
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE

From Stability to Hardness: High-Throughput First-Principles Screening Reveals Promising MAB Phases for Advanced Engineering Applications
1 School of Naval Architecture and Maritime, Zhejiang Ocean University, Zhoushan, China
2 Suzhou Laboratory, Suzhou, China
* Corresponding Authors: Jiexi Song. Email: ; Diwei Shi. Email:
(This article belongs to the Special Issue: Computational Modeling and Simulation of Energy and Environmental Materials)
Computers, Materials & Continua 2026, 87(3), 29 https://doi.org/10.32604/cmc.2026.078225
Received 26 December 2025; Accepted 27 February 2026; Issue published 09 April 2026
 View Full Text
View Full Text Download PDF
Download PDFAbstract
MAB phases are a class of layered ternary transition-metal borides, characterized by hard M-B slabs interleaved with softer A-element layers, and thus hold promise for wear-resistant and high-temperature structural applications. However, their compositional space and structural diversity remain insufficiently explored, limiting guidance for synthesis and property optimization. In this work, we perform a comprehensive exploration and screening of the MAB family using high-throughput first-principles calculations. We systematically identify 855 candidate MAB compounds with orthorhombic and hexagonal structures across multiple transition-metal families, which form the starting pool for subsequent stability and property evaluation. The workflow evaluates viability using three criteria: quantifying thermodynamic stability through formation energy and energy above the convex hull, confirming dynamical stability using phonon spectra, and evaluating mechanical stability via the Born criteria. We identify 336 MAB candidates that satisfy all three stability requirements, and further conduct a comprehensive evaluation and statistical analysis of their elastic constants and derived mechanical properties, including bulk, shear, and Young’s modulus as well as Vickers hardness, thereby elucidating how stoichiometry and composition influence hardness and brittle-ductile behavior. This study not only provides a curated set of experimentally viable MAB materials spanning diverse stoichiometries but also establishes a robust and transferable computational workflow for accelerating the discovery of layered ternary borides.Keywords
Supplementary Material
Supplementary Material FileAgainst the backdrop of the global energy transition to low-carbon technologies, next-generation devices such as fuel cells, solid-state batteries, and high-efficiency electrocatalysts are placing ever more rigorous and frequently competing demands on structural and functional materials. In practical operation, key components must tolerate high-temperature oxidative atmospheres and corrosive environments, while simultaneously operating under coupled thermal, electrical, and mechanical fields. Under these extreme conditions, conventional materials rarely combine high thermal and electrical conductivity with strong oxidation/corrosion resistance and sufficient damage tolerance. In this context, layered ternary transition-metal borides, namely MAB phases (where M denotes a transition metal, A represents a group III–VI element, and B stands for boron), have attracted growing interest due to their unusual combinations of ceramic-like and metallic characteristics [1–3]. MAB phases adopt a nanolaminated architecture in which stiff, covalently bonded M-B slabs provide ceramic-like rigidity, while the intervening A-element layers are comparatively compliant and metallic, together giving rise to this unique set of mechanical robustness and transport properties [4]. Benefiting from their layered crystal structure and mixed chemical bonding, MAB phases integrate ceramic-like thermal stability with metal-like electrical and thermal conductivity. They also exhibit promising catalytic performance, which has spurred extensive research. Their excellent conductivity and large specific surface area further render MAB phases attractive candidates for electrode materials in supercapacitors and high-power batteries, where the lamellar structure provides abundant active sites and efficient ion-transport pathways to enable rapid charge/discharge processes. Beyond energy storage systems, MAB materials have also been explored as high-performance electrocatalysts for the oxygen reduction reaction (ORR) in fuel cells. Moreover, their spacious interlayer regions are hypothesized to serve as favorable adsorption sites for reversible hydrogen storage [5,6].
First proposed in the 1940s but not experimentally synthesized until the 1960s [7], MAB phases attracted relatively little attention for decades owing to the limited early understanding of their chemistry and structure. However, the surge of interest in MAX phases (Mn+1AXn, X is C or N) and their two-dimensional derivatives, MXenes, has since stimulated renewed exploration of layered borides with analogous architectures, including MAB phases [8]. Despite their shared nanolaminated architecture, MAX and MAB phases differ fundamentally in their coordination motifs: MAX phases feature an M-X sublattice based on octahedral coordination, while MAB phases are constructed from BM6-type trigonal prisms as their fundamental structural units [9]. The conventional MAX-phase nomenclature is not directly applicable to MAB phases [10]. Ade and Hillebrecht proposed a unified structural description for MAB-type ternary borides using the general formula (MB)2Aly(MB2)x, which rationalizes a series of orthorhombic phases in the Cr-Al-B system, including MAlB, M2AlB2, M3AlB4 and M4AlB6 [11]. The experimental synthesis of Fe3Al2B2 reported in 2018 provided further evidence that the MAB family can extend beyond the initially established compositions [12]. Expanding on this descriptive approach, Kota et al. proposed a more general compositional framework, (MB)2zAx(MB2)y with z = 1–2, x = 1–2 and y = 0–2, which is applicable to both single-layer and double-layer configurations [13]. This generalized framework allows A-site intercalation and systematic variations in the M:B ratio. Wang et al. achieved the synthesis of the hexagonal MAB phase Ti2InB2 and its two-dimensional derivative h-MBene TiB in 2018 [14]. Carlsson et al. proposed theoretical predictions for hexagonal MAB stoichiometries including M3AB4 and M4AB6 in 2021 [15], which were experimentally corroborated by Yin et al.’s synthesis of V3PB4 in 2023 [16]. Collectively, these developments have broadened the scope of the MAB family and motivated exploration of their application-relevant properties. Compared with MAX phases, MAB phases span a wider range of structural motifs and compositions. However, the range of transition metals that have been successfully synthesized in experiments remains limited, constituting a key challenge and opportunity for future research.
High-throughput computations have become a powerful tool for the prediction and design of MAB phases. For instance, they have been used to predict a hexagonal MAB phase that was later synthesized, confirming a strong theory-experiment match [14]. Liu et al. performed ab initio high-throughput screening of Al-containing ternary MAB phases, identifying 23 stable candidates across two stoichiometries (MAB and M2AB2) [17]. Koutná et al. investigated several representative stoichiometries (MAB, M2AB2 and M3AB4), screening more than 600 orthorhombic/hexagonal structures and successfully identifying over 300 stable MAB phases [18]. Li et al. further combined high-throughput calculations with machine learning to explore layered M2AB2 and related MAX phases, identifying numerous stable candidates and calculating the lattice thermal conductivity [19]. Carlsson et al. evaluated the thermodynamic stability and structural preferences of 5 MAB stoichiometries (MAB, M2AB2, M3AB4, M4AB4 and M4AB6), where the M-site elements were group 3–6 transition metals, Mn, Fe, and Co, and the A-site elements were Al, Ga, and In [15]. Nevertheless, given the immense number of possible elemental combinations, much of the MAB chemical space is still unexplored.
While most high-throughput DFT studies on MAB phases have focused on discovering new phases or assessing stability via a single criterion, systematic property prediction and rigorous multi-criteria screening remain underexplored [19,20]. To address this gap, we conducted high-throughput first-principles calculations for a set of 855 designed MAB candidates. First, we evaluated thermodynamic stability based on formation energy (Ef) and energy above the convex hull (Ehull), identifying 495 candidates that meet this criterion. Subsequent high-throughput phonon calculations assessed dynamical stability, narrowing the pool to 384 candidates. Finally, mechanical stability validation confirmed 336 of these as mechanically stable. For this final set, we carry out a comprehensive mechanical characterization by calculating key parameters including the bulk modulus (B), shear modulus (G), Young’s modulus (E), Pugh ratio (B/G), Poisson’s ratio (ν), Cauchy pressure and Vickers hardness (Hv). Fig. 1 summarizes the complete workflow of this multi-criteria screening strategy.
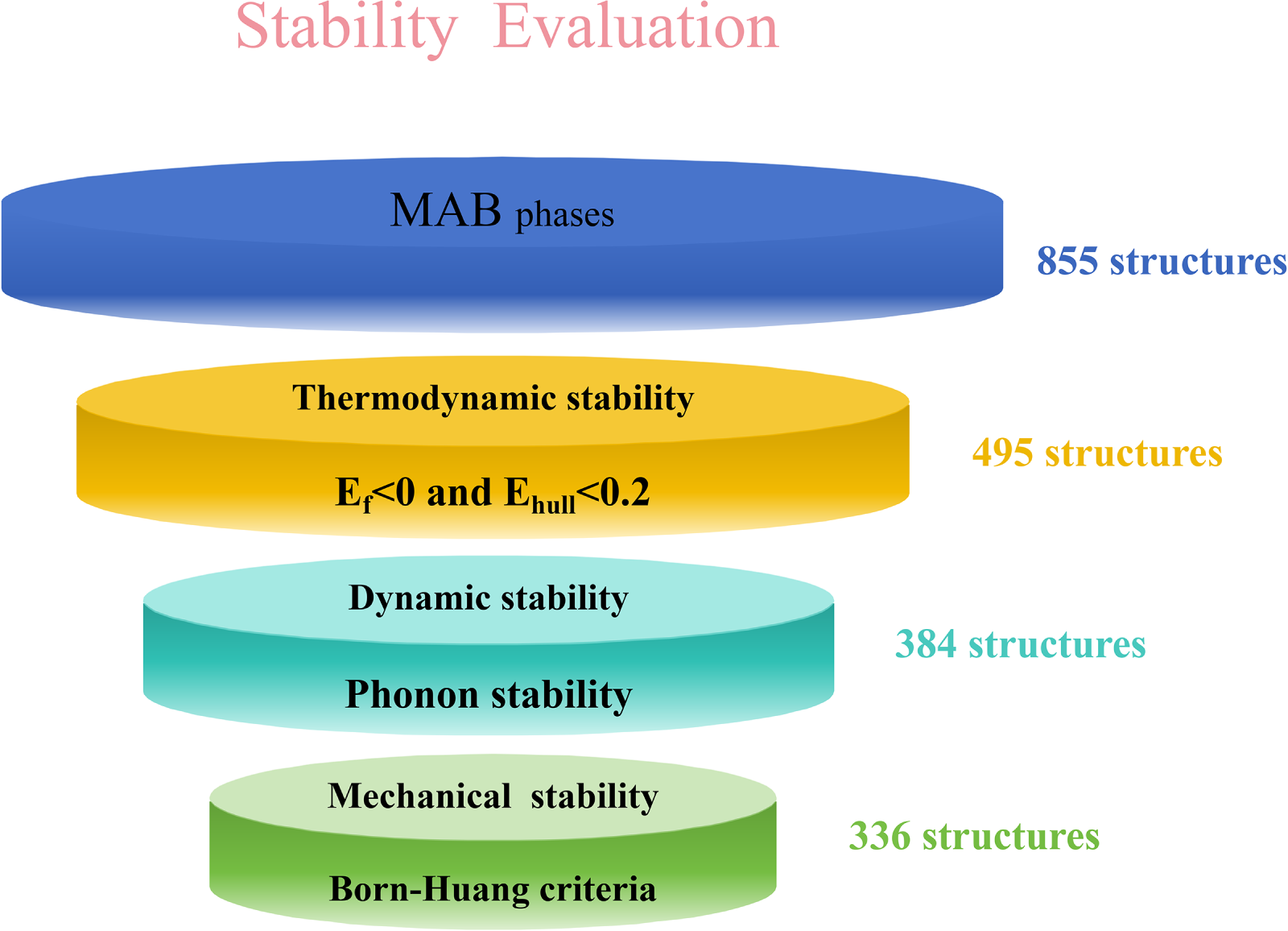
Figure 1: Schematic diagram of the multi-stability screening process for MAB candidate structures.
The high-throughput screening framework used in this work follows a workflow similar to that in our previous studies [21–23], and the specific procedures are described as follows. First, a set of simulation configurations containing the target elements was constructed, and MAB configurations were generated by substituting specific atomic sites. The designed configurations were then structurally relaxed to obtain stable ground-state structures. For thermodynamic stability assessment, we jointly considered energy descriptors including the formation energy and the energy above the convex hull derived from phase diagrams [24–26]. In particular, phase diagrams for the configurations generated by high-throughput substitutions were obtained using the Python Materials Genomics (pymatgen) toolkit and the PhaseDiagram module implemented in the Materials Project (MP) database [27–29]. With this comprehensive approach, we determined Ehull for each newly designed structure and used it as the criterion for thermodynamic stability. Dynamical stability is subsequently assessed by phonon dispersion calculations [30].
In conventional DFT-based phonon workflows, each candidate requires a set of displaced supercells followed by numerous single-point energy evaluations and iterative convergence [31], which makes the computational cost scale unfavorably with the candidate set size and becomes prohibitive for large-scale high-throughput screening [32]. To enable tractable screening at this scale, we compute phonon spectra for all candidates using the finite-displacement method with a 2 × 2 × 2 supercell, and we extract elastic constants using the energy-strain approach implemented in phonopy and VASPKIT [24,33]. Given the large number of structures, we adopt the universal machine-learning interatomic potential MatterSim-v1.0.0-5M as the baseline model for downstream calculations [34], which has been reported to achieve relatively small errors in predicted energies and forces when benchmarked against DFT-PBE [35]. Finally, mechanical stability is determined by analyzing the elastic stiffness tensor according to the Born stability criteria [36].
All density functional theory (DFT) based calculations in this work were performed using the Vienna Ab initio Simulation Package (VASP) [37–39], together with the projector augmented-wave (PAW) method and the generalized gradient approximation (GGA) in the Perdew-Burke-Ernzerhof (PBE) form [40,41]. The plane-wave kinetic energy cutoff was set to 520 eV. All structures were fully relaxed until the total energy and atomic forces converged within 10−5 eV and 0.005 eV/Å, respectively. Brillouin-zone sampling was carried out using Γ-centered uniform k-point meshes, which were automatically generated with a target k-point spacing of 0.03 Å−1 [33,42].
In this section, we systematically investigate the stability and mechanical properties of orthorhombic and hexagonal MAB candidates using high-throughput calculations. The study considers all possible elemental combinations within the selected M, A and B elements (see Fig. 2). Specifically, the M-site elements cover groups IIIB (Sc, Y), IVB (Ti, Zr, Hf), VB (V, Nb, Ta), VIB (Cr, Mo, W), VIIB (Mn, Tc, Re), and VIIIB (Fe, Ru, Co, Rh, and Ni). The A-site elements include Al, Ga, In, Si, and Tl, and the boron element occupies the B site. M:A:B stoichiometries (1:1:1, 2:1:2, 3:1:4, 3:2:2, 4:1:4, 4:1:6) are investigated, with both orthorhombic and hexagonal prototypes evaluated for the corresponding composition. The candidate pool integrates previously reported experimental MAB phases and manually designed structures for validation and comparison [14–16]. In total, 855 initial MAB candidates were constructed across the crystal systems, elemental combinations and stoichiometries, followed by full structural optimizations. The representative MAB configurations are shown in Fig. 3.

Figure 2: Elements and stoichiometric ratios of MAB candidates. The yellow-highlighted elements in the periodic table represent transition metals considered for the M-site, the green elements denote the A-site, and the blue element represents the B-site. Candidate compositions are expressed as MxAyBz with x = 1–4; y = 1–2; z = 1, 2, 4 or 6.
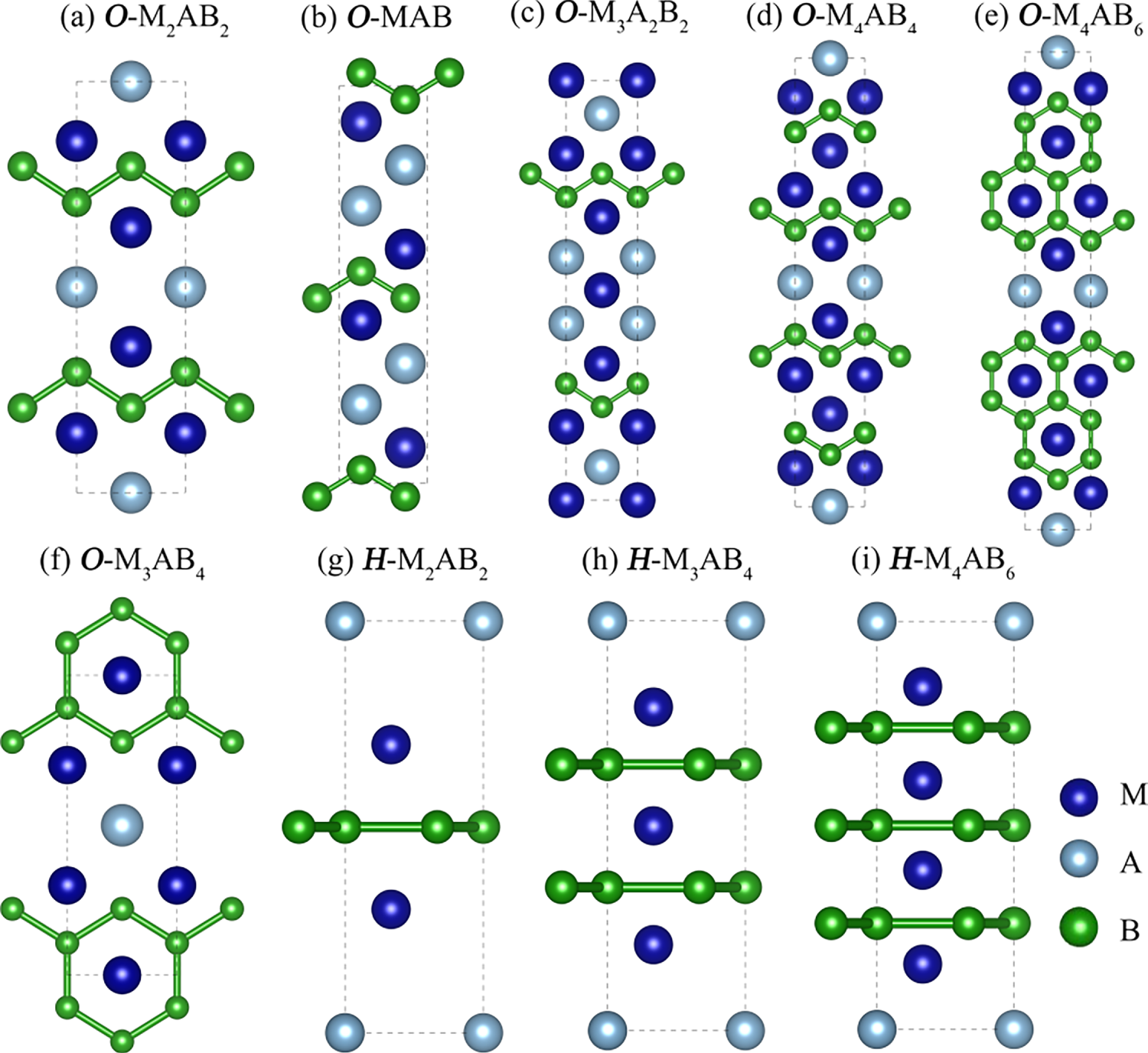
Figure 3: The crystal structures of representative MAB candidates. Panels (a–f) illustrate the stacking types in the orthorhombic crystal system, and panels (g–i) depict the corresponding hexagonal structures. The blue, gray, and green spheres represent M, A, and B atoms, respectively.
Thermodynamic stability was evaluated via phase-diagram analysis, implemented using pymatgen and the PhaseDiagram module from the Materials Project ecosystem. For each candidate, we calculated the Ehull as a quantitative index of stability, and generated convex-hull plots to facilitate visualization, as well as to distinguish thermodynamically stable phases from metastable candidates that may still be experimentally accessible. By definition, compounds with Ehull = 0 lie on the convex hull, are thermodynamically stable, whereas those with Ehull > 0, which lie above the hull are commonly regarded as thermodynamically metastable or unstable [43]. Notably, stability assessments are highly sensitive to small energetic differences. Bartel showed that an energy shift of 100 meV per atom can be sufficient to change conclusions about synthetic accessibility [26]. Therefore, high-throughput studies typically employ a “near-hull window” (usually 100–200 meV/atom) to avoid prematurely excluding metastable phases that may still be experimentally accessible [44,45]. Building upon prior studies, we note that compounds with Ehull < 50 meV/atom exhibit higher experimental realizability, while phases with Ehull < 200 meV/atom may still be synthesizable under practical conditions [46]. Considering numerical uncertainties in calculation, possible incompleteness of the competing-phase set, and finite-temperature effects, we adopt 200 meV/atom as the thermodynamic stability threshold in this work. Based on this criterion, 495 thermodynamically viable candidates were identified from the initial pool of 855 predicted structures and the distribution of Ehull values across the full MAB dataset is visualized in Fig. 4.
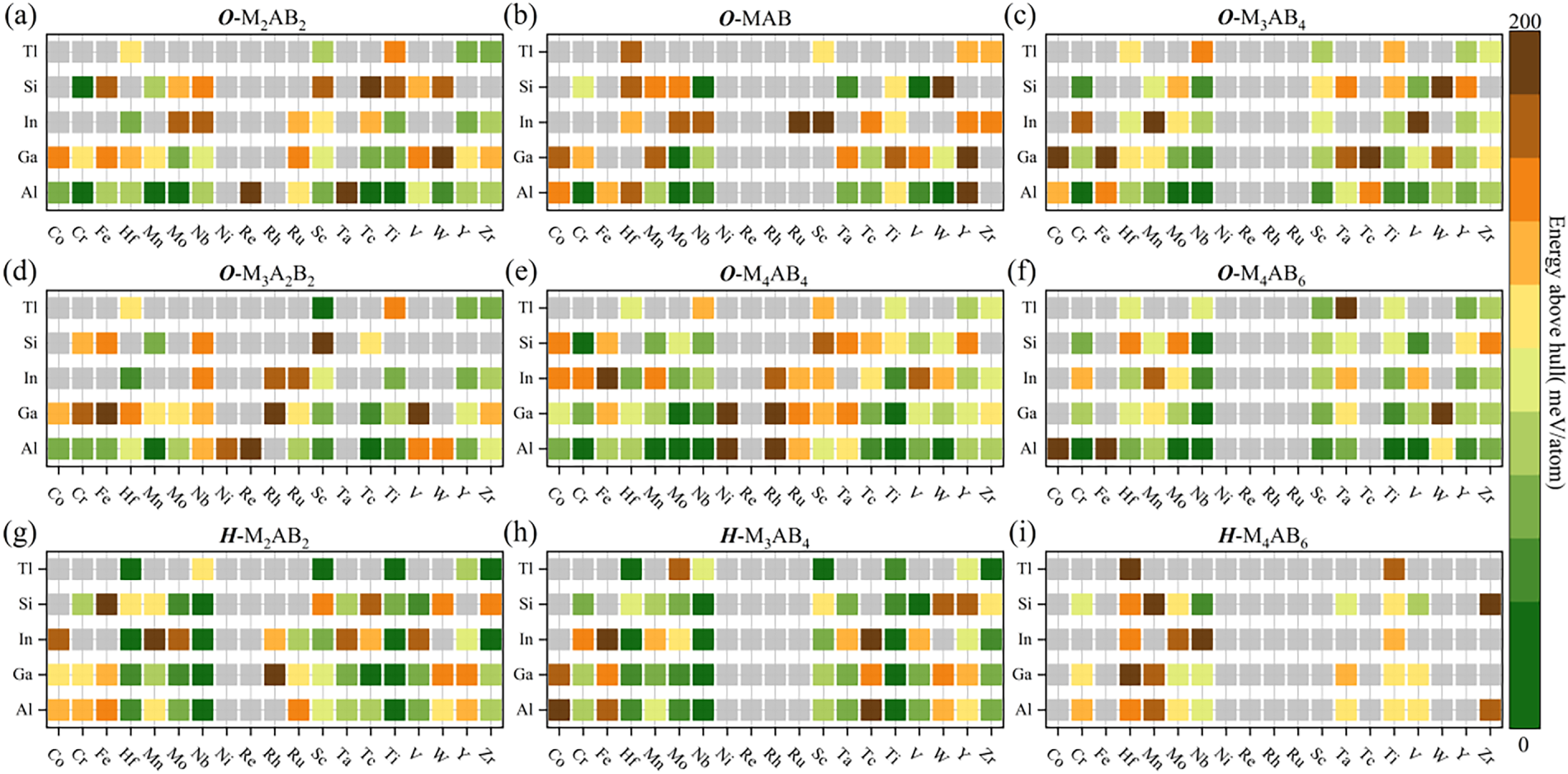
Figure 4: Distribution of the Ehull for (a) O-M2AB2, (b) O-MAB, (c) O-M3AB4, (d) O-M3A2B2, (e) O-M4AB4, (f) O-M4AB6, (g) H-M2AB2, (h) H-M3AB4 and (i) H-M4AB6 structures. In each panel, the vertical axis corresponds to the A-site element and the horizontal axis corresponds to the M-site element. The color bar indicates the magnitude of Ehull. A pre-screening threshold of Ehull < 200 meV/atom is adopted, and MAB phases with Ehull ≥ 200 meV/atom are shown as gray squares.
Among the 570 orthorhombic candidates, 335 structures (58.8%) satisfy Ehull < 200 meV/atom, while 160 of the 285 hexagonal candidates (56.1%) meet the same criterion, indicating similar retention under this thermodynamic threshold. In contrast, when subjected to the stricter condition of Ehull = 0, the hexagonal configuration shows a clear advantage with 6.3% of hexagonal candidates lying on the hull, approximately twice the fraction observed for the orthorhombic candidates (3.0%). This disparity directly indicates that hexagonal MAB phases occupy a larger proportion of thermodynamically stable configurations. Among the orthorhombic MAB candidates, the M4AB4 stoichiometry exhibits the highest thermodynamic pass rate under the established criterion, followed by M4AB6 with a pass rate of 57.9%. For the hexagonal MAB phases, M2AB2 and M3AB4 show relatively high pass rates of 69.5% and 65.3%, respectively. By contrast, hexagonal M4AB6 displays a notably lower pass rate of only 33.7%, indicating low thermodynamic viability for this stoichiometry in the hexagonal framework. Overall, these trends are consistent with previous reports that hexagonal MAB structures are thermodynamically stable only for specific stoichiometries, and that increasing boron content tends to destabilize hexagonal prototypes. In such cases, the energetic landscape favors the formation of orthorhombic variants or decomposition into competing phases with lower formation energies [18].
From the A-site element perspective, Al- and Ga-based orthorhombic MAB candidates exhibit the highest thermodynamic pass rates. Under the screening criterion of 200 meV/atom, their pass rates reach 82.5% and 75.4%, respectively. For the hexagonal MAB candidates, Al- and Ga-based candidates show an identical pass rate of 70.2%. From the perspective of M-site element, thermodynamically viable candidates are predominantly concentrated in systems containing early- to mid-transition metals. In the orthorhombic MAB candidates, Ti- and Nb-based MAB candidates exhibit very high pass rates of 93.3% and 90.0%, whereas Ni- and Re-based candidates show much lower pass rates of only 10.0% and 6.7%, respectively. A similar trend is observed for the hexagonal candidates: Ti- and Nb-based candidates reach pass rates of 100% and 93.3%, while Ni- and Re-based candidates show 0% in both cases. This trend may reflect the differences in M-B bonding characteristics where early- to mid-transition metals can form a more robust bonding framework with boron, which lowers the total energy and enhances the stability of the MAB structure. In contrast, when the M site is occupied by late transition metals, the energetic landscape tends to favor decomposition into competing boride phases with higher thermodynamic stability [47].
Next, we assess dynamical stability using phonon calculations. A structure is considered dynamically stable if no imaginary modes appear across the Brillouin zone [48]. Given the high computational cost of density functional perturbation theory (DFPT), we employ a machine-learning-based pre-screening strategy to predict phonon spectra and identify potential imaginary modes. The feasibility of this approach has been supported by prior studies. For instance, Ojih et al. reported that a pretrained CHGNet model can efficiently screen dynamically stable structures [49], and Hwang et al. further confirmed the reliability of combining machine learning predicted phonon dispersions with experimental validation [50]. Leveraging machine-learning-predicted phonon spectra, we screened the 495 thermodynamically viable candidates for imaginary modes and identified 384 dynamically stable structures, comprising 261 orthorhombic and 123 hexagonal candidates. Notably, dynamical stability and thermodynamic stability represent fundamentally different criteria [51]. Thermodynamically stable compounds with Ehull close to zero are generally less prone to imaginary phonon modes. Nevertheless, imaginary frequencies may still emerge due to atypical local coordination or interlayer mismatch. Conversely, candidates with much larger Ehull values may exhibit no imaginary phonon frequencies yet remain thermodynamically disfavored, and thus can decompose or transform into competing phases under realistic synthesis conditions [52–54]. Therefore, we adopt a two-stage strategy with an initial thermodynamic screen using Ehull < 200 meV/atom, followed by a phonon-based re-screening for dynamical stability.
The procedures above establish the viability of MAB candidates in terms of energetics and lattice dynamics [55], but they do not guarantee that the elastic strain energy is positive-definite under arbitrary infinitesimal strain perturbations. Therefore, it is necessary to further assess mechanical stability by evaluating the response of candidate structures to small static strains [56]. Hence, we calculated the elastic constants of all the MAB candidates and evaluated the mechanical stability according to the Born stability criteria [36]. Specifically, orthorhombic crystals must satisfy constraints associated with 9 independent elastic constants, including C11 > 0, C44 > 0, C55 > 0, and C66 > 0; C11C22 > (C12)2, and the higher-order condition C11C22C33 + 2C12C13C23 − C11(C23)2 − C22(C13)2 − C33(C12)2 > 0. For hexagonal crystals, constraints on 5 independent elastic constants are required, including C44 > 0; C11 > |C12|, and (C11 + C12)C33 − 2(C13)2 > 0. After applying the Born criteria, 336 mechanically stable structures were retained from the 384 dynamically stable candidates. The outcomes of thermodynamic, dynamical, and mechanical screening are summarized in Fig. 5. Overall, 336 MAB structures satisfy all the aforementioned criteria.
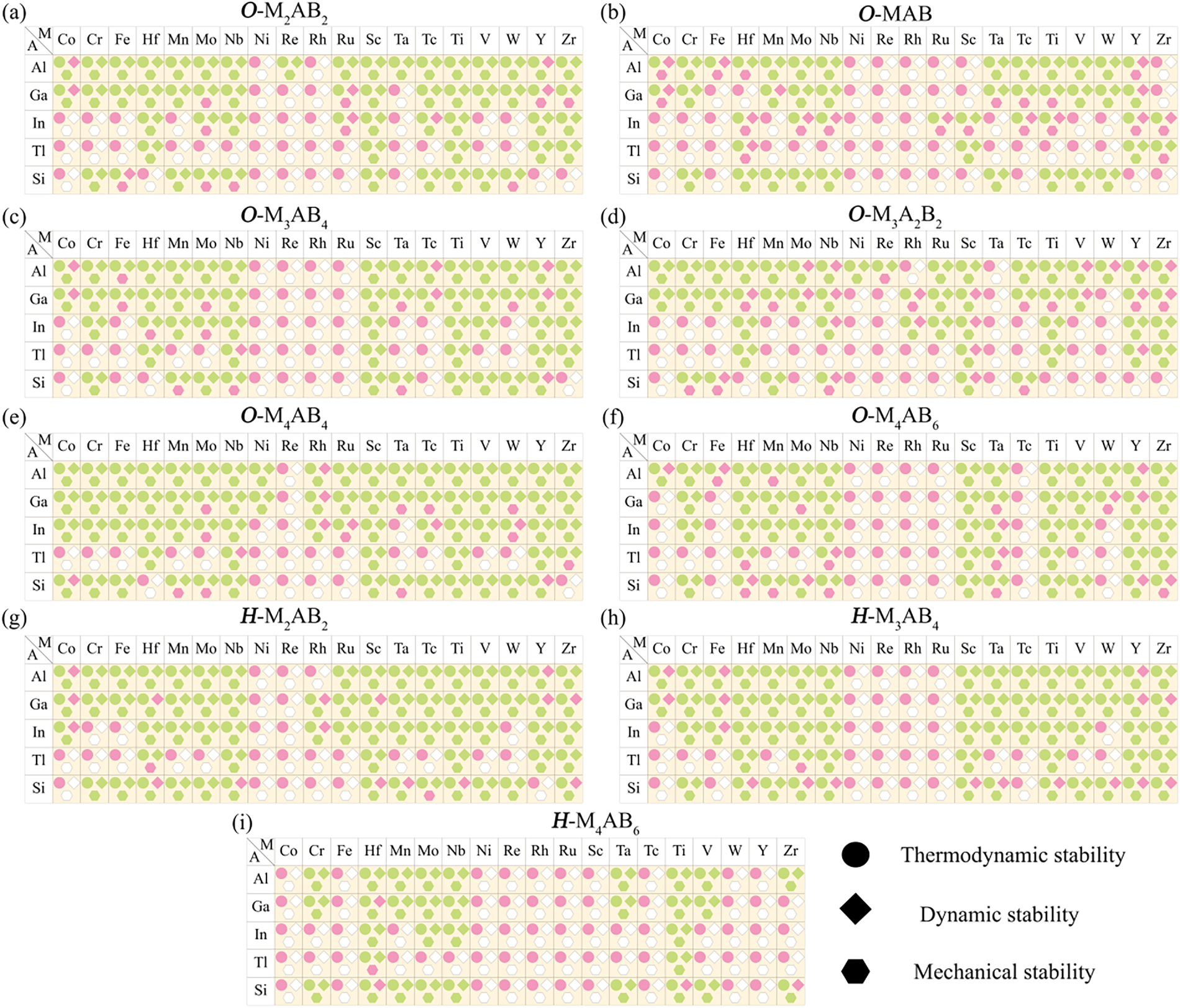
Figure 5: Screening results for the thermodynamic, dynamical, and mechanical stability of the candidate MAB phases of (a) O-M2AB2, (b) O-MAB, (c) O-M3AB4, (d) O-M3A2B2, (e) O-M4AB4, (f) O-M4AB6, (g) H-M2AB2, (h) H-M3AB4 and (i) H-M4AB6. In each panel, the horizontal and vertical axes represent the M-site and A-site elements, respectively. Green indicates stability, pink indicates instability, and blank cells denote uncalculated cases, because these candidates failed to meet the preceding stability criteria and were thus excluded from subsequent calculations. The 336 fully stable MAB phases correspond to structures with all three symbols colored green.
Following this, we employed the Voigt-Reuss-Hill (VRH) averaging scheme to estimate polycrystalline elastic moduli from the single-crystal elastic constants, enabling comparison with macroscopic experimental observables [57]. Using this approach, we systematically calculated the bulk modulus, shear modulus, Young’s modulus, Pugh ratio, Poisson’s ratio, and Vickers hardness for the 336 retained structures. To validate the reliability of our workflow, we compared the calculated moduli of representative MAB compounds (Cr4AlB4, Mo4AlB4, W4AlB4) against literature values [58]. For these compounds, our calculations (Tables S1 and S2) yield bulk moduli of 253.6, 270.3, and 295.7 GPa and shear moduli of 197.8, 164.9, and 174.4 GPa, respectively. These values show excellent agreement with reported literature data (255.2, 262.5, 288.7 GPa for B; 199.3, 159.6, 166.6 GPa for G), with mean relative deviations of ~2.0% for B and ~2.9% for G. This close agreement confirms the high fidelity of our workflow in reproducing the mechanical properties of MAB systems. Furthermore, the MAB phases were categorized based on boron content into three groups: B2 (M2AB2 and M3A2B2), B4 (M3AB4 and M4AB4), and B6 (M4AB6). A systematic increase in both bulk modulus and shear modulus was observed with higher boron content. Specifically, in the orthorhombic system, the average B values for B2, B4, and B6 were 173.85, 195.85, and 199.5 GPa, respectively, while the average G values were 92.56, 115.8, and 132.43 GPa. In the hexagonal system, the average B values for B2, B4, and B6 were 181.6, 197.31, and 248.6 GPa, respectively, and the average G values were 103.8, 124.92, and 164.78 GPa. The results indicate that the hexagonal system generally exhibits higher stiffness than the orthorhombic system, and increasing boron content is a key factor in enhancing the modulus.
Based on the Vickers hardness results, the orthorhombic subset contains 63 structures with Hv > 20 GPa, including 8 high-hardness candidates with Hv > 30 GPa. The hexagonal subset includes 46 structures with Hv > 20 GPa, among which 6 candidates exhibit Hv > 30 GPa. In the hexagonal system, the high-hardness candidates are primarily associated with the M4AB6 and M3AB4 prototypes. Specifically, 6 representative phases are Ti4AlB6, V4AlB6, Ti4GaB6, V4GaB6, Ti4InB6, and Ti3AlB4, with corresponding Vickers hardness values of 33.429, 33.010, 31.815, 31.583, 31.452, and 30.269 GPa, respectively. Similarly, high-hardness orthorhombic candidates are concentrated in the M3AB4, M4AB4, and M4AB6 families (where A denotes Al, Ga, or Si), 8 representative phases Ti4AlB6, Ti4GaB4, Sc4SiB6, Ti3AlB4, VSiB, Ti4GaB6, Sc3SiB4 and Sc4AlB6 exhibit Vickers hardness values of 34.897, 34.761, 32.994, 32.949, 31.446, 31.440, 30.083, and 30.001 GPa, respectively. The stable MAB structures identified in this work include a substantial number of candidates with Vickers hardness values comparable to those of conventional hard borides, such as MnB4 (34.6 GPa) and ReB2 (34.1 GPa), suggesting that certain MAB phases can reach the high-hardness regime and merit further exploration for hard-material applications [59–61]. Boron-rich compositions tend to exhibit higher hardness in MAB phases, and our dataset reveals a clear statistical increase in Vickers hardness with increasing boron content. For the orthorhombic phases, the average hardness values for B2 (M2AB2 and M3A2B2), B4 (M4AB4 and M3AB4) and B6 (M4AB6) compositions are 12.22, 15.95, and 19.91 GPa, respectively. For the hexagonal phases, the corresponding averages for B2 (M2AB2), B4 (M3AB4) and B6 (M4AB6) are 14.00, 17.44, and 22.27 GPa, respectively. Reported MAB compounds such as Cr4AlB4, Mo4AlB4, and W4AlB4 exhibit Vickers hardness values of 30.15, 18.78, and 18.02 GPa, respectively, which are broadly consistent with our calculated results.
T-distributed stochastic neighbor embedding (t-SNE) was employed to project high-dimensional structural representations onto a two-dimensional space [62], thus enabling the identification of clusters and local neighborhoods primarily based on structural similarity. We constructed a 14-dimensional geometric descriptor comprising unit-cell volume, mass density, total number of atoms, lattice parameters, statistical moments of the atomic coordinates, and maximum interatomic distance. Fig. 6 presents the resulting t-SNE map of the MAB dataset, along with the corresponding Vickers hardness, Pugh ratio, and Poisson’s ratio for each structure. Structures with the same stoichiometry tend to cluster together, whereas different stoichiometries exhibit distinct structural signatures, as exemplified by the M2AB2, M3AB4, and M4AB6 compositions. Notably, the orthorhombic M3A2B2, M4AB4, and M4AB6 groups cluster within region Q2 and show similar structural characteristics. This clustering behavior may stem from the fact that these stoichiometries possess analogous layered building units consisting of alternating M-B slabs and A layers (Fig. 3), which in turn give rise to comparable local coordination environments.
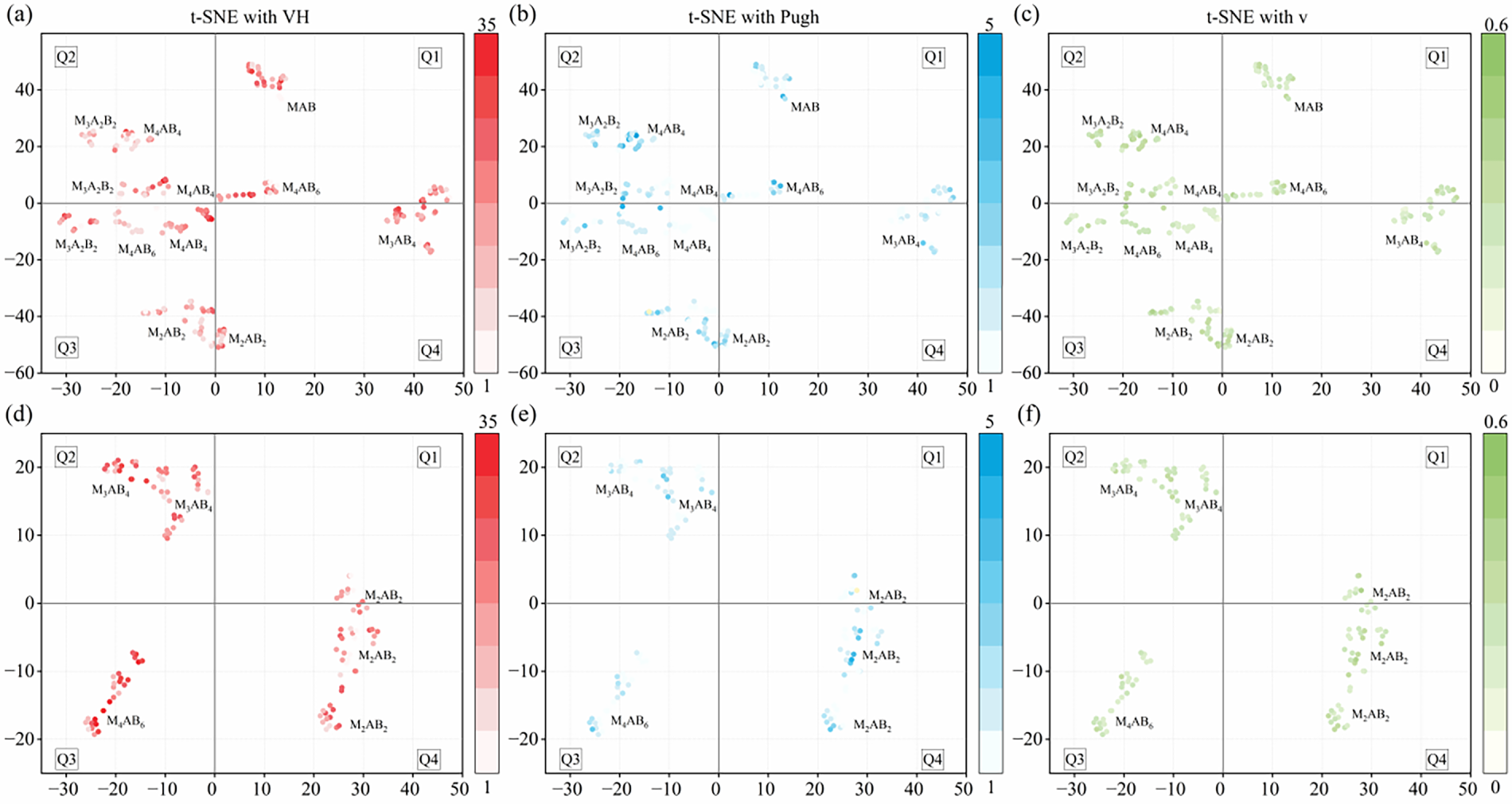
Figure 6: The two-dimensional, dimension-reduced visualization of the MAB structural distribution, overlaid with the corresponding Vickers hardness, Pugh ratio, and Poisson’s ratio. Panels (a–c) correspond to orthorhombic structures, and panels (d–f) correspond to hexagonal structures.
In this study, we adopt a Pugh ratio of 1.75 and a Poisson’s ratio of 0.25, and the sign of the Cauchy pressure, as empirical thresholds to distinguish brittle and ductile behavior [63,64]. A material is considered ductility-dominated when its Pugh ratio exceeds 1.75; otherwise, it tends to exhibit brittleness. For Poisson’s ratio, values above 0.25 indicate enhanced ductility, whereas those below this threshold suggest pronounced brittleness. In addition, a positive Cauchy pressure is commonly taken to indicate a ductile tendency, while a negative Cauchy pressure suggests brittle behavior. Based on these criteria, approximately 38.7% of the orthorhombic MAB candidates exhibit ductile characteristics, compared with 29.4% of the hexagonal subset. Overall, the screened structures span a continuous brittle-to-ductile spectrum, with many lying near the brittle-ductile transition rather than clustering exclusively in the strongly brittle regime. Tables S1 and S2 further indicate that reported values of Pugh ratios and Poisson’s ratios for representative MAB compounds (e.g., Cr4AlB4, Mo4AlB4, and W4AlB4) are consistent with our calculated results. Focusing further on high-hardness candidates with Hv > 20 GPa, the orthorhombic and hexagonal subsets show Pugh ratio ranges of 1.02–1.5 and 1.11–1.52, respectively, and Poisson’s ratio ranges of 0.129–0.277 and 0.154–0.231, respectively. In both cases, the majority of candidates do not surpass the empirical ductility thresholds of 1.75 and 0.25, and their Cauchy pressures are also predominantly negative, further indicating a brittle tendency. This result suggests that when hardness increases to levels approaching those of traditional hard borides, toughness-related indicators tend to decrease simultaneously, consistent with the trend observed in Fig. 6. As boron enrichment is known to boost hardness, the M4AB6 stoichiometry achieves the highest average hardness; conversely, its Pugh ratios and Poisson’s ratios are typically lower, and its Cauchy pressures tend to be more negative which directly reflects the inherent trade-off between hardness and toughness-related properties.
In summary, we have established a robust high-throughput computational framework to systematically explore the structural and compositional space of layered MAB phases. By integrating thermodynamic, dynamical, and mechanical stability criteria, we successfully identified 336 viable MAB candidates across orthorhombic and hexagonal prototypes. Our analysis reveals that both boron content and crystal symmetry critically govern mechanical performance: higher boron stoichiometries (e.g., M4AB6) and hexagonal structures consistently yield enhanced stiffness and hardness, with over a dozen candidates predicted to exceed 30 GPa in Vickers hardness. The excellent agreement between our computed elastic properties and available experimental data validates the reliability of our workflow. This work not only delivers a prioritized materials library for targeted synthesis but also provides fundamental insights into structure–property relationships in ternary transition-metal borides, paving the way for the rational design of next-generation hard, layered ceramics for structural and functional applications.
Acknowledgement: We gratefully acknowledge all individuals who provided assistance, support, and valuable discussions that contributed to this work.
Funding Statement: This work was supported by Opening Grant of Zhejiang Key Laboratory of Data-Driven High-Safety Energy Materials and Applications (OG2024008).
Author Contributions: Jiamin Xue: data analysis, preparation of paper. Jiexi Song: discussion, paper editing and review. Diwei Shi: discussion, paper editing and review, supervision, funding acquisition. All authors reviewed and approved the final version of the manuscript.
Availability of Data and Materials: The crystal structure files and phonon dispersion results supporting the findings of this study are available in the Zenodo repository with the DOI: 10.5281/zenodo.18512685.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare no conflicts of interest.
Supplementary Materials: The supplementary material is available online at https://www.techscience.com/doi/10.32604/cmc.2026.078225/s1.
References
1. Miao N, Yan Y, Wang J. A rising layered boride family for energy and catalysis applications: novel hexagonal MAB phases and MBenes. ChemSusChem. 2024;17(22):e202400229. doi:10.1002/cssc.202400229. [Google Scholar] [PubMed] [CrossRef]
2. Rezaie AA, Yan Z, Scheifers JP, Zhang J, Guo J, Fokwa BPT. Synthesis and Li-ion electrode properties of layered MAB phases Nin+1ZnBn (n = 1, 2). J Mater Chem A. 2020;8(4):1646–51. doi:10.1039/c9ta12937e. [Google Scholar] [CrossRef]
3. Amin MT, Hossain MS, Billah MM, Rahman MA. MBenes: a comprehensive review of synthesis techniques and energy storage capabilities. Glob Chall. 2025;9(10):e00245. doi:10.1002/gch2.202500245. [Google Scholar] [PubMed] [CrossRef]
4. Siriwardane EMD, Birol T, Erten O, Çakır D. Nanolaminated Fe2AB2 and Mn2AB2 (a = Al, Si, Ga, In) materials and the assessment of their electronic correlations. Phys Rev Materials. 2022;6(12):124005. doi:10.1103/physrevmaterials.6.124005. [Google Scholar] [CrossRef]
5. Kota S, Zapata-Solvas E, Ly A, Lu J, Elkassabany O, Huon A, et al. Synthesis and characterization of an alumina forming nanolaminated boride: MoAlB. Sci Rep. 2016;6(1):26475. doi:10.1038/srep26475. [Google Scholar] [PubMed] [CrossRef]
6. Dahlqvist M, Tao Q, Zhou J, Palisaitis J, Persson POÅ, Rosen J. Theoretical prediction and synthesis of a family of atomic laminate metal borides with in-plane chemical ordering. J Am Chem Soc. 2020;142(43):18583–91. doi:10.1021/jacs.0c08113. [Google Scholar] [PubMed] [CrossRef]
7. Jeitschko W. Die kristallstruktur von MoAlB. Monatshefte Für Chem Und Verwandte Teile Anderer Wiss. 1966;97(5):1472–6. (In Dutch). [Google Scholar]
8. Alam MS, Chowdhury MA, Khandaker T, Hossain MS, Islam MS, Islam MM, et al. Advancements in MAX phase materials: structure, properties, and novel applications. RSC Adv. 2024;14(37):26995–7041. doi:10.1039/d4ra03714f. [Google Scholar] [PubMed] [CrossRef]
9. Eklund P, Beckers M, Jansson U, Högberg H, Hultman L. The Mn+1AXn phases: materials science and thin-film processing. Thin Solid Films. 2010;518(8):1851–78. doi:10.1016/j.tsf.2009.07.184. [Google Scholar] [CrossRef]
10. Barsoum MW. The MN+1AXN phases: a new class of solids. Prog Solid State Chem. 2000;28(1–4):201–81. doi:10.1016/s0079-6786(00)00006-6. [Google Scholar] [CrossRef]
11. Ade M, Hillebrecht H. Ternary borides Cr2AlB2, Cr3AlB4, and Cr4AlB6: the first members of the series (CrB2)nCrAl with n = 1, 2, 3 and a unifying concept for ternary borides as MAB-phases. Inorg Chem. 2015;54(13):6122–35. doi:10.1021/acs.inorgchem.5b00049. [Google Scholar] [PubMed] [CrossRef]
12. Hirt S, Hilfinger F, Hillebrecht H. Synthesis and crystal structures of the new ternary borides Fe3Al2B2 and Ru9Al3B8 and the confirmation of Ru4Al3B2 and Ru9Al5B8-x (x≈2). Z Für Kristallogr Cryst Mater. 2018;233(5):295–307. doi:10.1515/zkri-2017-2095. [Google Scholar] [CrossRef]
13. Kota S, Sokol M, Barsoum MW. A progress report on the MAB phases: atomically laminated, ternary transition metal borides. Int Mater Rev. 2020;65(4):226–55. doi:10.1080/09506608.2019.1637090. [Google Scholar] [CrossRef]
14. Wang J, Ye TN, Gong Y, Wu J, Miao N, Tada T, et al. Discovery of hexagonal ternary phase Ti2InB2 and its evolution to layered boride TiB. Nat Commun. 2019;10(1):2284. doi:10.1038/s41467-019-10297-8. [Google Scholar] [PubMed] [CrossRef]
15. Carlsson A, Rosen J, Dahlqvist M. Theoretical predictions of phase stability for orthorhombic and hexagonal ternary MAB phases. Phys Chem Chem Phys. 2022;24(18):11249–58. doi:10.1039/d1cp05750b. [Google Scholar] [PubMed] [CrossRef]
16. Yin H, He X, Zhang J, Song G, Zheng Y, Bai Y. DFT-assisting discovery and characterization of a hexagonal MAB-phase V3PB4. J Materiomics. 2023;9(6):1141–50. doi:10.1016/j.jmat.2023.07.002. [Google Scholar] [CrossRef]
17. Liu Y, Jiang Z, Jiang X, Zhao J. New refractory MAB phases and their 2D derivatives: insight into the effects of valence electron concentration and chemical composition. RSC Adv. 2020;10(43):25836–47. doi:10.1039/d0ra04385k. [Google Scholar] [PubMed] [CrossRef]
18. Koutná N, Hultman L, Mayrhofer PH, Sangiovanni DG. Phase stability and mechanical property trends for MAB phases by high-throughput ab initio calculations. Mater Des. 2024;241:112959. doi:10.1016/j.matdes.2024.112959. [Google Scholar] [CrossRef]
19. Li S, Yang Z, Khaledialidusti R, Lin S, Yu J, Khazaei M, et al. High-throughput study and machine learning on MAX and MAB phases: new materials and fingerprints of superior lattice thermal conductivities. Acta Mater. 2023;254:119001. doi:10.1016/j.actamat.2023.119001. [Google Scholar] [CrossRef]
20. Lind H, Dahlqvist M, Rosen J. In-plane ordered quaternaryM4/3′M2/3′′AlB2 phases (i-MABelectronic structure and mechanical properties from first-principles calculations. J Phys Condens Matter. 2021;33(25):255402. doi:10.1088/1361-648X/abf9bc. [Google Scholar] [PubMed] [CrossRef]
21. Li D, Hu Q, Wu Q, Chang Y, Wang J, Xia Q, et al. High-throughput computational exploration of ternary M3A2X phases: stability, properties, and exfoliation potential. Phys Rev Materials. 2025;9(8):084002. doi:10.1103/stpc-qkpy. [Google Scholar] [CrossRef]
22. Jain A, Hautier G, Moore CJ, Ping Ong S, Fischer CC, Mueller T, et al. A high-throughput infrastructure for density functional theory calculations. Comput Mater Sci. 2011;50(8):2295–310. doi:10.1016/j.commatsci.2011.02.023. [Google Scholar] [CrossRef]
23. Rosado-Miranda AE, Posligua V, Sanz JF, Márquez AM, Nath P, Plata JJ. Design principles guided by DFT calculations and high-throughput frameworks for the discovery of new diamond-like chalcogenide thermoelectric materials. ACS Appl Mater Interfaces. 2024;16(22):28590–8. doi:10.1021/acsami.4c04120. [Google Scholar] [PubMed] [CrossRef]
24. Togo A, Tanaka I. First principles phonon calculations in materials science. Scr Mater. 2015;108:1–5. doi:10.1016/j.scriptamat.2015.07.021. [Google Scholar] [CrossRef]
25. Bartel CJ. Review of computational approaches to predict the thermodynamic stability of inorganic solids. J Mater Sci. 2022;57(23):10475–98. doi:10.1007/s10853-022-06915-4. [Google Scholar] [CrossRef]
26. Pandey S, Qu J, Stevanović V, St John P, Gorai P. Predicting energy and stability of known and hypothetical crystals using graph neural network. Patterns. 2021;2(11):100361. doi:10.1016/j.patter.2021.100361. [Google Scholar] [PubMed] [CrossRef]
27. Jain A, Ong SP, Hautier G, Chen W, Richards WD, Dacek S, et al. Commentary: the materials project: a materials genome approach to accelerating materials innovation. APL Mater. 2013;1(1):011002. doi:10.1063/1.4812323. [Google Scholar] [CrossRef]
28. Ong SP, Richards WD, Jain A, Hautier G, Kocher M, Cholia S, et al. Python Materials Genomics (pymatgena robust, open-source python library for materials analysis. Comput Mater Sci. 2013;68:314–9. doi:10.1016/j.commatsci.2012.10.028. [Google Scholar] [CrossRef]
29. Cao C, Song J, Qin Y, Wang Y, Bai X, Wang F, et al. A method and system for analyzing non-centrosymmetric superconducting topological electronic materials. China patent ZL202110993795.0. 2023 Dec 16. [Google Scholar]
30. Khandy SA, Islam I, Gupta DC, Khenata R, Laref A. Lattice dynamics, mechanical stability and electronic structure of Fe-based Heusler semiconductors. Sci Rep. 2019;9(1):1475. doi:10.1038/s41598-018-37740-y. [Google Scholar] [PubMed] [CrossRef]
31. Baroni S, de Gironcoli S, Dal Corso A, Giannozzi P. Phonons and related crystal properties from density-functional perturbation theory. Rev Mod Phys. 2001;73(2):515–62. doi:10.1103/revmodphys.73.515. [Google Scholar] [CrossRef]
32. Voss J, Vegge T. Gamma-point lattice free energy estimates from O(1) force calculations. J Chem Phys. 2008;128(18):184708. doi:10.1063/1.2919122. [Google Scholar] [PubMed] [CrossRef]
33. Wang V, Xu N, Liu JC, Tang G, Geng WT. VASPKIT: a user-friendly interface facilitating high-throughput computing and analysis using VASP code. Comput Phys Commun. 2021;267(1):108033. doi:10.1016/j.cpc.2021.108033. [Google Scholar] [CrossRef]
34. Yang H, Hu C, Zhou Y, Liu X, Shi Y, Li J, et al. MatterSim: a deep learning atomistic model across elements, temperatures and pressures. arXiv:2405.04967. 2024. doi:10.48550/arxiv.2405.04967. [Google Scholar] [CrossRef]
35. Loew A, Sun D, Wang HC, Botti S, Marques MAL. Universal machine learning interatomic potentials are ready for phonons. npj Comput Mater. 2025;11(1):178. doi:10.1038/s41524-025-01650-1. [Google Scholar] [CrossRef]
36. Mouhat F, Coudert FX. Necessary and sufficient elastic stability conditions in various crystal systems. Phys Rev B. 2014;90(22):224104. doi:10.1103/physrevb.90.224104. [Google Scholar] [CrossRef]
37. Hohenberg P, Kohn W. Inhomogeneous electron gas. Phys Rev. 1964;136(3B):B864–71. doi:10.1103/physrev.136.b864. [Google Scholar] [CrossRef]
38. Kohn W, Sham LJ. Self-consistent equations including exchange and correlation effects. Phys Rev. 1965;140(4A):A1133–8. doi:10.1103/physrev.140.a1133. [Google Scholar] [CrossRef]
39. Kresse G, Furthmüller J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys Rev B. 1996;54(16):11169–86. doi:10.1103/physrevb.54.11169. [Google Scholar] [PubMed] [CrossRef]
40. Perdew J, Burke K, Ernzerhof M. Generalized gradient approximation made simple. Phys Rev Lett. 1996;77(18):3865–8. doi:10.1103/PhysRevLett.77.3865. [Google Scholar] [PubMed] [CrossRef]
41. Blöchl P. Projector augmented-wave method. Phys Rev B. 1994;50(24):17953–79. doi:10.1103/physrevb.50.17953. [Google Scholar] [PubMed] [CrossRef]
42. Geng WT, Liu YC, Xu N, Tang G, Kawazoe Y, Wang V. Empowering materials science with VASPKIT: a toolkit for enhanced simulation and analysis. Nat Protoc. 2025;20(11):3143–69. doi:10.1038/s41596-025-01160-w. [Google Scholar] [PubMed] [CrossRef]
43. Bartel CJ, Trewartha A, Wang Q, Dunn A, Jain A, Ceder G. A critical examination of compound stability predictions from machine-learned formation energies. npj Comput Mater. 2020;6(1):97. doi:10.1038/s41524-020-00362-y. [Google Scholar] [CrossRef]
44. Riebesell J, Surta TW, Goodall REA, Gaultois MW, Lee AA. Discovery of high-performance dielectric materials with machine-learning-guided search. Cell Rep Phys Sci. 2024;5(10):102241. doi:10.1016/j.xcrp.2024.102241. [Google Scholar] [CrossRef]
45. Ye W, Lei X, Aykol M, Montoya JH. Novel inorganic crystal structures predicted using autonomous simulation agents. Sci Data. 2022;9(1):302. doi:10.1038/s41597-022-01438-8. [Google Scholar] [PubMed] [CrossRef]
46. Pöllmann PJ, Bogdanovski D, Lellig S, Schweizer P, Hans M, Azina C, et al. Metastable phase formation of (Mo, Cr)2AlB2 MAB phase thin films revealed by theory and experiments. Mater Res Lett. 2024;12(1):58–66. doi:10.1080/21663831.2023.2292054. [Google Scholar] [CrossRef]
47. Shen C, Gao Q, Fortunato NM, Singh HK, Opahle I, Gutfleisch O, et al. Designing of magnetic MAB phases for energy applications. J Mater Chem A. 2021;9(13):8805–13. doi:10.1039/d0ta11026d. [Google Scholar] [CrossRef]
48. Pallikara I, Kayastha P, Skelton JM, Whalley LD. The physical significance of imaginary phonon modes in crystals. Electron Struct. 2022;4(3):033002. doi:10.1088/2516-1075/ac78b3. [Google Scholar] [CrossRef]
49. Ojih J, Al-Fahdi M, Yao Y, Hu J, Hu M. Graph theory and graph neural network assisted high-throughput crystal structure prediction and screening for energy conversion and storage. J Mater Chem A. 2024;12(14):8502–15. doi:10.1039/d3ta06190f. [Google Scholar] [CrossRef]
50. Hwang J, Jin Y, Lee J. Machine learning force field based phonon dispersion prediction. Curr Appl Phys. 2024;66:76–80. doi:10.1016/j.cap.2024.07.001. [Google Scholar] [CrossRef]
51. Malyi OI, Sopiha KV, Persson C. Energy, phonon, and dynamic stability criteria of two-dimensional materials. ACS Appl Mater Interfaces. 2019;11(28):24876–84. doi:10.1021/acsami.9b01261. [Google Scholar] [PubMed] [CrossRef]
52. Petretto G, Dwaraknath S, Miranda PC, Winston H, Giantomassi D, van Setten M, et al. High-throughput density-functional perturbation theory phonons for inorganic materials. Sci Data. 2018;5(1):180065. doi:10.1038/sdata.2018.65. [Google Scholar] [PubMed] [CrossRef]
53. Sun W, Dacek ST, Ong SP, Hautier G, Jain A, Richards WD, et al. The thermodynamic scale of inorganic crystalline metastability. Sci Adv. 2016;2(11):e1600225. doi:10.1126/sciadv.1600225. [Google Scholar] [PubMed] [CrossRef]
54. Therrien F, Jones EB, Stevanović V. Metastable materials discovery in the age of large-scale computation. Appl Phys Rev. 2021;8(3):031310. doi:10.1063/5.0049453. [Google Scholar] [CrossRef]
55. Ohmer D, Qiang G, Opahle I, Singh HK, Zhang H. High-throughput design of 211-M2AX compounds. Phys Rev Materials. 2019;3(5):053803. doi:10.1103/physrevmaterials.3.053803. [Google Scholar] [CrossRef]
56. Born M. On the stability of crystal lattices. I Math Proc Camb Philos Soc. 1940;36(2):160–72. doi:10.1017/s0305004100017138. [Google Scholar] [CrossRef]
57. Chung DH, Buessem WR. The Voigt-reuss-hill approximation and elastic moduli of polycrystalline MgO, CaF2, β-ZnS, ZnSe, and CdTe. J Appl Phys. 1967;38(6):2535–40. doi:10.1063/1.1709944. [Google Scholar] [CrossRef]
58. Lu Y, Li T, Li K, Hao D, Chen Z, Zhang H. Theoretical prediction on the stability, elastic, electronic and optical properties of MAB-phase M4AlB4 compounds (M = Cr, Mo, W). RSC Adv. 2024;14(2):1186–94. doi:10.1039/d3ra06267h. [Google Scholar] [PubMed] [CrossRef]
59. Dovale-Farelo V, Tavadze P, Lang L, Bautista-Hernandez A, Romero AH. Vickers hardness prediction from machine learning methods. Sci Rep. 2022;12(1):22475. doi:10.1038/s41598-022-26729-3. [Google Scholar] [PubMed] [CrossRef]
60. Solozhenko VL, Gregoryanz E. Synthesis of superhard materials. Mater Today. 2005;8(11):44–51. doi:10.1016/S1369-7021(05)71159-7. [Google Scholar] [CrossRef]
61. Tian Y, Xu B, Zhao Z. Microscopic theory of hardness and design of novel superhard crystals. Int J Refract Met Hard Mater. 2012;33:93–106. doi:10.1016/j.ijrmhm.2012.02.021. [Google Scholar] [CrossRef]
62. van der Maaten L, Hinton G. Visualizing data using t-SNE. J Mach Learn Res. 2008;9(86):2579–605. [Google Scholar]
63. Pugh S.F. XCII. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Lond Edinb Dublin Philos Mag J Sci. 1954;45(367):823–43. doi:10.1080/14786440808520496. [Google Scholar] [CrossRef]
64. Su L, Duan Y, Su L, Ma L, Li S, Huang B, et al. Insights to structure stability, elastic properties, fracture toughness, and thermal conductivities of TM7B3 (TM = Tc, Ru, Rh and Re) borides. J Mater Res Technol. 2025;38:6198–208. doi:10.1016/j.jmrt.2025.09.048. [Google Scholar] [CrossRef]
Cite This Article
 Copyright © 2026 The Author(s). Published by Tech Science Press.
Copyright © 2026 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools