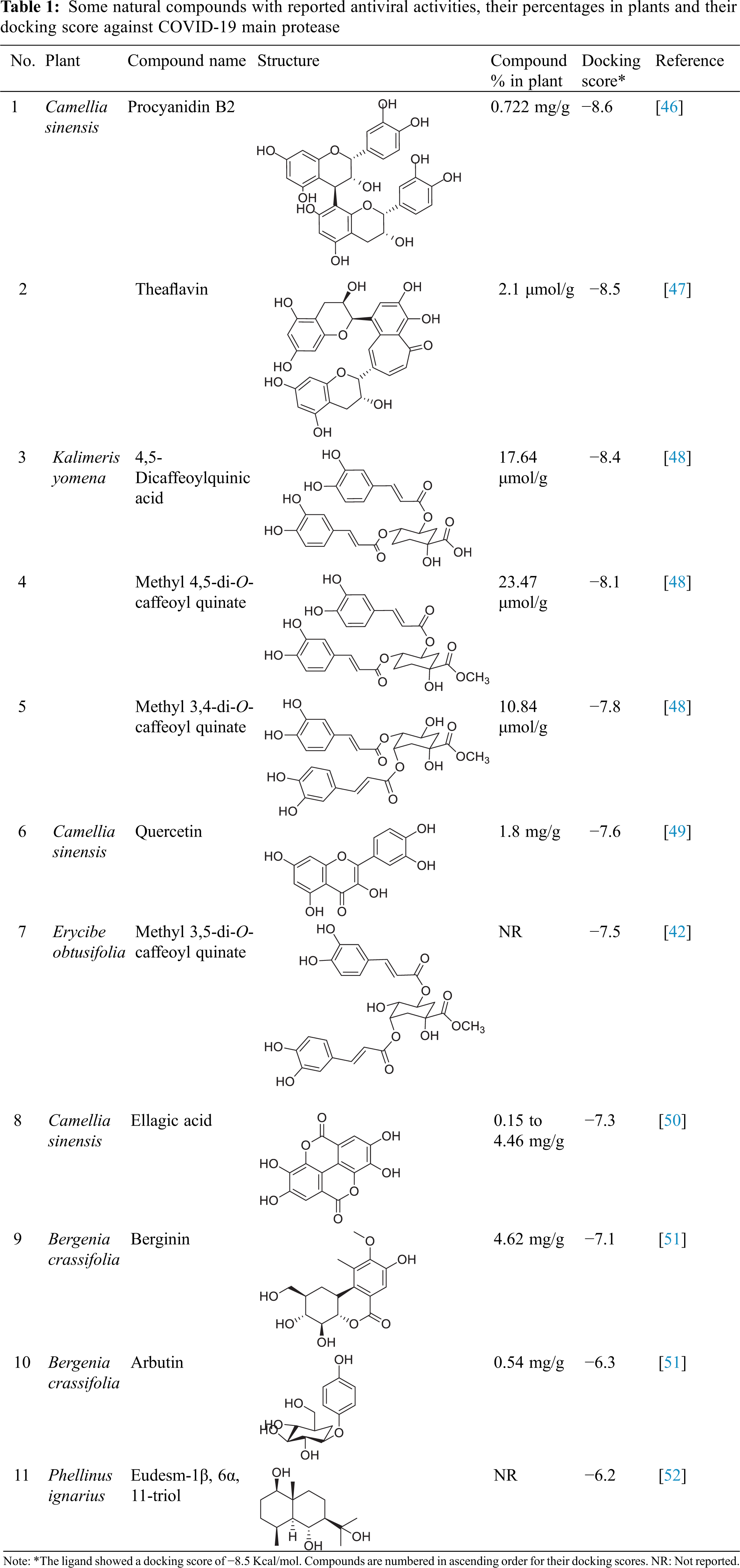
| Phyton-International Journal of Experimental Botany |
DOI: 10.32604/phyton.2022.018239
ARTICLE
Investigation of SARS-CoV-2 Main Protease Potential Inhibitory Activities of Some Natural Antiviral Compounds Via Molecular Docking and Dynamics Approaches
1Department of Pharmacognosy, Faculty of Pharmacy, Ain Shams University, Cairo, 11566, Egypt
2Department of Pharmaceutical Chemistry, Faculty of Pharmacy, The British University in Egypt, Cairo, 11837, Egypt
3Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Ain Shams University, Cairo, 11566, Egypt
4Department of Biochemistry, Faculty of Pharmacy, Ain Shams University, Cairo, 11566, Egypt
5Center for Drug Discovery and Development Research, Ain Shams University, Cairo, 11566, Egypt
*Corresponding Authors: Abdel Nasser B. Singab. Email: dean@pharma.asu.edu.eg; Omayma A. Eldahshan. Email: oeldahshan@pharma.asu.edu.eg
#These authors contributed equally to the work
Received: 09 July 2021; Accepted: 08 October 2021
Abstract: Coronaviruses caused an outbreak pandemic disease characterized by a severe acute respiratory distress syndrome leading to the infection of more than 200 million patients and the death of more than 4 million individuals. The primary treatment is either supportive or symptomatic. Natural products have an important role in the development of various drugs. Thus, screening of natural compounds with reported antiviral activities can lead to the discovery of potential inhibitory entities against coronaviruses. In the current study, an in-silico molecular docking experiment was conducted on the effects of some of these natural antiviral phytoconstituents, (e.g., procyanidin B2, theaflavin, quercetin, ellagic acid, caffeoylquinic acid derivatives, berginin, eudesm-1β, 6α, 11-triol and arbutin), on the crystal structure of SARS-CoV-2 main protease (PDB ID: 6w63) using AutoDock-Vina software. Many of the docked compounds revealed good binding affinity, with procyanidin B2 (–8.6 Kcal/mol) and theaflavin (–8.5 Kcal/mol) showing a better or similar binding score as the ligand (–8.5 Kcal/mol). Molecular dynamics simulations were carried out at 100 ns and revealed that procyanidin B2 forms a more stable complex with SARS-CoV-2 main protease than theaflavin. Procyanidin B2, theaflavin, and 4,5-dicaffeoylquinic acid were evaluated for toxicity by ProTox-II webserver and were non-toxic according to the predicted LD50 values and safe on different organs and pathways. Additionally, these phytoconstituents showed good ADME properties and acceptable lipophilicity, as evaluated using WLOGP. Amongst the tested compounds, procyanidin B2 showed the highest lipophilic value. It is worth mentioning that these natural inhibitiors of SARS-CoV-2 main protease are components of green and black tea that can be used as a supporting supplement for COVID patients or as potential nuclei for further drug design and development campaigns.
Keywords: Coronavirus; natural products; SARS-CoV-2 main protease; molecular docking; molecular dynamics; tea
Coronavirus pandemic (also known as coronavirus disease 2019, COVID-19), is an outbreak pandemic disease caused by a severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infecting more than 200 million people and causing the death of more than 4 million individuals worldwide [1]. While its common symptoms resemble those of common cold including cough, fever, and breath shortness, its complications can be fatal including pneumonia and acute respiratory distress syndrome [2,3].
The primary method of COVID-19 transmission is through small droplets produced by coughing or sneezing of an infected individual. Thus, wearing face masks and maintaining social distancing are recommended preventive measures [4]. Coronavirus pandemic has created a global socio-economic recession forcing millions of people to stay at home. The vaccination campaigns are intended to elevate the immunity of most of the population, yet the primary treatment is still either supportive or symptomatic [5].
Natural products represent fertile soil having different classes of compounds [6] and can develop inhibitors into successful drug candidates to treat such life-threatening pandemic. Some natural compounds were tested against various CoV species and revealed potential inhibitory activities with IC50 values of 12 to 143 nM. These compounds include lycorine, silvestrol, 7-methoxycryptopleurine, homoharringtonine, tylophorine and ouabain. In addition, plant extracts from Lycoris radiate, Rheum officinale and Polygonum multiflorum showed potential effects against SARS-CoV [7]. Rolta et al. [8] reported that both emodin and artemisinin inhibited the binding of SARS-CoV-2 spike protein to ACE2 receptor via a molecular docking approach with good binding affinities. Additionally, they predicted that the compounds tested are non-carcinogenic and non-toxic using in-silico toxicity studies. Amongst phytoconstituents of Rheum emodi evaluated in-silico for binding affinity to RNA-binding domain active sites of SARS-CoV-2, aloe-emodin, anthrarufin and alizarin form stable complexes (at 50 ns), showed good binding affinities and followed drug-likeness properties [9]. A recent molecular docking study investigated more than two thousand flavone analogs against coronavirus main protease and revealed that 371 of the tested analogs showed low docking scores and well-predicted potency [10]. A similar study revealed the potential inhibition of curcumin (−9.2 kcal/mol) and salvianolic acid A (−9.7 kcal/mol) on SARS-CoV Mpro [11].
Thus, the search for natural antiviral compounds can be a clue for dealing with such pandemic for the meantime, and since coronavirus is an RNA virus that is encapsulated within a viral envelope. This encapsulated genome contains eleven genes that encode the viral proteins and peptides [12], and relies on a chymotrypsin-like main protease (also known as Mpro and 3CLpro) for the processing of many proteins that are eventually translated from the viral RNA to allow these proteins to function properly [13]. Thus, finding new molecules with high molecular affinity to inhibit such protease can effectively cause interruption of the steps of viral replication by preventing the cleavage of the non-structural poly-proteins into small fragments and thus, represents a global challenge nowadays.
This can be accomplished through molecular docking that represents a way for predicting the preferred orientation of one molecule with regard to another when they are bound together forming a stable complex. Such orientation allows the anticipation of the association or binding affinity between such molecules [14–16]. The binding of biological molecules plays a major regulatory role in their signaling pathway causing either up-regulation or down-regulation in their signaling cascade. Therefore, docking represents a very powerful tool for predicting both the strength and type of signal produced [17,18]. Additionally, the high expenses and long duration required for the identification and isolation of natural products have guided the researchers to design small molecules-based libraries having molecules that mimic the natural products with improved stability and the least predicted toxicity [19]. Molecular docking is now commonly used in pharmaceutical drug design and in the elucidation of major fundamental biochemical processes.
Thus, the aims of the current study were to (1) search among reported antiviral natural compounds for potential inhibitory activity against coronavirus main protease enzyme via a molecular docking study, and (2) elucidate their different binding affinities and stabilities via molecular dynamics. Furthermore, the toxicity of the selected phyto-constituents was studied in-silico on different organs and pathways as well as ADME/physicochemical properties and drug-likeness activities. This was in an attempt to find a supporting supplement to the patients or find a high-affinity molecule that can be the core for the synthesis of drug candidates against coronavirus.
All screened compounds were imported and converted to 3D structures as SDF format via Open Babel [20], considering the different tautomers for each ligand with the appropriate protonation state at pH 7. All structures were then prepared for docking through converting them to the PDBQT format via AutoDock Tools [21]. The PDB file of the SARS-CoV-2 Main Protease co-crystallized with an inhibitor was obtained from the Protein Data Bank [22] (PDB ID: 6w63).
The protein was prepared by removing water molecules, adding hydrogen atoms, typing all atoms with Gasteiger charges, removing the co-crystallized, and finally saving the prepared protein in the PDBQT format to be ready for docking. The different protonation states of Histidine and Arginine amino acids were considered in the docking experiment.
Molecular docking was carried out using AutoDockVina [23]. Rotatable bonds of the ligands were assigned while the binding site residues were considered rigid during the docking calculations. The search box was set to have a center of −18.093, 18.097 and −26.884 for the X, Y and Z coordinates, respectively. The box side and grid spacing were set to 16 Å and 0.375 Å, respectively. The docking parameters were set to the default settings. The co-crystallized ligand was also re-docked to ensure the accuracy of the docking protocol. Standard exhaustiveness and randomized seed numbers were used. The output poses and log files were then inspected to determine the best docking poses and the nature of formed interactions. Root Mean Square Deviation (RMSD) between the docked and co-crystallized ligand was measured using DockRMSD software (https://jcheminf.biomedcentral.com/articles/10.1186/s13321-019-0362-7).
The main protease protein of SARS-CoV-2 (PDB ID: 6W63) was subjected to two runs of molecular dynamics simulations with the docking top-ranked two ligands, procyanidin B2 and theaflavin. The simulations were carried out at the NPT (constant number of molecules, pressure, and temperature) ensemble. Open MM setup utility [24] was used for the system preparation through cleaning up the protein-ligand complex file and adding missing atoms. Missing hydrogen atoms were added at pH 7.0, and then the complex was immersed in a cubic water box with a padding distance of 1 nm along each axis of the complex molecule. NaCl molecules were added at a concentration of 0.15 Molar. The protein molecule and water molecules were described using the AMBER 14 force field [25] and TIP3P-FB model [26], respectively. Small molecules were parametrized using the generalized amber force field (GAFF) [27]. OpenEye toolkits 2019.4 was used for generating the GAFF parameters by assigning ELF10 charges using the canonical AM1-BCC charging method of OpenEye toolkits [28]. The Particle Mesh Ewald (PME) method [29] was used for calculating long-range electrostatics assigning 0.0005 as the error tolerance for truncating the Ewald summation. A 1.2 nm cut-off was assigned for both the PME direct space interactions and Lennard-Jones interactions. The length of all bonds that involve a hydrogen atom, and the water molecules, involving their bond length and angles, were constrained. Langevin integrator with 2 fs step size, 310 K temperature, and 1 ps-1 friction coefficient was used for integration of force equations. Monte Carlo barostat [30] was used for maintaining pressure coupling assigning a constant temperature of 310 K, a constant pressure of 1 atm, and a pressure update every 25 steps (50 fs). The dynamics simulations were carried out using the OpenMM 7.4.2 application layer [24]. The simulation started with energy minimization with energy convergence criteria of 10 kJ/mole, then 500,000 steps (1 ns) of equilibration, and finally 50,000,000 steps (100 ns) for production. Trajectory reporting was set to one snapshot per 10,000 steps resulting in 5,000 snapshots in the final trajectory file. ProDy python library [31,32] was used for calculating protein RMSD, RMSF and radius of gyration. RMSD trajectory analysis and hydrogen bonds utilities of VMD were used for calculating Ligand RMSD and number of hydrogen bonds to the protein, respectively [33]. Figures were generated using matplotlib python library [34].
The elevating trends of people mobility for the purpose of urbanization or even migration have led to the spread of viruses among them. Treating viral infections with conventional drugs has been associated with many side effects and resistance cases for the microbes have been elevated, which triggered scientists to search for natural products [35].
Nature has provided us with a variety of compounds [36,37], that showed potential antiviral and antioxidant activities [7,38]. The current study indicates compounds having prominent antiviral activity, thus they were selected due to these reported activities. Tea polyphenols have many beneficial health effects, including antiviral activity. Theaflavin and procyanidin showed anti-influenza B virus activity [39]. Green tea catechins exhibited virucidal activity against influenza virus subtypes including A/H1N1, A/H3N2 and B virus [40]. Tea polyphenols also inhibited enterovirus 71 (EV71) replication, where they potently inhibited replication of E-lokV71 and reduced the titer of infectious progeny virus by 95% [41]. The new quinic acid derivatives isolated from the root and stem of Erycibe obtusifolia displayed potent in-vitro anti-respiratory syncytial virus effect [42]. In addition, quercetin and its derivatives displayed strong antiviral activity against arbovirus Mayaro compared to ribavirin [43]. Quercetin inhibited the synthesis of hepatitis B antigen compared to lamivudine [44]. Also, quercetin has shown clinical efficacy in the treatment of virus C infection and experimentally inhibited other RNA and DNA viruses such as hepatitis B, poliomyelitis, influenza, measles, and smallpox [45].
An in-silico molecular docking study was carried out by these selected nature compounds on the crystal structure of COVID-19 main protease (Table 1).
At first, a pose retrieval experiment was conducted, for the validation of the vina docking software, by redocking the co-crystallized ligand into the main protease binding site. RMSD between the co-crystallized and the docked poses was 0.963 Å. This RMSD value, which is less than 2 Å, gives a validation on the use of vina as our docking software. Additionally, the docked pose kept the same hydrogen bonding interactions with the same residues as the co-crystallized ligands, Gly143 and Glu166. The superposition and binding interactions of the co-crystallized and the docked poses are shown in Fig. 1.
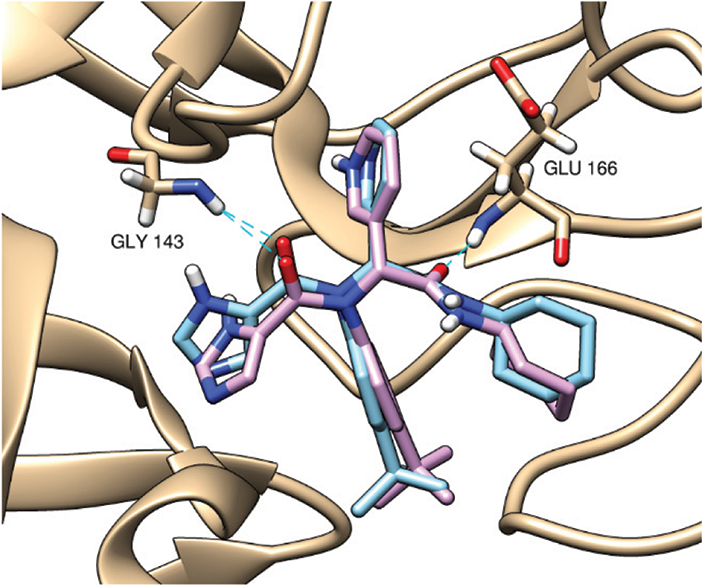
Figure 1: Superposition of the co-crystallized (cyan) and the docked (violet) poses inside the main protease binding site (PDB ID: 6w63). The docking validation showed an acceptable RMSD value of 0.963 Å (<2 Å). Both poses are keeping the same hydrogen bonding interactions (cyan dashed lines) with Gly143 and Glu166
Inspection of ligand interactions with COVID-19 main protease, revealed numerous hydrophobic residues in the protease moiety such as His 41, Gly 143, Glu 166, His 163 and many others. Among the tested compounds, procyanidin B2 presented the best docking score (a little lower than the co-crystallized ligand), the binding affinity of which to the enzyme was through hydrogen bonds with Ser 144 and Glu 166 moieties, Pi-Pi interaction with His-41 amino acid and some polar interactions. The binding mode of procyanidin B2 is shown in Fig. 2A. Theaflavin followed procyanidin B2 in docking score (equal to the ligand). The binding affinity of theaflavin was through different polar, hydrophobic interactions, H-bonds with both His 41 and Gln 189 moieties (Fig. 2B). 4, 5-dicaffeoylquinic acid came after theaflavin in the docking score, even though it showed the most interactions with the enzyme with six H-bonds with Arg 188, His 163, Ser 144, Asn 142, Cys 44 and Met 48 moieties, one Pi-Pi interaction with His-41 and many other polar and hydrophobic interactions, as shown in Fig. 2C.
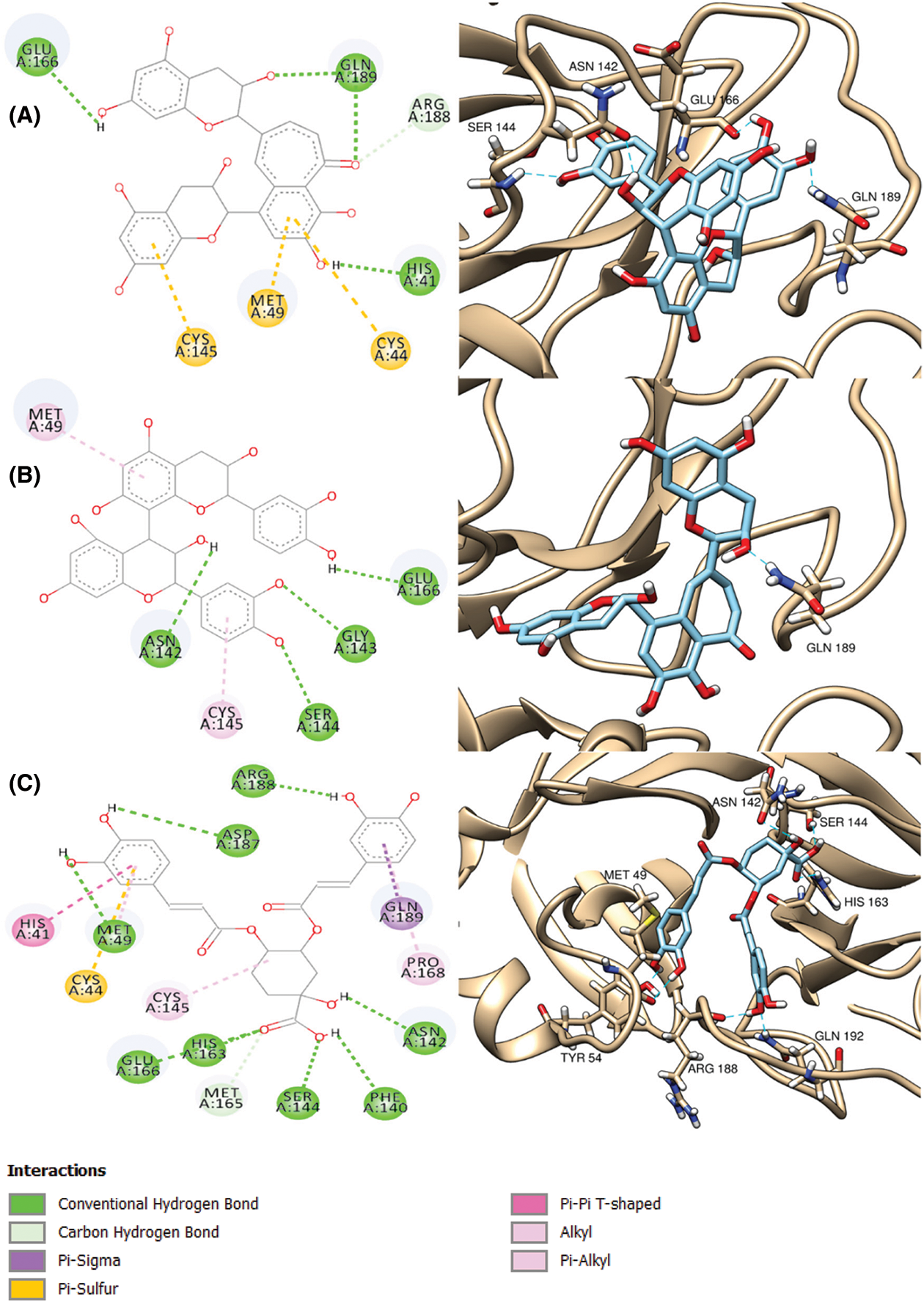
Figure 2: 2D (left column) and 3D (right column) binding diagrams of procyanidin B2 (A), theaflavin (B) and 4,5-dicaffeoylquinic acid (C)
Molecular dynamics (MD) simulations were carried out to compare the binding stability of the compounds, procyanidin B2 (Complex 1) and theaflavin (Complex 2), inside the main protease binding site. The radius of gyration (Rg) of Complex 2 showed higher fluctuation than Complex 1 (Fig. 3A). This can be attributed to a conformational change that took place in this complex and can also be observed in the slightly higher root mean square deviation (RMSD) of the backbone atoms of this complex (Fig. 3B) reflecting the protein structure stability during the simulation time. It reached stability for the two MD runs with the highest RMSD for Complex 2. The per residue root mean square fluctuation (RMSF) of the two MD runs is shown in Fig. 3C, as a measure of protein residue conformational change and stability. Low RMSF of the binding site residues, <2Å, was shown during the simulation time for both protein complexes indicating their relatively strong binding to their respective bound ligands.

Figure 3: Radius of gyration (Rg) of the protein molecules (A), RMSD of the protein backbone atoms (B), and per residue RMSF for the protein residues (C) in the two MD runs are shown (green: Complex 1 (Procyanidin B2), blue: Complex 2 (Theaflavin). The binding site amino acids are highlighted with a red background, depicting low RMSF, <2Å, showing the binding stability of the ligands
Ligand RMSD analysis showed that theaflavin had the most deviation from its starting pose indicating its weaker binding to the protein (Fig. 4A). Procyanidin-B2 showed the lowest RMSD during the simulation which reflects its better binding. Fig. 4B depicts the number of hydrogen bonds formed between each ligand and the protein. Both compounds show a relatively high number of formed hydrogen bonds with the protein indicating their strong binding, with a relatively higher number with theaflavin.

Figure 4: RMSD of each ligand (A) and number of hydrogen bonds formed between each ligand and the respective protein complex (B) (green: Complex 1 (Procyanidin-B2), blue: Complex 2 (Theaflavin))
In the pursuit of identifying the pharmacokinetic properties of the top 3 ranked compounds, we calculated the ADME/physicochemical properties using Swiss ADME online server (https://www.nature.com/articles/srep42717) and reported them in Table 2. As concluded from Table 2, the three compounds showed good ADME properties. The high Topological Polar Surface Area (TPSA) of the three compounds can be attributed to the high number of polar groups, like hydroxyl groups, present in the compounds. This high TPSA can affect GI absorption and Brain Blood Barrier crossing as depicted in Table 2. The lipophilicity was also measured using WLOGP which showed an acceptable lipophilicity, with Procyanidin B2 having the highest value among the three compounds. These compounds are not substrates for CYP inhibition which indicates their good predicted metabolic and excretory profiles.
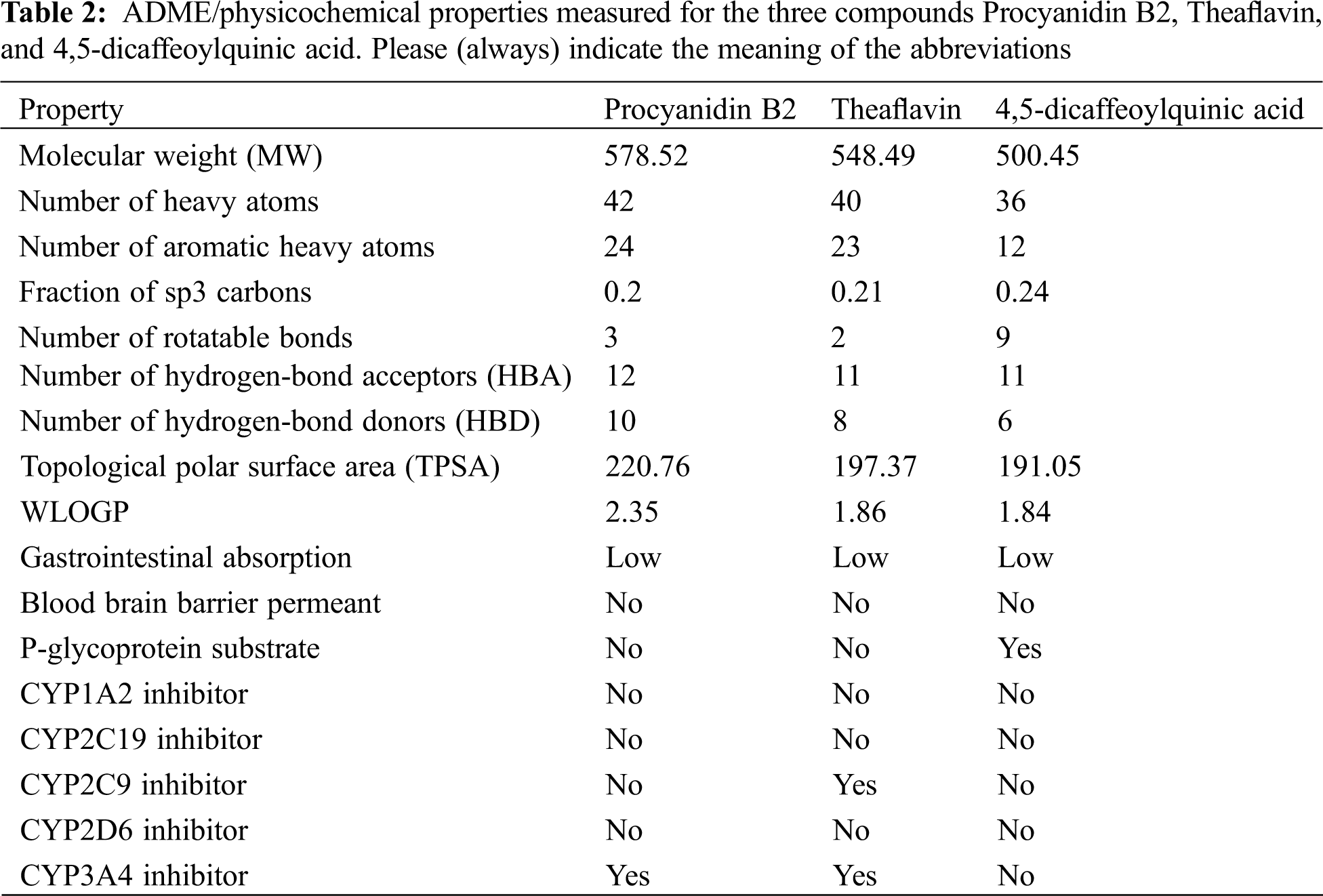
Drug likeness for the three compounds was also predicted using Lipinski, Ghose, Veber, Egan, and Muegge rules for drug likeness (Table 3). All rules were violated by the three compounds mainly due to their high polarity because of the presence of polar hydroxyl groups. This also contributed to a high number of hydrogen bond donors and acceptors present in the compounds which led to the violation of Lipinski’s rule. PAINS and BRENK alerts were also detected for the three compounds. PAINS alert is an indication of the presence of a chemical group that often gives false positive results in high-throughput screens. BRENK alert indicates a fragment with possible toxicity, chemical reactivity, or metabolic instability. The catechol group is predicted as the group responsible for these two alerts. However, this cannot be confirmed without biological evaluation. As a result, we encourage that this point of modification be investigated in future medicinal chemistry projects for the development of SARS-CoV-2 main protease inhibitors.

Toxicity of the studied compounds was predicted by ProTox-II webserver (https://academic.oup.com/nar/article/46/W1/W257/4990033). Predicted LD50 for Procyanidin B2, Theaflavin, and 4,5-Dicaffeoylquinic acid were calculated to be 2500, 2500, and 5000 mg/kg. The predicted toxicity class for the three compounds was Class VI. The toxicity classes were decided by the ProTox-II webserver depending on the toxic doses as per the following criteria: Class I: fatal if swallowed (LD50 ≤ 5), Class II: fatal if swallowed (5 < LD50 ≤ 50), Class III: toxic if swallowed (50 < LD50 ≤ 300), Class IV: harmful if swallowed (300 < LD50 ≤ 2000), Class V: may be harmful if swallowed (2000 < LD50 ≤ 5000), Class VI: non-toxic (LD50 > 5000). Therefore, the three compounds are predicted as non-toxic which indicates their promising safety profile. Fig. 5 depicts the toxicity radar charts for the three compounds Procyanidin B2, Theaflavin, and 4,5-Dicaffeoylquinic acid, showing their toxic potential on different toxicity organs and pathways. Both Procyanidin B2 and 4,5-Dicaffeoylquinic acid showed predicted immunotoxicity. However, the three compounds were predicted to be safe on the other organs and pathways.
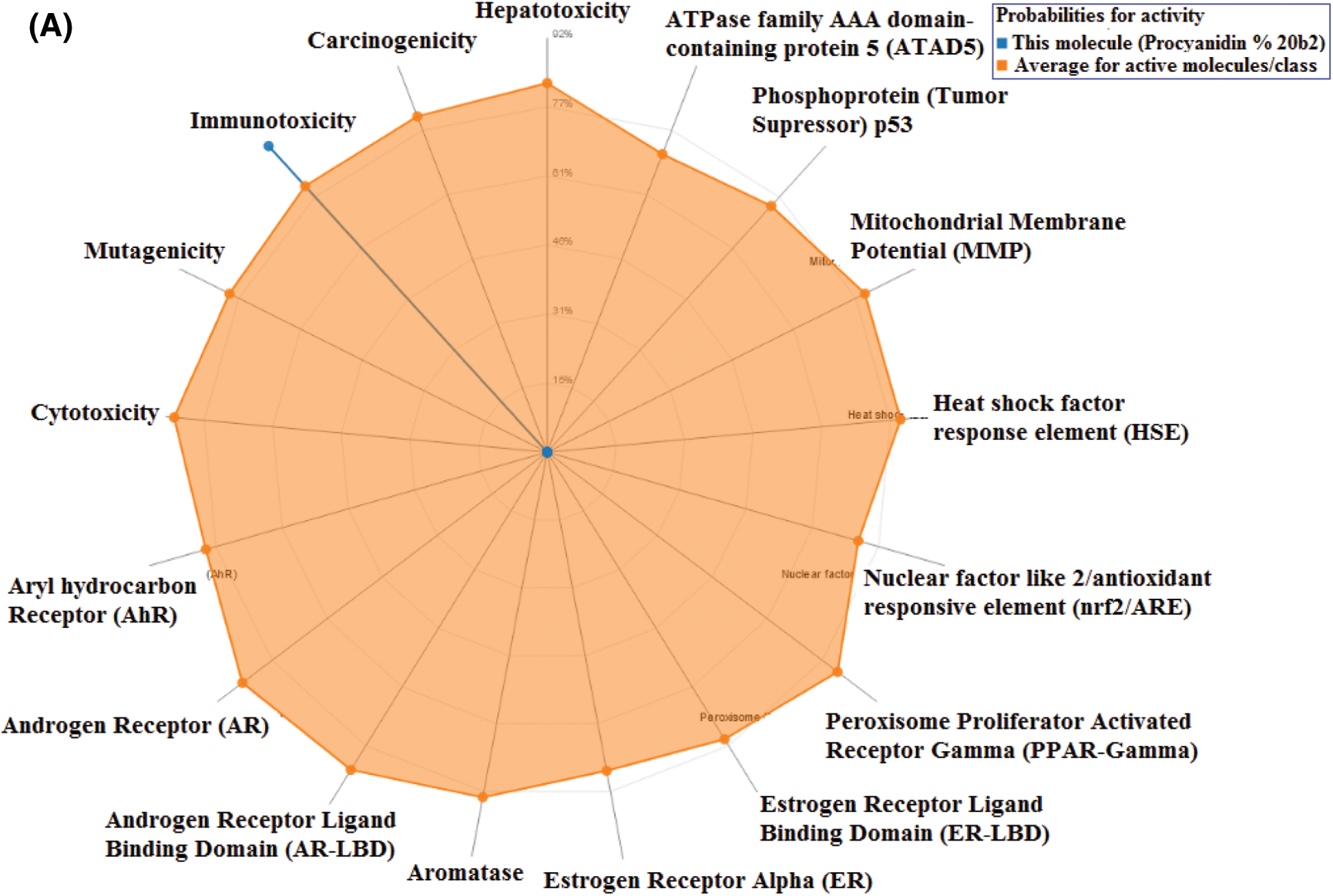
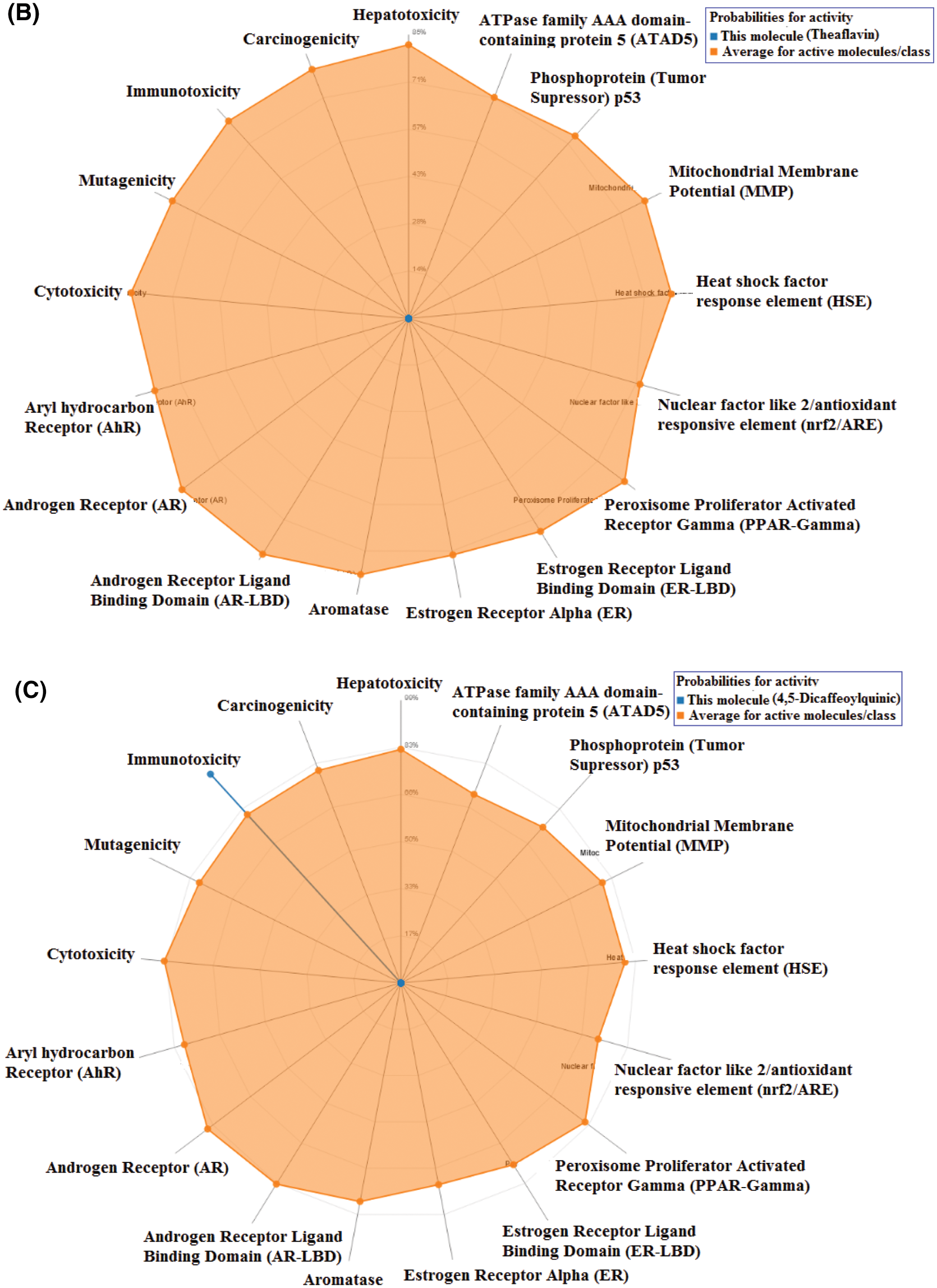
Figure 5: The toxicity radar charts for Procyanidin B2 (A), Theaflavin (B), and 4,5-Dicaffeoylquinic acid (C) illustrating the confidence of positive toxicity results compared to the average of each class
It is worth mentioning that the highest eight compounds in docking score and binding affinity to the enzyme (procyanidin B2, theaflavin, different caffeoylquinic acid derivatives, quercetin and ellagic acid) are components of green and black tea [39,53,54]. Chen et al. [55] have reported that some black tea components such as tannic acid, 3-isotheaflavin-3-gallate and theaflavin-3,3-digallate have shown inhibitory activities on SARS-CoV 3CLpro having IC50 values of 3, 7 and 9.5 μM, respectively. Epigallocatechin gallate, gallocatechin-3-gallate and epicatechin gallate are components of tea phenolics and were reported to be able to in-silico bind strongly to SARS-CoV main protease catalytic residues [56]. These results were further confirmed by Jang et al. [57]. 3-O-Caffeoylquinic acid and methyl 3,5-di-O-caffeoyl quinate showed better binding to SARS CoV-2 Mpro as compared to remdesivir [58]. Ravi [59] reported that the hot aqueous fruit pulp extract of Cassia fistula rich in Procyanidin B2 can be used to allievate COVID patients based on a molecular dynamics study. Moreover, Mesli et al. [60] reported that methyl-1, 4, 5-tri-O-caffeoyl quinate showed stronger affinity than the docked ligand of ACE2 enzyme receptor of COVID. Furthermore, Nguyen et al. [61] reported that quercetin inhibited SARS-CoV main protease in-vitro with an IC50 value of 73 µM. These results agree with our results, highlighting the importance of natural product supplements as those in tea for the inhibition of coronaviruses.
Coronavirus has led to a global health and economic burden which requires a quick provision of potential treatments. Natural products are considered a safe and available repository for potential dug target modulators. In this endeavor, this study presented an in-silico approach investigating the potential inhibition of SARS-CoV-2 main protease utilizing a natural products database with reported antiviral activities, implementing molecular docking and dynamics simulations. We are suggesting some natural products, present in green and black tea, to be a supporting supplement, or a nucleus for further drug design and development campaigns.
Funding Statement: The authors received no specific funding for this study.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
1. WHO (2021). WHO Coronavirus (COVID-19) dashboard. https://COVID19.who.int. [Google Scholar]
2. Huang, C., Wang, Y., Li, X., Ren, L., Zhao, J. et al. (2020). Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. The Lancet, 395, 497–506. DOI 10.1016/S0140-6736(20)30183-5. [Google Scholar] [CrossRef]
3. Jiang, S., Xia, S., Ying, T., Lu, L. (2020). A novel coronavirus (2019-nCoV) causing pneumonia-associated respiratory syndrome. Cellular and Molecular Immunology, 17(5), 554. DOI 10.1038/s41423-020-0372-4. [Google Scholar] [CrossRef]
4. Ji, Y., Ma, Z., Peppelenbosch, M. P., Pan, Q. (2020). Potential association between COVID-19 mortality and health-care resource availability. The Lancet Global Health, 8, 475–480. DOI 10.1016/S2214-109X(20)30068-1. [Google Scholar] [CrossRef]
5. Wang, C., Horby, P. W., Hayden, F. G., Gao, G. F. (2020). A novel coronavirus outbreak of global health concern. The Lancet, 395, 470–473. DOI 10.1016/S0140-6736(20)30185-9. [Google Scholar] [CrossRef]
6. Elkhawas, Y. A., Elissawy, A. M., Elnaggar, M. S., Mostafa, N. M., Al-Sayed, E. et al. (2020). Chemical diversity in species belonging to soft coral genus Sacrophyton and its impact on biological activity: A review. Marine Drugs, 18(1), 41–52. DOI 10.3390/md18010041. [Google Scholar] [CrossRef]
7. Islam, M. T., Sarkar, C., El-Kersh, D. M., Jamaddar, S., Uddin, S. J. et al. (2020). Natural products and their derivatives against coronavirus: A review of the non-clinical and pre-clinical data. Phytotherapy Research, 34, 2471–2492. DOI 10.1002/ptr.6700. [Google Scholar] [CrossRef]
8. Rolta, R., Salaria, D., Kumar, V., Sourirajan, A., Dev, K. (2021). Phytocompounds of Rheum emodi, Thymus serpyllum and Artemisia annua inhibit COVID-19 binding to ACE2 receptor: In silico approach. Current Pharmacology Reports, 7, 135–149. DOI 10.1080/07391102.2020.1850364. [Google Scholar] [CrossRef]
9. Rolta, R., Yadav, R., Salaria, D., Trivedi, S., Imran, M. et al. (2020). In silico screening of hundred phytocompounds of ten medicinal plants as potential inhibitors of nucleocapsid phosphoprotein of COVID-19: An approach to prevent virus assembly. Journal of Biomolecular Structure and Dynamics, 39(18), 1–18. DOI 10.1080/07391102.2020.1804457. [Google Scholar] [CrossRef]
10. Ibrahim, M. A., Mohamed, A. R., Abdelrahman, H. M., Allemailem, S., Moustafa, M. F. et al. (2021). Rutin and flavone analogs as prospective SARS-CoV-2 main protease inhibitors: In silico drug discovery study. Journal of Molecular Graphics and Modelling, 105, 1079–1084. DOI 10.1016/j.jmgm.2021.107904. [Google Scholar] [CrossRef]
11. Ibrahim, M. A., Abdelrahman, H. M., Hussien, T. A., Badr, A. A., Mohamed, T. A. et al. (2020). In silico drug discovery of major metabolites from spices as SARS-CoV-2 main protease inhibitors. Computers in Biology and Medicine, 126, 1040–1046. DOI 10.1016/j.compbiomed.2020.104046. [Google Scholar] [CrossRef]
12. Ibrahim, M. A., Abdelrahman, H. M., Mohamed, A., Atia, A. M., Al-Hammady, A. M. et al. (2021). In silico mining of terpenes from Red-Sea invertebrates for SARS-CoV-2 main protease (Mpro) inhibitors. Molecules, 26(7), 20–82. DOI 10.3390/molecules26072082. [Google Scholar] [CrossRef]
13. Zhang, L., Lin, D., Sun, X., Curth, U., Drosten, C. et al. (2020). Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science, 368, 409–412. DOI 10.1126/science.abb3405. [Google Scholar] [CrossRef]
14. Lengauer, T., Rarey, M. (1996). Computational methods for biomolecular docking. Current Opinion in Structural Biology, 6, 402–406. DOI 10.1016/S0959-440X(96)80061-3. [Google Scholar] [CrossRef]
15. Ashmawy, A., Mostafa, N. M., Eldahshan, O. (2019). GC/MS analysis and molecular profiling of lemon volatile oil against breast cancer. Journal of Essential Oil-Bearing Plants, 22, 903–916. DOI 10.1080/0972060X.2019.1667877. [Google Scholar] [CrossRef]
16. Mostafa, N. M. (2018). Antibacterial activity of ginger (Zingiber officinale) leaves essential oil nanoemulsion against the cariogenic Streptococcus mutans. Journal of Applied Pharmaceutical Science, 8, 34–41. DOI 10.7324/JAPS.2018.8906. [Google Scholar] [CrossRef]
17. Kahraman, A., Morris, R. J., Laskowski, R. A., Thornton, J. M. (2007). Shape variation in protein binding pockets and their ligands. Journal of Molecular Biology, 368(1), 283–301. DOI 10.1016/j.jmb.2007.01.086. [Google Scholar] [CrossRef]
18. Kitchen, D. B., Decornez, H., Furr, J. R., Bajorath, J. (2004). Docking and scoring in virtual screening for drug discovery: Methods and applications. Nature Reviews Drug Discovery, 3(11), 935–949. DOI 10.1038/nrd1549. [Google Scholar] [CrossRef]
19. Ibrahim, M. A., Abdeljawaad, A. A., Abdelrahman, H. M., Hegazy, F. (2020). Natural-like products as potential SARS-CoV-2 Mpro inhibitors: In-silico drug discovery. Journal of Biomolecular Structure and Dynamics, 39(15), 5722–5734. DOI 10.1080/07391102.2020.1790037. [Google Scholar] [CrossRef]
20. O’Boyle, N., Banck, M., James, C., Morley, C., Vandermeersch, T. et al. (2011). Open babel: An open chemical toolbox. Journal of Chemistry Information, 3(1), 33–38. DOI 10.1186/1758-2946-3-33. [Google Scholar] [CrossRef]
21. Morris, G. M., Huey, R., Lindstrom, W., Sanner, M. F., Belew, R. K. et al. (2009). AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. Journal of Computational Chemistry, 30(16), 2785–2791. DOI 10.1002/jcc.21256. [Google Scholar] [CrossRef]
22. Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N. et al. (2000). The protein data bank. Nucleic Acids Research, 28(1), 235–242. DOI 10.1093/nar/28.1.235. [Google Scholar] [CrossRef]
23. Trott, O., Olson, A. J. (2010). AutoDock vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry, 31, 455–461. DOI 10.1002/jcc.21334. [Google Scholar] [CrossRef]
24. Eastman, P., Swails, J., Chodera, J. D., McGibbon, R. T., Zhao, Y. et al. (2017). OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLoS Computational Biology, 13(7), 56–59. DOI 10.1371/journal.pcbi.1005659. [Google Scholar] [CrossRef]
25. Maier, J. A., Martinez, C., Kasavajhala, K., Wickstrom, L., Hauser, K. E. et al. (2015). ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. Journal of Chemical Theory and Computation, 11(8), 3696–3713. DOI 10.1021/acs.jctc.5b00255. [Google Scholar] [CrossRef]
26. Wang, L. P., Martinez, T. J., Pande, V. S. (2014). Building force fields: An automatic, systematic, and reproducible approach. Journal of Physical Chemistry Letters, 5(11), 1885–1891. DOI 10.1021/jz500737m. [Google Scholar] [CrossRef]
27. Wang, W. R., Wolf, R., Caldwell, J. W., Kollman, P. A., Case, D. A. (2004). Development and testing of a general amber force field. Journal of Computational Chemistry, 25(9), 1157–1174. DOI 10.1002/(ISSN)1096-987X. [Google Scholar] [CrossRef]
28. OpenEye Toolkits (2019). OpenEye scientific software. Santa Fe, NM. https://www.eyesopen.com. [Google Scholar]
29. Essman, U., Perera, L., Berkowitz, M., Darden, T., Lee, H. et al. (1995). A smooth particle mesh ewald potential. Journal of Chemical Physics, 103(19), 8577–8592. DOI 10.1063/1.470117. [Google Scholar] [CrossRef]
30. Chow, K. H., Ferguson, D. M. (1995). Isothermal-isobaric molecular dynamics simulations with monte carlo volume sampling. Computer Physics Communication, 91(1–3), 283–289. DOI 10.1016/0010-4655(95)00059-O. [Google Scholar] [CrossRef]
31. Bakan, A., Dutta, A., Mao, W., Liu, Y., Chennubhotla, C. et al. (2014). Evol and ProDy for bridging protein sequence evolution and structural dynamics. Bioinformatics, 30(18), 2681–2683. DOI 10.1093/bioinformatics/btu336. [Google Scholar] [CrossRef]
32. Bakan, A., Meireles, L. M., Bahar, I. (2011). ProDy: Protein dynamics inferred from theory and experiments. Bioinformatics, 27(11), 1575–1577. DOI 10.1093/bioinformatics/btr168. [Google Scholar] [CrossRef]
33. Humphrey, W., Dalke, A., Schulten, K. (1996). Visual molecular dynamics. Journal of Molecular Graphics, 14, 33–38. DOI 10.1016/0263-7855(96)00018-5. [Google Scholar] [CrossRef]
34. Price-Whelan, A. M., Sipőcz, B., Günther, H., Lim, P., Crawford, S. et al. (2018). The astropy project: Building an open-science project and status of the v2.0 core package. The Astronomical Journal, 2, 116–123. DOI 10.3847/1538-3881/aabc4f. [Google Scholar] [CrossRef]
35. Bhattacharya, R., Dev, K., Sourirajan, A. (2021). Antiviral activity of bioactive phytocompounds against coronavirus: An update. Journal of Virological Methods, 290, 1140–1170. DOI 10.1016/j.jviromet.2021.114070. [Google Scholar] [CrossRef]
36. Todirascu-Ciornea, E., El-Nashar, H. A., Mostafa, N. M., Eldahshan, O. A., Boiangiu, R. S. et al. (2019). Schinus terebinthifolius essential oil attenuates scopolamine-induced memory deficits via cholinergic modulation and antioxidant properties in a zebrafish model. Evidence Based Complementary and Alternative Medicine, 2, 344–349. DOI 10.1155/2019/5256781. [Google Scholar] [CrossRef]
37. Ayoub, N., Singab, A. N., Mostafa, N. M., Schultze, W. (2010). Volatile constituents of leaves of Ficus carica Linn grown in Egypt. Journal of Essential Oil-Bearing Plants, 13, 316–321. DOI 10.1080/0972060X.2010.10643827. [Google Scholar] [CrossRef]
38. Mostafa, N. M., Abd El-Ghffar, E. A., Hegazy, H. G., Eldahshan, O. A. (2018). New methoxyflavone from Casimiroa sapota and the biological activities of its leaves extract against lead acetate induced hepatotoxicity in rats. Chemistry and Biodiversity, 15, 17–28. DOI 10.1002/cbdv.201700528. [Google Scholar] [CrossRef]
39. Yang, Z. F., Bai, L. P., Huang, W. B., Li, X. Z., Zhao, S. S. et al. (2014). Comparison of in vitro antiviral activity of tea polyphenols against influenza A and B viruses and structure-activity relationship analysis. Fitoterapia, 93, 47–53. DOI 10.1016/j.fitote.2013.12.011. [Google Scholar] [CrossRef]
40. Song, J. M., Lee, K. H., Seong, B. L. (2005). Antiviral effect of catechins in green tea on influenza virus. Antiviral Research, 68, 66–74. DOI 10.1016/j.antiviral.2005.06.010. [Google Scholar] [CrossRef]
41. Ho, H. Y., Cheng, M. L., Weng, S. F., Leu, Y. L., Chiu, D. Y. (2009). Antiviral effect of epigallocatechin gallate on enterovirus 71. Journal of Agricultural and Food Chemistry, 57, 6140–6147. DOI 10.1021/jf901128u. [Google Scholar] [CrossRef]
42. Fan, L., Wang, Y., Liang, N., Huang, X. J., Li, M. M. et al. (2013). Chemical constituents from the roots and stems of Erycibe obtusifolia and their in vitro antiviral activity. Planta Medica, 79, 1558–1564. DOI 10.1055/s-0033-1350804. [Google Scholar] [CrossRef]
43. Dos Santos, A. E., Kuster, R. M., Yamamoto, K. A., Salles, T. S., Campos, R. et al. (2014). Quercetin and quercetin 3-O-glycosides from Bauhinia longifolia (Bong.) showing anti-mayaro virus activity. Parasites and Vectors, 7, 130–138. DOI 10.1186/1756-3305-7-130. [Google Scholar] [CrossRef]
44. Parvez, M. K., Rehman, M. T., Alam, P., Al-Dosari, M. S., Alqasoumi, S. I. et al. (2019). Plant-derived antiviral drugs as novel hepatitis B virus inhibitors: Cell culture and molecular docking study. Saudi Pharmceutical Journal, 27, 389–400. DOI 10.1016/j.jsps.2018.12.008. [Google Scholar] [CrossRef]
45. Davies, K., Schwinn, K. (2006). Molecular biology of flavonoid biosynthesis. Flavonoids: Chemistry, Biochemistry and Applications, 50, 143–218. [Google Scholar]
46. Zhang, L., Tai, Y., Wang, Y., Meng, Q., Yang, Y. et al. (2017). The proposed biosynthesis of procyanidins by the comparative chemical analysis of five Camellia species using LC-MS. Scientific Reports, 7(1), 1–10. DOI 10.1038/srep46131. [Google Scholar] [CrossRef]
47. Kim, Y., Goodner, K. L., Park, J. D., Choi, J., Talcott, S. T. (2011). Changes in antioxidant phytochemicals and volatile composition of Camellia sinensis by oxidation during tea fermentation. Food Chemistry, 129(4), 1331–1342. DOI 10.1016/j.foodchem.2011.05.012. [Google Scholar] [CrossRef]
48. Nugroho, A., Kim, K. H., Lee, K. R., Alam, M. B., Choi, J. S. et al. (2009). Qualitative and quantitative determination of the caffeoylquinic acids on the Korean mountainous vegetables used for chwinamul and their peroxynitrite-scavenging effect. Archives of Pharmacal Research, 32(10), 1361–1367. DOI 10.1007/s12272-009-2003-6. [Google Scholar] [CrossRef]
49. Perva-Uzunalić, A., Škerget, M., Knez, Ž., Weinreich, B., Otto, F. et al. (2006). Extraction of active ingredients from green tea (Camellia sinensisExtraction efficiency of major catechins and caffeine. Food Chemistry, 96(4), 597–605. DOI 10.1016/j.foodchem.2005.03.015. [Google Scholar] [CrossRef]
50. Yang, X., Tomás-Barberán, F. A. (2019). Tea is a ignificant dietary source of ellagitannins and ellagic acid. Journal of Agricultural and Food Chemistry, 67(19), 5394–5404. DOI 10.1021/acs.jafc.8b05010. [Google Scholar] [CrossRef]
51. Boros, B., Silvia, J., Tamás, M., Réka, M., Bertalan, G. et al. (2014). Validated HPLC method for simultaneous quantitation of bergenin, arbutin, and gallic acid in leaves of different Bergenia species. Chromatographia, 77, 1129–1135. DOI 10.1007/s10337-014-2624-x. [Google Scholar] [CrossRef]
52. Song, A. R., Sun, X. L., Kong, C., Zhao, C., Qin, D. et al. (2014). Discovery of a new sesquiterpenoid from Phellinus ignarius with antiviral activity against influenza virus. Archives of Virology, 159, 753–760. DOI 10.1007/s00705-013-1857-6. [Google Scholar] [CrossRef]
53. Cialdella-Kam, L., Ghosh, S., Meaney, M. P., Knab, A. M., Shanely, R. A. et al. (2017). Quercetin and green tea extract supplementation down regulates genes related to tissue inflammatory responses to a 12-week high fat-diet in mice. Nutrients, 9, 773–779. DOI 10.3390/nu9070773. [Google Scholar] [CrossRef]
54. Grujić-Letić, N., Rakić, B., Šefer, E., Rakić, D., Nedeljković, I. et al. (2015). Determination of 5-caffeoylquinic acid (5-CQA) as one of the major classes of chlorogenic acid in commercial tea and coffee samples. Vojnosanitetski Pregled, 72(11), 1018–1023. DOI 10.2298/VSP130915096G. [Google Scholar] [CrossRef]
55. Chen, C. N., Lin, C. P., Huang, K. K., Chen, W. C., Hsieh, H. P. et al. (2005). Inhibition of SARS-CoV 3C-like protease activity by theaflavin-3, 3’-digallate (TF3). Evidence Based Complementary and Alternative Medicine, 2(2), 22–29. DOI 10.1093/ecam/neh081. [Google Scholar] [CrossRef]
56. Ghosh, R., Chakraborty, A., Biswas, A., Chowdhuri, S. (2021). Evaluation of green tea polyphenols as novel corona virus (SARS CoV-2) main protease (Mpro) inhibitors-an in silico docking and molecular dynamics simulation study. Journal of Biomolecular Structure and Dynamics, 39(12), 4362–4374. DOI 10.1080/07391102.2020.1779818. [Google Scholar] [CrossRef]
57. Jang, M., Park, Y. I., Cha, Y. E., Park, R., Namkoong, S. et al. (2020). Tea polyphenols EGCG and theaflavin inhibit the activity of SARS-CoV-2 3CL-protease in vitro. Evidence-Based Complementary and Alternative Medicine, 2020, 5630–5638. DOI 10.1155/2020/5630838. [Google Scholar] [CrossRef]
58. Neupane, N. P., Abhishek, K. K., Imdad, H. M., Prateek, P., Praveen, K. et al. (2021). Molecular dynamics analysis of phytochemicals from ageratina adenophora against COVID-19 main protease (Mpro) and human angiotensin-converting enzyme 2 (ACE2). Biocatalysis and Agricultural Biotechnology, 32, 1019–1024. DOI 10.1016/j.bcab.2021.101924. [Google Scholar] [CrossRef]
59. Ravi, L. (2020). Procyanidin B2 of cassia fistula a potent inhibitor of COVID19 protease: A molecular dynamic simulation analysis. Asian Journal of Pharmaceutics, 14(2), 274–281. DOI 10.1016/j.bcab.2021.101924. [Google Scholar] [CrossRef]
60. Mesli, F., Ghalem, M., Daoud, I., Ghalem, S. (2021). Potential inhibitors of angiotensin converting enzyme 2 receptor of COVID-19 by Corchorus olitorius linn using docking, molecular dynamics, conceptual DFT investigation and pharmacophore mapping. Journal of Biomolecular Structure and Dynamics, 2(4), 1–13. DOI 10.1080/07391102.2021.1896389. [Google Scholar] [CrossRef]
61. Nguyen, T. H., Woo, H. J., Kang, H. K., Kim, Y. M., Kim, D. W. et al. (2012). Flavonoid-mediated inhibition of SARS coronavirus 3C-like protease expressed in Pichia pastoris. Biotechnology Letters, 34, 831–838. DOI 10.1007/s10529-011-0845-8. [Google Scholar] [CrossRef]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |