| Phyton-International Journal of Experimental Botany |
DOI: 10.32604/phyton.2022.019520
ARTICLE
Transcriptome Profiling of Barley Cultivar Hua 30 MDEC in Response to Agrobacterium Infection
1Biotech Research Institute, Shanghai Academy of Agricultural Sciences, Shanghai, 201403, China
2Shanghai Key Laboratory of Agricultural Genetics and Breeding, Shanghai, 201106, China
*Corresponding Authors: Chenghong Liu. Email: liuchenghong@saas.sh.cn; Longhua Zhou. Email: zhoulonghua@saas.sh.cn
#These authors contributed equally to this work
Received: 27 September 2021; Accepted: 16 November 2021
Abstract: Agrobacterium-mediated transformation has been widely used in plants. However, the mechanism in plant cells’ response to Agrobacterium infection was very complex. The mechanism of the determinants in host cell remains obscure, especially in barley, which is recalcitrant for Agrobacterium-mediated transformation. In the present study, microspore-derived embryogenic calli (MDEC) from barley elite cultivar were employed as unique subjects to characterize the mechanisms during the Agrobacterium infection process. Hua 30 MDEC can be successfully infected by Agrobacterium. RNA-sequencing at different infection points (0, 2, 6, 12, 24 hpi) was performed. The average expressional intensity of the whole genomics increased from 0 to 2 hpi, and then decreased subsequently. More upregulated than downregulated differentially expressed genes (DEGs) were counted at the same time. GO enrichment analysis showed that protein modification was significantly overrepresented in upregulated DEGs. Chromosome-related biological processes, gene expression and cellular metabolic processes were significantly overrepresented in downregulated DEGs. KEGG analysis showed that plant defense responses, phenylpropanoid biosynthesis and biosynthesis of amino acids were significantly enriched across the infection time course. Nine DEGs related to defense responses were identified. All DEGs were upregulated from 2 to 24 hpi. We speculate that these genes are possibly related to Agrobacterium infection. These findings will provide deep insights into the molecular events occurring during the process of Agrobacterium-mediated transformation.
Keywords: Barley; Agrobacterium-mediated transformation; microspore-derived embryogenic callus; transcriptome analysis
Agrobacterium tumefaciens possesses the ability to integrate self T-DNA into plant genomic [1]. The Agrobacterium tumefaciens-mediated genetic transformation system has been widely used in both basic research and breeding in plants, based on the low copy number of large segments of DNA into host genome DNA and low cost. The Agrobacterium-mediated transformation system has been established in many crops, such as soybean, rice, cotton, maize, wheat and barley [2]. However, the transformation efficiency is still a restricting factor in the Agrobacterium-mediated transformation. Based on the previous studies, the Agrobacterium infection process involves two steps: the attachment of the bacterium to host cells and then the transfer of the DNA-protein complex into the plant cell [3]. The plants have also evolved defense systems for the Agrobacterium infection, which may be an important factor influencing the infection efficiency of plant cells [4]. As a gram-negative bacterium, Agrobacterium tumefaciens possesses bacteria-derived compounds known as pathogen-associated molecular patterns (PAMP), which caused immune response termed as PAMP-triggered immunity (PTI) in host cells [5]. During infection, Agrobacterium transfer proteins (effectors) into the plant cell which activates a stronger defense response called effector-triggered immunity (ETI) [5]. The mechanisms in plant cells’ response to Agrobacterium infection were very complex [6,7]; any molecular interactions at these stages can affect the infection efficiency. Thus, it is critically important to get more knowledge on the molecular mechanisms involved in the plant-Agrobacterium interaction [8].
Microspores can be induced into an embryogenic callus in vitro. Meanwhile, the micropores-derived embryogenic callus (MDEC) possesses high regeneration ability [9]. The easily accessible populations of callus make microspore embryogenesis excellent for transformation. Barley is one of the important cereal crops, and it is also recognized as an ideal model plant. In previous reports, stable Agrobacterium-mediated transformations of barley microspore cultures have been reported [10]. But the technique is still genotypic dependent. This suggested that some factors affect the efficiency of Agrobacterium-mediated transformation in barley MDEC [11]. The improvement of the infection efficiency depends on further modification of the plant itself. Thus, a comprehensive understanding of the mechanisms underlying the transformation process is indispensable.
RNA-seq is one of the most important approaches to understand the molecular events that are involved in the transformation. Meanwhile, the high-quality reference genome assembly for barley makes it an even more efficient way to investigate the mechanism of the Barley-Agrobacterium interaction [12]. Transcriptome analyses have been performed to determine the mechanism in response to Agrobacterium in several studies. However, there is little information in barley MDEC response to Agrobacterium.
In this study, we isolated and cultured the microspores from the barley cultivar Hua 30 according to Lu’s protocol [13]. The Hua 30 MDEC can be successfully infected by the Agrobacterium strain LBA4404. With the aim to get a better understanding of the molecular mechanisms in barley MDEC response to Agrobacterium infection, RNA-seq was performed during Agrobacterium infection on the barley MDEC. Using time course transcriptome analysis to measure gene expressions throughout infection, different biological processes, pathways and genes were identified during the infection process. These findings will contribute to additional insights into the mechanisms during the Agrobacterium infection process in monocots plants.
2.1 Plant Materials and Microspore Culture
Barley (Hordeum vulgare L.) cv. Hua 30 was grown in the greenhouse under a 16 h/8 h light/dark cycle at 20 ± 2°C with 60% relative humidity and approximately 1000 mol m−2 s−1 light intensity. Microspores were isolated and cultured using the protocol described by Lu et al. [13]. After 21 days of culturing, the MDEC was used for Agrobacterium-mediated transformation.
2.2 Agrobacterium Infection on Barley MDEC
Agrobacterium strain LBA4404, harboring binary vector pCAMBIA1305.1, was used in the barley microspore induced callus transformation. When the OD600 of Agrobacterium was about 0.6–0.8, 200 µM acetosyringone was added to the solution and treated for 2 h. The Agrobacterium was centrifugated at 3270 g for 15 min, and the suspension was resuspended in the MDEC induction solution (with 200 µM acetosyringone) to an OD600 of about 0.6–0.8.
More than 100 barley MDEC were incubated with 50 ml Agrobacterium suspensions with a slight agitation (100 rpm) for 30 min and then dried by wind from the clean bench during 30 min. The MDEC was then placed on filter paper soaked with 5 ml liquid induction medium containing 200 µM acetosyringone in petri dishes, and co-cultivated at 23oC in a growth chamber. Samples were harvested for RNA seq (0, 2, 6, 12, 24 h) and GUS staining (3, 5 days). Three replicates were performed for each time point.
GUS staining was performed on the third and fifth day after co-cultivation with Agrobacterium. Callus pieces (more than 100) were randomly picked to stain them with X-Gluc solution at 37oC overnight. The number of blue staining calli was counted to calculate the efficiency. The frequency of transformation was calculated as follows: (GUS+ calli/Total calli) ×100%. The significant difference was analyzed by t-test.
2.4 RNA Extraction, Sequencing and Reads Mapping
The total RNA of each sample was isolated using TRIzol regent (Invitrogen, Carlsbad, CA, USA). The quality and integrity of RNA were determined by 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA). The cDNA libraries were constructed using IIIumina RNA Seq library kit (Illumina, Inc., USA). RNA data were acquired on Illumina HiSeqTM 4000 platform (Illumina) in Beijing Novogene Biotech Co., Ltd. (China). Raw data were filtered to obtain clean reads by removing adapters and excluding low-quality reads (Q < 30 and length < 50 bp). The clean reads were mapped onto the reference genome of barley. Differential expression analysis was performed using the R package DESeq. Genes with a P-value < 0.05, and (Log2 fold change) ≧1 were classed as significantly differentially-expressed genes (DEGs).
The GO and KEGG enrichment analysis were performed to assign possible functional categorization and metabolic pathways using AgriGO tool (http://systemsbiology.cau.edu.cn/agriGOv2/). Venn diagrams software (https://magic.novogene.com/) was used to sort the DEGs.
2.5 Quantitative RT-PCR Analysis
To validate the data of RNA-seq, 1 μg total RNA of each sample was used for qRT-PCR assay. First strand cDNA was synthesized according to the manufacturer’s recommended procedures (Toyobo). Primer pairs were designed using Primer 3 (http://www.ncbi.nlm.nih.gov/tools/primerblast/), and the selected gene name and primer information are listed in Supplementary Table 1. qRT-PCR was performed in the ABI 7500 fast system (Applied Biosystems, USA) using SYBR qPCR Mix (Toyobo). Hv-Actin was used as internal control [14]. ΔΔCT method was used to quantify the relative expression of identified genes [15].
3.1 Infection of Hua 30 MDEC by Agrobacterium
Microspores isolated from the anthers of Hua 30 were cultured under controlled aseptic conditions. After 21-day of culturing, the microspores derived embryogenic callus (MDEC) were obtained and subsequently used for Agrobacterium-mediated transformation (Fig. 1).
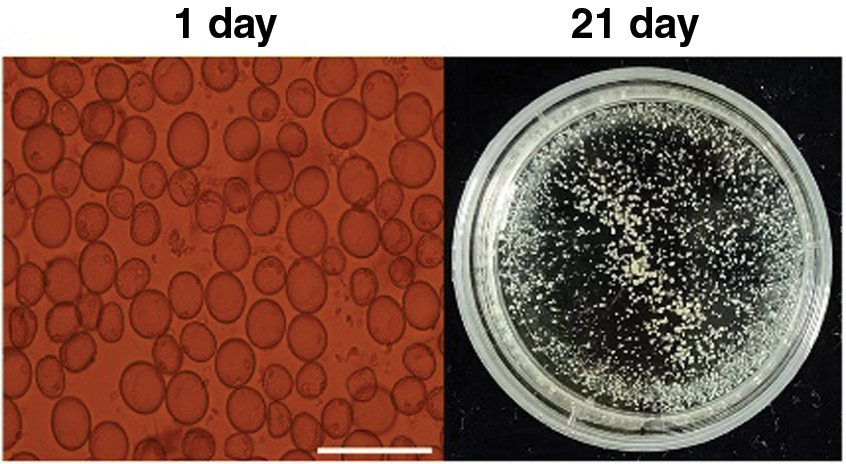
Figure 1: Microspores-derived embryogenic callus culturing process of Hua 30 at 1 day (single cells) and 21 days (calli). Bars = 125 μm
Infection efficiency was measured by counting blue spots after three- and five-day co-cultivation (Fig. 2A). As shown in Fig. 2B and Supplementary Table 2, the infection efficiency was 6.1% at 3 days post inoculation (dpi). At 5 dpi, infection efficiency increased to 32.3%. The results indicated that Hua 30 MDEC can be successfully infected by Agrobacterium strain LBA4404, but the infection efficiency was not high.
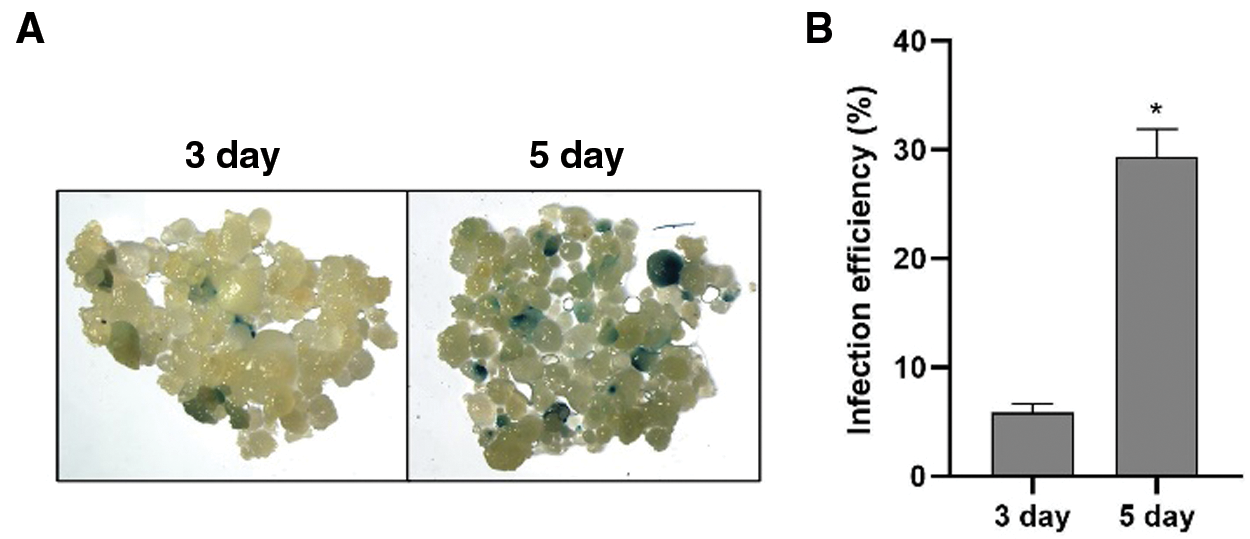
Figure 2: Infection efficiency of Hua 30 MDEC. A: GUS staining of Hua 30 MDEC at 3 and 5 dpi. B: Infection efficiency of Hua 30 MDEC at 3 and 5 dpi. The data are presented as average ± s.e. of three independent samples. * indicates significant differences at the level of P < 0.05
3.2 RNA Sequencing of Hua30 MDEC and Data Analysis
To investigate the mechanism of Hua 30 MDEC in response to Agrobacterium, RNA was extracted from Hua 30 MDEC inoculated with Agrobacterium at five time points (0, 2, 6, 12, 24 h). Fifteen digital gene expression libraries were constructed (Supplementary Table 3). On average, the raw reads generated from each library were 47.27 million. The clean-reads numbers generated from each library ranged from 44.28 to 51.55 million. The number of reads, ranging from 83.74% to 92.96% were mapped to the barley reference genome. ‘Phred value’ > 30 (Q30) of each library ranged from 93.39% to 94.64%. The transcriptome data were further used for bioinformatics analysis.
3.3 Identification of Differentially Expressed Genes (DEGs)
Whole genomic gene expression profiling of Hua 30 MDEC was analyzed during the Agrobacterium infection process. The results showed that the average expression level was highest at 2 hpi, and then decreased in the subsequently time points (Fig. 3).
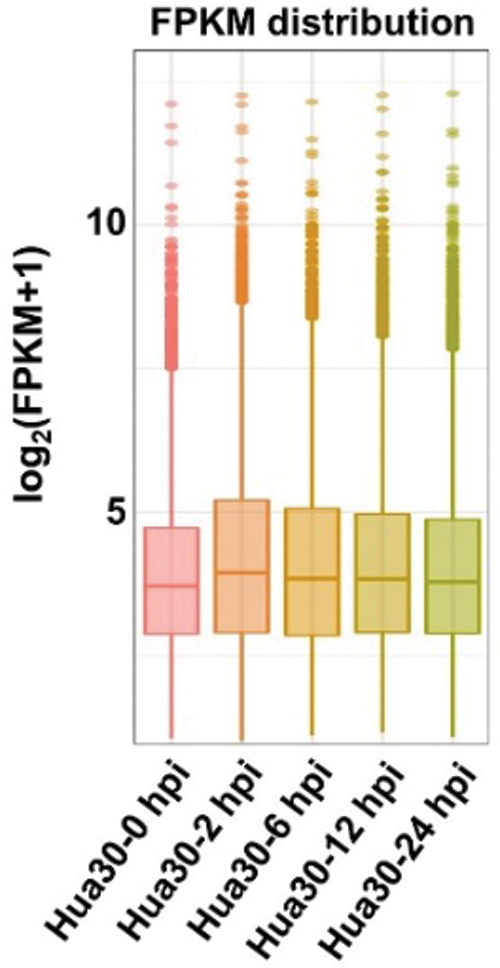
Figure 3: Expression profiling of the whole genomic gene expression under Agrobacterium infection at five time points (0, 2, 6, 12, and 24 hpi)
The number of DEGs at four infection time points was counted [0 hpi was used as control, P < 0.05, and log2(fold change) ≧ 1]. The DEGs were separated as upregulated- and downregulated- groups at each time point (Fig. 4). The number of the upregulated DEGs decreased from 2 to 24 hpi. The changing trend of downregulated DEGs was different from that of the upregulated DEGs. The number of downregulated DEGs increased from 2 to 6 hpi and then decreased from 6 to 24 hpi. More upregulated than downregulated DEGs were detected at any time. These results indicated that complex regulatory mechanisms were involved in Hua 30 MDEC response to Agrobacterium infection.
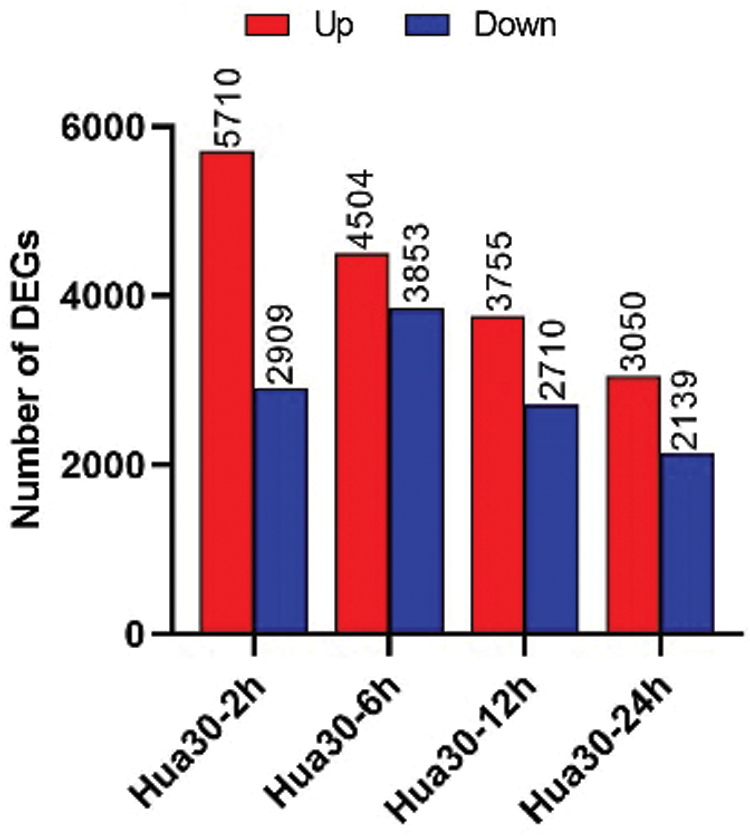
Figure 4: Number of DEGs at four time points (2, 6, 12, and 24 hpi). The y axis represents DEGs number. Red columns show the number of up-regulated DEGs, blue columns show the number of down-regulated DEGs
Venn diagrams were further constructed using DEGs at four time points (Fig. 5). 2382 upregulated DEGs were specially detected at 2 hpi, which were higher than those detected at any other time point (ranging from 360 to 664). At 6 hpi, more downregulated DEGs (1262) were specially detected as compared to those at any other time points. Meanwhile, 1353 up- and 502 down-regulated DEGs were detected at all four time points.
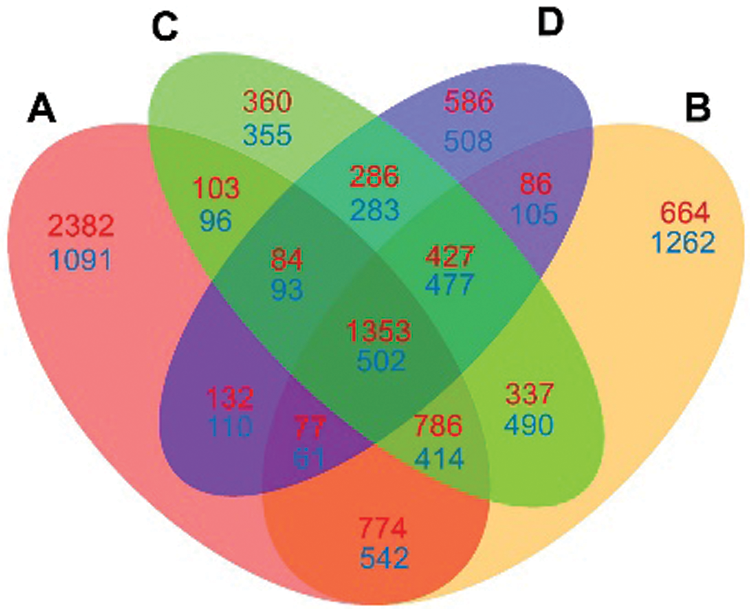
Figure 5: Venn diagram comparison of DEGs at four infection stages (2, 6, 12 and 24 hpi) compared each with 0 hpi. A: DEGs at 2 hpi; B: DEGs at 6 hpi; C: DEGs at 12 hpi; D: DEGs at 24 hpi. The numbers of up-regulated DEGs are shown in red, and the numbers of down-regulated DEGs are shown in blue
Nine DEGs were randomly selected to validate the RNA-seq data (Supplementary Table 4). There was a good coefficient (R2 = 0.80) between RNA-seq data and qRT-PCR analysis (Fig. 6), indicating that the RNA-seq data were reliable.
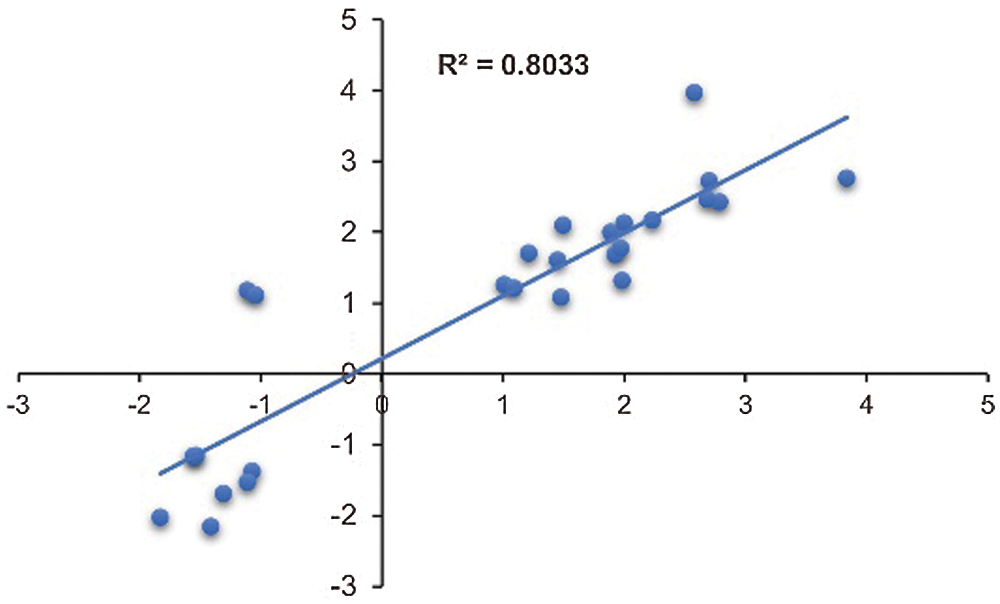
Figure 6: Validation of transcriptome data by qRT-PCR. Scatter plots indicate the transcriptional changes of qRT-PCR analysis and RNA-seq for 26 data points from 9 genes. The Pearson correlation coefficient (R2) was 0.80
3.4 Gene Ontology (GO) Enrichment Analysis of the DEGs in Hua 30
GO functional analysis was performed by assigning the DEGs across the infection time course. Biological processes were categorized into separate sets of upregulated and downregulated groups (Fig. 7, Supplementary Data s1). Most of the biological processes were enriched at 2 hpi among the upregulated groups. Protein ubiquitination and protein modification by small protein conjugation were significantly over-represented among the upregulated genes from 2 hpi to 24 hpi. In the downregulated group, function categories were enriched at 6 and 24 hpi. Chromosome-related biological processes (chromatin assembly, protein-DNA complex assembly and nucleosome assembly) were significantly enriched at 6 hpi. Categories related to gene expression (regulation of transcription, regulation of RNA biosynthetic process, regulation of gene expression and RNA biosynthetic-related process) and cellular metabolic processes (regulation of biosynthetic process, regulation of cellular metabolic process, regulation of nucleobase-containing compound and nitrogen compound, regulation of primary metabolic process) were significantly enriched at 24 hpi.

Figure 7: Selected overrepresented biological processes of GO categories across the time course. The categories are significantly enriched (P < 0.05) among either upregulated or downregulated genes. P-values from each category enrichment test have been negatively log10-transformed and plotted for each time point. Please increase the size of the text at the right of the figure
3.5 KEGG Enrichment Analysis of the DEGs in Hua 30
KEGG is a useful tool for the analysis of the roles of genes in various biological functions. We further performed KEGG enrichment using the DEGs from 4 infection time points. As shown in Table 1, the upregulated pathways were more than those downregulated. Five pathways were significantly enriched through the time course. One of them was plant-pathogen interaction, which was upregulated from 2 to 24 hpi, except at 12 hpi. Plant hormone signal transduction was downregulated at 2 hpi and upregulated from 12 to 24 hpi. Phenylpropanoid biosynthesis and phenylalanine, tyrosine and tryptophan biosyntheses were upregulated from 2 to 24 hpi. Biosynthesis of amino acids was upregulated from 6 to 24 hpi. Glutathione metabolism was upregulated from 6 to 12 hpi. Seven pathways, including DNA replication, carbon metabolism, arginine biosynthesis, citrate cycle (TCA cycle), cysteine and methionine metabolism, pyruvate metabolism and glycine, serine and threonine metabolisms were upregulated at 24 hpi. Protein processing at the endoplasmic reticulum was downregulated at 12 hpi. The results suggested that more upregulated than downregulated pathways were induced during Agrobacterium infection in barley MDEC.

3.6 DEGs Involved in Plant Defense Response
Plant immune responses play important roles during Agrobacterium- mediated transformation [16,17]. Plant defense was also significantly enriched in Hua 30 MDEC response to Agrobacterium. We further screened defense response genes that upregulated at all four infection time points (2, 6, 12, 24 hpi). Nine DEGs were identified in the plant-pathogen interaction pathway. The identified genes include two CDPK (HORVU1Hr1G071060 and HORVU4Hr1G019530), CML (HORVU5Hr1G066260), FLS2 (HORVU7Hr1G085790), FRK1 (HORVU6Hr1G001520), WRKY22 (HORVU7Hr1G080950), PR1 (HORVU5Hr1G106020), RIN4 (HORVU7Hr1G053300) and KCS (HORVU4Hr1G030810). Heat maps of these genes were constructed, and the transcript accumulation of all genes was more from 2 to 24 hpi than that at 0 hpi (Fig. 8).
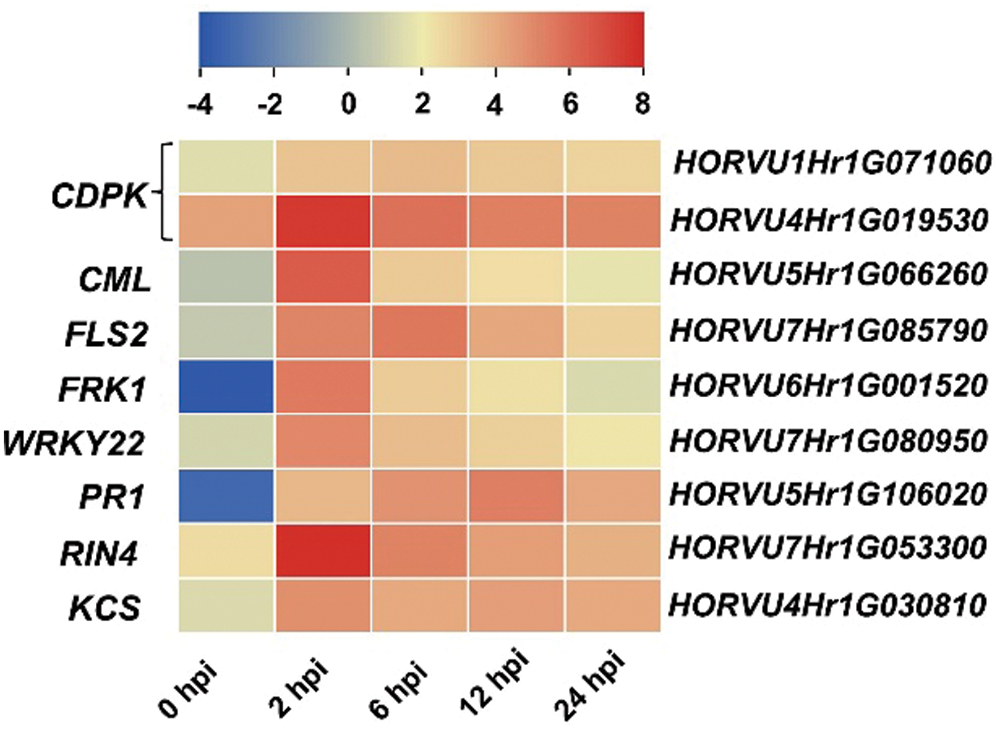
Figure 8: Heat maps of the nine identified genes across the infection process. The bottom color bars represent the log2 of FPKM for each gene, ranging from blue (–4) to red (8). A deeper color indicates more transcript accumulation. Please increase the size of the text at the right of the figure
Genotypic dependency is still the main limiting factor that influences the Agrobacterium infection efficiency. In the present study, the infection efficiency of Hua 30 MDEC increased from 3 to 5 dpi. This suggested that the genotype was compatible with the Agrobacterium strain LBA4404. Thus, RNA-seq was performed on Hua 30 MDEC during the infection process to detect more information on the molecular mechanism.
Our results showed that the Agrobacterium infection brought rapid changes in gene expression at the early stages of infection (2 hpi). This was similar to previous reports. The defense response genes were activated as early as 3–6 h in tobacco BY2 cell suspensions upon Agrobacterium infection [4]. Host defense response was stimulated by Agrobacterium as early as 3 hpi in Arabidopsis [16]. One study in rice embryogenic calli showed gene expression changes as early as 1 hpi upon Agrobacterium infection [18]. In addition, plant defense response was also stimulated quickly in the Hua 30 MDEC under Agrobacterium infection. These studies suggested that the MDEC cell senses the Agrobacterium immediately and subsequently influences the transcripts.
GO enrichment analysis showed that DEGs involved in protein ubiquitination and protein modification processes by small protein conjugation were upregulated across the infection process. This was consistent with a previous report in wheat, which showed that ubiquitin protein ligase was upregulated during the Agrobacterium infection process [19]. Protein ubiquitination was necessary for the Agrobacterium-mediated transformation [20]. Chromosome-related biological processes were overrepresented among downregulated DEGs at 6 hpi. Chromosome-related biological processes were important for Agrobacterium infection. In Arabidopsis, the overexpression of one histone gene (H2A-1) results in increased Agrobacterium-mediated transformation [21]. In rice, Tie et al. [18] showed that chromatin modification was repressed at the early time of infection in a recalcitrant genotype. There was a successful infection but low infection efficiency in Hua 30 MDEC. Our results suggested that the chromosome-related biological processes may be one influencing factor on the infection efficiency in barley MDEC.
Most plant secondary metabolites might play important roles during the Agrobacterium infection process. Phenylpropanoid biosynthesis was upregulated across the infection process. Zhou et al. showed that genes involved in this metabolic process are significantly upregulated upon Agrobacterium infection in wheat calli [19]. In soybean, genes involved in this metabolic process are also upregulated. But these genes’ expression was reduced when a-aminooxyacetic acid (AOA, transformation promotion factor) was added in tissue culture following transformation [22]. This suggested that the upregulation of phenylpropanoid biosynthesis pathway may negatively affect the Agrobacterium infection.
KEGG enrichment analysis showed that the plant-pathogen interaction was significantly upregulated during the infection process. As a soilborne pathogenic bacterium, Agrobacterium tumefaciens can activate plant defense and immune responses [23]. Defense genes were rapidly induced in tobacco BY2 cells upon Agrobacterium infection. But these defense genes were repressed at later stages of transformation by the transfer-competent instead of transfer-deficient Agrobacterium strains [4]. Similar results were reported in rice. Some defense genes were downregulated in the compatible genotype and upregulated in the recalcitrant genotype at the later stages of the Agrobacterium infection process [18]. In our results, the defense response (‘Plant-pathogen interaction’) was upregulated at the later stage (24 hpi) in Hua 30 MDEC. This suggested that the pathway may negatively affect the Agrobacterium infection.
Nine DEGs related defense responses were identified. All the genes exhibited high transcript accumulation after Agrobacterium infection. CDPK and CML (calcium-binding protein) were functional in the Ca2+ signal transduction [24]. Ca2+ was recognized as a conserved second messenger and principal mediator in the plant immune response. Six genes (Fls2, FRK1, WRKY22, PR1, RIN4 and KCS) were reported to function in the flg22-dependent defense response. The defense responses were always considered as PTI [25], which includes reactive oxygen species (ROS) burst, calcium flux and transcriptional reprogramming [5]. One PTI loss mutant showed increased susceptibility to Agrobacterium in Arabidopsis. This indicated that PTI plays an important role during the Agrobacterium transformation process. Thus, we speculate that the identified genes are related to Agrobacterium infection. The function of these genes needs further investigation.
In conclusion, this study showed the response of barley MDEC under Agrobacterium infection. Upon Agrobacterium infection, more upregulated than downregulated DEGs were counted at the same time point. Genes involved in protein modification, defense response, phenylpropanoid biosynthesis and biosynthesis of amino acids were upregulated. Meanwhile, genes involved in chromosome-related biological processes, gene expression and cellular metabolic processes were downregulated. Nine genes related to defense response were identified, which may affect the Agrobacterium infection. The finding will provide additional insights of the molecular events occurring during the process of Agrobacterium-mediated transformation.
Availability of Data and Materials: The datasets generated and analyzed during the current study are available in the National Center for Biotechnology Information: Submission ID: SUB10402909; BioProject ID: PRJNA764780.
Funding Statement: This work was supported by the Shanghai Agriculture Applied Technology Development Program, China (Grant No. G2016060102), National Key Research and Development Program of China (2018YFD1000702-5), and Earmarked Fund for China Agriculture Research System (CARS-05-01A-02).
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
1. Matveeva, T. V. (2018). Agrobacterium-mediated transformation in the evolution of plants. Current Topics in Microbiology and Immunology, 418(2), 421–441. DOI 10.1007/82_2018_80. [Google Scholar] [CrossRef]
2. Wang, K. (2015). Agrobacterium protocols. In: Methods in molecular biology, vol. 1224. DOI 10.1007/978-1-4939-1658-0. [Google Scholar] [CrossRef]
3. Willig, C. J., Duan, K., Zhang, Z. J. (2018). Transcriptome profiling of plant genes in response to Agrobacterium tumefaciens-mediated transformation. Current Topics in Microbiology and Immunology, 418, 319–348. DOI 10.1007/82_2018_115. [Google Scholar] [CrossRef]
4. Veena, J. H., Doerge, R. W., Gelvin, S. B. (2003). Transfer of T-DNA and Vir proteins to plant cells by Agrobacterium tumefaciens induces expression of host genes involved in mediating transformation and suppresses host defense gene expression. Plant Journal, 35(2), 219–236. DOI 10.1046/j.1365-313x.2003.01796.x. [Google Scholar] [CrossRef]
5. Yuan, M., Ngou, B. P. M., Ding, P., Xin, X. F. (2021). PTI-ETI crosstalk: An integrative view of plant immunity. Current Opinion in Plant Biology, 62, 102030. DOI 10.1016/j.pbi.2021.102030. [Google Scholar] [CrossRef]
6. Gelvin, S. B. (2017). Integration of Agrobacterium T-DNA into the plant genome. Annual Review of Genetics, 51(1), 195–217. DOI 10.1146/annurev-genet-120215-035320. [Google Scholar] [CrossRef]
7. Hwang, H. H., Yu, M., Lai, E. M. (2017). Agrobacterium-mediated plant transformation: Biology and applications. Arabidopsis Book, 15, e0186. DOI 10.1199/tab.0186. [Google Scholar] [CrossRef]
8. Altpeter, F., Springer, N. M., Bartley, L. E., Blechl, A., Brutnell, T. P. et al. (2016). Advancing crop transformation in the era of genome editing. Plant Cell, 28, 1510–1520. DOI 10.1105/tpc.16.00196. [Google Scholar] [CrossRef]
9. Ferrie, A., Caswell, K. L. (2011). Isolated microspore culture techniques and recent progress for haploid and doubled haploid plant production. Plant Cell, Tissue and Organ Culture, 104(3), 301–309. DOI 10.1007/s11240-010-9800-y. [Google Scholar] [CrossRef]
10. Kumlehn, J., Serazetdinova, L., Hensel, G., Becker, D., Loerz, H. (2006). Genetic transformation of barley (Hordeum vulgare L.) via infection of androgenetic pollen cultures with Agrobacterium tumefaciens. Plant Biotechnology Journal, 4(2), 251–261. DOI 10.1111/j.1467-7652.2005.00178.x. [Google Scholar] [CrossRef]
11. Otto, I., Müller, A., Kumlehn, J. (2015). Barley (Hordeum vulgare L.) transformation using embryogenic pollen cultures. Methods in Molecular Biology, 1223, 85–99. DOI 10.1007/978-1-4939-1695-5_7. [Google Scholar] [CrossRef]
12. Mascher, M., Gundlach, H., Himmelbach, A., Beier, S., Twardziok, S. O. et al. (2017). A chromosome conformation capture ordered sequence of the barley genome. Nature, 544(7651), 427–433. DOI 10.1038/nature22043. [Google Scholar] [CrossRef]
13. Lu, R. J., Wang, Y. F., Sun, Y. F., Shan, L. L., Huang, J. (2008). Improvement of isolated microspore culture of barley (Hordeum vulgare L.The effect of floret co-culture. Plant Cell, Tissue and Organ Culture, 93(1), 21–27. DOI 10.1007/s11240-008-9338-4. [Google Scholar] [CrossRef]
14. Goodall, A. J., Kumar, P., Tobin, A. K. (2013). Identification and expression analyses of cytosolic glutamine synthetase genes in barley (Hordeum vulgare L.). Plant and Cell Physiology, 54(4), 492–505. DOI 10.1093/pcp/pct006. [Google Scholar] [CrossRef]
15. Livak, K. J., Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods, 25(4), 402–408. DOI 10.1006/meth.2001.1262. [Google Scholar] [CrossRef]
16. Lee, C. W., Efetova, M., Engelmann, J. C., Kramell, R., Wasternack, C. et al. (2009). Agrobacterium tumefaciens promotes tumor induction by modulating pathogen defense in Arabidopsis thaliana. Plant Cell, 21(9), 2948–2962. DOI 10.1105/tpc.108.064576. [Google Scholar] [CrossRef]
17. Nonaka, S., Sugawara, M., Minamisawa, K., Yuhashi, K. I., Ezura, H. (2008). 1-Aminocyclopropane-1-carboxylate deaminase enhances Agrobacterium tumefaciens-mediated gene transfer into plant cells. Applied and Environmental Microbiology, 74(8), 2526–2528. DOI 10.1128/AEM.02253-07. [Google Scholar] [CrossRef]
18. Tie, W. W., Fei, Z., Lei, W., Xie, W. B., Hao, C. et al. (2012). Reasons for lower transformation efficiency in indica rice using Agrobacterium tumefaciens-mediated transformation: Lessons from transformation assays and genome-wide expression profiling. Plant Molecular Biology, 78(1–2), 1–18. DOI 10.1007/s11103-011-9842-5. [Google Scholar] [CrossRef]
19. Zhou, X., Wand, K., Lv, D., Wu, C., Ye, X. (2013). Global analysis of differentially expressed genes and proteins in the wheat callus infected by Agrobacterium tumefaciens. PLoS One, 8(11), e79390. DOI 10.1371/journal.pone.0079390. [Google Scholar] [CrossRef]
20. Anand, A., Rojas, C. M., Tang, Y., Mysore, K. S. (2012). Several components of SKP1/Cullin/F-box E3 ubiquitin ligase complex and associated factors play a role in Agrobacterium-mediated plant transformation. New Phytologist, 195(1), 203–216. DOI 10.1111/j.1469-8137.2012.04133.X. [Google Scholar] [CrossRef]
21. Tenea, G. N., Spantzel, J., Lee, L. Y., Zhu, Y. M., Lin, K. et al. (2009). Overexpression of several Arabidopsis histone genes increases Agrobacterium-mediated transformation and transgene expression in plants. Plant Cell, 21(10), 3350–3367. DOI 10.1105/tpc.109.070607. [Google Scholar] [CrossRef]
22. Zhang, Y. M., Liu, Z. H., Yang, R. J., Li, G. L., Guo, X. L. et al. (2016). Improvement of soybean transformation via Agrobacterium tumefaciens methods involving α-aminooxyacetic acid and sonication treatments enlightened by gene expression profile analysis. Plant Cell Reports, 35, 1259–1271. DOI 10.1007/s00299-016-1958-2. [Google Scholar] [CrossRef]
23. Gohlke, J., Deeken, R. (2014). Plant responses to Agrobacterium tumefaciens and crown gall development. Frontiers in Plant Science, 5(e3306), 155. DOI 10.3389/fpls.2014.00155. [Google Scholar] [CrossRef]
24. Kudla, J., Becker, D., Grill, E., Hedrich, R., Hippler, M. et al. (2018). Advances and current challenges in calcium signaling. New Phytologist, 218(2), 414–431. DOI 10.1111/nph.14966. [Google Scholar] [CrossRef]
25. Peng, Y., van Wersch, R., Zhang, Y. (2018). Convergent and divergent signaling in PAMP-triggered immunity and effector-triggered immunity. Molecular Plant-Microbe Interactions, 31(4), 403–409. DOI 10.1094/MPMI-06-17-0145-CR. [Google Scholar] [CrossRef]
Supplementary Table 1: Primer information for qRT-PCR
Supplementary Table 2: Infection efficiency of Hua 30 MDEC
Supplementary Table 3: An overview of sequencing and assembly of Hua 30 MDEC
Supplementary Table 4: Validation of the transcriptome data by qRT-PCR
Supplementary Data s1: Data of significant enriched Biological Process of GO categories across time-course comparisons in Hua 30 MDEC. The categories are separated as upregulate and downregulate groups
Supplementary Data s2: Data of pathways by KEGG enrichment in Hua 30 MDEC at 2, 6, 12 and 24 hpi. The pathways are separated as upregulate and downregulate groups
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |