. Thus, induction of these two compounds is expected to affect existence of other pathogenic bacteria in the rhizosphere soil of the two wild plants.
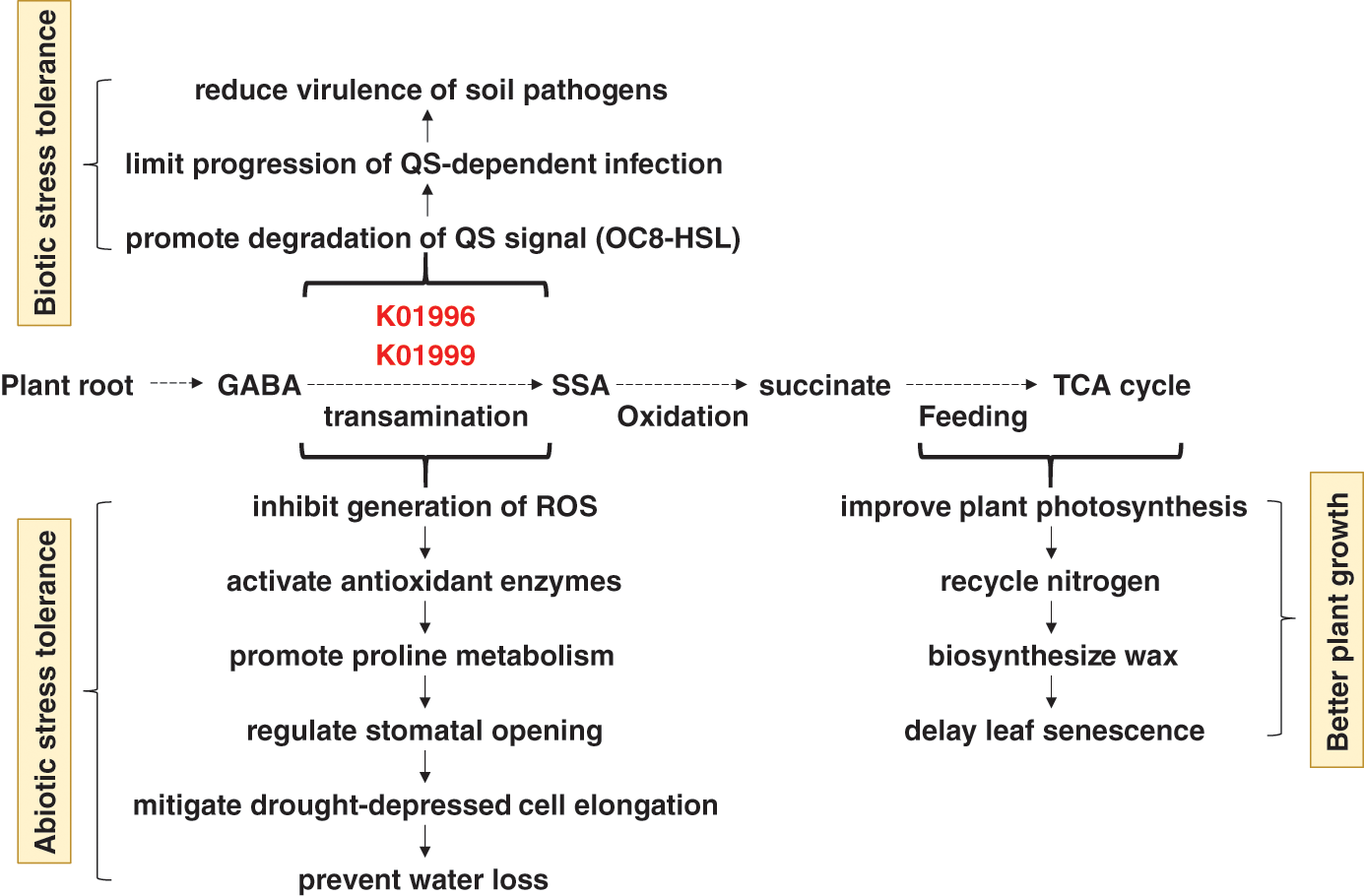
Figure 8: Consequences of high enrichment of the bacterial K01996 and K01999 enzymes in rhizosphere soil on the ability of the wild plants A. fruticosum and N. vermiculata to tolerate biotic and abiotic stresses and on overall plant growth performance. K01996 = branched-chain amino acid transport system ATP-binding protein, K01999 = branched-chain amino acid transport system substrate-binding protein, QS = quorum sensing, OC8-HSL = N-(3-oxooctanoyl) homoserine lactone, GABA = γ aminobutyric acid, SSA = succinic semialdehyde, TCA = tricarboxylic acid, ROS = reactive oxygen species
In terms of abiotic stress tolerance in plants, GABA was also proved to accumulate during plant response to environmental stresses [41]. High concentration of GABA was proven to elevate plant stress tolerance by inhibiting the generation of reactive oxygen species (ROS) in the plant cells, activating antioxidant enzymes, promoting proline metabolism, regulating stomatal opening, mitigating drought-depressed cell elongation, thus, preventing water loss [40,42]. GABA also improves the plant’s ability to make photosynthesis, nitrogen recycling, wax biosynthesis, and delay of leaf senescence [43]. We speculate that high enrichment of the compounds K01996 and K01999 is induced by GABA production in root cells of either wild plant as a model of symbiotic host-bacterial interaction (Fig. 8).
The two compounds K01996 and K01999 also participate in “ABC transporters” pathway that is affiliated to the bacterial membrane transport system through coupling ATP hydrolysis to the uptake and efflux of solutes across the cell membrane [44]. ABC transporters are known to be powered by the energy from ATP to transport substrates across bacterial cellular membranes [45]. This action helps bacteria to scavenge essential minerals, like phosphate, and compounds, like branched-chain amino acids, for their survival within the plant host [45]. We speculate that the two compounds act in transporting branched-chain amino acids to bacteria that respond to GABA release in the soil rhizosphere of plant roots. If these bacteria are commensal, then it is likely that branched-chain amino acids will also be available for plant root cells. On the other hand, phosphate transport system substrate-binding protein (K02040) encoded by pstS gene was less enriched in rhizosphere soil of the two wild plants (Figs. S14 & S15). Accordingly, the chance of phosphate to be transported to bacterial rhizospheres of the two wild plants is low. Therefore, there should be other alternative systems required for the bacterial and plant cells to fulfil proper transfer of phosphate across their membranes.
Peptide/nickel transport system permease protein (K02033 & K02034) encoded by dppB and/or dppC genes, respectively, was also highly enriched in rhizosphere soil microbiomes of the two wild plants (Figs. S14 & S15) and participates in “Quorum sensing” pathway (Table S2). Bacterial dipeptide permeases (Dpp) comprise an operon of a membrane-associated complex of five proteins namely DppA-E that are involved in the uptake of essential amino acids [46]. In particular, DppB and DppC are transmembrane proteins that are involved in the transport of substrates across the cytoplasmic membrane, an action similar to that of K01996 and K01999 compounds in transporting branched-chain amino acids. Nevertheless, expression of the virulence factor SpeB (major cysteine protease) is expected to be regulated by Dpp family, hence, enrichment of di-(Dpp) and/or oligo-(Opp) peptides in bacterial cell due to enrichment of K02033 and/or K02034 (https://www.genome.jp/pathway/map02024) is required for speB regulation [46]. Thus, K02033 and K02034 compounds seem to promote pathogenic infection of the plant roots. In return, there should be a defense mechanism(s) in plant cells to reduce virulence of rhizosphere pathogens. There are beneficial features of Dpp family for commensal microbes in plant root rhizosphere, like the participation in haem synthesis and chemotaxis in E. coli [47,48] and the induction of sporulation in B. subtilis, a commensal microbe [49,50].
The 3-Oxoacyl-[ACP] reductase (K00059) or OAR was highly enriched in rhizosphere of the two wild plants (Figs. S14 & S15). The enzyme is also called FabG and is encoded by fabG gene. OAR functions in fatty acid elongation cycles, in the biosynthesis of unsaturated fatty acids and in the biotin biosynthesis pathway [51,52]. This enzyme is a member of the short-chain alcohol dehydrogenase/reductase (SDR) superfamily, which catalyzes a wide range of oxidation/reduction reactions using NADH/NADPH as the cofactor [53]. OAR has homotetramer structure and each monomer is complexed with on molecule of NADPH and eight Ca2+ ions. These bindings result in a conformational change of the enzyme that helps in initiating allosteric structure, important for its function [52]. Contribution of SDR enzymes and their orthologs to plant-microbe symbiotic relationships as well as in saprotrophy of the microbe was proven [54]. The results of the latter report demonstrated the interaction of the gram-negative Sinorhizobium meliloti of phylum α-Proteobacteria with its plant hosts (ex., alfalfa) via the role of SDR. The bacteria forms root nodules in plant rhizosphere when atmospheric nitrogen (N2) is converted to ammonia (nitrogen fixation), a form that is beneficial to the plant. This process is important for conservation of ecosystems. When fabG gene was knocked out, symbiotic relationship with the plant is severely reduced [54]. Results of symbiotic relationships involving several plant species as well as SDR sequence identity in other species of genus Sinorhizobium indicated the wide host range of these symbionts [54]. Other earlier reports indicated that several other taxa of the Rhiziobiales order, including Bradyrhizobium japonicum, have intimate interactions with eukaryal host cells as symbionts [55]. In the present study, taxa of this order that exist in rhizosphere of one or of the two wild plants include Neorhizobium huautlense, Rhizobium petrolearium, and unidentified species of genera Bradyrhizobium sp. and Mesorhizobium sp. (data provided upon request). We speculate that these taxa harbor orthologs of SDR-encoding gene, e.g., fabG, and make symbiotic relationships with the two wild plants.
The enoyl-CoA hydratase (K01692) was also highly enriched in rhizosphere of the two wild plants. AcuH is a well-known enzyme of the enoyl-CoA hydratase family that was first identified in γ-Proteobacteria with the ability to grow on acrylate as a sole carbon source [56]. The enzyme is encoded by paaF gene and seems to, nonetheless, have a reverse action to that of 3-Oxoacyl-[ACP] reductase (K00059) as it participates in “Fatty acid degradation” pathway [57], particularly in the second step of the β-oxidation [58]. The enoyl-CoA hydratase (sometime called crotonase) also converts 4-coumaroyl-CoA, caffeoyl-CoA and feruloyl-CoA to the corresponding hydroxybenzaldehydes that results in subverting the plant “Phenylpropanoid” pathway and channel carbon flux [58]. Expression of paaF gene in Pseudomonas fluorescens caused phenotypic abnormalities in tobacco plants including plant stunting, interveinal chlorosis/senescence and male sterility [59]. Other abnormalities include the development of distorted cells in the xylem and phloem bundles, and reduction of lignification. We speculate that the differential enrichment of the two enzymes enoyl-CoA hydratase (K01692) and enoyl-CoA hydratase (K01692) attenuate the level of fatty acids in rhizosphere of the two wild plants.
The methyl-accepting chemotaxis protein or MCP (K03406) was also highly enriched in rhizosphere of the two wild plants (Figs. S14 & S15). The MCPs are environmental signals that sense “Bacterial chemotaxis” pathway [60]. In turn, chemotactic signal transduction span the chemoreceptor cluster to the flagellar motors through “Two-component system” pathway [61]. The two components involve CheA, the histidine kinase that binds to the cytoplasmic domain of the MCPs, and CheY, the response regulator that interacts with the flagellar motor complex to control its rotation [62]. Chemotaxis helps bacteria to rapidly respond to their immediate environmental niche and move toward favorable ones. Influence of bacterial motility and chemotaxis was also recently documented in establishing symbiosis with plant roots for taxa of the Rhizobiaceae family [63]. As indicated earlier, several taxa of this family are abundant in the rhizosphere of the two wild plants. It was recently proposed that active bacterial migration through motility and chemotaxis facilitates recruitment/colonization of microbial partners (symbionts) in the vicinity of plant rhizosphere [64].
The dual-specific aspartyl-tRNA (Asn)/glutamyl-tRNA (Gln) amidotransferase subunit A (K02433) was highly enriched in rhizosphere soil 24 h after watering of the two wild plants, while showed less enrichment at the same time point for the wild plant N. vermiculata (Fig. 7). This enzyme participates in “Aminoacyl-tRNA biosynthesis” pathway. This pathway crosstalk with “Alanine, aspartate and glutamate metabolism” pathway. The enzyme is encoded by gatA gene that acts in synthesizing both aspartyl-tRNA (Asn) and glutaminyl-tRNA (Gln) amidotransferase subunit A (485 aa). The dual-specific enzyme allows the formation of correctly charged Gln-tRNA (Gln) and/or Asn-tRNA (Asn) through the transamidation of misacylated Glu-tRNA (Gln) and/or Asn-tRNA (Asn) in organisms which lack glutaminyl-tRNA and/or asparaginyl-tRNA synthetases. Such aminoacyl-tRNA synthetases are well-documented for their role in translation of the genetic code as they are responsible for attaching the correct amino acid to its cognate tRNA species, while hydrolyzing incorrectly attached amino acid; the latter action is called editing. The in vitro and in vivo studies in E. coli indicated that ROS arisen from oxidative stress can result in increased level of misfolded proteins and reduced fidelity of translation. Examples of stress responses include impairment of editing activity of threonyl-tRNA synthetase leading to protein mistranslation and impairment of bacterial growth [65]. The gene is part of gatCAB operon-like structure that exists in many microorganisms including genera Bacillus and Acidithiobacillus [66,67]. The latter taxon is known for its action in bioleaching of soil minerals, an important process for the plant roots in close vicinity [68]. As aspartyl-tRNA (Asn)/glutamyl-tRNA (Gln) amidotransferase subunit A was highly enriched 24 h after watering, this might indicate that this favorable condition allowed the bacteria to avoid protein mistranslation, albeit, justification of the involvement of this enzyme in plant-microbe interaction remains to be deciphered.
In conclusion, the present study scopes the light on the predicted differential enrichment of pathways and compounds in rhizosphere soil microbiomes across the two wild plants Abutilon fruticosum and Nitrosalsola vermiculata as compared with those in microbiomes of the surrounding bulk soil. Differential enrichment involved a number of microbial compounds participating in symbiotic relationship with both wild plants. Excretion of plant root exudates and consequent enrichment of microbial compounds mediate such a relationship. Influence of watering assured the high enrichment of dual-specific aspartyl-tRNA (Asn)/glutamyl-tRNA (Gln) amidotransferase subunit A in rhizosphere soil after 24 h of watering. A question still remains to be answered is whether plant root exudates initiates the relationship with the microbes in its rhizosphere through promoting enrichment of specific microbial compounds or vice versa. The study of these two wealthy genetic resources contributes to our understanding of the host-microbe interactions and the possible improvement of plant breeding programs in Saudi Arabia.
Authorship: The authors confirm contribution to the paper as follows: study conception and design: L.B., A.A.A., M.A.T., H.M.B., M.Y.R., A.A.B., A.S., H.W.A., R.A.A., R.S.J.; data collection: L.B., M.A.T., M.Y.R., A.S., R.A.A.; analysis and interpretation of results: A.A.A., H.M.B., A.A.B., H.W.A., R.S.J.; draft manuscript preparation: L.B., A.A.A., M.A.T., H.M.B., M.Y.R., A.A.B., A.S., H.W.A., R.A.A., R.S.J. All authors reviewed the results and approved the final version of the manuscript.
Data Availability: Supplemental data can be accessed at: https://drive.google.com/drive/folders/1aCQOt3zbgjz9mF0P6DatZsYNgP1GNEu-?usp=sharing.
Acknowledgement: This work was supported by Princess Nourah bint Abdulrahman University Researchers Supporting Project No. (PNURSP2022R31), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia.
Funding Statement: This work was supported by Princess Nourah bint Abdulrahman University Researchers Supporting Project No. (PNURSP2022R31), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia.
Conflicts of Interest: The authors declare no conflicts of interest regarding the publication of the present manuscript.
1. Al-Eisawi, D. M., Al-Ruzayza, S. (2015). The flora of holy Mecca district, Saudi Arabia. International Journal of Biodiversity and Conservation, 7(3), 173–189. DOI 10.5897/IJBC2014.0773. [Google Scholar] [CrossRef]
2. Husain, S., Baquar, S. (1974). Biosystematic studies in genus Abutilon from Pakistan. 1. Taxonomy. Proceedings of the Phyton-Annales Rei Botanicae, 15(3–4), 219. Austria: Ferdinand Burger Soehne. [Google Scholar]
3. Patel, M. K., Rajput, A. P. (2013). Therapeutic significance of Abutilon indicum: An overview. American Journal of PharmTech Research, 3(2), 20–35. DOI 10.11648/j.ajbio.s.2015030201.12. [Google Scholar] [CrossRef]
4. Al-Tabini, R., Al-Khalidi, K., Al-Shudiefat, M. (2012). Livestock, medicinal plants and rangeland viability in Jordan’s Badia: Through the lens of traditional and local knowledge. Pastoralism: Research, Policy & Practice, 2(4), 1–16. [Google Scholar]
5. Nadal-Sala, D., Grote, R., Birami, B., Knuver, T., Rehschuh, R. et al. (2021). Leaf shedding and non-stomatal limitations of photosynthesis mitigate hydraulic conductance losses in scots pine saplings during severe drought stress. Frontiers in Plant Science, 12, 715127. DOI 10.3389/fpls.2021.715127. [Google Scholar] [CrossRef]
6. Wei, Z., Hu, X., Li, X., Zhang, Y., Jiang, L. et al. (2017). The rhizospheric microbial community structure and diversity of deciduous and evergreen forests in Taihu Lake area, China. PLoS One, 12(4), e0174411. DOI 10.1371/journal.pone.0174411. [Google Scholar] [CrossRef]
7. Afonso, A. C., Gomes, I. B., Saavedra, M. J., Giaouris, E., Simoes, L. C. et al. (2021). Bacterial coaggregation in aquatic systems. Water Research, 196, 117037. DOI 10.1016/j.watres.2021.117037. [Google Scholar] [CrossRef]
8. Lazar, V., Holban, A. M., Curutiu, C., Chifiriuc, M. C. (2021). Modulation of quorum sensing and biofilms in less investigated gram-negative ESKAPE pathogens. Frontiers in Microbiology, 12, 676510. DOI 10.3389/fmicb.2021.676510. [Google Scholar] [CrossRef]
9. Berg, G., Grube, M., Schloter, M., Smalla, K. (2014). Unraveling the plant microbiome: Looking back and future perspectives. Frontiers in Microbiology, 5(165), 148. DOI 10.3389/fmicb.2014.00148. [Google Scholar] [CrossRef]
10. Batool, T., Ali, S., Seleiman, M. F., Naveed, N. H., Ali, A. et al. (2020). Plant growth promoting rhizobacteria alleviates drought stress in potato in response to suppressive oxidative stress and antioxidant enzymes activities. Scientific Reports, 10(1), 16975. DOI 10.1038/s41598-020-73489-z. [Google Scholar] [CrossRef]
11. Glick, B. R. (2012). Plant growth-promoting bacteria: Mechanisms and applications. Scientifica, 2012(5), 963401. DOI 10.6064/2012/963401. [Google Scholar] [CrossRef]
12. Lucy, M., Reed, E., Glick, B. R. (2004). Applications of free living plant growth-promoting rhizobacteria. Antonie van Leeuwenhoek, 86(1), 1–25. DOI 10.1023/B:ANTO.0000024903.10757.6e. [Google Scholar] [CrossRef]
13. Naylor, D., deGraaf, S., Purdom, E., Coleman-Derr, D. (2017). Drought and host selection influence bacterial community dynamics in the grass root microbiome. The ISME Journal, 11(12), 2691–2704. DOI 10.1038/ismej.2017.118. [Google Scholar] [CrossRef]
14. Xu, L., Naylor, D., Dong, Z., Simmons, T., Pierroz, G. et al. (2018). Drought delays development of the sorghum root microbiome and enriches for monoderm bacteria. Proceedings of the National Academy of Sciences, 115(18), E4284–E4293. DOI 10.1073/pnas.1717308115. [Google Scholar] [CrossRef]
15. Timm, C. M., Carter, K. R., Carrell, A. A., Jun, S. R., Jawdy, S. S. et al. (2018). Abiotic stresses shift belowground populus-associated bacteria toward a core stress microbiome. mSystems, 3(1), e00070–17. DOI 10.1128/mSystems.00070-17. [Google Scholar] [CrossRef]
16. Naylor, D., Coleman-Derr, D. (2017). Drought stress and root-associated bacterial communities. Frontiers in Plant Science, 8, 2223. DOI 10.3389/fpls.2017.02223. [Google Scholar] [CrossRef]
17. Potts, M. (1994). Desiccation tolerance of prokaryotes. Microbiological Reviews, 58(4), 755–805. DOI 10.1128/mr.58.4.755-805.1994. [Google Scholar] [CrossRef]
18. Chodak, M., Gołębiewski, M., Morawska-Płoskonka, J., Kuduk, K., Niklińska, M. (2015). Soil chemical properties affect the reaction of forest soil bacteria to drought and rewetting stress. Annals of Microbiology, 65(3), 1627–1637. DOI 10.1007/s13213-014-1002-0. [Google Scholar] [CrossRef]
19. Langille, M. G., Zaneveld, J., Caporaso, J. G., McDonald, D., Knights, D. et al. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nature Biotechnology, 31(9), 814–821. DOI 10.1038/nbt.2676. [Google Scholar] [CrossRef]
20. Aßhauer, K. P., Wemheuer, B., Daniel, R., Meinicke, P. (2015). Tax4Fun: Predicting functional profiles from metagenomic 16S rRNA data. Bioinformatics, 31(17), 2882–2884. DOI 10.1093/bioinformatics/btv287. [Google Scholar] [CrossRef]
21. Louca, S., Parfrey, L. W., Doebeli, M. (2016). Decoupling function and taxonomy in the global ocean microbiome. Science, 353(6305), 1272–1277. DOI 10.1126/science.aaf4507. [Google Scholar] [CrossRef]
22. Douglas, G. M., Maffei, V. J., Zaneveld, J., Yurgel, S. N., Brown, J. R. et al. (2020). PICRUSt2: An improved and customizable approach for metagenome inference. bioRxiv, 672295, 497. DOI 10.1101/672295. [Google Scholar] [CrossRef]
23. Ling, N., Zhu, C., Xue, C., Chen, H., Duan, Y. et al. (2016). Insight into how organic amendments can shape the soil microbiome in long-term field experiments as revealed by network analysis. Soil Biology & Biochemistry, 99, 137–149. DOI 10.1016/j.soilbio.2016.05.005. [Google Scholar] [CrossRef]
24. Geng, L. L., Shao, G. X., Raymond, B., Wang, M. L., Sun, X. X. et al. (2018). Subterranean infestation by Holotrichia parallela larvae is associated with changes in the peanut (Arachis hypogaea L.) rhizosphere microbiome. Microbiological Research, 211, 13–20. DOI 10.1016/j.micres.2018.02.008. [Google Scholar] [CrossRef]
25. Dai, L., Zhang, G., Yu, Z., Ding, H., Xu, Y. et al. (2019). Effect of drought stress and developmental stages on microbial community structure and diversity in peanut rhizosphere soil. International Journal of Molecular Sciences, 20(9), 2265. DOI 10.3390/ijms20092265. [Google Scholar] [CrossRef]
26. Hurt, R. A., Qiu, X., Wu, L., Roh, Y., Palumbo, A. V. et al. (2001). Simultaneous recovery of RNA and DNA from soils and sediments. Applied Environmental Microbiology, 67(10), 4495–4503. DOI 10.1128/AEM.67.10.4495-4503.2001. [Google Scholar] [CrossRef]
27. McLean, E. (1983). Soil pH and lime requirement. Methods of soil analysis: Part 2. Chemical and Microbiological Properties, 9, 199–224. DOI 10.2134/agronmonogr9.2.2ed.c12. [Google Scholar] [CrossRef]
28. Magoč, T., Salzberg, S. L. (2011). FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics, 27(21), 2957–2963. DOI 10.1093/bioinformatics/btr507. [Google Scholar] [CrossRef]
29. Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D. et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nature Methods, 7(5), 335–336. DOI 10.1038/nmeth.f.303. [Google Scholar] [CrossRef]
30. Bokulich, N. A., Subramanian, S., Faith, J. J., Gevers, D., Gordon, J. I. et al. (2013). Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nature Methods, 10(1), 57–59. DOI 10.1038/nmeth.2276. [Google Scholar] [CrossRef]
31. Edgar, R. C. (2004). MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research, 32(5), 1792–1797. DOI 10.1093/nar/gkh340. [Google Scholar] [CrossRef]
32. Edgar, R. C. (2013). UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nature Methods, 10, 996–998. DOI 10.1038/nmeth.2604. [Google Scholar] [CrossRef]
33. Altschul, S. F., Gish, W., Miller, W., Myers, E. W., Lipman, D. J. (1990). Basic local alignment search tool. Journal of Molecular Biology, 215(3), 403–410. DOI 10.1016/S0022-2836(05)80360-2. [Google Scholar] [CrossRef]
34. Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T. et al. (2012). The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Research, 41, D590–D596. DOI 10.1093/nar/gks1219. [Google Scholar] [CrossRef]
35. Bahieldin, A., Atef, A., Sabir, J. S., Gadalla, N. O., Edris, S. et al. (2015). RNA-Seq analysis of the wild barley (H. spontaneum) leaf transcriptome under salt stress. Comptes Rendus Biologies, 338(5), 285–297. DOI 10.1016/j.crvi.2015.03.010. [Google Scholar] [CrossRef]
36. Chevrot, R., Rosen, R., Haudecoeur, E., Cirou, A., Shelp, B. J. et al. (2006). GABA controls the level of quorum-sensing signal in Agrobacterium tumefaciens. Proceedings of the National Academy of Sciences of the United States of America, 103(19), 7460–7464. DOI 10.1073/pnas.0600313103. [Google Scholar] [CrossRef]
37. Bouyahya, A., Dakka, N., Et-Touys, A., Abrini, J., Bakri, Y. (2017). Medicinal plant products targeting quorum sensing for combating bacterial infections. Asian Pacific Journal of Tropical Medicine, 10(8), 729–743. DOI 10.1016/j.apjtm.2017.07.021. [Google Scholar] [CrossRef]
38. Bouché, N., Lacombe, B., Fromm, H. (2003). GABA signaling: A conserved and ubiquitous mechanism. Trends in Cell Biology, 13(12), 607–610. DOI 10.1016/j.tcb.2003.10.001. [Google Scholar] [CrossRef]
39. Musk, D. J.Jr, Hergenrother, P. J. (2006). Chemical countermeasures for the control of bacterial biofilms: Effective compounds and promising targets. Current Medicinal Chemistry, 13(18), 2163–2177. DOI 10.2174/092986706777935212. [Google Scholar] [CrossRef]
40. Li, L., Dou, N., Zhang, H., Wu, C. (2021). The versatile GABA in plants. Plant Signaling & Behavior, 16(3), 1862565. DOI 10.1080/15592324.2020.1862565. [Google Scholar] [CrossRef]
41. Bor, M., Seckin, B., Ozgur, R., Yılmaz, O., Ozdemir, F. et al. (2009). Comparative effects of drought, salt, heavy metal and heat stresses on gamma-aminobutryric acid levels of sesame (Sesamum indicum L.). Acta Physiologiae Plantarum, 31(3), 655–659. DOI 10.1007/s11738-008-0255-2. [Google Scholar] [CrossRef]
42. Yong, B., Xie, H., Li, Z., Li, Y. P., Zhang, Y. et al. (2017). Exogenous application of GABA improves PEG-induced drought tolerance positively associated with GABA-shunt, polyamines, and proline metabolism in white clover. Frontiers in Physiology, 8, 1107. DOI 10.3389/fphys.2017.01107. [Google Scholar] [CrossRef]
43. Li, Z., Huang, T., Tang, M., Cheng, B., Peng, Y. et al. (2019). iTRAQ-based proteomics reveals key role of gamma-aminobutyric acid (GABA) in regulating drought tolerance in perennial creeping bentgrass (Agrostis stolonifera). Plant Physiology & Biochemistry, 145, 216–226. DOI 10.1016/j.plaphy.2019.10.018. [Google Scholar] [CrossRef]
44. Zhou, X., Wang, J. T., Zhang, Z. F., Li, W., Chen, W. et al. (2020). Microbiota in the rhizosphere and seed of rice from China, with reference to their transmission and biogeography. Frontiers in Microbiology, 11, 995. DOI 10.3389/fmicb.2020.00995. [Google Scholar] [CrossRef]
45. Tanaka, K. J., Song, S., Mason, K., Pinkett, H. W. (2018). Selective substrate uptake: The role of ATP-binding cassette (ABC) importers in pathogenesis. Biochimica et Biophysica Acta (BBA)-Biomembranes, 1860(4), 868–877. DOI 10.1016/j.bbamem.2017.08.011. [Google Scholar] [CrossRef]
46. Podbielski, A., Leonard, B. A. (1998). The group A streptococcal dipeptide permease (Dpp) is involved in the uptake of essential amino acids and affects the expression of cysteine protease. Molecular Microbiology, 28(6), 1323–1334. DOI 10.1046/j.1365-2958.1998.00898.x. [Google Scholar] [CrossRef]
47. Verkamp, E., Backman, V. M., Bjornsson, J. M., Soll, D., Eggertsson, G. (1993). The periplasmic dipeptide permease system transports 5-aminolevulinic acid in Escherichia coli. Journal of Bacteriology, 175(5), 1452–1456. DOI 10.1128/jb.175.5.1452-1456.1993. [Google Scholar] [CrossRef]
48. Manson, M. D., Blank, V., Brade, G., Higgins, C. F. (1986). Peptide chemotaxis in E. coli involves the Tap signal transducer and the dipeptide permease. Nature, 321(6067), 253–256. DOI 10.1038/321253a0. [Google Scholar] [CrossRef]
49. Mathiopoulos, C., Mueller, J. P., Slack, F. J., Murphy, C. G., Patankar, S. et al. (1991). A Bacillus subtilis dipeptide transport system expressed early during sporulation. Molecular Microbiology, 5(8), 1903–1913. DOI 10.1111/j.1365-2958.1991.tb00814.x. [Google Scholar] [CrossRef]
50. Slack, F., Mueller, J., Sonenshein, A. (1993). Mutations that relieve nutritional repression of the Bacillus subtilis dipeptide permease operon. Journal of Bacteriology, 175(15), 4605–4614. DOI 10.1128/jb.175.15.4605-4614.1993. [Google Scholar] [CrossRef]
51. Lai, C. Y., Cronan, J. E. (2004). Isolation and characterization of beta-ketoacyl-acyl carrier protein reductase (fabG) mutants of Escherichia coli and Salmonella enterica serovar Typhimurium. Journal of Bacteriology, 186(6), 1869–1878. DOI 10.1128/JB.186.6.1869-1878.2004. [Google Scholar] [CrossRef]
52. Guo, Q. Q., Zhang, W. B., Zhang, C., Song, Y. L., Liao, Y. L. et al. (2019). Characterization of 3-oxacyl-acyl carrier protein reductase homolog genes in Pseudomonas aeruginosa PAO1. Frontiers in Microbiology, 10, 1028. DOI 10.3389/fmicb.2019.01028. [Google Scholar] [CrossRef]
53. Oppermann, U., Filling, C., Hult, M., Shafqat, N., Wu, X. et al. (2003). Short-chain dehydrogenases/reductases (SDRThe 2002 update. Chemico-Biological Interactions, 143–144, 247–253. DOI 10.1016/s0009-2797(02)00164-3. [Google Scholar] [CrossRef]
54. Jacob, A. I., Adham, S. A., Capstick, D. S., Clark, S. R., Spence, T. et al. (2008). Mutational analysis of the Sinorhizobium meliloti short-chain dehydrogenase/reductase family reveals substantial contribution to symbiosis and catabolic diversity. Molecular Plant-Microbe Interactions, 21(7), 979–987. DOI 10.1094/MPMI-21-7-0979. [Google Scholar] [CrossRef]
55. Williams, K. P., Sobral, B. W., Dickerman, A. W. (2007). A robust species tree for the alphaproteobacteria. Journal of Bacteriology, 189(13), 4578–4586. DOI 10.1128/JB.00269-07. [Google Scholar] [CrossRef]
56. Todd, J. D., Curson, A. R., NikolaidouKatsaraidou, N., Brearley, C. A., Watmough, N. J. et al. (2010). Molecular dissection of bacterial acrylate catabolism-unexpected links with dimethylsulfoniopropionate catabolism and dimethyl sulfide production. Environmental Microbiology, 12(2), 327–343. DOI 10.1111/j.1462-2920.2009.02071.x. [Google Scholar] [CrossRef]
57. Cao, H. Y., Wang, P., Xu, F., Li, P. Y., Xie, B. B. et al. (2017). Molecular insight into the acryloyl-CoA hydration by AcuH for acrylate detoxification in dimethylsulfoniopropionate-catabolizing bacteria. Frontiers in Microbiology, 8, 225. DOI 10.3389/fmicb.2017.02034. [Google Scholar] [CrossRef]
58. Nelson, D. L., Lehninger, A. L., Cox, M. M. (2008). Lehninger principles of biochemistry. London: Macmillan. [Google Scholar]
59. Mayer, M. J., Narbad, A., Parr, A. J., Parker, M. L., Walton, N. J. et al. (2001). Rerouting the plant phenylpropanoid pathway by expression of a novel bacterial enoyl-CoA hydratase/lyase enzyme function. Plant Cell, 13(7), 1669–1682. DOI 10.1105/TPC.010063. [Google Scholar] [CrossRef]
60. Parkinson, J. S. (2003). Bacterial chemotaxis: A new player in response regulator dephosphorylation. Journal of Bacteriology, 185(5), 1492–1494. DOI 10.1128/JB.185.5.1492-1494.2003. [Google Scholar] [CrossRef]
61. Wadhams, G. H., Armitage, J. P. (2004). Making sense of it all: Bacterial chemotaxis. Nature Reviews Molecular Cell Biology, 5(12), 1024–1037. DOI 10.1038/nrm1524. [Google Scholar] [CrossRef]
62. Porter, S. L., Wadhams, G. H., Armitage, J. P. (2011). Signal processing in complex chemotaxis pathways. Nature Reviews Microbiology, 9(3), 153–165. DOI 10.1038/nrmicro2505. [Google Scholar] [CrossRef]
63. Zatakia, H. M., Arapov, T. D., Meier, V. M., Scharf, B. E. (2018). Cellular stoichiometry of methyl-accepting chemotaxis proteins in Sinorhizobium meliloti. Journal of Bacteriology, 200(6), e00614–00617. DOI 10.1128/JB.00614-17. [Google Scholar] [CrossRef]
64. Raina, J. B., Fernandez, V., Lambert, B., Stocker, R., Seymour, J. R. (2019). The role of microbial motility and chemotaxis in symbiosis. Nature Reviews Microbiology, 17(5), 284–294. DOI 10.1038/s41579-019-0182-9. [Google Scholar] [CrossRef]
65. Ling, J., Söll, D. (2010). Severe oxidative stress induces protein mistranslation through impairment of an aminoacyl-tRNA synthetase editing site. Proceedings of the National Academy of Sciences of the United States of America, 107(9), 4028–4033. DOI 10.1073/pnas.1000315107. [Google Scholar] [CrossRef]
66. Curnow, A. W., Hong, K., Yuan, R., Kim, S., Martins, O. et al. (1997). Glu-tRNAGln amidotransferase: A novel heterotrimeric enzyme required for correct decoding of glutamine codons during translation. Proceedings of the National Academy of Sciences of the United States of America, 94(22), 11819–11826. DOI 10.1073/pnas.94.22.11819. [Google Scholar] [CrossRef]
67. Ibba, M., Becker, H., Stathopoulos, C., Tumbula, D., Söll, D. (2000). The adaptor hypothesis revisited. Trends in Biochemical Sciences, 25(7), 311–316. DOI 10.1016/S0968-0004(00)01600-5. [Google Scholar] [CrossRef]
68. Salazar, J. C., Zúñiga, R., Raczniak, G., Becker, H., Söll, D. et al. (2001). A dual-specific Glu-tRNAGln and Asp-tRNAAsn amidotransferase is involved in decoding glutamine and asparagine codons in Acidithiobacillus ferrooxidans. FEBS Letters, 500(3), 129–131. DOI 10.1016/s0014-5793(01)02600-x. [Google Scholar] [CrossRef]
Appendix
Figure S1. Relative abundance of the top 10 phyla of microbiomes of the two the wild plant species A. fruticosum and N. vermiculata.
Figure S2. Predicted KEGG pathways at levels I and II of microbiomes of A. fruticosum.
Figure S3. Predicted KEGG pathways at levels I and II of microbiomes of N. vermiculata.
Figure S4. Abundance of the top 10 predicted level-III KEGG pathways of microbiomes of A. fruticosum.
Figure S5. Abundance of the top 10 predicted level-III KEGG pathways of microbiomes of N. vermiculata.
Figure S6. Heat map referring to the top 35 KEGG level III pathways of microbiomes of A. fruticosum.
Figure S7. Heat map referring to the top 35 KEGG level III pathways of microbiomes of N. vermiculata.
Figure S8. Predicted level-III KEGG pathways of microbiomes of A. fruticosum.
Figure S9. Predicted level-III KEGG pathways of microbiomes of N. vermiculata.
Figure S10. Heat map referring to the top 35 enriched compounds of microbiomes of A. fruticosum.
Figure S11. Heat map referring to the top 35 enriched compounds of microbiomes of N. vermiculata.
Figure S12. Enriched compounds of predicted level-III KEGG pathways of microbiomes of A. fruticosum.
Figure S13. Enriched compounds of predicted level-III KEGG pathways of microbiomes of N. vermiculata.
Figure S14. Enriched compounds in one or more KEGG pathways in microbiomes of A. fruticosum.
Figure S15. Enriched compounds in one or more KEGG pathways in soil microbiomes of N. vermiculata.
Figure S16. Results of real time PCR of the gatA gene in microbiomes of A. fruticosum and N. vermiculata.
Table S1. KO hierarchy and description of enriched compounds in microbiomes of the two wild plants A. fruticosum and N. vermiculata across soil types and watering time points.
Table S2. Enriched compounds (8) with KEGG reference and pathway in microbiomes of the two wild plants A. fruticosum and N. vermiculata in terms of soil types and watering time points.
Table S3. KO hierarchy and description of differentially enriched compounds due to watering in microbiomes of the two wild plants A. fruticosum and N. vermiculata collected from surrounding bulk (subgroup style ABC) and rhizosphere (subgroup style DEF) soils after 0 (groups A & D, respectively), 24 (groups B & E, respectively) and 48 h (groups C & F, respectively) of watering.
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |