| Phyton-International Journal of Experimental Botany |
DOI: 10.32604/phyton.2022.021334
ARTICLE
Molecular Interaction Study to Explore the Nigella sativa Bioactive Components as an Inhibitor of Peptide Deformylase to Inhibit the Xanthomonas oryzae pv. oryzae via Applying Computational Approach
Department of Biology, College of Sciences and Humanities, Prince Sattam bin Abdulaziz University, Al-Kharj, 11942, Saudi Arabia
*Corresponding Author: Pravej Alam. Email: alamprez@gmail.com
Received: 09 January 2022; Accepted: 21 February 2022
Abstract: Bacterial leaf blight (BLB) caused by Xanthomonas oryzae pv. oryzae (Xoo) is one of the most damaging diseases to rice across the world. Various chemicals have been employed so far for the management of bacterial leaf blight. On the other hand, these compounds are damaging to the ecosystem and have an impact on non-target species such as humans and animals. As a result, there is a need to create a new natural inhibitor for BLB management. Deformylase (PDF) enzyme is present in all eubacteria and its necessity in bacterial protein synthesis reveals it as an attractive target for drug development. In this study, the active components of Nigella sativa have been selected based on their previously reported antimicrobial activity and screened on the active site of bacterial PDF by the in silico art of techniques. Among these compounds, dithymoquinone and p-cymene strongly bind with the PDF enzyme with binding energy values of 7.77 kcal/mol and 7.26 kcal/mol, respectively, which is comparatively higher than the control compound (−6.73 kcal/mol). Hence, the “dithymoquinone-PDF” and “p-cymene-PDF” complexes were selected for further study, and their stability was assessed by molecular dynamic (MD) simulation. In MD simulation, both selected compounds exhibited steady-state interaction with PDF for 20 ns. It has been hypothesized that p-cymene and dithymoquinone inhibit peptide deformylase and could be used as antibacterials or pesticides against Xoo against the BLB disease.
Keywords: Nigella sativa; peptide deformylase; molecular dynamic; dithymoquinone
Abbreviations
| Xoo | Xanthomonas oryzae pv. oryzae |
| BLB | bacterial leaf blight |
| DPF | peptide deformylase |
| BE | binding energy |
| MD | molecular dynamic |
| RMSD | root mean square deviation |
| Rg | radius of gyration |
| SASA | solvent accessible surface area |
| RMSF | root mean square fluctuation |
With the population increase, catering of food needs and satisfying hunger are fast emerging as the biggest challenge worldwide, especially in the backdrop of the scarcity of cultivable and fertile lands. Compounding the problem further is the loss of huge amounts of crops due to diseases caused by various microorganisms. Rice bacterial leaf blight (BLB) is caused by Xanthomonas oryzae pv. oryzae (Xoo). It is characterized by the presence of a white yellow rust in the crop along the veins, leaf margins, and leaf blades, which can also spread to the sheath and cause a decline in total dry weight, and is responsible for a 50% annual crop yield loss [1].
For bacteria to survive, the removal of the N-formyl moiety from the N-terminal methionine results in the formation of a mature protein. This step is catalyzed by the peptide deformylase (PDF) enzyme encoded by the def gene and is present in all bacterial pathogens [2]. PDF blockade inhibits protein synthesis (similar to the mechanism of action of antimicrobial agents like tetracyclines) [3].
Studies have documented that blocking PDF enzymes hinders bacterial survival and growth [4–6]. Therefore, the BLB disease of rice can be controlled by inhibiting the PDF. Various chemicals like 30% chlorine-containing bleaching powder have been employed for the management of BLB [7]. However, long-term use of these chemicals has deleterious and adverse effects not only on the environment but also on humans and animals, thereby warranting their cautious use [8]. Thus, keeping in view environmental safety and avoiding harm to humans and animals, the current need is to develop and research new and preferentially natural inhibitors in the management of BLB.
The primary active ingredients of Nigella sativa include carvacrol, thymoquinone, thymol, limonene, p-cymene, carvone, trans-anethole, thujene, pinene, thymohydroquinone, longifoline, dithymoquinone, and pinene [9,10]. N. sativa and its components have been thoroughly investigated for their pharmacological potential, including antibacterial ability against a variety of bacterial infections [11–14]. However, the potential of these compounds to inhibit the PDF enzyme has yet to be investigated. In the present study, we screened the N. Sativa active components for their potential interaction against the PDF enzyme by the molecular docking and molecular dynamic simulations. This study may help in the possible design of pharmacological compounds against the PDF to overcome the BLB of crops and also contribute in the novel antibacterial/pesticides development.
2.1 Structures of Ligands and Proteins are Retrieved
3D structures of N. sativa active components, including the reference compound actinonin, were obtained from the PubChem database. However, the 3-D structure of PDF from Xoo was retrieved from the Protein Data Bank (PDB ID: 5CY8).
2.2 Molecular Docking Interaction Study
The molecular interaction of compounds on the binding site of PDF was performed by ‘Autodock4.2’ individually [15]. The energy of each compound was minimized by applying the MMFF94 force field. AutoDock tools were used to add the hydrogen atoms, solvation constraints, and Kollman charges. Grid points were kept at 404040 with 0.375 spacing. However, the co-ordinates were set as 7.589 (x), −15.291 (y), and −2.049 (z), respectively. The Solis and Wets local search and LGA (Lamarckian genetic algorithm) approaches were used for docking each of the compounds with DPF. A hundred different runs were used for each docking, with an end limit of 2,500,000 energy calculations. For further analysis, the compounds having lower binding energy (BE) relative to actinonin (the reference molecule) were selected. Molecular interactions (hydrophobic interactions and hydrogen bonds) between the compounds and PDF complexes were analyzed by LIGPLOT + Version v.1.4.5 [16].
2.3 Molecular Dynamic (MD) Simulation Study
On the GROMACS 5.1.5 platform [17], MD simulations were run on a native protein structure (PDB ID: 5CY8) in innate and docked complex form with ligand structure, using the CHARMM27 all atom force field [18]. The ligand topology files were created using the CHARMM all atoms force field [19] via the SwissParam service (http://www.swissparam.ch/). The system was simulated in a triclinic box with protein atoms separated by 1.0 to 1.5 nm from the box wall dimensions while preserving periodic boundary conditions [20]. The system was equilibrated by placing position restraints on the complex and running simulations using conventional NVT and NPT ensembles [21].
BLB caused by Xoo is one of the most detrimental diseases to crops globally. More than 30 compounds, including antibacterial agents, have been employed to manage the BLB of crops to protect them from these pathogens. Among them, some antibiotics have broad-spectrum activity and hinder the growth of bacteria (Xoo) [22], and have exhibited variable efficacy against Xoo. Hence, to find possible antibacterial agents against Xoo, the active components of N. sativa have been selected based on their antibacterial properties.
The choice of PDF was based on the fact that it is found in all eubacteria and is required for protein synthesis, making it a suitable target for therapeutic development [4,23,24]. In this study, dithymoquinone and p-cymene strongly interact with the PDF enzyme more strongly than the other compounds. P-cymene interacted with PDF through 10 amino acid residues, namely, G44, V45, G46, R68, Y69, G98, C99, L100, G104, and L105, while R68, Y69, W96, E97, G98, G104, L105, R106, and D164 residues of PDF interacted with dithymoquinone (Figs. 1 and 2). The binding energy (BE) of p-cymene and dithymoquinone with PDF was found to be 7.77 kcal/mol and 7.26 kcal/mol, respectively (Table 1). The active site residues of PDF have been predicted as G44, V45, G46, Q51, R68, Y69, W96, E97, G98, C99, L100, I102, P103, G104, L105, R106, F134, R137, V138, H141, E142, H145, and D164. Consistent with this, in the present study, these residues of PDF were also found to interact with p-cymene and dithymoquinone.

Figure 1: Interaction of p-cymene (stick representation) with peptide deformylase

Figure 2: Interaction of dithymoquinone (stick representation) with peptide deformylase

The amino acid residues E42, G44, V45, Q51, R68, Y69, E97, G98, C99, L100, G104, L105, R106, F134, R137, V138, H141, H145, and D164 of PDF interacted with the control compound actinonin (Fig. 3). R68, Y69, G98, G104, and L105 were the common interacting residues of PDF with p-cymene, dithymoquinone, and actinonin (Figs. 1–3). The BE of actinonin with PDF was found as −6.12 kcal/mol (Table 1).

Figure 3: Interaction of actinonin (stick representation) with peptide deformylase
The hydrophobic interaction assists in explaining the potency of the inhibitor to the target protein and has a vital role in the stability of the inhibitor-protein complex. In this study, LigPlot analysis showed that R68 and G98 were the common hydrophobic interacting residues of PDF with p-cymene, dithymoquinone, and actinonin (Figs. 4a–4c). Interestingly, in a study, these residues of PDF have also been reported to form hydrophobic interactions with the inhibitors [24–26].
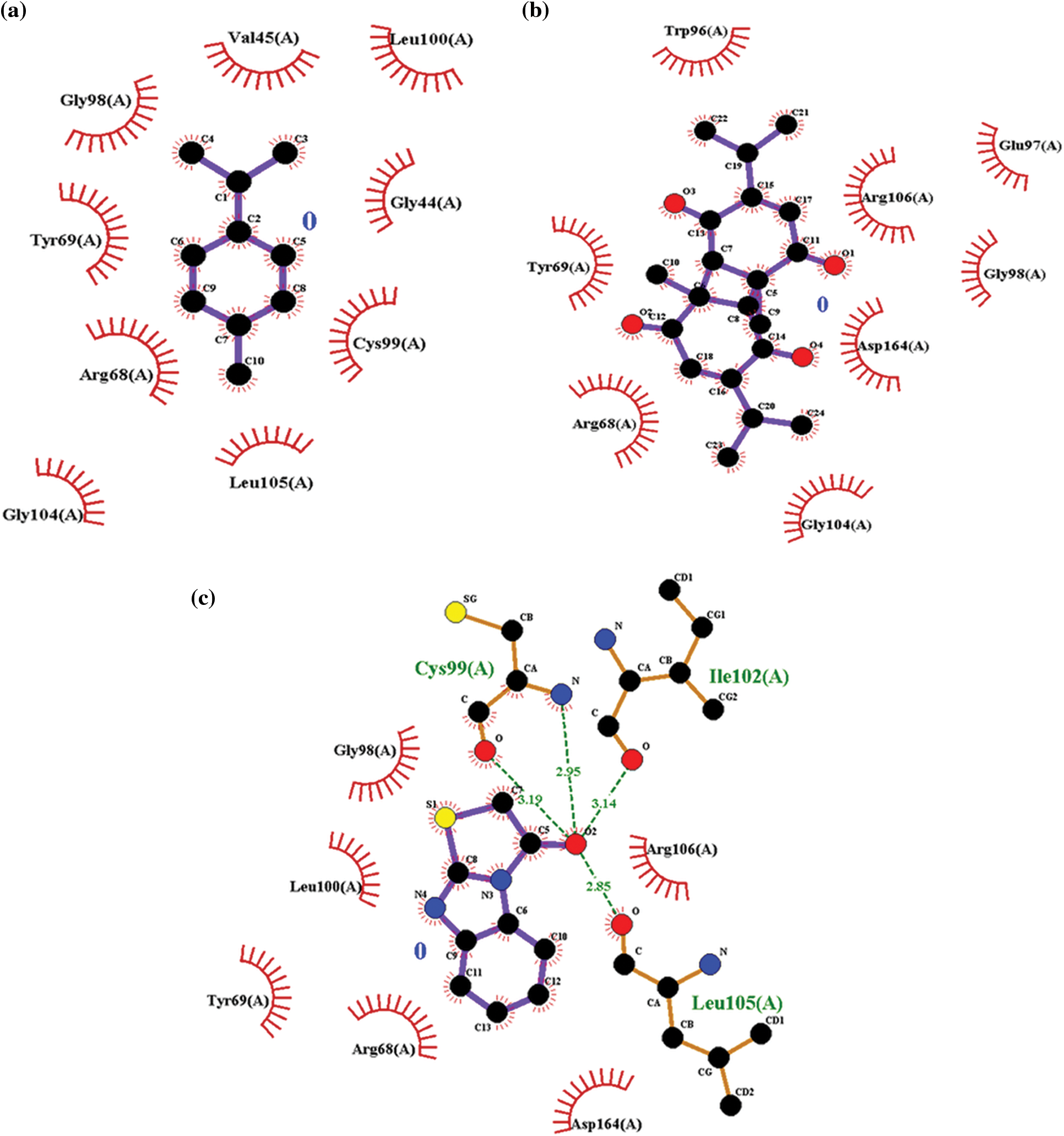
Figure 4: (a) Ligplot analysis showing the hydrogen and hydrophobic interacting residues of peptide deformylase with p-cymene. (b) Ligplot analysis showing the hydrogen and hydrophobic interacting residues of peptide deformylase with dithymoquinone. (c) Ligplot analysis showing the hydrogen and hydrophobic interacting residues of peptide deformylase with actinonin
Recently, a computational approach has been described to develop a potential PDF inhibitor to combat drug-resistant bacterial pathogens [27]. Computational docking is a useful tool which is employed in the drug discovery process to find out the binding potency of inhibitors to their target proteins [28,29], and a high (negative) BE measures the strength of binding between inhibitor-enzyme complexes [30,31]. According to the BE, dithymoquinone and p-cymene bind to PDF more strongly than the other compounds and significantly better than the reference compound 56 V, indicating that these compounds might be used as antibacterials or pesticides against the Xoo.
In addition to molecular docking, MD simulation experiments were conducted in addition to molecular docking to explain the dynamic behavior of the “dithymoquinone-PDF” and “p-cymene-PDF” complexes over time in a solvated environment. The root mean square deviation (RMSD), radius of gyration (Rg), solvent accessible surface area (SASA), number of hydrogen bonds, and root mean square fluctuation (RMSF) maintained throughout the simulation time, as well as the variance of the secondary structure pattern between the protein and its complexes, are all examined in the simulation study. Three simulations were run independently using the native protein alone and in complex with ligands for a total of 20 ns (dithymoquinone and p-cymene). The primary reason for running MD simulations is to better understand the binding affinities and time-bound stability of ligands to proteins bound complexes. It was evident from the RMSD plot (Figs. 5a and 6a) that the ‘dithymoquinone-PDF’ and ‘p-cymene-P DF’ complexes reached equilibrium approximately at 4.5 to 5.0 ns time and the remaining were shown to have a stable trajectory with minimal deviation in the 0.10 to 0.15 nm RMSD range, representing structural flexibility of protein is being reserved while in complex with ligands rather than in free form. Throughout the simulation run the unbound protein shows stable trajectories, whereas, ligand bound proteins reached equilibrium only after initial fluctuations. Rg considers the varied masses calculated to root mean square distances considering the central axis of rotation. The Rg figure (Figs. 5b and 6b) examines the capability, shape, and folding of the whole trajectory during the simulation at each time step. The Rg values of all protein entries and their related ligand complexes are comparable, ranging from 0.20 to 0.25 nm. The region surrounding the hydrophobic core produced between protein-ligand complexes is measured by SASA (Figs. 5c and 6d). SASA values were found to be consistent, ranging between 10 and 20 nm2 areas. The H-bonds that occur during the molecular docking research are examined during the course of the simulation. All intermolecular H-bonds between ligands and proteins were only taken into account during the analysis and shown appropriately (Fig. 6c); no hydrogen bonding between p-cymene and PDF protein was identified, hence none was traced throughout the simulation run. The plot shows that the number of H-bonds formed throughout the simulation runs is consistent with the molecular docking research, and that only a few bonds were broken and repaired at the same time. The RMSF figure (Figs. 5d and 6e) shows residue-by-residue changes, with essential interacting amino acids rigidified in the complex form as compared to the innate state of protein.
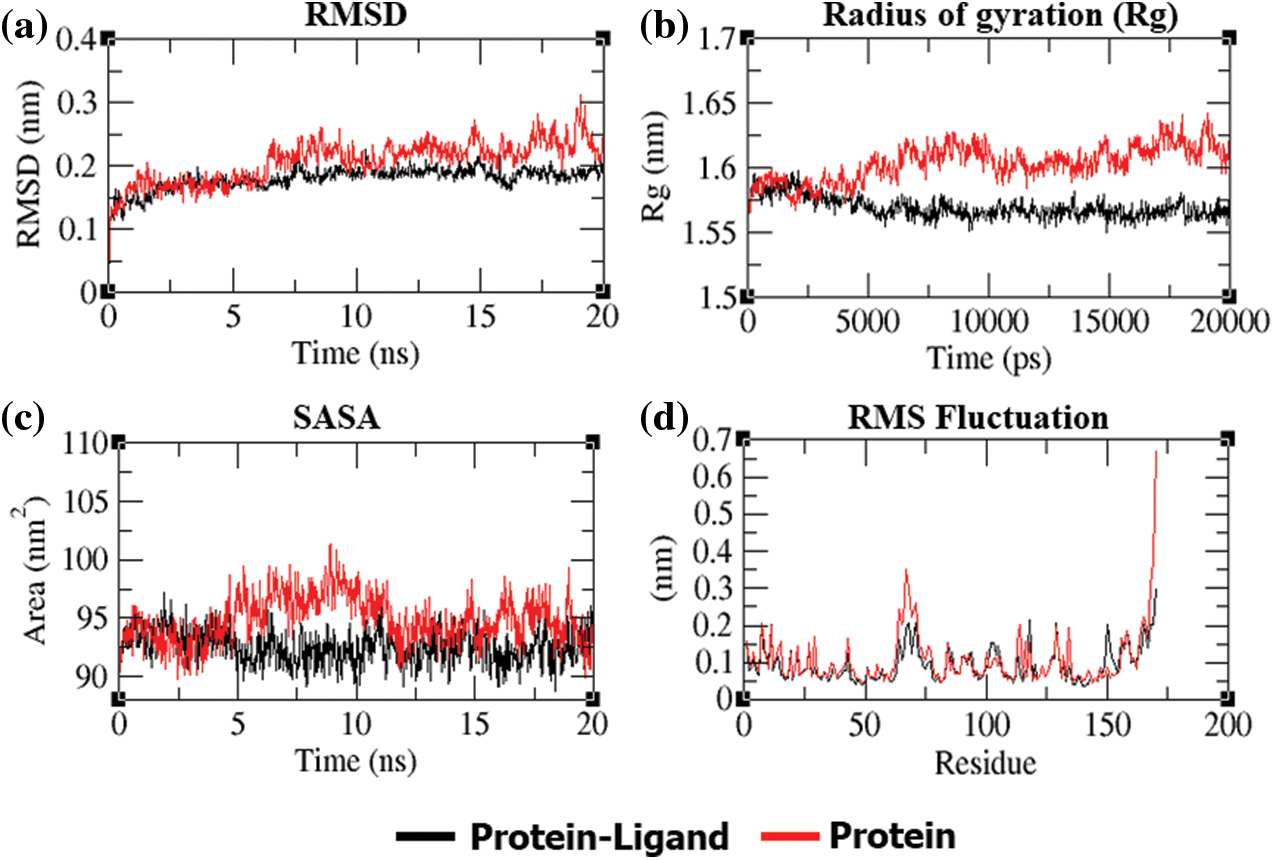
Figure 5: (a) RMSD plot of PDF backbone without p-cymene complex variation throughout a 20-ns period. (b) Rg of the PDF backbone in its free and p-cymene complex states during the course of the simulation, where nm = nanometer and ps = picosecond. (c) SASA is defined, with SASA (nm) as the ordinate and time as the abscissa (ns). (d) Average RMSF plot of PDF and p-cymene by residue
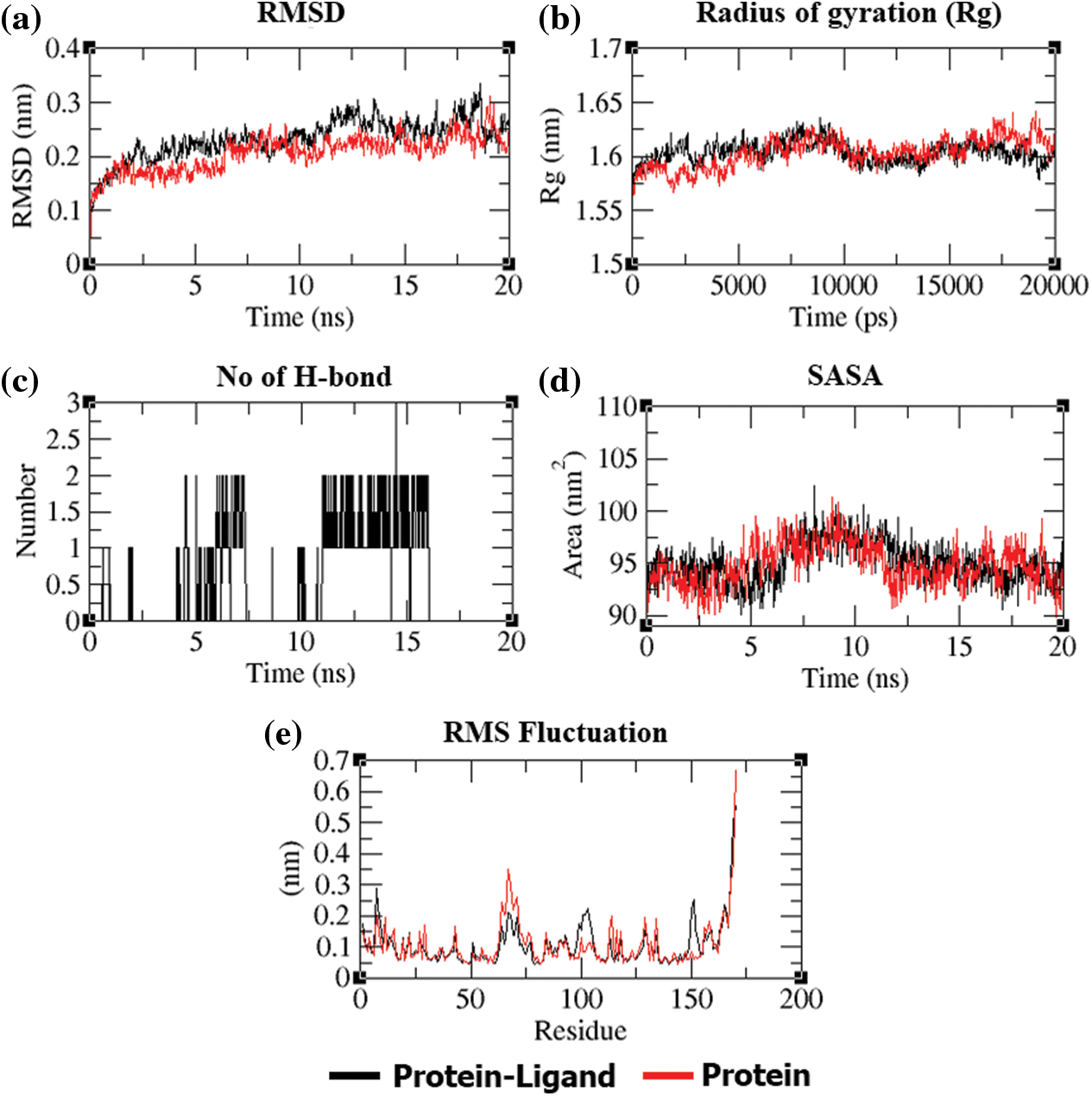
Figure 6: (a) Over a 20-ns period, the RMSD plot of the PDF backbone alone and with dithymoquinone complex deviation. (b) Rg of the PDF backbone in its free and dithymoquinone complex states during the course of the simulation, where nm = nanometer and ps = picosecond. (c) Over the simulation duration, hydrogen bonds form between PDF and dithymoquinone. (d) SASA is defined, with SASA (nm) as the ordinate and time as the abscissa (ns). (e) Average RMSF plot of PDF and dithymoquinone by residue
In summary, probably to the best of our knowledge, this is the first time that the bioactive constituents of N. sativa have been screened against the PDF enzyme. Dithymoquinone and p-cymene were found to bind strongly with PDF in comparison to other studied compounds as well as the reference compound (actinonin). The MD simulation studies revealed that dithymoquinone and p-cymene have steady-state interactions with the PDF. It is proposed that p-cymene and dithymoquinone could inhibit the PDF and thus could be utilized as potential antibacterial or pesticide agents against Xoo to overcome the BLB disease of the crops.
Availability of Data and Materials: The data presented in this study are available in this article.
Funding Statement: The authors extend their appreciation to the Deputyship for Research & Innovation, Ministry of Education in Saudi Arabia for funding this research work through the Project No. (IF-PSAU- 2021/01/18732).
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
1. Jiang, N., Yan, J., Liang, Y., Shi, Y., He, Z. et al. (2020). Resistance genes and their interactions with bacterial blight/Leaf streak pathogens (Xanthomonas oryzae) in rice (Oryza sativa L.)–An updated review. Rice, 13(1), 3. DOI 10.1186/s12284-019-0358-y. [Google Scholar] [CrossRef]
2. Piatkov, K. I., Vu, T. T., Hwang, C. S., Varshavsky, A. (2015). Formyl-methionine as a degradation signal at the N-termini of bacterial proteins. Microbial Cell, 2(10), 376–393. DOI 10.15698/mic. [Google Scholar] [CrossRef]
3. Guay, D. R. (2007). Drug forecast - The peptide deformylase inhibitors as antibacterial agents. Therapeutics and Clinical Risk Management, 3(4), 513–525. [Google Scholar]
4. Gao, J., Liang, L., Zhu, Y., Qiu, S., Wang, T. et al. (2016). Ligand and structure-based approaches for the identification of peptide deformylase inhibitors as antibacterial drugs. International Journal of Molecular Sciences, 17(7), 1141. DOI 10.3390/ijms17071141. [Google Scholar] [CrossRef]
5. O’Dwyer, K., Hackel, M., Hightower, S., Hoban, D., Bouchillon, S. et al. (2013). Comparative analysis of the antibacterial activity of a novel peptide deformylase inhibitor, GSK1322322. Antimicrobial Agents Chemother, 57(5), 2333–2342. DOI 10.1128/AAC.02566-12. [Google Scholar] [CrossRef]
6. Goemaere, E., Melet, A., Larue, V., Lieutaud, A., Alves de Sousa, R. et al. (2012). New peptide deformylase inhibitors and cooperative interaction: A combination to improve antibacterial activity. Journal of Antimicrobial Chemotherapy, 67(6), 1392–400. DOI 10.1093/jac/dks058. [Google Scholar] [CrossRef]
7. Chand, T., Singh, N., Singh, H., Thind, B. S. (1979). Field efficacy of stable bleaching powder to control bacterial blight of rice. International Rice Research Newsletter, 4(4), 12–13. [Google Scholar]
8. Meena, R. S., Kumar, S., Datta, R., Lal, R., Vijayakumar, V. (2020). Impact of agrochemicals on soil microbiota and management: A review. Land, 9, 34. DOI 10.3390/land9020034. [Google Scholar] [CrossRef]
9. Srinivasan, K. (2018). Cumin (Cuminum cyminum) and black cumin (Nigella sativa) seeds: Traditional uses, chemical constituents, and nutraceutical effects. Food Quality and Safety, 2, 1–16. DOI 10.1093/fqsafe/fyx031. [Google Scholar] [CrossRef]
10. Shokri, H. (2016). A review on the inhibitory potential of Nigella sativa against pathogenic and toxigenic fungi. Avicenna Journal of Phytomedicine, 6(1), 21–33. [Google Scholar]
11. Mouwakeh, A., Kincses, A., Nové, M., Mosolygó, T., Mohácsi-Farkas, C. et al. (2019). Nigella sativa essential oil and its bioactive compounds as resistance modifiers against Staphylococcus aureus. Phytotherapy Research, 33(4), 1010–1018. DOI 10.1002/ptr.6294. [Google Scholar] [CrossRef]
12. Chaieb, K., Kouidhi, B., Jrah, H., Mahdouani, K., Bakhrouf, A. (2011). Antibacterial activity of thymoquinone, an active principle of Nigella sativa and its potency to prevent bacterial biofilm formation. BMC Complementary & Alternative Medicine, 11, 29. DOI 10.1186/1472-6882-11-29. [Google Scholar] [CrossRef]
13. Hayatdavoudi, P., Khajavi Rad, A., Rajaei, Z., Hadjzadeh, M. A. (2016). Renal injury, nephrolithiasis and Nigella sativa: A mini review. Avicenna Journal of Phytomedicine, 6(1), 1–8. [Google Scholar]
14. Dhanasekaran, S. (2019). Alteration of multi-drug resistance activities by ethanolic extracts of Nigella sativa against urinary pathogens. International Journal of Pharmacology, 15, 962–969. DOI 10.3923/ijp.2019.962.969. [Google Scholar] [CrossRef]
15. Rizvi, S. M., Shakil, S., Haneef, M. (2013). A simple click-by-click protocol to perform docking: AutoDock 4.2 made easy for non-bioinformaticians. EXCLI Journal, 12, 831–857. [Google Scholar]
16. Laskowski, R. A., Swindells, M. B. (2011). LigPlot+ multiple ligand-protein interaction diagrams for drug discovery. Journal of Chemical Information & Modeling, 51, 2778–2786. DOI 10.1021/ci200227u. [Google Scholar] [CrossRef]
17. Hess, B., Kutzner, C., van der Spoel, D., Lindahl, E. (2008). GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. Journal of Chemical Theory and Computation, 4(3), 435–447. DOI 10.1021/ct700301q. [Google Scholar] [CrossRef]
18. Bjelkmar, P., Larsson, P., Cuendet, M. A., Hess, B., Lindahl, E. (2010). Implementation of the CHARMM force field in GROMACS: Analysis of protein stability effects from correction maps, virtual interaction sites, and water models. Journal of Chemical Theory and Computation, 6(2), 459–466. DOI 10.1021/ct900549r. [Google Scholar] [CrossRef]
19. Zoete, V., Cuendet, M. A., Grosdidier, A., Michielin, O. (2011). SwissParam: A fast force field generation tool for small organic molecules. Journal of Chemical Theory and Computation, 32(11), 2359–2368. DOI 10.1002/jcc.21816. [Google Scholar] [CrossRef]
20. Mark, P., Nilsson, L. (2001). Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. Journal of Physical Chemistry A, 105, 9954–9960. DOI 10.1021/jp003020w. [Google Scholar] [CrossRef]
21. Mandal, S. P., Garg, A., Prabitha, P., Wadhwani, A. D., Adhikary, L. et al. (2018). Novel glitazones as PPARγ agonists: Molecular design, synthesis, glucose uptake activity and 3D QSAR studies. Chemistry Central Journal, 12(1), 141. DOI 10.1186/s13065-018-0508-0. [Google Scholar] [CrossRef]
22. Kim, S. I., Song, J. T., Jeong, J. Y., Seo, H. S. (2016). Niclosamide inhibits leaf blight caused by Xanthomonas oryzae in rice. Scientific Reports, 6, 21209. DOI 10.1038/srep21209. [Google Scholar] [CrossRef]
23. Sharma, A., Khuller, G. K., Sharma, S. (2009). Peptide deformylase--A promising therapeutic target for tuberculosis and antibacterial drug discovery. Expert Opinion on Therapeutic Targets, 13, 753–765. DOI 10.1517/14728220903005590. [Google Scholar] [CrossRef]
24. Joshi, T., Joshi, T., Sharma, P., Chandra, S., Pande, V. (2021). Molecular docking and molecular dynamics simulation approach to screen natural compounds for inhibition of Xanthomonas oryzae pv. Oryzae by targeting peptide deformylase. Journal of Biomolecular Structure and Dynamics, 39(3), 823–840. DOI 10.1080/07391102.2020.1719200. [Google Scholar] [CrossRef]
25. Rizvi, S. M. D., Shaikh, S., Khan, M., Biswas, D., Hameed, N. et al. (2014). Fetzima (levomilnaciprana drug for major depressive disorder as a dual inhibitor for human serotonin transporters and beta-site amyloid precursor protein cleaving enzyme-1. CNS & Neurological Disorder-Drug Targets, 13(8), 1427–1431. DOI 10.2174/1871527313666141023145703. [Google Scholar] [CrossRef]
26. Kuppusamy, A., Arumugam, M., George, S. (2017). Combining in silico and in vitro approaches to evaluate the acetylcholinesterase inhibitory profile of some commercially available flavonoids in the management of Alzheimer’s disease. International of Journal of Biological Macromolecules, 95, 199–203. DOI 10.1016/j.ijbiomac.2016.11.062. [Google Scholar] [CrossRef]
27. Rampogu, S., Zeb, A., Baek, A., Park, C., Son, M. et al. (2018). Discovery of potential plant-derived peptide deformylase (PDF) inhibitors for multidrug-resistant bacteria using computational studies. Journal of Clinical Medicine, 7(12), 563. DOI 10.3390/jcm7120563. [Google Scholar] [CrossRef]
28. Shaikh, S., Rizvi, S. M. D., Suhail, T., Shakil, S., Abuzenadah, A. M. (2016). Prediction of anti-diabetic drugs as dual inhibitors against acetylcholinesterase and beta-secretase: A neuroinformatics study. CNS & Neurological Disorder-Drug Targets, 15(10), 1216–1221. DOI 10.2174/1871527315666161003125752. [Google Scholar] [CrossRef]
29. Hawkins, P. C., Skillman, A. G., Nicholls, A. (2007). Comparison of shape-matching and docking as virtual screening tools. Journal of Medicinal Chemistry, 50(1), 74–82. DOI 10.1021/jm0603365. [Google Scholar] [CrossRef]
30. Mashraqi, M. M., Chaturvedi, N., Alam, Q., Alshamrani, S., Bahnass, M. M. et al. (2021). Biocomputational prediction approach targeting FimH by natural SGLT2 inhibitors: A possible Way to overcome the uropathogenic effect of SGLT2 inhibitor drugs. Molecules, 26(3), 582. DOI 10.3390/molecules26030582. [Google Scholar] [CrossRef]
31. Prasad, S. K., Pradeep, S., Shimavallu, C., Kollur, S. P. (2021). Evaluation of annona muricata acetogenins as potential anti-SARS-CoV-2 agents through computational approaches. Frontier in Chemistry, 8, 624716. DOI 10.3389/fchem.2020.624716. [Google Scholar] [CrossRef]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |