
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW

Feature Selection Optimisation for Cancer Classification Based on Evolutionary Algorithms: An Extensive Review
1 Faculty of Computing and Meta-Technology, Universiti Pendidikan Sultan Idris, Tanjong Malim, 35900, Malaysia
2 Faculty of Science and Technology, Universitas Islam Negeri Sultan Syarif Kasim Riau, Pekanbaru, 28293, Indonesia
3 Centre for Global Sustainability Studies, Universiti Sains Malaysia, Penang, 11800, Malaysia
4 Premier Integrated Lab, Pantai Hospital Ampang, Kuala Lumpur, 59100, Malaysia
5 Sunway Medical Centre, Subang Jaya, Selangor, 47500, Malaysia
6 Fakultas Teknologi Informasi, Universitas Islam Kalimantan Muhammad Arsyad Al-Banjar, Banjarmasin, 70123, Indonesia
7 Data Intelligence and Knowledge Management Special Interest Group, Universiti Pendidikan Sultan Idris, Tanjong Malim, 35900, Malaysia
* Corresponding Author: Shir Li Wang. Email:
(This article belongs to the Special Issue: Advanced Computational Intelligence Techniques, Uncertain Knowledge Processing and Multi-Attribute Group Decision-Making Methods Applied in Modeling of Medical Diagnosis and Prognosis)
Computer Modeling in Engineering & Sciences 2025, 143(3), 2711-2765. https://doi.org/10.32604/cmes.2025.062709
Received 25 December 2024; Accepted 19 May 2025; Issue published 30 June 2025
 View Full Text
View Full Text Download PDF
Download PDFAbstract
In recent years, feature selection (FS) optimization of high-dimensional gene expression data has become one of the most promising approaches for cancer prediction and classification. This work reviews FS and classification methods that utilize evolutionary algorithms (EAs) for gene expression profiles in cancer or medical applications based on research motivations, challenges, and recommendations. Relevant studies were retrieved from four major academic databases–IEEE, Scopus, Springer, and ScienceDirect–using the keywords ‘cancer classification’, ‘optimization’, ‘FS’, and ‘gene expression profile’. A total of 67 papers were finally selected with key advancements identified as follows: (1) The majority of papers (44.8%) focused on developing algorithms and models for FS and classification. (2) The second category encompassed studies on biomarker identification by EAs, including 20 papers (30%). (3) The third category comprised works that applied FS to cancer data for decision support system purposes, addressing high-dimensional data and the formulation of chromosome length. These studies accounted for 12% of the total number of studies. (4) The remaining three papers (4.5%) were reviews and surveys focusing on models and developments in prediction and classification optimization for cancer classification under current technical conditions. This review highlights the importance of optimizing FS in EAs to manage high-dimensional data effectively. Despite recent advancements, significant limitations remain: the dynamic formulation of chromosome length remains an underexplored area. Thus, further research is needed on dynamic-length chromosome techniques for more sophisticated biomarker gene selection techniques. The findings suggest that further advancements in dynamic chromosome length formulations and adaptive algorithms could enhance cancer classification accuracy and efficiency.Graphic Abstract

Keywords
Supplementary Material
Supplementary Material FileCancer is a leading global health concern, characterised by the uncontrolled proliferation of cells, which can metastasise to other body parts [1,2]. Cancer classification is a critical area of research as it directly impacts early diagnosis, prognosis, and the development of personalised treatment plans. Over the years, advancements in statistics and computer engineering have facilitated the integration of computational methods [3], such as healthcare statistics and artificial intelligence (AI), to improve cancer prediction [4]. The accuracy of cancer prediction has significantly improved in recent years, largely due to the widespread adoption of AI, particularly evolutionary algorithms (EAs), machine learning (ML), and deep learning (DL), in clinical cancer research [5,6]. However, cancer datasets are often high-dimensional, posing challenges in identifying the most relevant features for classification [7,8]. Traditional ML methods struggle to process high-dimensional cancer datasets where the number of features far exceeds the number of available samples [9]. To address these issues, feature selection (FS) methods are essential for reducing the complexity of these datasets, improving classification accuracy, and increasing the interpretability of the models [10]. EAs have shown significant potential in enhancing cancer classification and diagnosis by optimising FS in high-dimensional gene expression profiles (GEPs). In EAs-based approaches, FS is crucial in identifying the most relevant genes for cancer classification, distinguishing key biomarkers from irrelevant data. Various EAs, including genetic algorithms (GAs), differential evolution (DE), and particle swarm optimisation (PSO), have been widely employed for FS, aiming to address the challenges associated with high-dimensional data and small sample sizes.
However, while the application of EAs to FS has gained traction, recent literature reviews have primarily focused on the successes of these algorithms in selecting relevant features without critically examining their limitations or potential gaps. For instance, many studies fail to account for the dynamic nature of cancer data and the necessity of adaptive chromosome length formulations through the FS process. The use of static chromosome lengths, a prevalent limitation in many GA-based FS models, may lead to suboptimal solutions when applied to cancer datasets as gene subsets evolve, especially across different cancer stages. Recent advancements in FS methods, such as ensemble FS approaches, have introduced strategies that integrate multiple FS techniques to improve robustness and adaptability across various cancer types [11]. Hybrid models integrating EAs with other ML methods, such as support vector machines (SVM) and DL, have also gained attention in recent studies [12]. However, compared to standalone EA-based FS models, hybrid and ensemble FS methods that integrate EAs with advanced ML techniques, particularly DL for feature extraction, remain relatively underexplored in the context of cancer classification [13]. These methods hold significant potential by leveraging the strengths of both heuristic search techniques and data-driven learning algorithms. While EAs excel in optimising feature subsets, DL methods can automatically learn hierarchical features from large-scale datasets, potentially overcoming the challenge of selecting optimal features in heterogeneous cancer datasets [14]. Although some recent works have explored such hybrid methods, integrating dynamic chromosome length formulations with DL models for enhanced cancer classification remains underexplored. This research seeks to bridge this gap by proposing a dynamic and adaptive FS parameter-setting approach to complement ensemble and hybrid models, thereby improving scalability, generalisation, and classification accuracy across various cancer types.
EAs, inspired by natural processes, solve optimisation by emulating the behaviours or actions of living organisms within populations. The application of EAs for FS in GEPs from high-dimensional microarray datasets aims to identify the optimal combination of genes that maximises relevant information while minimising redundancy. These algorithms optimise the gene selection for classification tasks, improving cancer diagnostics’ accuracy [15]. They are particularly effective in optimising cancer classification based on high-dimensional datasets with small sample sizes [16–18] leveraging FS techniques applied to ribonucleic acid (RNA) sequences or GEPs. Effective FS facilitates the identification of key features that enable the differentiation of relevant genes from essential biomarkers in extensive microarray datasets. Employing an appropriate FS strategy for marker gene identification allows researchers to focus on critical marker genes, thereby enhancing the accuracy of cancer classification [19–21].
Additionally, FS operational parameters and strategies influence the performance of classification models [22,23]. While many studies have applied EAs to FS for cancer classification, a significant research gap remains regarding the integration of dynamic chromosome length formulations and adaptive parameter-setting in FS. Most existing studies rely on fixed-length chromosome representation, which limits the adaptability and effectiveness of FS as the dataset evolves.
The main contribution of this paper is to review FS and classification methods that utilise EAs for GEP analysis in cancer and medical applications. The specific contributions of this paper are as follows:
1. Reviewing and categorising the literature: This paper systematically categorises and analyses existing research on FS and classification for cancer GEP data, drawing from four major databases.
2. Analysing trends and challenges: The paper identifies and discusses various key research trends, challenges, and recommendations associated with the use of evolutionary algorithms in FS and cancer classification, providing insights into future research directions.
3. Optimisation in cancer classification: The paper highlights underexplored areas in previous studies, such as the dynamic formulation of chromosome length and the adaptive selection of genes in the context of biomarker gene selection.
4. Development of FS models and algorithms: A substantial portion of the reviewed studies focuses on the development of novel FS models and algorithms for cancer prediction and classification, contributing to a deeper understanding of optimisation strategies within this field.
GEP data often have high dimensionality and small sample sizes, posing classification and feature selection challenges. EAs commonly employ a representative fixed chromosomal length to address optimisation problems [24]. However, using a fixed-length chromosome in EAs may lead to suboptimal solutions because the chromosome length remains unchanged throughout the evolutionary process. In cases where the domain space is unknown a priori when the number of variables needed to solve a problem is not predetermined–only a small subset of genes in microarray GEPs substantially corresponds to the target disease [25]. To address this limitation, several studies have focused on using dynamic-length chromosomes instead of fixed-length ones to solve optimisation problems. For instance, variable-length PSO, variable-length GA, and variable-length black hole optimisation integrate dynamic-length chromosomes by employing feature ranking and length adjustment mechanisms [26]. Dynamic-variable length settings and adaptive mechanisms exhibit remarkable final classification accuracy, computational efficiency, and robustness in optimising various objective functions [27]. This study reviews the use of FS techniques for GEPs to optimise the identification of potential biomarkers under current technological conditions, focusing on FS methods, variable-length chromosome strategies, and their impact on performance. Additionally, it examines contemporary research on FS efficiency, gene length adaptation, and the concept of dynamic variability. This study provides an extensive systematic review using four levels of eligibility criteria to filter articles from four major academic databases. The PRISMA Checklists are available in the supplementary materials.
The adaptation approach also utilises FS because high stagnation values may indicate that the search process is trapped in local minima. This review investigates the role of FS in biomarker gene identification and parameter optimisation within adaptive settings. Prioritising candidates or marker genes is crucial, as these methods enable biomedical researchers to focus on a limited set of potentially valuable genes for in-depth analysis. Selecting a subset of discriminative features from high-dimensional, low-sample-size microarray GEPs is essential for bioinformatics tasks in cancer diagnosis [28]. In DE, ensemble FS is implemented through self-adaptive ensemble-based DE (SAEDE) [29], which adjusts relevant parameters in each generation to guide the search toward optimal solutions. Additional efforts are required to enhance the exploratory capabilities of most DE algorithms and PSO [30]. This paper is structured into five main sections. Section 1 discusses the importance of early cancer detection and the current research supporting it. Section 2 details identifying and selecting eligible articles from four high-indexed databases. Section 3 categorises key areas of FS research into a structured taxonomy. Section 4 provides an in-depth analysis of motivations, challenges, and future research recommendations in FS. Finally, Section 5 summarises the study’s findings and acknowledges its limitations.
This review investigates FS optimisation for cancer classification using GEPs within the framework of EAs. The focus is on studies related to cancer classification, FS, and GEPs. English-language papers were prioritised to provide insights into the relationship between gene expression patterns and cancer phenotypes, facilitating the identification of potential biomarkers in disease progression. Research studies published in English are often more widely accessible, and their inclusion helps mitigate any potential translation errors that could compromise the integrity of the review. To minimise any bias arising from this language selection, the researcher ensured that the inclusion of English-language papers followed a transparent and systematic selection process. Papers were selected based on clearly defined inclusion criteria, and non-English studies were uniformly excluded across the entire search. Moreover, the manual screening process was conducted by a trained reviewer with subject matter expertise. To ensure consistency and reliability, the reviewer initially screened all papers independently. Any disagreements regarding the inclusion or exclusion of papers were resolved through discussion, leading to a final consensus to maintain objectivity. This dual-review process ensures that subjective biases are minimised, and the process is reproducible.
Additionally, the reliability of the manual screening process was further reinforced using a predefined set of exclusion criteria (e.g., studies unrelated to cancer, irrelevant methodologies) to guide reviewers. This structured approach ensures a transparent and reproducible selection process, aligning with best practices in systematic reviews.
The search was conducted across four main digital databases, prioritising high-indexed journals, particularly those indexed in SSCI/SCIE within the Q1–Q3 categories, as outlined below:
1. IEEE Xplore: Covers research in electronics, electrical engineering, and computer science.
2. Scopus Digital Journal Library: A comprehensive database for scientific, technical, and medical content.
3. ScienceDirect: Provides access to a wide range of scientific and technical articles.
4. Springer: Includes journals, books, and reference works across science, technology, and medicine.
These databases were selected to ensure a diverse and comprehensive range of studies, particularly those focusing on FS optimisation in cancer classification, EAs, and related technical disciplines.
Strict eligibility criteria were applied at every stage of the selection process to ensure consistency and reliability. Following the initial filtering, a manual screening process was conducted to further refine the selection, ensuring the inclusion of the most relevant studies on FS optimisation in cancer classification.
In August 2022 and November 2023, a range of keywords was employed to identify studies on FS and optimisation in cancer classification for GEPs. These keywords comprised ‘cancer classification’, ‘optimisation’, and ‘FS’. The search query combined these terms using the conjunction ‘AND’, followed by ‘gene expression profile’, as depicted in Fig. 1. The advanced search option was utilised to exclude book chapters and short communications, ensuring a focus on the latest peer-reviewed studies. Papers were retrieved, and irrelevant studies were filtered out through manual screening, particularly those unrelated to cancer classification, EAs, or FS.
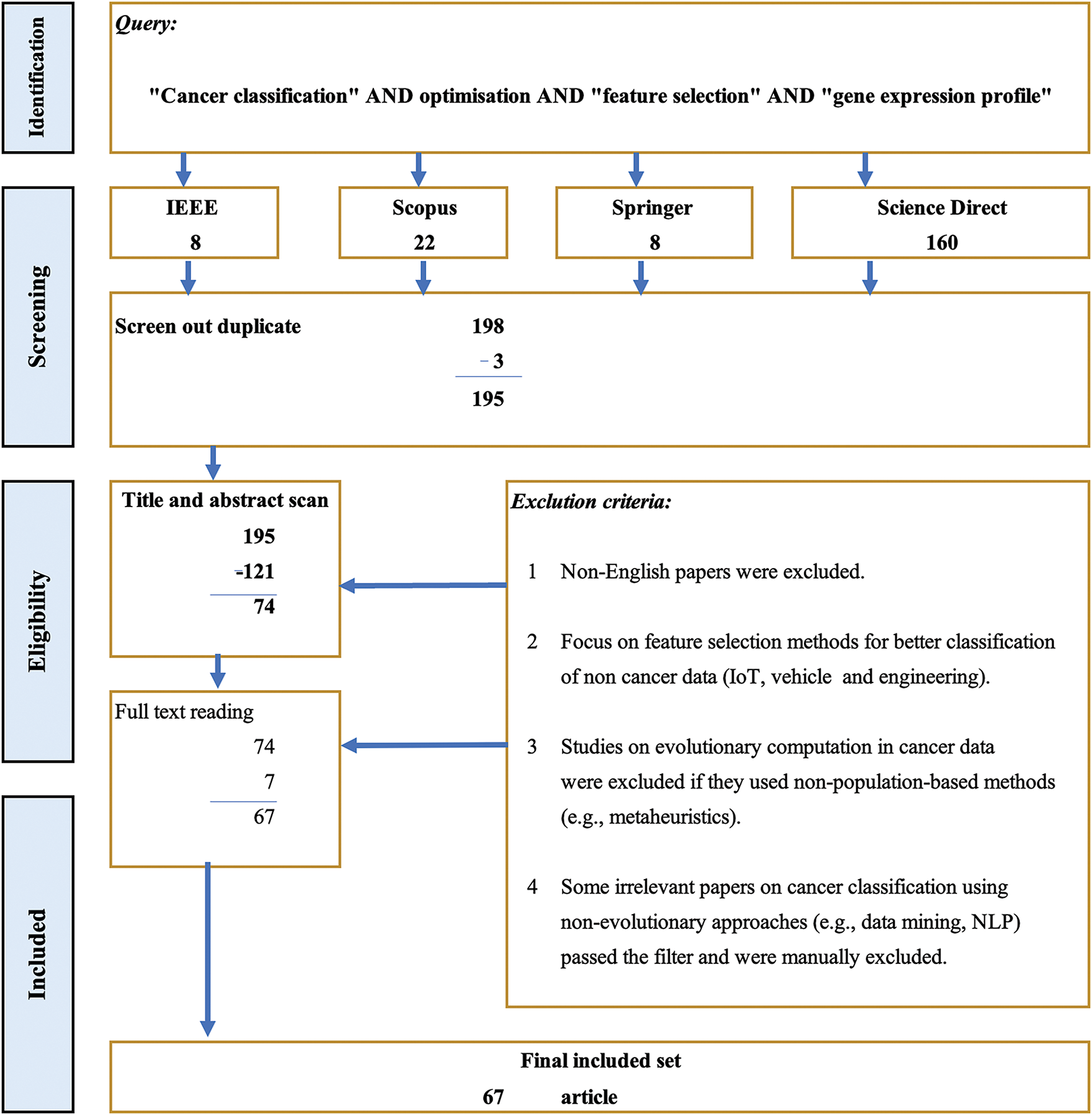
Figure 1: Diagram of research selection (including search query and exclusion criteria)
All relevant papers were extracted from the selected databases for initial classification using predefined objectives. Irrelevant references were manually excluded, specifically those unrelated to cancer or lacking an EA-based approach. However, despite applying the query criteria shown in Fig. 1, some FS studies that did not pertain to cancer classification, EAs, or population-based methods were retrieved. These papers were deemed irrelevant to this research and were manually excluded.
All relevant studies conforming to the predefined standards were included. Further refinement was conducted through manual screening to exclude studies that did not meet the criteria for cancer classification, FS optimisation, or EAs, as shown in Fig. 1. Subsequently, in accordance with the predefined classification objectives, all relevant papers were retrieved from the databases for preliminary classification. After retrieving papers that satisfied the search criteria, those meeting the exclusion criteria were excluded after two rounds of manual screenings.
Fig. 1 illustrates the systematic research selection process, including filter studies’ search query and exclusion criteria. Through manual screenings, duplicate and irrelevant references were excluded, resulting in a final selection of 67 articles that met the inclusion criteria. The key steps in the selection process included identifying relevant databases, screening for duplicates, and applying exclusion criteria based on language, relevance, and methodology used. Identifying research articles from four major databases (IEEE, Scopus, Springer, and Science Direct) is outlined, culminating in the final inclusion set after rigorous screening to ensure alignment with the review’s objectives. Fig. 2 presents a flowchart detailing the key steps in selecting articles for this review.
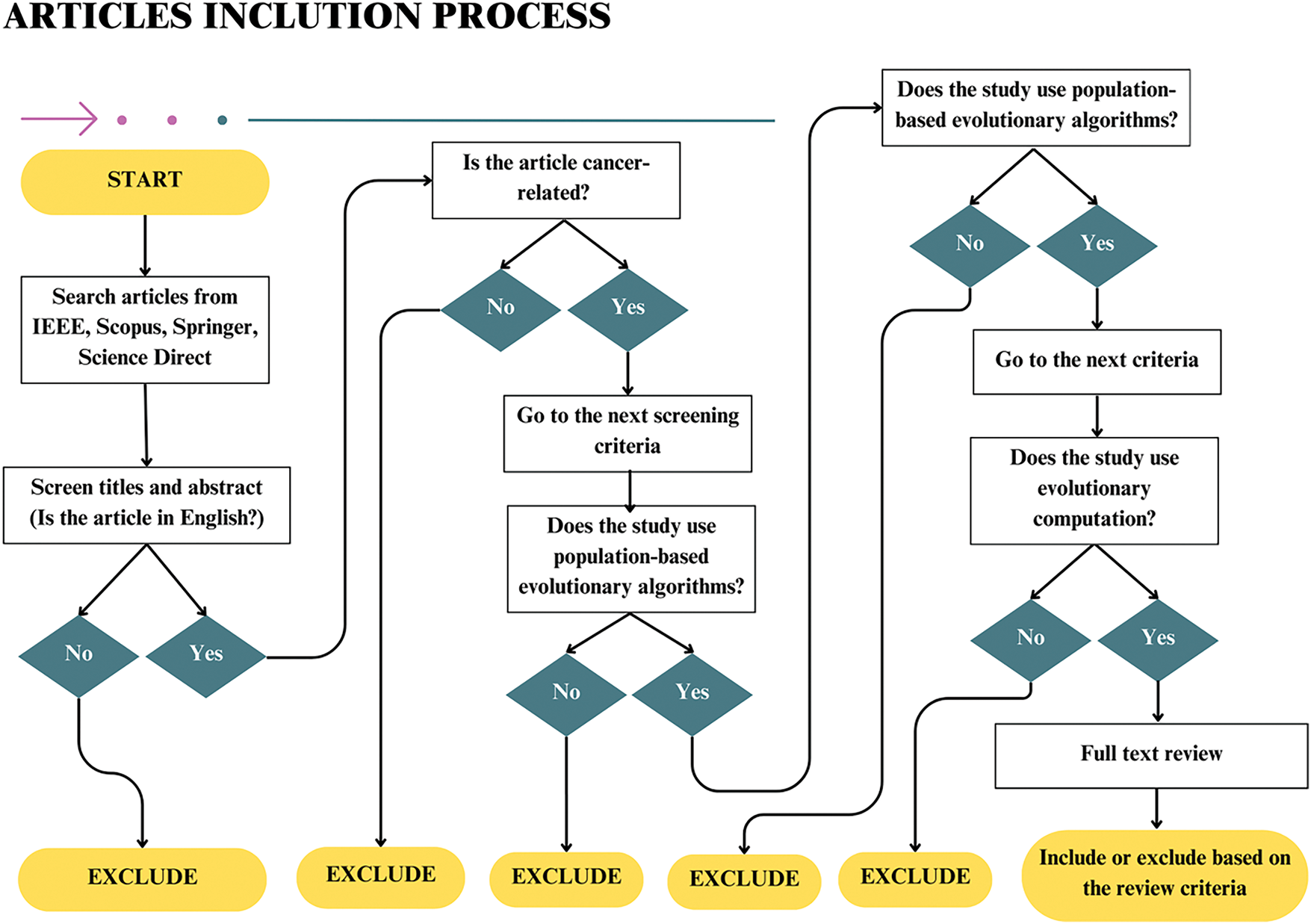
Figure 2: Flowchart of the inclusion and exclusion process for article selection
The flowchart in Fig. 2 visually represents the process of article inclusion based on specific criteria, such as language, cancer relevance, algorithm type, and methodology. It illustrates each decision point, including whether the paper is in English, focuses on cancer research, uses population-based evolutionary algorithms, and employs evolutionary computation. Additionally, the flowchart shows how papers are excluded at various stages, particularly those that do not meet the language, cancer relevance, or algorithmic criteria. This visual representation complements the systematic research selection process shown in Fig. 2, offering a detailed breakdown of each decision made during the screening process.
“Irrelevant references” were defined as research related to FS and optimisation but did not align with the cancer classification context, as shown in the exclusion criteria below. Despite adhering to the search query conditions shown in Fig. 2, some FS research papers that did not focus on cancer classification or EAs were retrieved and subsequently excluded. The exclusion criteria were as follows:
1. Non-English papers: Only English-language studies were included to ensure clarity and accessibility for a global audience. The primary goal of the review is to provide a comprehensive synthesis of relevant studies, ensuring that the collected data can be uniformly understood and disseminated across various international research communities. This decision was also driven by practical considerations, as English is the dominant language in academic publishing. Including studies in other languages would have significantly increased the complexity of data extraction and analysis, potentially leading to inconsistencies in interpretation.
2. Non-cancer-related FS studies: To maintain the specificity and relevance of the review, only studies focusing on cancer-related FS were included. FS methods in other domains, such as the Internet of things (IoT), vehicles, or engineering applications, often involve distinct feature spaces, algorithms, and evaluation metrics. Their inclusion could have introduced heterogeneity that would dilute the focus of the study. By excluding non-cancer-related FS studies, the review remains strictly aligned with the primary objective of analysing FS methods within cancer classification.
3. Non-population-based evolutionary algorithms: Studies that used non-population-based evolutionary algorithms were excluded because this review specifically assesses the role of population-based methods, which are fundamental to many evolutionary algorithms in cancer classification. Non-population-based methods, such as certain metaheuristics, do not adhere to the same principles as population-based evolutionary algorithms. Their inclusion would not contribute meaningfully to the objectives of this review.
4. Non-evolutionary computation approaches: This criterion was applied to exclude studies that, while retrieved based on the search query, did not align with the central theme of the review–namely, the use of evolutionary algorithms in cancer classification. Papers focusing on alternative computational techniques, such as data mining or natural language processing, were excluded as they used fundamentally different methodologies. Their inclusion would not directly contribute to the analysis of evolutionary computation approaches within the context of cancer classification. This review excludes these studies and focuses on the specific computational paradigms under investigation.
In this study, inter-reviewer reliability was assessed to measure the level of agreement between two reviewers responsible for screening and categorising articles based on their relevance. Cohen’s Kappa was employed to quantify the degree of agreement between the two evaluators. A total of 74 articles were assessed for relevance, with each article classified as either ‘relevant’ or ‘irrelevant’ according to the predefined inclusion criteria. Of these, 67 were deemed relevant, while 7 were excluded as irrelevant. Table 1 presents the number of agreements and disagreements between the two reviewers.

Where:
• A (67): Number of articles both evaluators agreed were relevant.
• B (7): Number of articles where Evaluator 1 considered relevant, but Evaluator 2 did not.
• C (4): Number of articles where Evaluator 1 considered irrelevant, but Evaluator 2 considered relevant.
• D (7): Number of articles both evaluators agreed were irrelevant.
The proportion of observed agreement (
The expected agreement (
The computation of the Cohen’s Kappa score is provided below:
The Cohen’s Kappa score for inter-reviewer reliability was calculated as 0.49, indicating a moderate level of agreement between the two evaluators. According to the standard interpretation scale, a Kappa score of between 0.41–0.60 suggests moderate agreement, implying that the evaluators were consistent and reliable in their classification of articles.
The researchers conducted multiple thorough readings of the full texts of the selected papers, carefully extracting key insights and classifying the studies based on detailed criteria. Each study was systematically analysed to ensure accurate classification and comprehensive understanding.
All relevant documents were catalogued under key categories using Microsoft Excel to facilitate efficient and orderly follow-up work. This approach enabled seamless tracking and organisation of the research findings. The major results of the studies were then summarised, tabulated, and explained in a structured manner. Relevant data, including paper titles, authors, publication dates, objectives, experimental data sources, research models, methodologies, challenges, and future directions, were systematically recorded to ensure smooth tracking and easy reference.
3 Review Results and Statistical Information
Relevant papers from the past five years (2018–2023) were identified using search engines. The initial query yielded 198 papers distributed across four major databases: eight from IEEE Explore, eight from Springer, 22 from Scopus, and 160 from Science Direct. During the screening stage, five duplicate papers were excluded. Subsequently, 195 papers were eliminated based on their titles and abstracts. A further 74 articles were excluded after a full-text review. As a result, the final number of included papers was 67.
The categorisation of these papers revealed several key research areas:
• Thirty papers (44.8%) focused on developing algorithms and models for FS and classification.
• Ten papers examined computational complexity and robust classification.
• Ten papers explored FS optimisation performances.
• Twenty papers (30%) investigated biomarker identification using EAs.
• Nine papers discussed challenges related to high-dimensional data and the identification of potential biomarkers from gene expression data.
• Two papers investigated the formulation of chromosome lengths for FS in GEPs.
• Nine papers (14%) applied FS techniques in decision support systems and prediction tools.
• Only three papers (4.5%) were comparative reviews or investigations on FS in cancer classification.
The taxonomy was refined and summarised, with a classification diagram presented in Fig. 3. Although some subcategories overlap, they were clearly distinguished to facilitate further discussion.
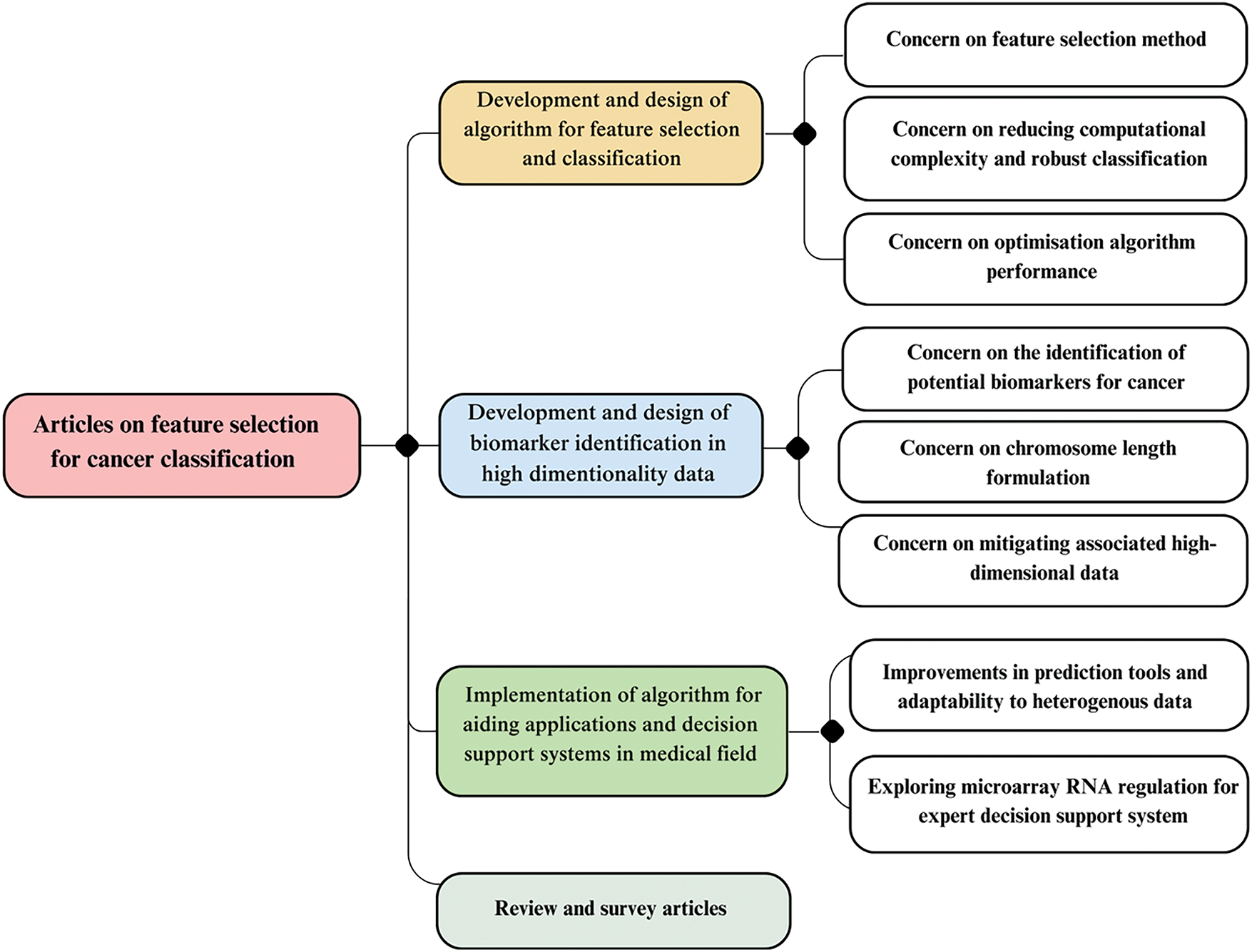
Figure 3: Taxonomy of research papers on FS and chromosome length
Fig. 3 presents the taxonomy categorising research papers in FS and optimising chromosome length. Taxonomy is crucial in identifying gaps in research methods and assisting users in making informed decisions. For example, while various algorithms are available for the clinical application of FS tools in cancer diagnosis and prognosis, selecting the most suitable one requires expertise. By classifying studies, taxonomy helps researchers avoid unnecessary detours and facilitates communication within the research community. Researchers adopting similar classifications establish a common language for discussing algorithms, models, decision support systems, and prediction tools. These are currently the key topics in the EA-based FS of GEPs.
3.1 Review of the Development and Design of FS
FS is a critical step in building effective models for cancer classification using genomic data. It involves identifying the most informative genes to distinguish between cancer classes. These papers explore FS methods, computational complexity reduction, robust classification, and algorithm performance optimisation using AI technologies in GEPs. The FS algorithm or method employs primary strategies–filter, wrapper, and embedding–to extract significant features for the subsequent phase [31].
Figs. 4 and 5 show the logical relationship between filter, embedded, and wrapper approaches. For bioinformatics analytic tasks, such as identifying disease-associated genes and developing classifiers for cancer detection, selecting a subset of discriminant characteristics from high-dimensional, low-sample-size microarray GEPs remains crucial. A primary research focus in this field is developing and designing algorithms and models. FS algorithm development is a key area of interest, with 30 papers categorised into three subcategories.
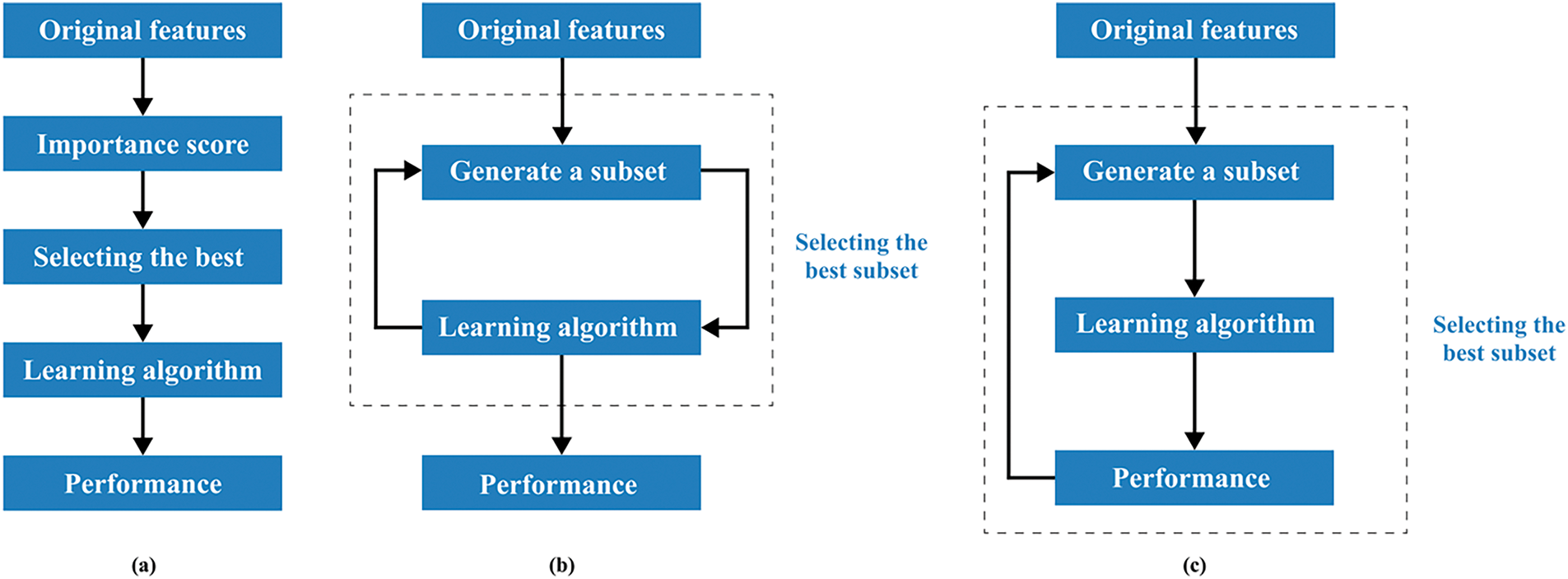
Figure 4: Three types of feature selection (FS): (a) Filter, (b) Wrapper, and (c) Embedded
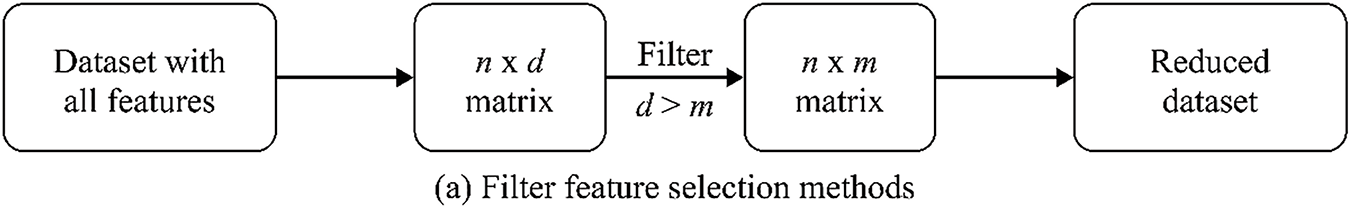
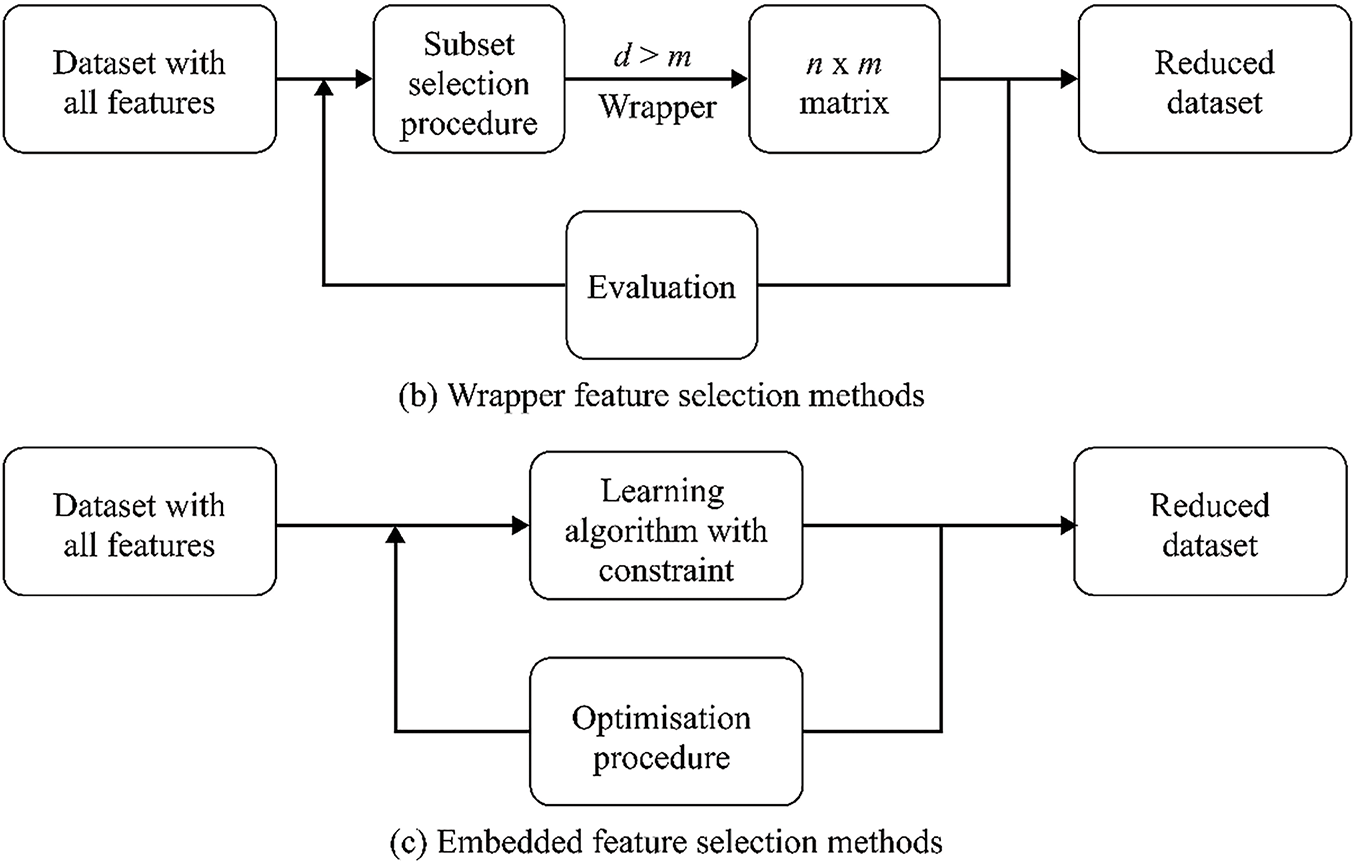
Figure 5: The logical flowchart of filter, wrapper, and embedded FS method
3.1.1 Evolutionary Algorithms for FS and Classification in Cancer Research
The first research subclass focuses on FS, specifically its integration with EAs for both FS and classification. Many researchers in this area are dedicated to refining and innovating FS methods within the EA framework, which has become a central focus for improving the effectiveness and efficiency of FS processes in classification tasks. This subclass explores various techniques and strategies within the EA framework to enhance the efficacy and efficiency of FS processes in classification.
The cross-referencing relationships between the keywords in the literature have been visualised in Fig. 6, providing insights into the field of FS. This network analysis highlights the pivotal role of evolutionary algorithms in cancer classification research, especially in handling the complexity of high-dimensional datasets. Their connection to DL and advanced optimisation techniques underscores the ongoing evolution of FS methodologies. However, challenges like parameter optimisation, algorithm efficiency, and issues like dataset imbalance and variability in GEPs persist. These challenges create opportunities for future research, especially in improving the scalability and generalisability of EA-based FS in cancer classification.
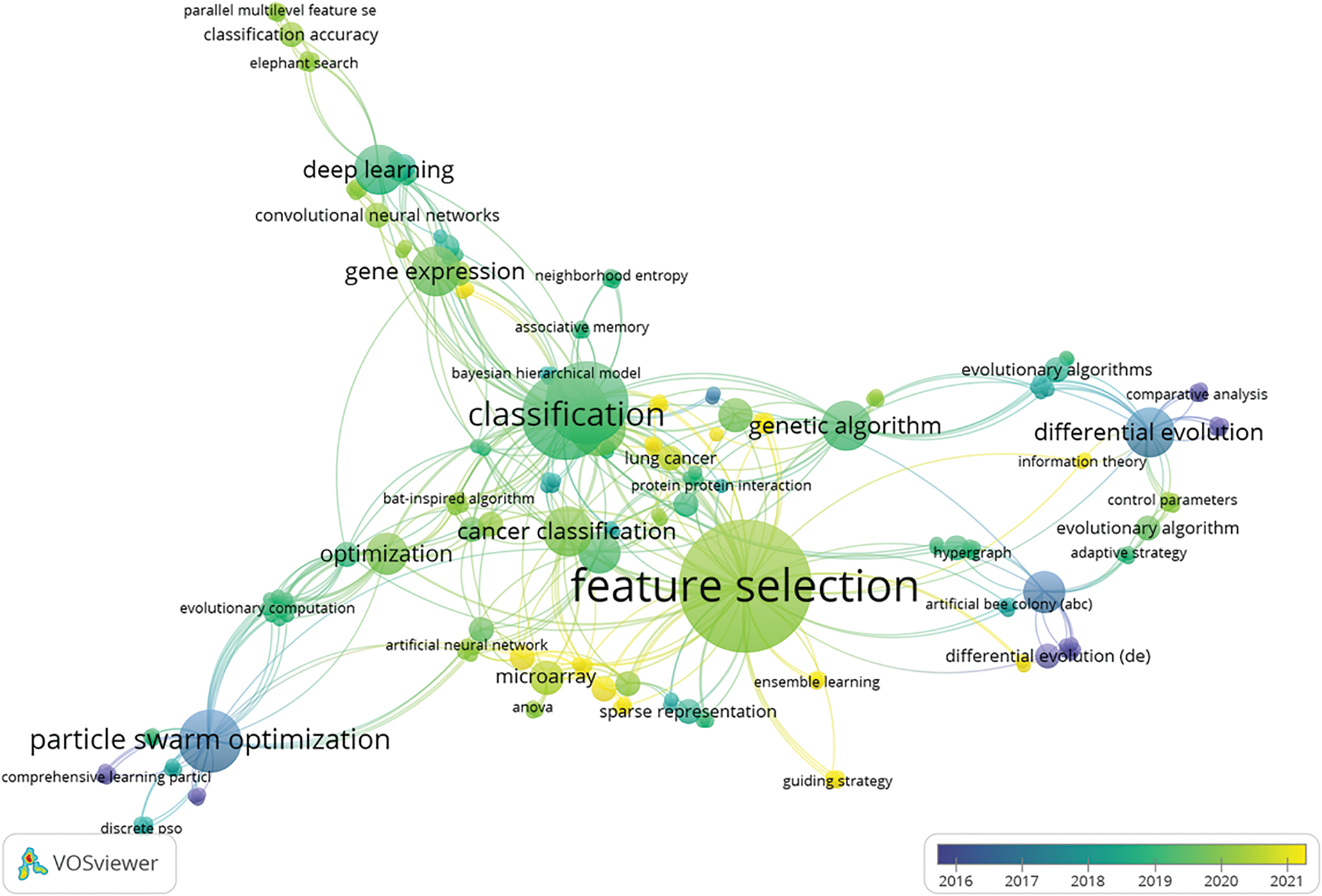
Figure 6: Interrelationships between research interests in FS
A new swarm intelligence technique, the spider monkey optimisation (SMO) algorithm, employs an SVM to classify minimum features, aiming to minimise the number of features in cancer classification while maximising classification accuracy in EAs [32]. Another approach, a bi-stage hierarchical swarm-based gene selection technique, uses multiple fitness functions (MFDPSO) as a multi-filtering-based gene selection procedure [33] and an artificial bee colony algorithm (ABC) for automatic clustering. A framework for multiple gene selection across all tumour subtypes has been developed to address current algorithms’ limitations in capturing each subtype’s unique characteristics. This algorithm combines the obtained subtype-specific gene subsets into a single gene subset [34]. By implementing three specific gene selection algorithms within the two frameworks, the algorithm categorises disease-specific genes and classifies different tumour subtypes at the molecular level.
The Harris Hawks optimisation (HHO) uses variable neighbourhood learning (VNLHHO) to balance the global exploration and local exploitation of HHO. Additionally, it employs a mutation operation to increase diversity, preventing convergence to a local optimum and optimising algorithm performance [35]. A unified learning framework, unsupervised FS with adaptive residual preserving, maintains the residual relationship between data samples and aids in reconstructing the original data. During graph construction, numerous data relationships, such as residual relationships, may need to be recovered [36]. The local manifold structure can be effectively preserved in data reconstruction by considering local residuals. A multilayer perceptron (MLP) selects genes from the pathway information of microarray data. After employing DE to select a relevant and minimal gene subset, MLP designs and builds a suitable and efficient vector representation for DE to improve the sensitivity, accuracy, and F-score in classification tasks [37].
A new cell separation algorithm (CSA) uses the centrifugal principle to enforce the separation solution based on objective functions. Multiple centrifugation separation steps result in genes highly correlated with the class attribute, affecting classification accuracy [38]. A novel hybrid filter and differential evolution FS employed by keeping the highest-ranked feature and eliminating superfluous and irrelevant ones, producing noticeably better classification results with only less features of the microarray datasets [39]. Logistic regression is used to optimise the accurate prediction of hepatocellular carcinoma (HCC), the most common liver cancer in adults, through a GA [40]. The process starts with the genetic selection of the logistic regression parameters, which extends to include genetic FS and subsequently trains the logistic regression model.
3.1.2 Reduction of Computational Complexity and Robust Classification
Reducing computational complexity is essential for improving classification performance. Various approaches, such as two-stage heteroassociative memory (HAM) neural networks and extended PSO models, optimise FS processes by eliminating irrelevant genes and improving algorithm speed. Other methods, such as the parallel multilevel FS algorithm, use data partitioning techniques to reduce processing time, while ensemble classifiers like SVM, Naïve Bayes (NB), and k-nearest neighbour (KNN) further enhance classification accuracy.
The computational effort is remarkably reduced by discarding non-informative genes, and the biological interpretability produced by a two-stage HAM neural network algorithm is enhanced [41]. HAMs consist of more than one layer, each fully connected to all other layers. Determining a random population size and dividing it into two groups promotes exploration and reduces the likelihood of stagnation. The process produced by the extended PSO model (EPSO) potentially enhances the PSO search process, reducing processing time for selecting the optimal feature set and improving classification accuracy [42]. Specific performance-dependent penalty parameters can be tailored to the problem structure, capturing inherent patterns in the data when used as a filter-wrapper hybrid ensemble FS [43]. This algorithm outperforms feature subspace selection, preserving predictive accuracy while eliminating noise and curtailing the high computational cost of training.
The parallel multilevel FS algorithm reduces overall execution time by employing vertical data partitioning along the feature space and horizontal partitioning along samples, facilitating data parallelism [44]. An ensemble of four classifiers–SVM, NB, C4.5, and KNN–serves as the fitness function within PSO to reduce dimensionality and comprehensively covers all search space fields. Improved binary PSO (iBPSO) addresses the problem of early convergence to local optima in traditional binary PSO (BPSO) by implementing a two-phase hybrid model for cancer classification, integrating correlation-based FS with an NB classifier [45]. Krill herd (KH) optimisation and the signal-to-noise ratio (SNR) technique produce compact, low-dimensional data and select relevant features. This method subsequently uses an entropy-based graph classifier to establish classification by discarding outliers from redundant and irrelevant features, aided by Euclidean distance calculations [46]. Following data normalisation, a feature-clustering SVM combined with an artificial neural network (ANN) classifier produces classification results through a clustering-centred FS approach [47].
The effectiveness and consistency of the minimum redundancy maximal relevancy (MRMR) method are enhanced by combining it with multiple filters, thereby addressing the shortcomings of individual filters [48]. This combination is achieved through the robust minimum redundancy maximum relevancy–hybrid bat-inspired algorithm (rMRMR–HBA), which utilises robust minimum redundancy and maximum relevancy principles alongside a hybrid bat-inspired algorithm incorporating beta hill climbing. This approach reduces computational complexity and ensures robust classification [49]. A framework for data pre-processing, FS, and classification–capable of automatically selecting the best classifier with optimised hyperparameters based on the input dataset–has been analysed to improve classification accuracy and computational time [50]. FS is performed in two steps using recursive feature elimination (RFE) and a metapool of diverse ensemble classifiers. Subsequently, a customised algorithm selects the best classifier from the metapool based on classification accuracy and computational efficiency.
3.1.3 Algorithm Optimisation Performance
This research’s third subclass focuses on optimising algorithm performance in FS and classification tasks. The effectiveness of an optimisation algorithm often involves experimentation, as the best-performing algorithm can vary depending on the specific characteristics of the data and model. This subclass focuses on optimising FS algorithms for classification tasks by implementing key optimisation strategies. One strategy is maintaining diversity through hybrid models, such as the multi-objective spotted hyena optimiser (MOSHO) and the barnacle mating optimiser (BMO). These approaches balance exploration and exploitation to enhance FS and classification accuracy. Other models, including the hybrid filter-wrapper method and various ML algorithms, also contribute significantly to achieving optimal algorithm performance for cancer classification.
Algorithm optimisation focuses on maintaining diversity and enhancing search capabilities. A hybrid novel technique, CSSMO-based gene selection for cancer classification alternative to the fitness of spider monkey optimization (SMO) with the cuckoo search algorithm (CSA) and CSSMO for feature selection. This method involves a cleaning process mRMR, classified using DL to identify different groups or classes related to a particular cancer disease, and achieves much better classification accuracy [51]. By increasing diversity, bilayer mutated PSO (BLMPSO) demonstrates high accuracy and precision in cancer cell classification [52]. The hybrid wrapper–filter comprehensive learning particle swarm optimisation with local search (COMP-PSO-LS) algorithm employs a randomised dependence coefficient (RDC)-based filter to streamline the PSO search process, resulting in consistent classification outcomes [53].
When combined with SVM, BMO augmented with SVM (BMO-SVM) imitates the mating behaviour of barnacles in nature to solve optimisation problems, exhibiting accuracy comparable to GA, PSO, ABC, and tunicate swarm algorithm (all combined with SVM) [54]. An ensemble of three filter methods–symmetrical uncertainty (SU), chi-square, and relief–reduces feature dimensions in the first phase of the process [55]. In the second phase, local search-based FS (LSFS) is applied using GA, followed by classification with SVM, KNN, and random forest (RF) classifiers, where SVM achieves 99% accuracy. A hybrid filter-wrapper method based on multi-objective simplified swarm optimisation (MOSSO) is integrated with SVM to identify an optimal and minimal gene subset from selected genes [56].
ML algorithms and their dependence on suitable FS methods are crucial for accurate prediction. Research on optimising three key parameters–learning algorithm, FS method, and rejection rate–for robust cancer prediction demonstrates that the predictive accuracies of rejection option (RO) classifiers vary depending on the FS methods for each dataset. FS methods such as t-test, Las Vegas filter (LVF), relief, and information gain (IG) reduce dataset features, while RO classifiers with different rejection thresholds enhance robustness in cancer prediction. In [57], a transformation-based three-level FS using wavelets is employed for prostate cancer classification. At the first level, wavelet-based initialisation selects essential features. At the second level, standardised gene selection techniques–including Relief-F, Fisher score, IG, and SNR–are applied. The third level implements FS using optimisation techniques before classification, incorporating methods such as marriage in honeybee optimisation, the migrating bird optimisation algorithm, the salp swarm optimisation algorithm, and the whale optimisation algorithm (WOA). The best classification performance is obtained when SNR with WOA is classified using ANN.
The accuracies of sequence learning models based on long short-term memory (LSTM) and bidirectional gated recurrent units (GRUs) using MLP, RF, decision tree, and KNN on unsupervised and supervised data reduction methods are compared to investigate the performance accuracy of GRU. This comparison aims to potentially aid medical professionals in cancer diagnostics, treatment, and prevention [58]. Additionally, functional annotations of genes are used to predict the class function of time-series gene expression without labels and cannot be considered gene features. GA uses these functional annotations to assign weights to gene expression levels (genetic algorithm-augmented weighted gene expression feature analysis, GAAWGEFA), ensuring appropriate weighting at each time point [59]. This method allows the similarity in gene expression levels to reflect their functional similarity.
3.2 Review of the Development and Design of Biomarker Identification for High-Dimensionality Data
The second major research focus on FS of GEP for cancer classification is the development of biomarker identification. This category, comprising 20 papers, is divided into three subcategories: (1) novel biomarker FS methods, (2) validation and clinical utility assessment, and (3) integration with multimodal data sources. Studies in these areas aim to deepen the understanding of cancer data, enhance diagnostic accuracy, and improve patient outcomes by identifying and validating clinically relevant biomarkers. The primary goal of this research is to discover genetic markers or biomarkers that exhibit strong associations with specific cancer subtypes, disease progression, or treatment responses.
3.2.1 Identification of Potential Biomarkers for Cancer
The second subclass of this research focuses on identifying potential biomarkers for cancer classification. A hybrid multilayer recursive FS (multilayer recursive feature elimination, MGRFE) in EAs leverages the evolutionary computation of GA, integrating the maximal information coefficient (MIC) and t-test with the explicit feature elimination of RFE. This approach aims to obtain the minimum discriminative gene subset with optimal classification ability [60]. Additionally, the firefly-based gene selection (FF–SVM) method shows high classification accuracy using a small number of selected genes [61]. This approach highlights the importance of efficient gene selection techniques in improving the discriminatory power of classification models, thereby improving cancer diagnosis and prognosis with increased accuracy.
Correlation patterns and causal connectivity have been largely overlooked in gene expression literature. To address this, Fisher’s test and Zou’s confidence intervals are employed to detect differences in correlation coefficients, while graph modelling visualises between-group differences in gene structures of two subgroups [62]. Additionally, Fisher linear discriminant (FLD) and neighbourhood rough set (NRS) methods, combined with an SVM classifier, reduce preliminary genetic data and calculate neighbourhood dependency to select a small yet well-classified gene subset. This approach improves classification performance [63]. By integrating these techniques, researchers can streamline the analysis of complex genetic data, leading to more accurate cancer classification models and ultimately contributing to precise diagnostic and prognostic outcomes in clinical settings.
Detecting stochastically independent variables that effectively capture all the essential information for modelling output variables requires a thorough analysis of structural dependencies among all considered features. This process is crucial for detecting linear and nonlinear dependencies between different components in ML [64]. Sequential reinforcement active feature learning (SRAFL) dynamically selects genes in each sample to automatically identify gene signatures for each subtype [65]. A binary artificial bee colony algorithm, coupled with SVM and a two-stage cascading classifier, identifies relevant genes and cancer using RNA-Seq data, improving feature reduction and increasing classification accuracy. By leveraging advanced ML algorithms and incorporating domain-specific knowledge, researchers are making substantial strides in unravelling the complexities of cancer data and developing personalised treatment strategies tailored to the needs of individual patients.
An ensemble FS (EFS) approach incorporating perturbation at the data level-either homogeneous (Hom-EFS) or level method (i.e., heterogeneous (Het-EFS)) is employed to investigate the adequacy of microarray data. This investigation facilitates an extensive comparison with other ensemble and single FS approaches to improve the stability and predictive power of candidate biomarkers. Other ensemble FS frameworks utilise sampling techniques to obtain multiple sample datasets and use two aggregation strategies to combine feature subsets into a single set, accommodating both binary and multiclass case datasets [66]. These strategies are designed to compare and improve classification performance [67]. By leveraging ensemble-based methodologies, researchers aim to mitigate the impact of noise and variability inherent in microarray data, ultimately strengthening the robustness and generalisability of predictive models for cancer classification.
A multi-metaheuristic FS (MHFS) model presents a promising approach to addressing feature selection challenges. This system uses two parallel algorithms–swarm optimisation and bat algorithm–to search for the best number of features [68]. Each algorithm independently searches for the best solutions, which are subsequently shared to refine the selection process. MHFS has demonstrated superior performance in achieving accurate classification results while utilising fewer features than PSO and the bat algorithm. By integrating swarm optimisation and the bat algorithm within a unified framework, MHFS enhances classification accuracy and improves efficiency in identifying the most informative feature subsets for cancer classification. This underscores the potential of multi-metaheuristic approaches in advancing the performance and scalability of feature selection models.
3.2.2 Chromosome Length Formulation
The second subcategory focuses on novel research regarding chromosome length formulation in GEPs. The extended mutual congestion (EMC) discrete weighted evolution strategy (DWES) aims to maximise classification accuracy while minimising the length of the selected subset [69]. Initially, EMC discards irrelevant and redundant features using a frequency-based filter ranking approach. Subsequently, DWES clusters features and applies mutation to simultaneously select the most relevant feature in each cluster. It assigns greater weights to the most informative clusters than to less significant ones to prevent redundancy. GAs employing variable-length chromosomes are extensively used, with same-point (SP) crossover being the most popular crossover mechanism [70]. Notably, there is no universally consistent chromosome length applicable across optimisation problems.
3.2.3 Mitigation of High-Dimensional Data
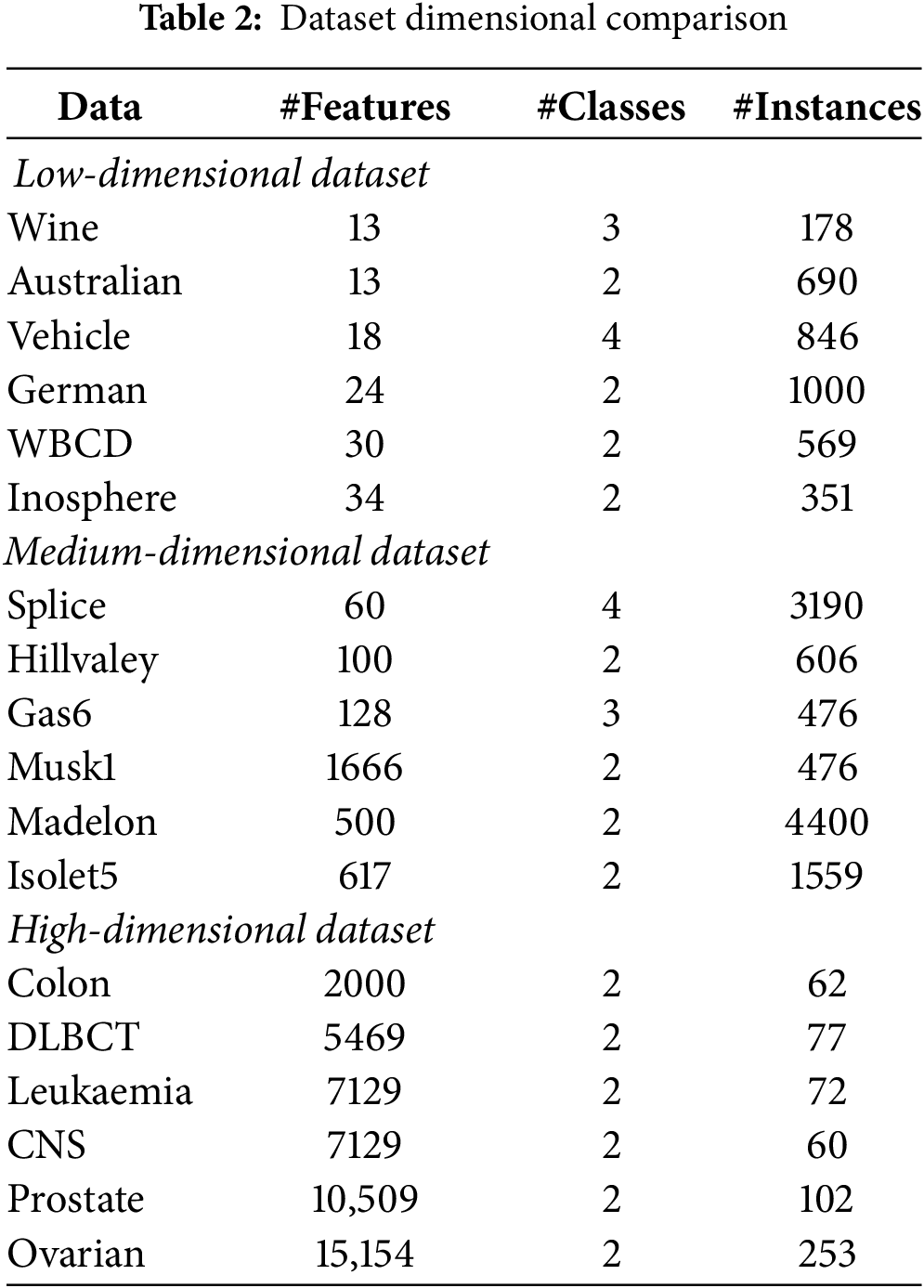
This subcategory focuses on efforts to mitigate challenges associated with high-dimensional data. Table 2 presents the characteristics of microarray datasets, which are categorised as high-dimensional, aligning with the third category in Table 1. Cancer datasets contain a vast number of features, ranging from 2000 to over 10,000, while the number of instances remains relatively small, typically between 72 and approximately 200 [71]. The problem of the ‘curse of dimensionality’ is addressed by utilising EAs. An evolutionary wrapper-based approach utilises the principles of the Enhanced Jaya (EJaya) algorithm and the forest optimisation algorithm (FOA) [72]. EJaya tunes two important parameters–FOA’s local and global seeding adjustments–to improve classification accuracy while reducing the number of selected features.
A hybridised harmony search and pareto optimisation (AHSGS) approach is recommended for FS in high-dimensional databases. This method involves an adaptive harmony search algorithm to select and rank the optimal gene [73]. Additionally, an elephant search algorithm (ESA)-based optimisation has been proposed to select the best gene expression from large-scale microarray data [74]. The firefly search (FFS) is used as a benchmark or comparison to evaluate the feature selection method’s efficiency and understand the effectiveness of the ESA method in the feature selection process. Moreover, efficient computation of multiple alternative classification models is achieved through the BIGBIOCL (a software tool) algorithm, which integrates a multiple tree-based classifier for big biological data [75]. This approach enables the extraction of alternative and equivalent classification models by iteratively removing selected features from large DNA methylation datasets.
The Bayesian evolutionary hypernetwork (BEHN) learns a high-order graphical model comprising a large population of hyperedges that encode high-order relationships among features in high-dimensional data. Sequence learning models based on LSTM and GRU for unsupervised data have demonstrated the potential to address challenges associated with the high dimensionality and sparsity of electronic health record (EHR) data [76]. This advancement aids medical professionals in cancer diagnosis, treatment, and prevention. The study of DNA methylation requires processing hundreds of thousands of features for every patient. DNA methylation is characterised by high dimensionality and a limited sample size, showing well-documented challenges in FS and data generation [77]. Additionally, Autoencoders (AEs) for nonlinear feature fusion can identify genes associated with breast cancer recurrence, highlighting their potential in mitigating issues related to high dimensionality.
3.3 Implementation of Algorithms for Aiding Medical Applications and Decision Support Systems
The third research focus on FS for GEPs in cancer classification involves developing and implementing algorithms to support medical applications and decision support systems. This category encompasses 14 papers and is divided into two subcategories. The first subcategory examines the integration of FS algorithms into medical decision support systems, while the second explores their real-world application in clinical settings and clinical practices. Collectively, these studies highlight the role of FS algorithms in enhancing medical decision-making and improving clinical practices.
3.3.1 Improvement in Prediction Tools and Adaptability to Heterogeneous Data
The subclass involves prediction tools and the adaptability of algorithms to heterogeneous data. The traditional rough set model is limited to discrete data. However, a novel approach involving NRS, entropy-based gene selection, and the Fisher score for tumour classification can effectively process real-valued data while preserving the original gene classification information. For certain cancer types, differentially expressed mqTrans features do not exhibit differential expression between early- and late-stage samples. Predictive models based on transcription factor expression levels have been developed to address this limitation [78]. These models formulate quantitative transcriptional regulatory relationships of metabolism-related genes using a multi-input multi-output (MIMO) regression framework implemented via the GRU network.
Various classifiers, including logistic regression, ridge classifiers, and Gaussian Naive Bayes, are employed to analyse risk factors in cancer prediction. These classifiers evaluate multiple risk factors and assess their performance on unseen data [79]. The results show that ML-assisted analysis of cytokine gene variants and sociodemographic characteristics can serve as a decision-support tool by automating the integration of bioinformatics tools from raw data [80], which focuses on selecting the most representative genes in multiclass problems and classifying new patients, thereby enhancing expert decision-making.
DL approaches are also employed to analyse complex two-dimensional images, utilising data reduction and visualisation techniques. These methods generate visual representations, such as expression heatmaps and hotspot maps, to illustrate the spatial distribution of disease features and their correlations [81]. A technique known as genomic data and pathological images on multiple kernel learning (GPMKL), based on multiple kernel learning (MKL), effectively integrates heterogeneous data sources, including genomic data (gene expression, copy number alteration, gene methylation, and protein expression) alongside pathological images, to improve the accuracy of breast cancer survival prediction [82]. This integrative approach provides a holistic understanding of the molecular mechanisms underlying cancer progression and enables accurate prognostic assessments.
3.3.2 Exploration of Microarray RNA Regulation for Expert Decision Support Systems
The second subcategory explores the role of microarray RNA regulation in expert decision support systems. In intelligent decision support systems (IDSSs), the most significant features are selected using IG. The selected genes are reduced through the grey wolf optimisation (GWO) algorithm and subsequently classified using SVM [83]. Quantum-inspired DE (QDE), integrated with a classification method, is employed to select a subset of genes from 12 well-known single-cell RNA sequencing (scRNA-seq) transcriptomic datasets for cell type identification. Specifically, QDE combines different ML classifiers, including logistic regression, decision tree, SVM with linear and radial basis function kernels, extreme learning machine, and QDE–SVM, demonstrating high accuracy in cell type classification. Additionally, a bio-inspired binary grey wolf optimisation algorithm (BGWOA) has been applied to cancer classification using eight microarray datasets. By integrating MRMR with a novel binary grey wolf, this algorithm effectively reduces data dimensionality while maintaining high classification performance [84]. Notably, it selects a minimal yet highly relevant subset of genes, a crucial factor in cancer diagnosis and detection.
Two sets of RNA and methylation datasets from early- and late-stage liver HCC are analysed using NB with different key feature sets, including Matthew’s correlation coefficient, the area under the receiver operating characteristic curve and multiclass classification metrics [85]. For serious ovarian carcinoma (SOC) prediction, RNA microarray expression regulation is utilised. Individualised pair analysis of gene expression (deiPAGE) is identified from noncoding RNA (ncRNA) regulation and the competing endogenous RNA (ceRNA) network. This approach defines competing endogenous gene pairs (ceGPs) derived from the ceRNA network and ncRNA interactions [86]. Furthermore, differential expression analyses using DESeq2 and edgeR classify cancer based on differential microRNA (miRNA) expression patterns between tumour and non-tumour samples in a single cancer type [87].
The integrative analysis of histopathological images and genomic data offers significant potential for improving cancer diagnosis and prognosis. A target gene screening (TGS) system, combined with pattern recognition matching, parameter detection genetic algorithm, and association rule mining, is employed to screen out a small subset of genes and predict cancer stages [88]. Multitask and multimodal FS use the relationship learning framework to discover the relationships between diagnosis and prognosis tasks automatically [89]. Additionally, the network game theory maker (NGTM) is implemented to discover HCC biomarkers by integrating scRNA-seq, gene regulatory networks (GRNs), and RF classifier to improve classification performances and enhance RFE performance [90].
3.4 Review and Survey of FS and Classification Based on EA
Reviews and surveys aim to provide an overview of the current state of research, identify key challenges, propose new hypotheses, and highlight under-researched or overlooked areas. This category includes four studies. Significant reviews have been conducted on the performance of FS across various cancer data types. However, existing reviews have not sufficiently addressed the effects and optimisation of FS concerning chromosome formulation length and the role of convolutional neural networks (CNNs) in biomarker identification and detection systems.
Following the principle of classification, our review systematically classifies FS, biomarker detection, and cancer classification based on EAs. It provides an in-depth analysis of the current status and development trends in FS and gene subset selection within the context of EAs while forecasting future research directions. This structured approach distinguishes our review from previous studies.
The challenges posed by high-dimensional data and small sample sizes affect the performance of data mining and ML algorithms. Evolutionary methods, which have the widest application, should be further promoted within FS field to reduce the complexity of microarray-based systematic mapping studies. Surveys assessing classification accuracy and the number of selected genes in cancer classification have identified GAs as the most widely applied wrapper method in the literature. This review is structured around six perspectives: methodology, classifiers, datasets, dataset dimensionality, performance metrics, and achieved results.
The majority of the literature (34.9%) has focused on the development of hybrid FS method (FSM). Moreover, wrapper-based FSM methods constitute a moderate proportion of published studies, whereas filter-based and parallel FS methods have garnered comparatively less attention.
Table 3 summarises the key research areas in the application of EAs for FS in cancer classification. These areas are categorised into several primary research domains, including algorithm and model development, biomarker identification, decision support systems, and review articles. The table shows that a significant number of studies focus on refining algorithms and models for FS and classification (10 articles), with many addressing challenges such as reducing computational complexity and improving classification robustness. Research on biomarker identification is also of critical importance, with nine studies dedicated to identifying potential biomarkers and two studies exploring chromosome length formulation to optimise the identification process. Furthermore, studies on high-dimensional data mitigation (nine articles) highlight the challenge of managing vast datasets with relatively small sample sizes, a prevalent issue in cancer genomics.

Research on decision support systems includes six studies on prediction tools and eight studies on expert decision support systems, both of which play a vital role in improving cancer diagnostics. Lastly, three review articles provide a synthesis of existing methods and identify key areas for future research in this field. Figs. 7 and 8 further illustrate the distribution of collected papers across different research hotspots and database sources.
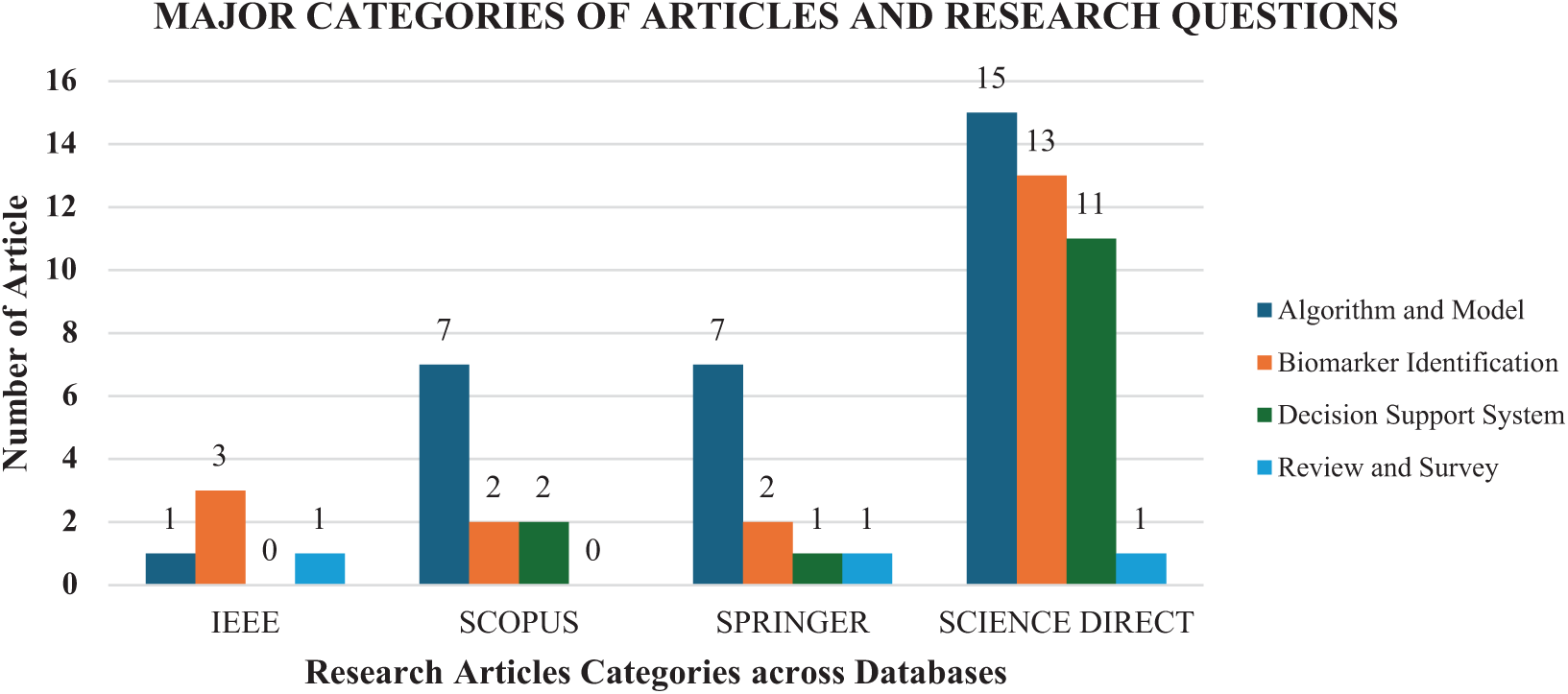
Figure 7: Number of papers classified by major research categories
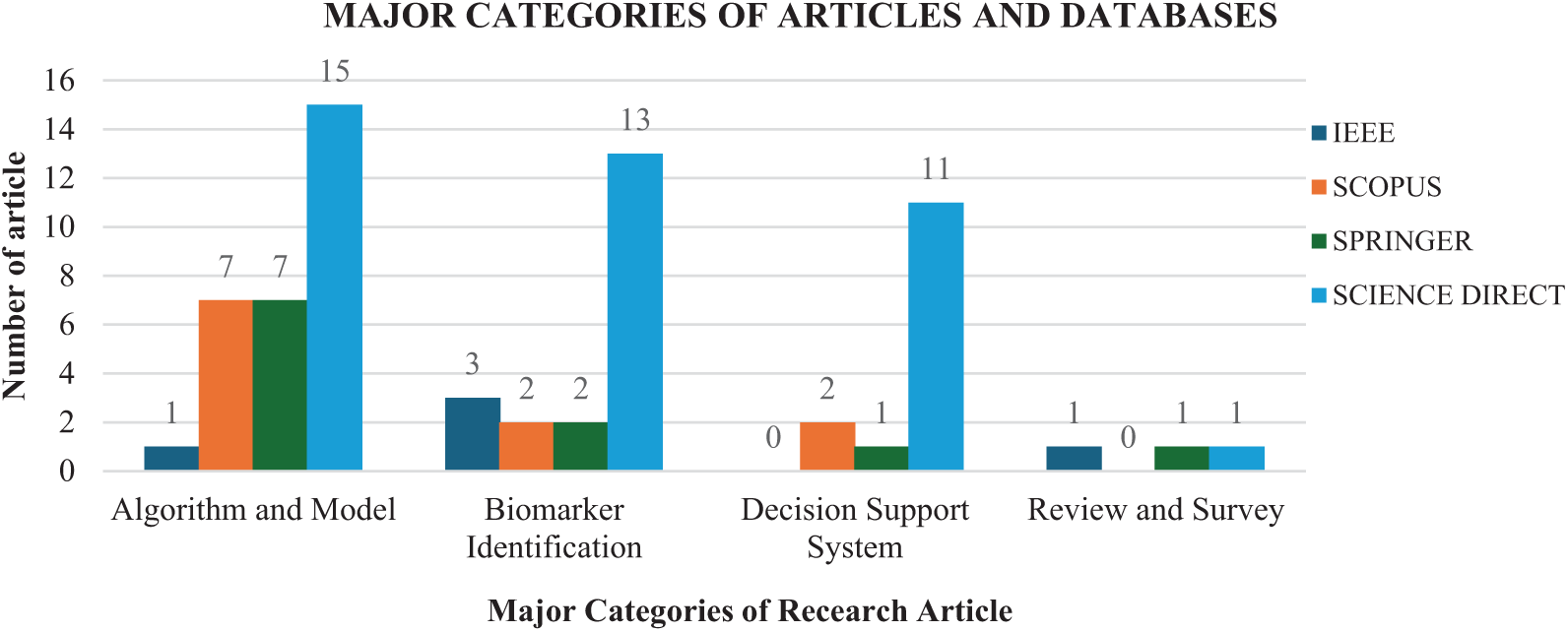
Figure 8: Number of papers classified by database sources
Fig. 7 shows the distribution of papers across different categories, such as algorithms and models, biomarker identification, decision-support systems, and review articles. This figure provides a clear representation of research trends in EA-based FS for cancer classification, with a strong focus on algorithm and model development. The bars in the figure represent the number of papers in each category across four major academic databases: IEEE, Scopus, Springer, and Science Direct. The total number of papers analysed is 67. As depicted in the chart, 30 papers focus on algorithm and model development, 20 on biomarker identification, 14 on decision-support systems, and three are review articles. These findings indicate a predominant research focus on algorithm and model development for FS in cancer classification.
Fig. 8 illustrates the distribution of papers across four major academic databases: IEEE, Scopus, Springer, and Science Direct. Each database is categorised based on research areas, including algorithms and models, biomarker identification, decision support systems, and review articles. The figure highlights the relative volume of research in FS for cancer classification, with Science Direct contributing the highest number of papers in the algorithms and models and biomarker identification categories. Additionally, the figure demonstrates the increasing research interest in this field across these databases, reflecting the growing significance of FS in cancer classification.
Fig. 9 presents the annual publication trends from 2018 to 2023, tracking the number of research papers published each year on EA-based FS for cancer classification. The figure highlights the contributions of papers indexed in ScienceDirect, Springer, Scopus, and IEEE, illustrating the role of these databases in advancing research in this field. Furthermore, the figure underscores the diverse sources contributing to the development of cancer diagnosis and treatment technologies, which reflects the increasing research interest in EA-based FS methodologies over time.
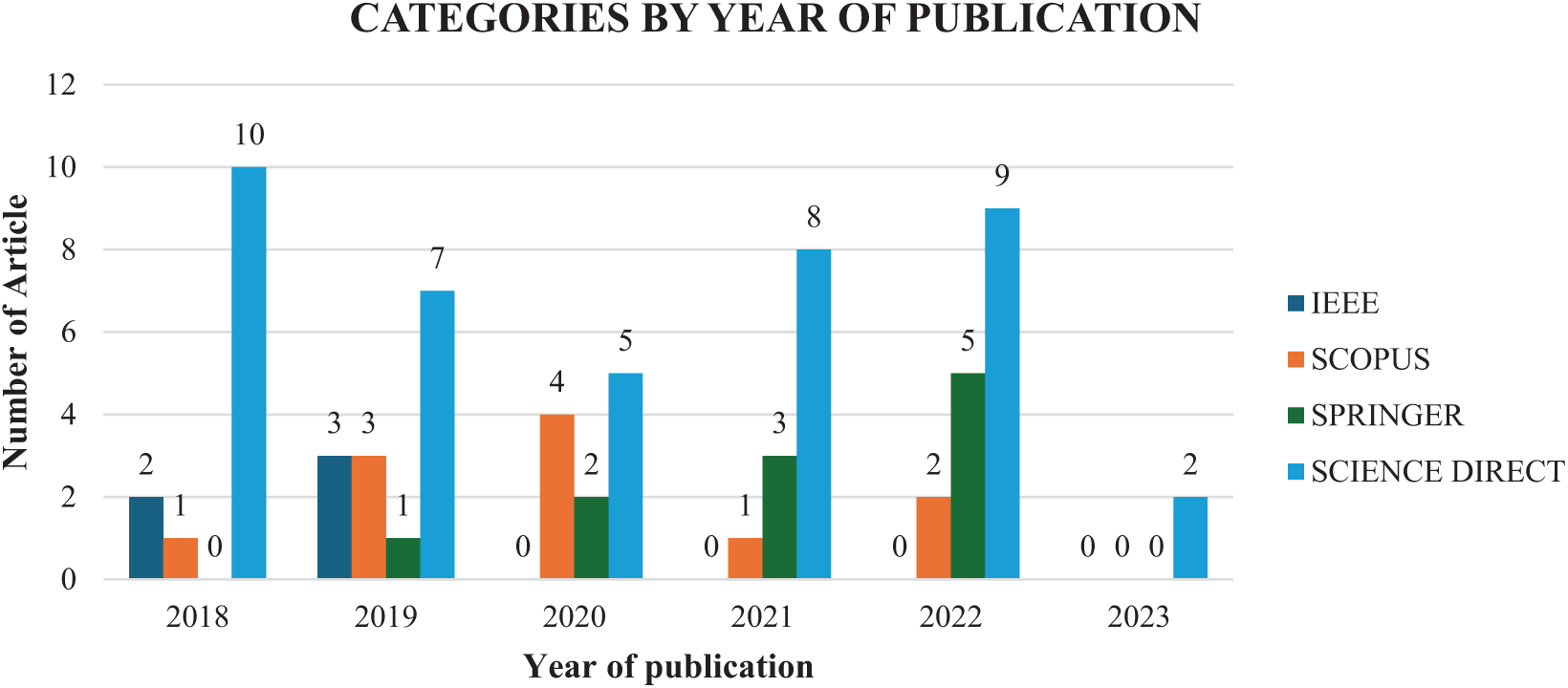
Figure 9: Number of papers under each category by year of publication
3.5 Comparison of FS Approaches in Terms of Accuracy, Efficiency, and Scalability
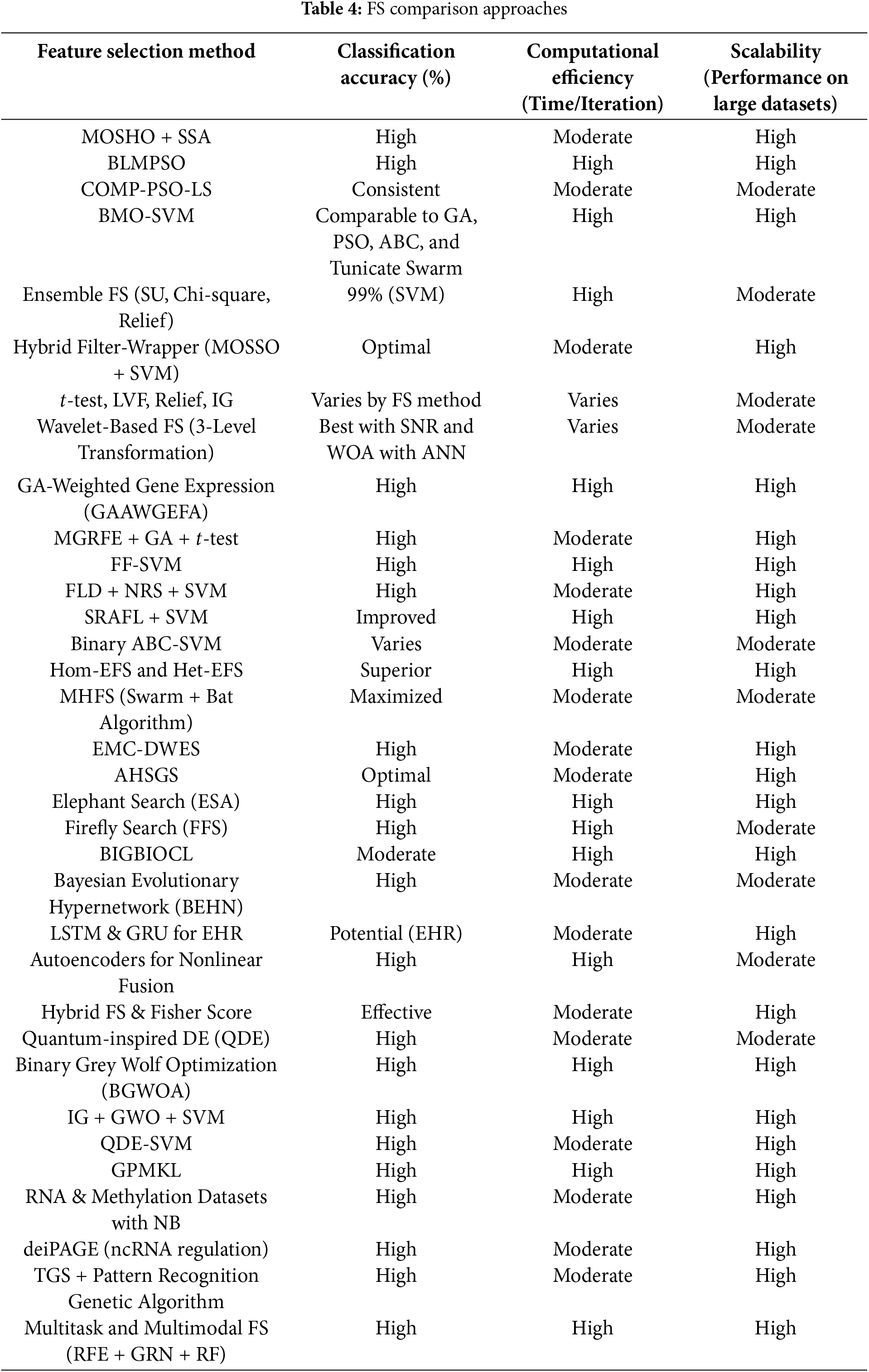
Table 4 presents a comprehensive comparison of various FS methods, focusing on key performance metrics such as classification accuracy, computational efficiency, and scalability. Each method is evaluated based on its ability to classify data accurately, the computational time required per iteration, and its effectiveness when applied to large datasets. The comparison highlights the strengths and limitations of each method, offering valuable insights into their suitability for different data types and applications. The accuracy levels, categorised as ‘High’ to ‘Moderate’, indicate the classification performance of each method across various tasks. Additionally, computational efficiency and scalability metrics offer a deeper understanding of the practical applicability of these methods in real-world scenarios.
In the context of classification accuracy, the terms ‘High,’ ‘Moderate,’ and ‘Consistent’ are used to represent the performance of feature selection methods in machine learning tasks:
• High accuracy generally refers to classification accuracy exceeding 85% or 90%. This indicates excellent performance, producing results that are both reliable and highly accurate. Models achieving this level of accuracy are well-suited for real-world applications where precision is critical.
• Moderate accuracy represents an acceptable level of performance, typically ranging between 70% and 85%. While sufficient for many applications, this range suggests potential for improvement. Methods in this category may perform well but could benefit from optimization or the incorporation of more sophisticated techniques.
• Consistent accuracy refers to methods that yield stable and reliable results across different datasets or iterations. Although the exact accuracy may vary, consistency indicates that the method can be trusted to perform similarly across diverse conditions. This range generally starts from approximately 60%, depending on the specific application.
Fig. 10 presents a bar chart comparing various FS methods regarding classification accuracy, computational efficiency, and scalability. Methods such as ensemble FS (SU, Chi-square, Relief) and hybrid filter-wrapper (MOSSO + SVM) demonstrate superior classification accuracy, whereas BLMPSO and FF-SVM exhibit high computational efficiency. Regarding scalability, approaches such as BMO-SVM, GPMKL, and hybrid FS (MOSSO + SVM) perform well, making them particularly suitable for large datasets. However, methods like BIGBIOCL and binary ABC-SVM show moderate performance across these metrics, suggesting potential for further optimisation. Notably, none of the evaluated methods explicitly incorporates dynamic-length feature selection; instead, most rely on fixed-length approaches. This presents a promising research area, as dynamic-length feature selection could enhance flexibility and efficiency in handling high-dimensional data.
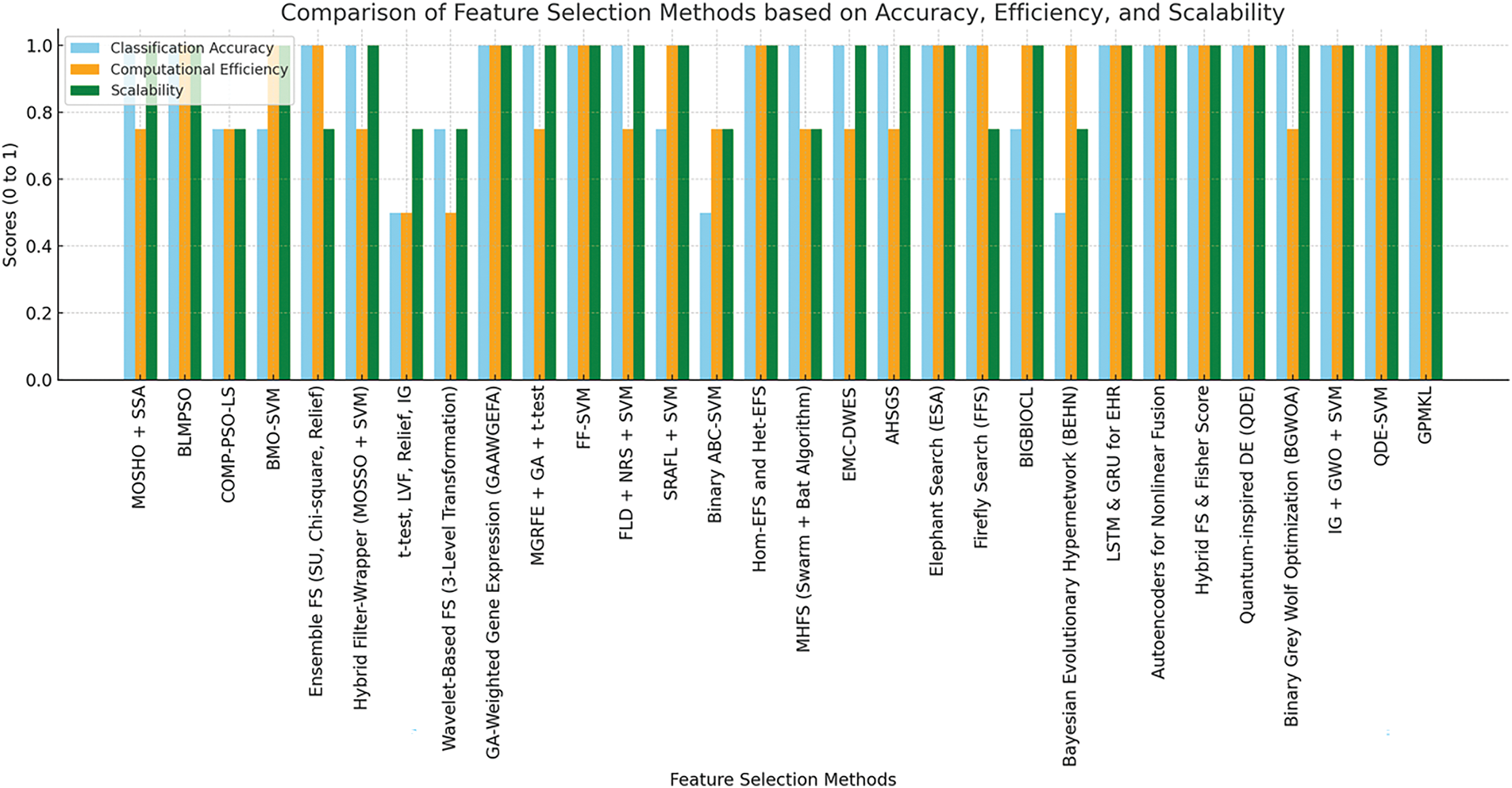
Figure 10: Comparison of FS methods based on accuracy, efficiency and scalability
Future research could focus on improving moderately performing methods by hybridising them with more efficient algorithms. A key research gap lies in developing FS methods that effectively balance accuracy, efficiency, and scalability, ensuring broader applicability. Additionally, addressing challenges related to high-dimensional data and conducting real-world validation of these methods could improve their robustness in practical scenarios. Hybrid approaches that integrate multiple algorithms may offer more reliable and efficient solutions for complex data problems.
This work aims to review the application and current research status of FS and classification based on EAs, identifying emerging trends in this research domain. The classification of research papers was undertaken for several reasons. Firstly, effective organisation enables a meaningful and systematic categorisation of numerous studies, allowing researchers to efficiently comprehend the ongoing developments in the field. Secondly, classifying papers helps summarise potential research directions, providing valuable insights for future studies. In the subsequent sections, the reviewed papers are examined from three perspectives: (i) the motivation behind the use of FS for GEPs for cancer classification, (ii) the challenges associated with the effective implementation of FS, (iii) recommendations for future research and advancements in FS methodologies.
Fig. 11 presents the distribution of the included papers based on their research objectives, categorised into distinct thematic areas. This classification helps clarify the focus areas of the research, including the motivations for applying FS to GEPs in cancer classification, the challenges encountered, and recommendations for future work. The distribution reflects the diversity of research directions in this field, providing insights into current trends and potential future advancements in FS and cancer classification using EAs.
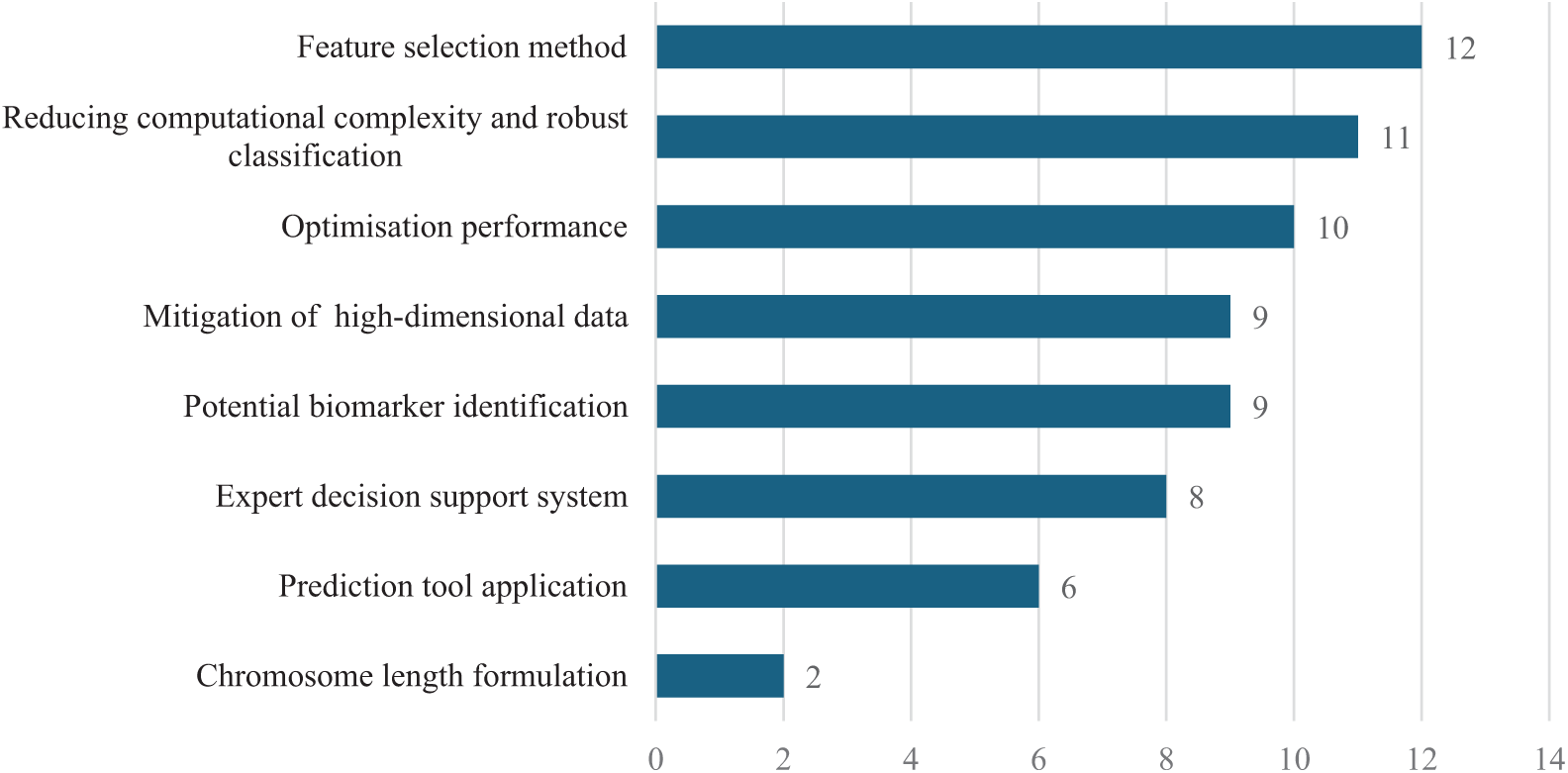
Figure 11: Number of included papers by research objective
In clinical settings, the optimisation of FS for cancer classification using EAs has proven highly effective in identifying optimal gene combinations. As highlighted in the literature, this approach has significantly transformed cancer diagnosis and prognosis, paving the way for more precise and personalised treatment strategies. However, despite numerous studies employing literature review methodologies to evaluate FS techniques in cancer research, notable gaps remain, which this study aims to address.
This review provides a comprehensive analysis of current FS methods that utilise EAs for cancer classification, with a particular focus on overcoming challenges related to high-dimensional data, dynamic chromosome length formulations, and biomarker identification for personalised treatment. By synthesising the latest advancements and identifying existing research gaps, this review offers valuable insights for future studies.
1) Benefits related to algorithms and models
Recent studies have focused on algorithmic advancements and model development for FS in cancer classification. Many of these studies integrate traditional FS methods with EAs, such as GA, PSO, and ABC, which have demonstrated effectiveness in identifying minimal yet relevant gene subsets. However, the potential of dynamic chromosome length formulations to enhance the FS process remains underexplored, representing an emerging area of innovation. Our research builds upon this foundation by specifically addressing the challenge of dynamic chromosome length in achieving more efficient FS and optimisation. For instance, a study [91] emphasised the success of GA in achieving high classification accuracy through optimal gene subset selection. Despite its widespread adoption, GA-based FS models often fail to account for the evolving nature of gene subsets across different cancer stages, a critical factor in improving the accuracy of personalised treatment plans [92]. Similarly, studies such as [93] and [94] those explored feature reduction for identifying independent variables, including the application of a hybrid multilayer recursive FS (MGRFE) and the use of SNR values combined with KH optimisation for selecting relevant features. However, these studies did not fully explore how dynamic changes in chromosome length could impact FS during the optimisation process. This study seeks to address this gap by incorporating dynamic chromosome length formulations that adapt throughout the selection process, thereby enhancing FS efficiency and improving cancer classification outcomes.
While traditional EAs have significantly contributed to FS, emerging non-evolutionary approaches, particularly DL-based FS techniques, are gaining attention. These methods excel in automatically learning hierarchical features from raw data. DL methods, such as CNNs and Autoencoders (AEs), have shown remarkable potential in high-dimensional feature extraction. When combined with transfer learning, DL models can effectively identify complex patterns and relationships in cancer data without the need for manually engineered features, offering advantages over conventional FS approaches. For example, a study by [95] demonstrated that DL techniques could automatically identify and select cancer-related features, yielding high accuracy across various cancer types. Furthermore, DL methods are particularly adept at handling heterogeneous datasets, where traditional EAs like PSO or GAs often struggle to generalise across different data types or cancer subtypes. Thus, while prior studies have made substantial contributions to FS and cancer classification, there remains a clear need for more sophisticated and adaptive FS techniques that account for the dynamic nature of cancer data. This research aims to bridge these gaps by proposing a more flexible and dynamic FS approach using EAs, focusing on dynamic chromosome length formulations and their application to personalised cancer classification models.
2) Benefits related to cancer disease prediction accuracy
FS has shown significant potential in improving the classification performance and accuracy of cancer prediction models by identifying the most informative genes. Various FS techniques have been explored to enhance prediction accuracy. For instance, robust minimum redundancy maximum relevancy (rMRMR-HBA) has been introduced to address the limitation of single-filter methods, which often exhibit high variability in classification results due to their limited scope [96]. Several FS methods, including t-test, LVF, Relief, and IG, have been used to reduce dataset dimensionality. Additionally, Rejection Objective (RO) classifiers with different rejection thresholds have been applied to investigate the robustness of cancer prediction models. In integrated IDSS, FS methods such as IG combined with GWO for feature reduction and SVM for classification have enhanced classification stability, ultimately contributing to better cancer prediction and clinical decision-making [97]. Similarly, the application of SSA with a low-computational hybrid multi-objective salp-henry optimisation (C-HMOSHSSA) has outperformed existing state-of-the-art techniques [98]. Furthermore, integrating FLD and NRS with an SVM classifier has led to notable enhancements in classification accuracy, further highlighting the critical role of FS techniques in addressing cancer classification challenges [99].
Swarm intelligence-based classification methods, specifically the feature clustering SVM with ANN classifier (FCSVM–ANN), have demonstrated notable improvements in classification performance through clustering-centred FS. Both low- and top-ranked features achieve promising predictive capabilities. A prediction model-based ranked feature subset, known as RIFS2D, has demonstrated that even low-ranked features may exhibit promising predictive accuracy [100]. In PSO, a bilayer mutation model has been introduced to increase diversity, resulting in the development of bi-layer mutated PSO (BLPSO), which improves classification accuracy and precision [101]. While these studies have contributed significantly to FS advancements in cancer classification, they often fail to address the dynamic nature of chromosome length adjustments and adaptive FS methodologies. Most existing research relies on fixed-length chromosome models, which may lead to suboptimal solutions when applied to diverse and high-dimensional cancer datasets. Moreover, current studies predominantly focus on traditional FS approaches, with limited emphasis on dynamic FS techniques that evolve throughout the optimisation process. Additionally, despite the potential of multi-objective optimisation in FS for cancer classification, its application remains underexplored, particularly in the integration of dynamic chromosome lengths. A more adaptive approach could better capture biomarker variability across different cancer subtypes, enhancing both classification accuracy and the reliability of FS methods in personalised cancer diagnosis.
3) Benefits related to reduced computational time and resources
Reducing the number of features through gene-related FS enhances predictive accuracy while significantly lowering computational time and resource consumption. ML models have demonstrated effectiveness in predicting cervical cancer using various risk factors. For instance, ML-based models incorporating logistic regression, ridge classifiers, and Gaussian NB have shown promise in minimising computational demands by focusing on the most relevant features. However, these studies often overlook advanced techniques such as dynamic FS models, which adapt throughout the optimisation process and could further enhance computational efficiency. Additionally, recent studies have employed pipeline methods that incorporate Bayesian optimisation to improve computational efficiency. These methods select the most suitable classifier from a meta pool by optimising hyperparameters and addressing class imbalance using the synthetic minority oversampling technique (SMOTE) [102]. While effective, these methods do not typically incorporate dynamic FS mechanisms that evolve with the dataset, which could further optimise resource allocation and enhance convergence speed.
Bipartite graph theory has also been applied in unsupervised FS (efficient unsupervised feature selection, EUFS) to accelerate algorithm performance by leveraging high-quality discrete indicators and row-space matrices, resulting in reduced computational complexity [103]. However, these approaches primarily focus on clustering and classification without considering dynamic FS techniques that could optimise FS over time and further reduce computational costs. A promising method integrates HHO with variable neighbourhood learning to balance global exploration and local exploitation, thereby improving classification accuracy while maintaining low computational costs [104,105]. Nonetheless, similar studies have yet to fully explore the benefits of adaptive chromosome length formulations within EAs, which could further enhance computational efficiency without compromising accuracy.
4) Benefits related to specific motivation
Despite the extensive research on FS for cancer classification, specific motivations—such as improving classification accuracy across different cancer stages—are often not explicitly addressed. Many studies, particularly those analysing various RNA sets as well as early- and late-stage methylation patterns, rely on traditional filter-based methods and classifiers such as NB [106]. While these methods have shown effectiveness, they often rely on fixed feature sets, limiting their adaptability to the complexity of cancer datasets. Cancer data can vary significantly based on cancer type, stage, and molecular data sources. Trilevel FS research has shown that the highest classification accuracy is achieved when SNR combined with WOA is classified using an ANN. While this method is promising, it has yet to explore the role of dynamic FS and adaptive algorithms, which could better accommodate the evolving nature of cancer data as new biomarkers are discovered. Additionally, iBPSO has been utilised to enhance diversity and improve classification accuracy with NB, effectively classifying biological samples from both binary and multiclass cancer datasets. However, despite these advancements, much of the existing research remains focused on static models, failing to fully leverage the dynamic nature of FS across multiple cancer subtypes and stages.
Recent studies have focused on specific cancer types, such as SOC, where microRNA profiling has been applied to predict cancer stages and identify key molecular signatures. Analysing different RNA regulatory mechanisms has demonstrated the SOC index’s discriminative capability in investigating ovarian cancer mechanisms and potential therapeutic strategies [107]. FS enhances algorithm performance by leveraging data characteristics, whether discrete or real-valued. Traditional rough set models primarily address discrete data. Recent studies have introduced novel gene selection techniques that combine entropy measures and Fisher scores to improve classification accuracy for real-valued data while retaining critical gene classification information [108]. However, these approaches still face challenges related to scalability and adaptability, especially when dealing with high-dimensional datasets typical in cancer research. Furthermore, an ensemble FS framework that integrates multiple FS methods has been proposed to improve stability scores for both binary and multiclass classification. However, the lack of dynamic adaptation across different cancer stages continues to limit the real-world applicability of these methods.
While multitask multimodal FS has shown potential in analysing histopathological images and genomic data for cancer diagnosis and prognosis, the development of automated tools for selecting the most representative genes in multiclass problems remains an ongoing challenge. For instance, a tool capable of generating comprehensive HTML reports on the FS process can assist experts in making more informed clinical decisions. Another promising development is the use of a tool generation system (TGS) for identifying critical biomarkers through the integration of regulatory networks [109]. However, much of this research still relies on static and predefined gene sets that may not reflect the latest molecular insights into cancer progression.
5) Benefits related to evolutionary chromosome length design
GAs with variable-length chromosomes have found widespread applications in optimisation problems. Traditionally, single-point (SP) crossover has been the most commonly used crossover mechanism. However, recent advancements have introduced novel mechanisms such as the same adjacency (SA) crossover, which has demonstrated superior performance over traditional SP crossover [110]. Studies show that GAs utilizing SA crossover offer superior search capabilities, improved solution quality, and rapid convergence, particularly in optimising large and complex networks. These improvements are especially relevant in high-dimensional domains like cancer classification, where feature spaces are vast and complex.
Despite extensive research on GAs with SP and SA crossovers, the dynamic nature of chromosome lengths in evolving systems remains largely unaddressed. Many existing studies rely on fixed-length chromosome models, which are limited in their adaptability to the changing FS process. This is particularly restrictive in high-dimensional datasets, such as GEPs used for cancer classification. While EAs have been explored in the context of dimensionality reduction, most approaches still rely on static chromosome lengths, which cannot adjust dynamically as feature sets expand or contract. This limitation reduces efficiency, particularly in data-rich environments such as cancer genomics.
A notable study in this area introduced a hybrid FS method that integrates both filter- and wrapper-based techniques to reduce dimensionality and improve classification performance. This method applies an ANOVA statistical filter to select the most relevant genes from the original gene set, followed by an evolutionary wrapper approach combining EJaya and FOA principles [111]. By fine-tuning key parameters such as local and global seeding within FOA, this method enhances classifier performance. When used alongside SVM classifiers, it has demonstrated superior accuracy with fewer selected features compared to traditional FS methods. However, despite these improvements, this approach still relies on static chromosome lengths, which may not be optimal for managing the evolving nature of genomic data.
6) Benefits related to handling large and high-dimensional data
An AHSGS is recommended for FS in high-dimensional databases. FS remains a pivotal task in high-dimensional datasets, especially in genomic and clinical applications. AHSGS incorporates multiple heuristic search algorithms (such as GA, DE, PSO), harmony search (HS), and ant colony optimisation (ACO) to reduce computational costs while ensuring optimal FS. As the process of identifying cancer marker genes in high-dimensional datasets involves varying individual lengths (as shown in Fig. 8), the population initialisation starts by randomly generating individual lengths. In this context, Wang et al. first proposed a hybrid DE approach known as differential evolution with CNN (DECNN) classification, designed to evolve CNN model architectures [112].
In the DECNN, as shown in Fig. 12, the length is randomly sampled from a Gaussian distribution, with the standard deviation and length determined by the complexity of the classification task. At the start of the evolutionary process, the lengths of individuals are approximately 10. However, as the CNN architectures evolve, their lengths reduce to between 3 and 5, depending on the complexity of the dataset. This demonstrates that DECNN can effectively develop CNN architectures of varying lengths, adapting to different classification challenges. DECNN operations are similar to the standard DE mutation and crossover, as shown in Fig. 13. However, it introduces an additional step to trim longer vectors before applying any operations, as DECNN candidates have varying lengths [113]. This demonstrates DECNN’s capability to evolve the length of architectures, whereas traditional DE operations are only applicable to fixed-length vectors.
Figure 12: Second crossover of the proposed DECNN algorithm
Figure 13: Modified differential mutation and crossover for dynamic-length chromosomes
PSO has also been adapted for the automatic optimisation of CNN architectures for internet protocols, eliminating the need for manual intervention. This approach, termed Internet Protocol-based PSO (IPPSO), introduces three key improvements over traditional PSO [114]. These methods have shown promise in selecting the minimal number of top-ranked genes while balancing classification accuracy and efficiency. Previous studies have explored AHSGS in various domains, emphasising its ability to manage large feature spaces without excessively burdening computational resources [115]. However, many of these studies have only implemented static models, prioritising FS accuracy without considering the evolving nature of biological datasets. Another algorithm, BIGBIOCL, has been specifically developed for big data in genomics, particularly to address the complexities of methylated features within the genome. BIGBIOCL iteratively removes selected features, effectively extracting alternative and equivalent classification models that better represent the underlying relationships in large-scale biological datasets [116]. While this method has advanced genomic feature reduction, its application has been limited to specific datasets. Much of the existing literature continues to focus on traditional static FS techniques, which may not fully account for the complexities and interactions of dynamic genomic features.
Furthermore, data reduction techniques combined with data visualisation methods have gained traction as a means of automating the diagnosis of high-dimensional gene expression and clinical data. Techniques such as 2D image generation, including heatmaps and hotspot maps, encapsulate complex data into easily interpretable visual formats, illustrating disease feature proportions and spatial correlations [117]. These visualisation methods have been widely applied in cancer research to improve diagnostic accuracy and streamline the analysis of gene expression patterns. However, despite their potential, there remains a gap in understanding how data visualisation techniques can be adapted and integrated with dynamic FS methods to better handle high-dimensional data as they evolve.
While this study has highlighted significant advancements, it also identifies critical limitations that need to be addressed to enhance the real-world applicability of FS techniques, particularly in clinical settings:
a. Addressing high dimensionality
High-dimensional data remains a major challenge in cancer classification, where the number of features (genes) far exceeds the number of samples. One promising solution is the integration of dynamic chromosome length formulations in EAs. These dynamic formulations enable the chromosome size to adapt as the FS process evolves, effectively mitigating high dimensionality. In this study, we propose the development of adaptive evolutionary models that can dynamically adjust both population size and chromosome length based on the complexity of the data. This approach has the potential to significantly improve the efficiency of FS techniques by reducing unnecessary computations and focusing on the most relevant features throughout the optimization process.
Additionally, dimensionality reduction techniques such as principal component analysis (PCA), t-distributed stochastic neighborhood embedding (t-SNE), and autoencoders could be integrated as preprocessing steps [118]. These methods help reduce data dimensionality while retaining critical information for cancer classification, making FS more manageable and efficient.
b. Addressing Data Imbalance
Data imbalance, where certain classes (e.g., cancer vs. non-cancer) are underrepresented, can lead to biased models. Several resampling techniques could be applied to address this issue. Oversampling of underrepresented classes using techniques such as synthetic minority over-sampling technique (SMOTE) or undersampling of overrepresented classes could be incorporated into the FS pipeline to prevent classifiers from being biased toward the majority class [119]. Another promising approach involves cost-sensitive learning, where algorithms are adjusted to assign higher penalties for misclassification of the minority class, thereby improving classification fairness. Additionally, multi-objective optimization can be integrated into the FS process, allowing the algorithm to optimize both feature relevance and class balance simultaneously. This approach helps reduce bias while improving overall classification performance.
c. Improving interpretability of FS models
Interpretability is particularly critical in clinical applications, where transparent decision-making is essential. A viable solution involves integrating EAs with explainable AI (XAI) techniques. Approaches such as shapley additive explanations (SHAP) and Local interpretable model-agnostic explanations (LIME) can be applied to FS models to provide deeper insights into how selected features influence prediction outcomes [120,121].
Furthermore, ensemble methods that combine multiple FS models are recommended to generate more interpretable and robust feature subsets. By leveraging a diverse range of FS methods (e.g., filter, wrapper, and embedded methods), the interpretability of the final selected features can be enhanced, enabling clinicians to gain clearer insights into the key factors contributing to cancer classification.
d. Practical recommendation for overcoming limitations
While the theoretical foundations of FS using EAs have advanced significantly, practical implementations can be further enhanced by integrating EAs with DL models. Hybrid models which integrate DL methods for feature extraction with EAs for feature selection have the potential to process large-scale, high-dimensional datasets efficiently [122,123]. Additionally, the adoption of multi-level FS frameworks that dynamically adapt to evolving data characteristics such as variations in cancer biomarkers over time can contribute to the development of more accurate and scalable models.
By implementing these strategies, future research can lead to the development of more adaptive, robust, and interpretable FS techniques for cancer classification, effectively addressing challenges related to the high-dimensionality and data imbalance while enhancing clinical applicability.
ML offers different techniques and methodologies for FS, with classification results dependent on datasets, each having its distribution and feature. Research on optimising three parameters—learning algorithm, FS method, and rejection rate–for robust cancer prediction highlights that the predictive accuracies of RO classifiers vary with different FS methods for each dataset. Multiple gene selection methods are employed in numerous tumour and cancer classification studies. The cross-referencing relationships between the keywords in the literature have been visualised in Figs. 2 and 3, providing insights and guiding further research.
However, most of these methods aim to identify a common subset of genes across all tumour subtypes, failing to capture the unique characteristics of each subtype. Based on the collected papers, the following sections outline the crucial challenges in the field, categorising them by nature.
4.2.1 New Variable-Length Chromosome Design
MGRFE utilises variable-length integer encoding, an approach that has not been extensively explored in prior studies, which results in a limited number of references on its application in cancer classification. This algorithm employs a variable-length integer encoding technique within an embedded GA framework, recursively refining the feature set by dynamically adjusting the encoding length during the RFE process. By iteratively removing irrelevant and redundant features, MGRFE converges on a minimal yet informative subset, thereby enhancing the discriminative power of the selected features [124]. This makes MGRFE an effective method for optimising cancer prediction and prognosis, particularly in high-dimensional datasets where feature reduction is a critical challenge.
Despite its potential, several limitations must be considered. First, the performance of the algorithm in real-world clinical applications may be affected by dataset imbalance. Cancer datasets often exhibit significant class imbalances, where certain cancer types or stages are underrepresented. As with other evolutionary algorithms, MGRFE may struggle with imbalanced datasets without proper handling techniques, such as oversampling or weighting adjustments. This may lead to biased FS, where the model favours the more prevalent class thereby reducing predictive accuracy for rare cases.
Additionally, while MGRFE effectively prunes redundant features and selects relevant genes, its scalability to larger datasets remains a concern. As cancer datasets grow increasingly large and complex, the computational cost of iterative feature elimination may become prohibitive. In contrast, recent advancements in DL models, such as CNNs and recurrent neural networks (RNNs), have demonstrated the ability to process high-dimensional data and extract complex patterns without explicit FS. Notably, a DL-based framework employing enhanced chimp optimisation (ECO) algorithm for FS, combined with a depth-wise separable convolutional neural network (DSCNN) for classification, has outperformed existing approaches in addressing challenges such as high dimensionality, overfitting, and data redundancy [125]. While EAs, such as MGRFE and ECO-DSCNN, offer advantages in feature interpretability and flexibility, their performance could be improved by integrating them with more scalable DL approaches to handle large and imbalanced datasets efficiently.
Furthermore, the variability in GEPs across different cancer subtypes introduces additional complexity. While MGRFE is designed to optimise gene selection, it may not fully capture intra-class variability, potentially affecting its generalisability across different patient populations and cancer types. This is a notable limitation, as personalised cancer treatment requires highly adaptable models that can accommodate individual patient variability.
The use of diverse datasets presents a challenge in cancer classification. Binary and multiclass microarray datasets have been applied in BMO–SVM algorithm research. Although its classification accuracy remains relatively low, it achieves a high informational superiority ratio [126]. By applying this approach to gene expression datasets, small gene sets can be used to distinguish cancer from control cases; however, their similarity across datasets is limited. While the model identifies higher-order gene interactions, the real-world challenge lies in understanding and validating these interactions in biological terms. This method also faces challenges regarding time efficiency and scalability when processing large datasets, particularly in clinical settings where fast predictions are often needed and would require further biological insights into how these interactions influence cancer progression, treatment response, and recurrence risk.
The effectiveness of classification algorithms is largely dependent on dataset characteristics. Methods such as EJaya-based FOA for FS and classification of microarray data have demonstrated mixed results. In the brain tumor, classification outcomes were obtained using the depth-first search (DFS) algorithm that distinguishes healthy cells from the cells affected by using segmentation technique [127]. Similarly, in research employing rMRMR-HBA with B-hill climbing for gene selection, only two out of 10 datasets achieved high classification accuracy with a minimal number of selected genes, suggesting that the method is not universally effective [128]. The availability of adequate sample populations is another critical factor in gene expression studies. Normal ovarian samples are crucial for expression profiling comparisons between malignant and healthy ovarian tissues. However, these samples are rare due to the invasive procedures required to obtain them. Prior to this study, only 33 normal ovarian samples for gene expression analysis were publicly available in the cancer genome atlas (TCGA) and the gene expression omnibus (GEO). Profiling the expression of eight normal ovarian samples from patients with cervical cancer that has not metastasised to the ovaries offers necessary support and complements ovarian research.
Expanding dataset size is also a significant factor in improving predictive accuracy. For instance, research on breast cancer survival prediction could be extended and validated by employing a large population of patients with breast cancer [129]. However, the availability of genomic and pathological image data remains a limiting factor. The filter-wrapper hybrid ensemble approach for optimising high-dimensional biomedical data requires large datasets to perform effectively; however, dataset availability remains a bottleneck. Additionally, vital penalty parameters must be pre-optimised through training to ensure optimal algorithmic performance. This highlights a persistent challenge in cancer classification: the models are highly dependent on the size and quality of the datasets. Without access to both large and well-curated datasets, these methods cannot be comprehensively evaluated or optimised.
To address these limitations, future work should prioritise the development of scalable models capable of processing large datasets efficiently. Additionally, advanced methodologies must be developed to address class imbalance, a prevalent cancer research issue, while accounting for gene expression variability to enhance consistency and generalisability across different studies and data sources.
4.2.3 Optimisation of FS for Biomarker Extraction
The optimisation of FS algorithms plays a crucial role in cancer diagnosis by identifying predictive biomarkers. While recent advancements have led to improvements in classification accuracy, significant challenges remain, particularly in applying these methods to real-world genomic datasets. For example, the firefly-based gene selection algorithm, a wrapper FS method, has demonstrated promising classification accuracy. However, hybrid algorithms that integrate multiple FS methods have been shown to outperform this approach in classification accuracy and the number of selected genes [130]. Similarly, the IFS strategy for low-ranked biomarkers is limited by its reliance on continuously ranked features from filter algorithms [131]. As researchers navigate these challenges, they are tasked with devising innovative solutions to enhance the efficacy and reliability of FS methodologies, ultimately advancing the ability to extract meaningful insights from complex genomic datasets.
One of the primary limitations of FS methods, including IFS, is the risk of disregarding biologically significant biomarkers due to rigid ranking criteria. Moreover, some FS techniques fail to maintain the biological relevance of selected biomarkers across different datasets, a problem particularly evident in the QDE–SVM method. While QDE–SVM performs well in some cases, it is less effective than more recent wrapper-based methods, such as fuzzy self-configuring attention model (FSCAM) for scRNA-seq gene selection. Specifically, QDE–SVM tends to select a large number of features, necessitating further reduction to obtain a more informative subset of marker genes for downstream analyses. This trade-offs between feature quantity and biological relevance underscore the need for enhanced FS and dimensionality reduction techniques [132,133]. Additionally, methods like FLD face challenges in accurately identifying the most informative biomarkers, sometimes discarding genes with substantial classification influence as irrelevant, thereby reducing overall model accuracy [134]. The scalability and robustness of FS algorithms remain significant concerns, particularly in clinical applications where large, high-dimensional datasets are prevalent. For instance, while fractal feature selection models can be effective for small to medium-sized datasets, this method faced scalability issues with very large datasets due to the computational complexity of evaluating many high-dimensional feature subsets [135]. As the dataset grows, simulation time increases exponentially, limiting the practicality of FS techniques in large-scale clinical genomics studies where processing speed is crucial.
Moreover, dataset imbalance presents another significant challenge. Many FS methods, including a quantum-inspired differential evolution (QDE) wrapped with a classification method to select a subset of genes from twelve well-known scRNA-seq transcriptomic datasets to identify cell types. QDE–SVM, operates under the assumption of balanced datasets, which is rarely the case in cancer research [136]. Class imbalance can lead to biased biomarker selection, often underrepresenting minority classes, such as rare cancer types, or excluding them entirely. This method shows significant differences in cell type classification accuracy. Addressing this imbalance while ensuring the selection of relevant and generalisable biomarkers across diverse patient populations remains a critical area for future research.
4.2.4 In Terms of Algorithm and Model Combinations
The integration of FS and classifier combinations requires a tailored approach. In the application of FS with a reject option classifier for robust cancer prediction, a classifier that performs well without rejection may, in some cases, also perform well with rejection. However, decision-making in this context involves trade-offs, as there are inherent costs associated with correct classifications, misclassifications, and instances where no decision is made. In BLMPSO, variations in cellular and tissue characteristics influence classification outcomes. Specifically, when the threshold value is adjusted to 0.17, 0.2, or higher, classification accuracy declines compared to the optimal result obtained with a threshold value of zero. While accuracy improves when no threshold IG value is applied, implementing a threshold significantly reduces the number of features from 7129 to 32, thereby lowering computational costs and memory usage.
While the algorithm has demonstrated effectiveness across different datasets, its real-world applicability remains uncertain. For instance, studies on gene expression levels in subgroups of patients with chronic lymphocytic leukaemia have yet to compare correlation-based FS methods with alternative approaches, such as data mining techniques, which offer greater flexibility in handling high-dimensional data. This gap highlights a key limitation in FS research—many studies do not sufficiently explore alternative methodologies or benchmark their performance using clinical datasets. A more comprehensive comparative analysis of FS and data mining techniques would provide deeper insights into their respective strengths and weaknesses in real-world clinical settings. Similarly, challenges arise in autoencoder-based DNA methylation analysis for predicting breast cancer recurrence, particularly due to the nonlinear feature fusion process [137]. While autoencoders have proven effective in handling high-dimensional data, their performance is constrained by dataset size. Without sufficiently large patient datasets, the generalisability of findings across populations is limited. Moreover, parameterisation issues, such as dependence on the initial random selection of weights, contribute to variability in results, complicating validation across diverse datasets. The lack of robust validation protocols and comprehensive data fusion further limits the broader clinical application of autoencoder-based FS techniques. Addressing these limitations requires improvements in dataset size, parameter optimisation, and validation protocols to ensure scalability and robustness in clinical settings.
Thus, while the integration of FS with classifiers and autoencoder-based FS methods holds considerable promise, several critical challenges must be addressed. These include dataset imbalance, variability in gene expression, and scalability to larger datasets. Furthermore, a more comprehensive comparative analysis with other ML and data mining approaches could help clarify the strengths and limitations of these techniques in real-world clinical environments.
4.2.5 In Terms of Classification Accuracy and Computation Time
The absence of a universal FS method capable of handling all datasets presents a significant challenge in cancer classification. In the EMC–DWES algorithm, one of the primary limitations is the high computational cost associated with EMC when applied to multilabel datasets with an excessive number of labels or observations [138]. This constraint reduces its suitability for high-dimensional datasets, where scalability is essential. Additionally, classifier selection remains a critical factor, as the choice of classifier significantly influences both the accuracy and efficiency of the classification process. For instance, RF which is commonly employed alongside the two-stage HAM approach, exhibits low sensitivity in detecting positive cases-arguably the most critical aspect of cancer classification despite maintaining high specificity. This imbalance suggests that while RF classifiers effectively identify negative cases, they struggle to detect cancerous samples, ultimately limiting their clinical utility, where accurate positive case detection is essential.
Furthermore, although the NRS algorithm effectively selects a small subset of genes, it often discards relevant genes crucial for accurate classification, thereby reducing overall accuracy. This limitation has been partially addressed by extending NRS to include FLD-NRS, LLE-NRS, and RelieF+NRS, each introducing complementary strategies to enhance classification performance [139]. For example, FLD-NRS integrates FLD to refine gene selection and improve the discriminatory power of the selected subset. LLE-NRS incorporates locally linear embedding (LLE) to preserve the manifold structure of the data, thereby offering a more accurate representation of gene relationships. Meanwhile, RelieF+NRS combines the NRS framework with the ReliefF algorithm to identify relevant features while mitigating the risk of discarding critical genes.
Despite these improvements, several challenges persist, particularly concerning real-world applicability. Dataset imbalance remains a significant issue in cancer classification, as most datasets contain substantially more negative samples than positive ones. This imbalance can lead to classifier overfitting, particularly in models such as RF, which struggle to detect minority-class instances. Additionally, variability in GEPs across different datasets further complicates the generalisation of predictive models.
4.2.6 Algorithms for Medical Applications
Researchers often struggle to interpret the extracted features from FS methods, which substantially limits their practical application in identifying drug targets and locating disease-associated genes. The ability to predict the clinical staging of liver hepatocyte carcinoma, as demonstrated in various studies, underscores the clinical potential of FS in cancer prediction and prognosis [140]. However, while such methodologies show promise, they often fall short in addressing real-world challenges. One of the key issues is that clinical datasets, such as those derived from blood, serum, and urine, are highly variable, and the presence of dataset imbalance where non-cancer cases outnumber cancer cases can distort prediction models, potentially leading to biased results and reduced performance when applied to clinical practice. A similar strategy could be employed to develop non-invasive biomarkers, provided sufficient blood, serum, urine, and cell-free DNA data are available. One study introduced the KnowSeq R-Bioc package as a tool to assist expert decision-making in cancer prediction [141]. However, this tool has notable limitations, including the absence of gene set enrichment analysis (GSEA) within its three functional enrichment methods and a lack of support for retrieving pathway information from Reactome.
Findings suggest that different miRNAs contribute to a limited number of cancer-associated molecular processes by regulating distinct gene targets, highlighting the need for more comprehensive regulatory network models [142]. However, miRNA-based models often fail to capture spatial and temporal variability in gene expression data. For instance, the GAAWGEFA method assigns weights to each time point in gene expression data using a GA framework to improve cancer prognosis and prediction [143]. While this approach enhances predictive accuracy, it oversimplifies the dynamic nature of gene expression by neglecting spatial relationships within tissue samples, which are critical for accurate cancer staging and prognosis.
Another significant challenge is the application of sequential ML models in predicting lung, breast, cervical, and liver cell cancers using EHR data. The inherent sparsity and noisiness of EHR databases hinder detailed medical data analysis and reduce the efficacy of ML algorithms in clinical settings [144]. EHR data are often incomplete and lack key medical features, such as medical notes and laboratory results, which are essential for making accurate predictions. Furthermore, the temporal aspect of patient visits such as the time intervals between visits is often not captured by RNNs, which primarily focus on learning sequential patterns without understanding the temporal gaps between data points [145]. This gap in modelling temporal information limits the scalability and applicability of RNNs in clinical decision support systems, where understanding the time-sensitive nature of medical conditions is critical for early detection and treatment planning.
Finally, this review summarises the relevant recommendations from the collected papers to alleviate the challenges faced by researchers and to enhance FS optimisation for cancer classification through EA-based GEPs. These recommendations are categorised based on the target groups, including users, developers or providers, healthcare and federation authorities, and researchers. For users, the emphasis is on the importance of user-friendly interfaces and intuitive tools that facilitate the seamless integration of FS algorithms into existing workflows. It outlines the process of selecting the most relevant genes for cancer classification using an evolutionary-based framework, as shown in Fig. 13.
The Evolutionary Algorithm (EA)-based Optimization Framework for Cancer Classification in Fig. 14 is designed to enhance the accuracy of cancer classification by selecting the most relevant genes from a high-dimensional microarray dataset. The process starts with an adaptive parameter setting, which dynamically adjusts evolutionary parameters to improve the efficiency of gene selection. A key feature of this framework is the use of a dynamic-length chromosome representation, allowing the number of selected genes to be adjusted throughout the optimization process. This adaptability helps reduce computational complexity while ensuring that only the most significant genes are classified. The framework then evaluates different gene subsets and identifies the best-performing set, which is later validated for accuracy and reliability. The selected genes serve as biomarkers, helping to distinguish between different cancer types more effectively. By leveraging an adaptive evolutionary approach, this framework improves feature representation, optimizes gene subset selection, and enhances classification performance. As a result, it provides a more efficient and accurate method for cancer classification, addressing the challenges of high-dimensional gene expression data. Furthermore, Fig. 14 provides a more detailed explanation of this framework, illustrating key processes such as dynamic chromosome formulation, imbalance data handling, and performance evaluation to ensure optimal gene expression profile selection.
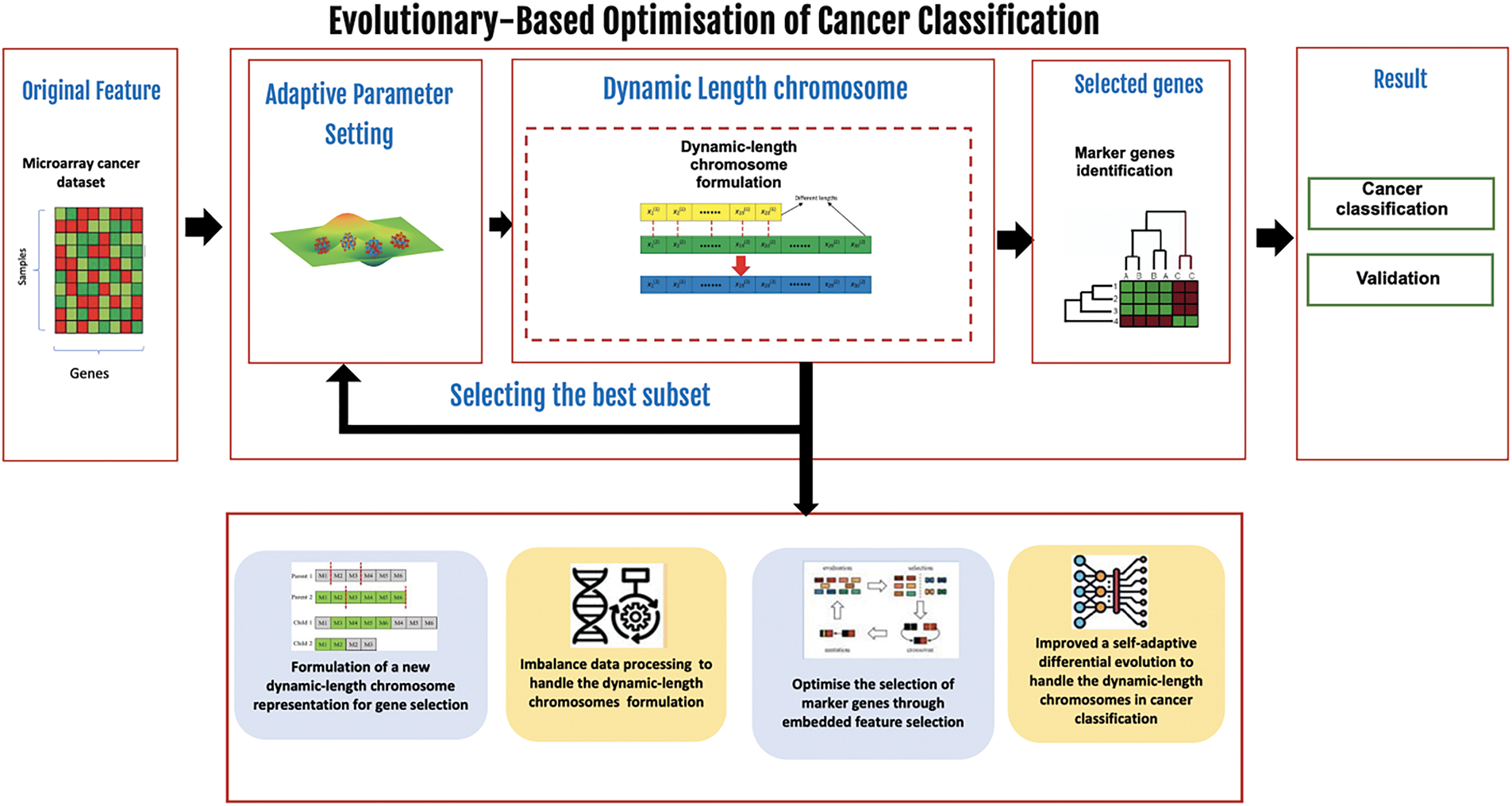
Figure 14: EAs-based optimisation framework
Fig. 15 illustrates a gene selection framework for cancer classification based on evolutionary algorithms and machine learning techniques. It begins with benchmark microarray datasets containing gene expression profiles used for cancer classification. Next, a dynamic-length chromosome representation is formulated to optimize gene selection, allowing flexibility in selecting relevant genes. The framework also incorporates imbalanced data processing to handle challenges associated with dynamic-length chromosomes, ensuring better data distribution. An adaptive differential evolution algorithm is applied to further refine the selection process, improving classification accuracy. The selected genes are then tested through a cancer classification model, followed by a performance evaluation to assess the model’s effectiveness. If the classification is successful, an optimal gene expression profile is identified; otherwise, the selection process is re-evaluated and refined.
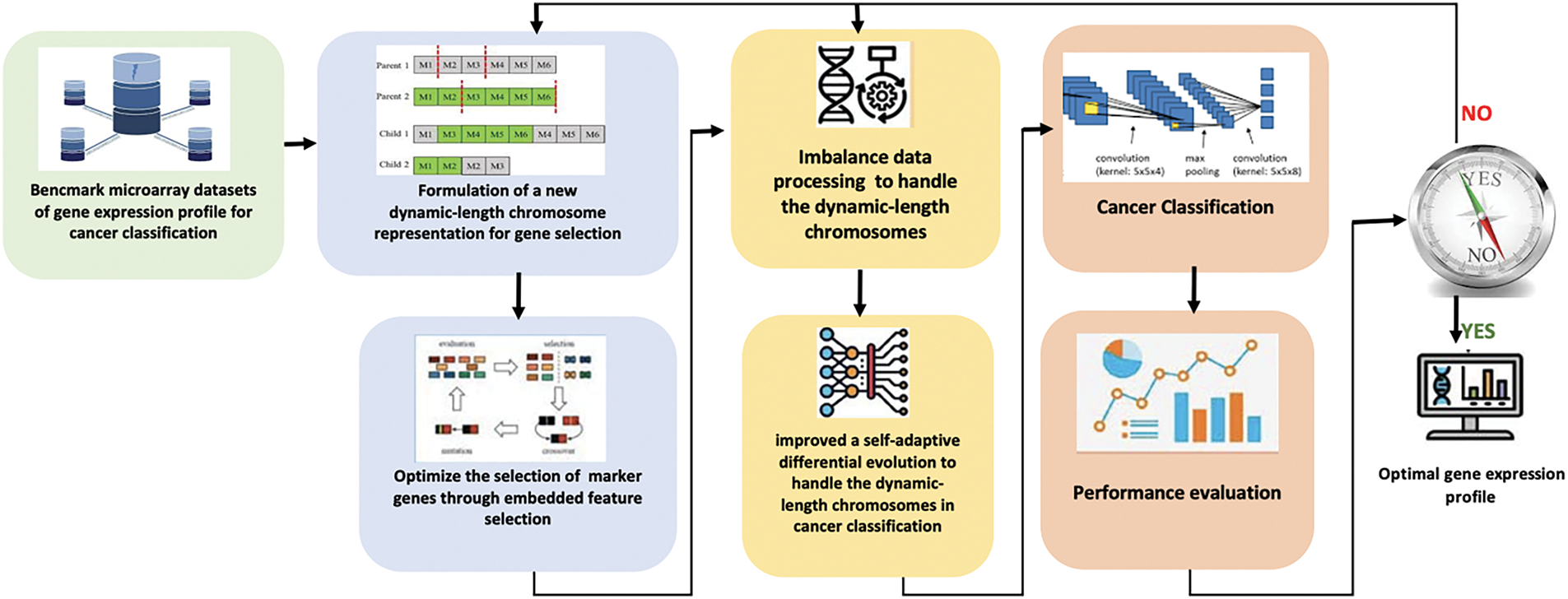
Figure 15: Gene selection framework for cancer classification
Developers and providers are encouraged to prioritise interoperability and standardisation efforts to ensure compatibility across different platforms and systems. Healthcare and federation authorities play a pivotal role in promoting the adoption of the FS framework in clinical practice. Finally, researchers are urged to prioritise interdisciplinary collaboration and data-sharing initiatives to accelerate progress in FS optimisation for cancer classification.
4.3.1 Combination of Algorithms and Approaches
The integration of FCSVM-ANN in GEP classification, with ANN serving as an optimal pre-processing technique, is recommended to expedite and improve SVM-RFE feature reduction while minimising computational resource requirements. To evaluate the effectiveness of this approach, future work could apply FCSVM-ANN to publicly available cancer gene expression datasets such as TCGA or GEO. Additionally, incorporating this algorithm with swarm optimisation techniques such as PSO, ABC, ACO, or bacterial foraging optimisation could further enhance performance. Applying these algorithms to gene expression datasets, such as those for breast cancer, could provide deeper insights into their capacity to identify more relevant features while reducing computational costs.
The integrated gene expression data with protein-protein interaction (IGEPPIN) can be applied to any classification algorithm beyond GAs to achieve optimal results. For future studies, evaluating IGEPPIN with SVMs or RF on cancer datasets, such as ovarian cancer, could provide valuable insights into its effectiveness in improving classification accuracy [146]. Additionally, C-HMOSHSSA, which requires low computational efforts, has been utilised to retain essential information [147]. This algorithm has the potential to identify disease-specific biomarkers and their interactions with other genes. Future work could explore its performance on pan-cancer datasets to assess its applicability across various cancer types and its potential in addressing multi-class classification problems.
A combination of logistic regression with GA efficiently trains the logistic regression model for HCC liver cancer in adults. This approach classifies data into two classes present in the dataset survival or mortality, while improving classification accuracy [148]. Future work could focus on using ensemble methods combined with DL, such as CNNs alongside GA, to detect HCC in large, heterogeneous datasets. Testing on large-scale clinical datasets would be crucial to evaluate its scalability and practical applicability. The transformation-based trilevel FS approach, incorporating wavelets and swarm computing with advanced ML techniques, could be applied to classify prostate cancer efficiently. A hybrid model combining Teaching-Learning-Based Optimisation (TLBO) with GA, known as TLBOG, to enhance the reliability of evolutionary algorithms in FS for breast cancer prediction [149]. Evaluation results demonstrate that this approach outperforms conventional wrapper techniques in terms of accuracy, sensitivity, precision, and F-measure on the WBCD and WDBC databases. Future work could use the cancer genome atlas prostate cancer (TCGA-PRAD) dataset to evaluate its effectiveness in improving prostate cancer diagnosis and treatment prediction. An EJaya-based FOA achieves better classification accuracy with fewer features compared to benchmark schemes. It is recommended to test this on high-dimensional datasets such as the GEO Series GSE6099 to refine gene selection and classification. Additionally, integrating EJaya-FOA with DL and reinforcement learning algorithms could further enhance its potential in improving classification accuracy.
Hybrid FS methods, including a filter-wrapper approach, have shown promise in cancer classification with microarray GEP [55]. Future work should continue exploring novel hybrid or parallel FS methods, especially those integrating filtering methods with wrapper-based algorithms such as SVM-RFE to handle noisy data in datasets like GSE19829. Another promising future direction is to develop a heuristic ensemble filter-based approach to classify cancerous gene expressions by predicting tumour and normal genes. Future research could test this algorithm on cancer datasets such as GSE1009 and explore its integration with protein secondary structure prediction and DL-based methods for enzyme function classification. Additionally, expanding the application of these methods to neurodegenerative disease datasets–using resources like Alzheimer’s Disease Neuroimaging Initiative (ADNI)–could facilitate the discovery of genes associated with neurological disorders.
Finally, the filter-wrapper hybrid greedy ensemble approach optimised using GA has demonstrated its ability to reduce dimensionality in high-dimensional biomedical datasets. Future work could explore metaheuristics techniques such as cuckoo search, firefly optimisation, and GA to balance intensification and diversification in FS. Testing these metaheuristics on single-cell RNA sequencing data could provide valuable insights into their potential for cancer biomarker discovery. Furthermore, optimising the automatic selection of ensemble components during model training-using frameworks such as Hom-EFS or Het-EFS for biomarker discovery from transcriptome profiles-holds significant promise for improving real-world applications. This research direction could lead to enhanced performance and broader applicability of FS methods in cancer classification.
4.3.2 Analysis of Algorithms Utilising Other Classifiers
A two-stage hybrid adaptive model (HAM) of neural networks has demonstrated competitiveness with standard classification models in terms of overall accuracy, sensitivity, and specificity [150]. Future research could explore the application of the HAM algorithm as a FS method for various classifiers. Specifically, researchers could evaluate its performance on cancer gene expression datasets such as GSE10245 for breast cancer or the cancer genome atlas breast cancer (TCGA-BRCA) dataset to determine its effectiveness across different cancer types. Additionally, testing the HAM algorithm with ML models such as RF and logistic regression could provide valuable insights into its adaptability and robustness in diverse classification tasks. EUFS algorithm, inspired by bipartite graph theory, has shown exceptional clustering and classification performance while maintaining low computational complexity [151]. An experimental comparison of EUFS with five state-of-the-art algorithms on 10 benchmark datasets has validated its efficiency and superiority. Future research could apply EUFS to gene expression datasets such as GEO Series GSE50081 for lung cancer or GSE45383 for colorectal cancer to assess its effectiveness in clustering and classifying cancerous tissues. Additionally, researchers should compare EUFS with other unsupervised clustering algorithms, such as k-means and density-based spatial clustering of applications with noise (DBSCAN), on these datasets to evaluate its relative performance in handling high-dimensional data.
BMO–SVM is an efficient and effective strategy for tackling complex optimisation scenarios and contributes to the theoretical foundation of intelligent optimisation. This algorithm could be tested as a filter FS method for use with other classifiers, such as neural networks, RF, and logistic regression. Future studies might apply BMO–SVM to datasets like TCGA-COAD (Colon Cancer) or GSE48313 for gastric cancer to evaluate its effectiveness in feature reduction and classification tasks. Expanding this approach to additional cancer types and incorporating other classifiers could help assess the generalisability and accuracy of the proposed method [152]. Additionally, this algorithm can be proposed as a filter FS method for use with other classifiers, such as neural networks, RF, and logistic regression. Cancer detection through relevant gene identification uses discrete filtering based on a binary artificial bee colony coupled with SVM and a two-stage cascading classifier to identify relevant genes and cancer using RNA-Seq data [153]. Further research should also consider integrating additional classifiers such as DL models (e.g., CNNs) and compare their performance against the SVM-based model on RNA-Seq data, providing a comprehensive evaluation of the method’s robustness across different cancer types.
4.3.3 Algorithm Application to Multimodal Data and Methods
Bayesian hyperparameter optimisation is performed as a two-step pipeline, where RFE and a diverse metapool are applied to multimodal data for disease diagnosis and prediction [154]. Future work could explore applying this optimisation pipeline to publicly available datasets, such as breast cancer microarray data and glioma datasets, to evaluate the robustness of the method in diagnosing different cancer types. This method is recommended for employing DL models for multimodal data fusion and feature extraction such as GEO Series GSE68086, which combines transcriptomics and proteomics data for a more comprehensive cancer classification. A procedure has been confirmed to summarise DNA methylation by AEs in relation to breast cancer recurrence [155]. To mitigate these limitations, researchers could test the AE method on larger and more diverse datasets, such as extensive breast cancer cohorts, and explore the impact of different weight initialisations and parameters on model performance across a wider range of ML techniques. Additionally, multiomics data integration could be further explored using datasets that combine genomics and clinical data to validate the AE-based approach.
A multiobjective social spider optimisation (MOSSO) algorithm is integrated with an SVM as a wrapper method to identify an optimal and minimal gene subset from a pool of selected genes. This algorithm holds promise for extending MOSSO’s applicability to other multiobjective problems [156]. Future work could explore its scalability by applying it to datasets such as lung cancer microarray data to assess its generalisability. Additionally, comparing this approach with other multiobjective optimisation methods, such as the non-dominated sorting genetic algorithm II (NSGA-II), could provide valuable insights into its performance in gene selection tasks. A model employing dynamic scaling factor-based DE with MLP for gene selection could be expanded to future multiclass datasets, such as ovarian cancer, which includes a variety of tumour subtypes [157]. Leveraging pathway information from microarray data, this approach suggests a potential model for designing future multiclass data sets. Additionally, researchers should explore further optimisation techniques by integrating various EAs, such as PSO, GA, and multi-objective evolutionary algorithms based on decomposition (MOEA/D), for more effective multi-objective gene selection. Evaluating these techniques on large-scale multiomics datasets, such as cancer drug sensitivity data, could provide valuable insights into their applicability and efficiency in real-world scenarios.
4.3.4 Algorithm Validation with Variation in Cancer Data
The potential biomarkers of early and late-stage liver HCC, derived from genomics and epigenomics data of patients, require validation on external datasets from diverse studies to fully understand their clinical utility. Future work could test these biomarkers on publicly available liver cancer datasets such as liver hepatocellular carcinoma to evaluate their generalisability across different patient populations and platforms. The implementation of NGTM to discover HCC biomarkers through the integration of scRNA-seq and GRN demonstrates strong classification performances [158]. This approach provides a competitive alternative for identifying cancer biomarkers, and further validation using external datasets including HCC microarray data, as well as breast and prostate cancer datasets—could enhance its reliability for high-throughput data analysis. The construction of diagnostic systems and GRN-based microarray analysis offers a novel strategy for identifying critical genes or biomarkers and their regulatory networks. Future work could apply this method to multi-disease datasets, such as pan-cancer, to identify common biomarkers across different cancer types. Similarly, identifying subtype-dependent biomarkers using benchmark microarray datasets, such as for breast cancer subtypes, could improve the prediction of molecular biomarkers specific to tumour subtypes. Future investigations could also explore the method’s effectiveness in other applications, such as protein structure and function prediction for diseases like neuroblastoma.
An ensemble of classifiers, including SVM, NB, C4.5, and KNN, can be used in combination with PSO to reduce dimensionality in cancer datasets. Future work could test this ensemble approach on semi-supervised datasets, such as those for lung cancer, to evaluate its performance in expanding sample sizes while maintaining the generalisation ability. The method could also be compared with other dimensionality reduction techniques like t-SNE or uniform manifold and approximation and projection (UMAP) to assess its robustness [159]. In the case of GWO and SVM, future studies could apply this technique to other binary-class datasets, such as lung or colon cancer datasets, to validate its reliability in repeated tissue sampling from the same patient. Spider monkey optimisation and cancer classification using SVM have been shown to minimise the number of features needed in cancer data. Testing this approach on datasets such as gastric cancer could help determine the optimal number of features required for accurate classification across various cancer types.
For gastric and colon cancer detection, future research could focus on a direct cancer classification method using original GEPs, leveraging an entropy-based graph classifier on datasets like gastric or colon cancer. Additionally, frequency-based FS using EMC-DWES has shown promise in selecting the most informative features. In COVID-19 datasets, automatic frequency-based FS using EMC-DWES as a frequency-based filter ranker discards irrelevant and redundant features. It achieves this by leveraging the intrinsic statistics of features and assigning greater weights simultaneously to the most informative clusters. This method could be tested on datasets like COVID-19 RNA-Seq data to assess its effectiveness in handling various biological datasets. GAAWGEFA, which improves gene similarity using functional annotations, could be tested on time-series datasets, such as breast cancer progression data, to explore its ability to capture gene expression changes over time. Similarly, cell separation algorithm (CSA), which regulates the trade-off between exploration and exploitation, could be tested on leukaemia and prostate cancer datasets, respectively, to evaluate its classification performance.
For multiomics tumour gene mining, GEP classification using VNLHHO is recommended for application on TCGA datasets, such as prostate cancer or glioma, to assess its applicability to large-scale, multiomics cancer datasets. Additionally, C-HMOSHSSA, which minimises computational effort, can be expanded for use on large healthcare datasets like MIMIC-III—a critical care database–aiming to address large-scale challenges in the healthcare data analysis. ESA-based optimisation could be further evaluated on datasets like lung cancer to assess its performance in gene selection tasks for high-dimensional datasets. Finally, SRAFL can automatically identify different gene signatures for subtypes, ensuring accurate classification of renal cell carcinoma subtypes. Future studies could test this method on renal cancer datasets to evaluate its robustness in subtype classification and explore its potential in cancer evolution studies.
4.3.5 Algorithm Application in Clinical Practice
Multipoint generic routing encapsulation (MGRE), based on an embedded GA, is valuable for understanding molecular mechanisms related to disease phenotypes and developing potential early detection methods and molecularly targeted cancer therapies [160]. Its applicability extends to FS challenges in high-dimensional data, often characterised by the ‘large p small n’ paradigm, where the number of features (p) exceeds the number of samples (n). Future work could apply MGRE to publicly available cancer datasets, such as lung adenocarcinoma or pan-cancer gene expression data, to evaluate its performance in distilling the essential biological signals embedded in high-dimensional data. Researchers could also compare MGRE with other FS methods, such as L1-regularization or Boruta, to assess its efficiency and effectiveness in extracting biologically relevant features.
Given its practical significance, a gene selection approach based on the FLD and NRS should be applied to clinical cancer diagnosis. Future studies could test this approach on clinical cancer datasets, such as breast cancer or colon adenocarcinoma, to evaluate its ability to select relevant genes for early cancer detection. Researchers could also compare its performance with other dimensionality reduction techniques, such as t-distributed stochastic neighborhood embedding (t-SNE) or UMAP, to assess the stability and accuracy of the gene selection process across different cancer types [161]. In a microarray study profiling the whole transcriptomes of eight human serous ovarian cancers (SOCs) and eight controls, a ceRNA network was constructed, including mRNAs, long non-coding RNAs (lncRNAs), and circular RNAs (circRNAs) experimentally validated to be involved in ovarian cancer development. Future work could expand on this study by applying the ceRNA network analysis to larger ovarian cancer datasets to explore the role of noncoding RNAs in disease progression. The integration of RNA-Seq data and single-cell RNA sequencing (scRNA-seq) could provide a more detailed understanding of the regulatory roles of these noncoding RNAs across various cancer subtypes.
Data reduction and visualisation for automatic diagnosis using gene expression and clinical data have demonstrated good performance, with data reduced via PCA and visualised as 2D images. This diagnostic method leverages DL approaches to develop a tool that offers valuable insights for automatic diagnosis and supports clinicians in actual clinical practice [162]. Future research could test this method on clinical cancer datasets, such as ovarian cancer or pan-cancer datasets, to evaluate its effectiveness. Additionally, integrating DL algorithms, such as CNNs, could enhance classification accuracy and feature identification. Applying this approach to real-time clinical data from healthcare institutions could further support the development of diagnostic tools that offer actionable insights for clinicians in practice.
4.3.6 Model Performance Improvement
A prediction model called restarted incremental FS by integrating multiple blocks (RIFS2D) is constructed to evaluate the individual correlation of each feature with the class label. It employs the incremental FS (IFS) strategy to compare the low-complexity and top-ranked features for the best predictive performance [163]. This possibility, which has implications for reducing computational requirements, should be explored further by applying RIFS2D to publicly available cancer datasets, such as breast cancer or liver cancer, to assess the effectiveness of low-ranked features in improving predictive accuracy. Future work could involve testing RIFS2D on these datasets to validate the potential of low-ranked biomarkers in classification tasks and evaluating the computational efficiency compared to top-ranked feature subsets. An ML approach on cytokine gene variants and sociodemographic characteristics as predictors of cervical cancer has conferred logistic regression with the highest average accuracy. Future studies could apply this approach to the cervical cancer dataset and explore the inclusion of additional risk factors like HPV infection status, smoking history, and genetic variants to improve prediction accuracy. Integrating a wider range of sociodemographic and clinical factors, possibly through ensemble learning methods, could further improve classification performance. For optimisation problems, evolutionary particle swarm optimisation (EPSO) and MHFS models may enhance the search process by adapting the variable search volume in most optimisation techniques [164]. Future work could test EPSO and MHFS models on multi-omics datasets, such as pan-cancer data, to explore their effectiveness in identifying biomarkers for different types of cancers and improving the search for optimal solutions. Comparing EPSO and MHFS with other metaheuristic algorithms like PSO, DE, and GA on large-scale datasets could provide insights into their relative strengths for FS in high-dimensional data.
An ordered search with a large margin classifier for FS, known as GAAWGEFA, finds a combination of weights for expressions that maximises the fitness of chromosomes in GA. This method can be applied to the functional time-series data of any species to improve similarity when the functional annotations of some genes and expression values for different time points are available [59,165]. Testing GAAWGEFA on datasets from longitudinal studies, such as colorectal cancer, could improve its ability to detect similarities over time. BEHN constructs a higher-order graphical model for high-dimensional data, including DL models, which offers interpretability through visualisation, unlike kernel-based models such as SVMs and neural networks. This method is particularly valuable when combined with probabilistic models of evolutionary computation, making it useful for investigating large populations. Future work could apply BEHN to complex genomic datasets, such as ovarian or breast cancer, to examine its interpretability in identifying gene networks and their roles in disease progression. Combining BEHN with probabilistic models of evolutionary computation could enhance its ability to model large populations and uncover gene interactions that conventional methods may overlook.
Multitask multimodal learning for cancer diagnosis and prognosis utilises inherent correlations across different tasks to guide FS, enhancing performance. Future studies could test this approach on datasets like lung or prostate cancer, applying multitask learning to predict both cancer stages and clinical outcomes. Replacing shallow morphological image features with high-level features learned by deep neural networks (DNNs) could further improve diagnosis and prognosis accuracy, particularly when applied to multi-modal datasets [166]. Sequential ML models based on LSTM and GRU have been compared with traditional ML methods based on multilayer perceptron, RF, decision tree, and KNN in predicting common cancers such as lung, breast, cervical, and liver cell cancers [167]. Among these models, GRU demonstrates superior accuracy. For future work, researchers could explore improving RNN model cells by incorporating patient visit timing data to make models more sensitive to temporal trends. Testing LSTM and GRU models on longitudinal datasets such as lung cancer progression or breast cancer recurrence could further validate their effectiveness. Additionally, the inclusion of factors, such as gender and disease type, to better discriminate between patient subgroups in datasets like chronic lymphocytic leukaemia could improve model performance.
4.3.7 Ethical Implications and Data Privacy in FS-Based Cancer Classification
The advancement of FS techniques in cancer classification has demonstrated significant potential, offering promising solutions for personalized treatment and improved diagnostic accuracy. However, it is essential to address the ethical implications and data privacy concerns associated with their application in medical and genomic research [168]. The increasing reliance on large-scale datasets, particularly genomic data, raises important questions regarding the collection, processing, and utilization of sensitive patient information. While FS techniques provide crucial insights into cancer biomarkers, they also highlight the need to uphold ethical standards in data handling, mitigating risks related to misuse and unintended harm to patients.
One of the most pressing concerns is patient privacy data. Genomic information, being highly sensitive, poses considerable risks concerning confidentiality and re-identification. Given that FS-based cancer classification models require access to detailed patient data, stringent data anonymization methods must be implemented to protect patient identities. Anonymization ensures that personal identities are removed, reducing the risk of exposure during research processes [169]. Additionally, encryption protocols should be integrated into the data storage and transfer processes to prevent unauthorized access, further safeguarding patient confidentiality and minimizing the likelihood of data breaches.
Moreover, informed patient consent is a fundamental ethical consideration. Patients must be fully informed of how their data will be utilized in research, including the potential access and analysis of their genomic information [170]. Consent should be obtained through a transparent process, clearly outlining the intended research purposes and associated risks, such as the possibility of re-identification in future studies. Ethical research requires that participants not only provide consent but also retain the right to withdraw their data at any stage without adverse consequences. Upholding informed consent procedures fosters trust between researchers and participants while safeguarding patients’ right and ensuring ethical compliance in FS-based cancer classification.
4.3.8 Potential Biases in Biomarker Selection and Reproducibility Challenges
Beyond the technical challenges associated with FS in cancer classification, a significant ethical concern is the potential bias in biomarker selection. FS methods are inherently dependent on the datasets they are trained on, and if these datasets lack diversity, the resulting models may exhibit biased outcomes. For example, genomic datasets that are predominantly composed of individuals from specific ethnic groups may lead FS models to prioritize biomarkers that are more relevant to the overrepresented population [171]. Consequently, these models may exhibit reduced accuracy or even become ineffective for underrepresented groups, thereby contributing to disparities in cancer diagnosis and treatment.
Failure to identify relevant biomarkers across diverse populations can have serious consequences, potentially leading to inappropriate or suboptimal treatment recommendations. Cancer therapies that are designed based on biomarkers specific to one demographic group may not be as effective for individuals with different genetic or ethnic backgrounds [172]. To mitigate this issue, it is crucial to ensure that training datasets for FS models incorporate genetic and ethnic diversity reflective of real-world clinical populations. Expanding datasets to include a broader range of patient demographics and continuously updating FS models with new and diverse data can help reduce bias and improve the generalizability of cancer classification models.
Furthermore, another pressing challenge in FS-based cancer classification is the issue of reproducibility. Reproducibility is critical for ensuring the reliability and robustness of FS models, particularly when applied across different institutions or cancer types [173]. However, variations in datasets, experimental conditions, and model parameters can result in inconsistent findings, reducing the credibility and applicability of FS methods in real-world clinical settings. If research results cannot be replicated across multiple institutions or cancer datasets, the utility of FS methods in clinical settings may be significantly compromised. This issue not only affects the scientific validity of FS models but also undermines confidence in their potential to provide accurate and personalized treatment recommendations.
To address reproducibility challenges, researchers must document their methodologies transparently, including data sources, model parameters, and experimental conditions. This transparency allows for independent validation and replication of findings, which is essential for building trust in FS models. Additionally, the adoption of standardized protocols and the use of open-source platforms for FS research can further enhance reproducibility. By ensuring that FS models are reproducible and transparent, the scientific community can foster greater confidence in their clinical application, ultimately advancing the field of cancer classification.
This article presents a comprehensive review of the research on FS for cancer classification, focusing on the evolution of algorithms, models, and techniques used in the field. All viewpoints presented in this review are derived from selected references. Many studies have predominantly focused on algorithm optimisation and robust classification using fixed-length chromosomes. However, this approach poses challenges distinguishing informative biomarkers while minimising selected features. The study includes an analysis of 67 papers from 2018–2024, categorised into several key research areas: algorithm and model development for FS and classification, computational complexity reduction, biomarker identification, high-dimensional data handling, and decision support system implementation. Key findings from the review include:
a. Feature selection and classification algorithms: A significant number of studies have focused on EAs for FS, such as GA, PSO, and ABC, which have been integrated with classification tasks to identify minimal yet relevant gene subsets for cancer classification.
b. Challenges with high-dimensional data: One of the major challenges identified is the high-dimensionality of cancer datasets, which contain thousands of features but relatively few samples. Techniques such as multi-metaheuristics, EAs with variable-length chromosomes, and hybrid models have been explored to improve computational efficiency and classification accuracy.
c. Biomarker Identification: Several studies focused on the identification of biomarkers for cancer, using FS methods to select relevant genes associated with different cancer subtypes. These studies have contributed to improving diagnostic and prognostic models by targeting genes critical to disease progression and treatment response.
d. Decision support systems (DSS): FS methods are also integrated into DSS to enhance clinical decision-making by improving prediction accuracy and robustness in identifying cancer-related biomarkers.
e. Future Directions: The article highlights several research gaps, including the need for dynamic chromosome length formulations to adapt to the evolving nature of cancer data, the integration of deep learning-based FS techniques for improved feature extraction, and the fusion of multi-omics data to gain a more comprehensive understanding of cancer mechanisms.
Overall, while this research provides promising solutions for cancer classification based on EAs and FS, there are several significant challenges that still need to be addressed, particularly related to using broader datasets, more efficient dimensionality reduction, and addressing ethical and data privacy issues in clinical applications. Here are the limitations of the research that should be considered:
a. Limited dataset usage: Many FS algorithms and GEP are applied to limited datasets, which may constrain their generalizability and applicability to different types of cancer or larger datasets. This limitation may lead to biased results if the datasets used are not representative of diverse patient populations.
b. Computational complexity: Some algorithms, while effective in improving accuracy, require significant computational resources, especially when dealing with high-dimensional genetic datasets. The use of methods that minimize computational requirements is crucial for real-world applications, but this remains a significant challenge in the research.
c. Challenges with imbalanced data: Cancer datasets are often imbalanced, with certain cancer types having fewer samples. This imbalance presents challenges for FS models in effectively addressing class imbalance, which may affect the reliability and accuracy of the results.
d. Reproducibility issues: Reproducibility of results in FS-based cancer classification remains a significant challenge. Variations in datasets, experimental conditions, and model parameters often lead to inconsistent findings, reducing the reliability and applicability of FS methods in clinical settings.
e. Ethical issues and data privacy: The use of sensitive patient data, particularly genomic data, raises ethical concerns about privacy and data security. FS-based cancer classification models heavily depend on patient data, requiring strict data protection measures to prevent misuse and ensure patient confidentiality.
f. Dependency on limited data: The research may be limited to data available from specific sources, and its findings might not be fully representative when compared to data from a broader or more diverse population. This is also related to bias issues, where datasets that lack diversity could lead to models that are less accurate for different patient groups.
g. Challenges in handling high-dimensional data: Although FS offers essential dimensionality reduction, challenges persist in managing high-dimensional datasets. Proper dimensionality reduction heavily depends on the methods and algorithms used, but in practice, more complex or intricate approaches sometimes compromise results that could be achieved using simpler but effective methods.
Future research should shift towards adaptive models that address the limitations of fixed-length chromosomes. Specifically, there is a clear need for more dynamic chromosome length formulations that can adapt throughout the optimisation process to handle high-dimensional data better and overcome the limitations of static approaches. Researchers should focus on embedding adaptive FS techniques into existing algorithms, integrating dynamic-length chromosomes to improve scalability and robustness, particularly in the context of biomarker identification for cancer. A promising area for future research is the exploration of interdisciplinary approaches, combining insights from genetics, ML, and bioinformatics, to enhance the precision and personalisation of cancer treatments.
Finally, ethical considerations will continue to play a crucial role in the future of FS research, particularly in the context of genomic and clinical applications. Ensuring transparency, reproducibility, and data security is essential when handling sensitive patient data. Ethical frameworks must be developed to safeguard patient privacy, ensure data security, and promote transparency in how research findings are reported and validated. Maintaining reproducibility in FS models is particularly vital, as independent validation by other research groups is essential for scientific integrity in cancer research.
In summary, while evolutionary algorithm-based FS has shown substantial progress in cancer classification, several key areas require further research. Future research should focus on integrating dynamic chromosome lengths, deep learning-based FS, multi-omics data fusion, as well as ensuring the real-world implementation of FS models into clinical workflows. By addressing these challenges, FS models can reach their full potential in advancing personalised cancer diagnostics and treatment, ultimately improving patient outcomes.
Acknowledgement: This project is supported by the Fundamental Research Grant Scheme (FRGS/1/2022/ICT02/UPSI/02/1) under the Ministry of Higher Education of Malaysia. The authors would like to thank Universiti Pendidikan Sultan Idris for its invaluable support in managing the grant.
Funding Statement: This research was funded by the Ministry of Higher Education of Malaysia, grant number FRGS/1/2022/ICT02/UPSI/02/1.
Author Contributions: Conceptualization, Siti Ramadhani, Lestari Handayani and Shir Li Wang; methodology, Siti Ramadhani; software, Siti Ramadhani and Haldi Budiman; validation, Lestari Handayani, Roziana Ariffin and Shir Li Wang; formal analysis, Siti Ramadhani; investigation, Siti Ramadhani; writing—original draft preparation, Siti Ramadhani; writing—review and editing, Lestari Handayani, Theam Foo Ng, Sumayyah Dzulkifly and Shir Li Wang; supervision, Lestari Handayani, Theam Foo Ng and Shir Li Wang; project administration, Sumayyah Dzulkifly and Shir Li Wang; funding acquisition, Shir Li Wang. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: The datasets analyzed during the current study are available from the corresponding author on reasonable request.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare no conflicts of interest to report regarding the present study.
Supplementary Materials: The supplementary material is available online at https://www.techscience.com/doi/10.32604/cmes.2025.062709/s1.
References
1. Brown JS, Amend SR, Austin RH, Gatenby RA, Hammarlund EU, Pienta KJ. Updating the definition of cancer. Mol Cancer Res. 2023;21(11):1142–7. doi:10.1158/1541-7786.MCR-23-0411. [Google Scholar] [PubMed] [CrossRef]
2. Ramadan SZ. Methods used in computer-aided diagnosis for breast cancer detection using mammograms: a review. J Healthc Eng. 2020; 1:1–21. doi:10.1155/2020/9162464. [Google Scholar] [PubMed] [CrossRef]
3. Khalifa NEM, Taha MHN, Ezzat Ali D, Slowik A, Hassanien AE. Artificial intelligence technique for gene expression by tumor RNA-seq data: a novel optimized deep learning approach. IEEE Access. 2020;8:22874–83. doi:10.1109/ACCESS.2020.2970210. [Google Scholar] [CrossRef]
4. Khalsan M, MacHado LR, Al-Shamery ES, Ajit S, Anthony K. A survey of machine learning approaches applied to gene expression analysis for cancer prediction. IEEE Access. 2022;10(4):27522–34. doi:10.1109/ACCESS.2022.3146312. [Google Scholar] [CrossRef]
5. Shigao H, Jie Y, Simon F, Qi Z. Artificial intelligence in cancer diagnosis: opportunities and challenges. Pathol Res Pract. 2019;253:154996. doi:10.1016/j.prp.2023.154996. [Google Scholar] [PubMed] [CrossRef]
6. Waseem MH, Nadeem MSA, Abbas A, Shaheen A, Aziz W, Anjum A. On the feature selection methods and reject option classifiers for robust cancer prediction. IEEE Access. 2019;7:141072–82. doi:10.1109/ACCESS.2019.2944295. [Google Scholar] [CrossRef]
7. Vahmiyan M, Kheirabadi M, Akbari E. Feature selection methods in microarray gene expression data: a systematic mapping study. Neural Comput Appl. 2022;34(22):19675–702. doi:10.1007/s00521-022-07661-z. [Google Scholar] [CrossRef]
8. Colombelli F, Kowalski TW, Recamonde-Mendoza M. A hybrid ensemble feature selection design for candidate biomarkers discovery from transcriptome profiles. Knowl Based Syst. 2022 Oct;254:109655–71. doi:10.1016/j.knosys.2022.109655. [Google Scholar] [CrossRef]
9. Wilson A, Anwar MR. The future of adaptive machine learning algorithms in high-dimensional data processing. Int Transact Artif Intell. 2024;3(1):97–107. doi:10.33050/italic.v3i1.656. [Google Scholar] [CrossRef]
10. Mohamed M, Abdullah A, Zaki AM, Rizk FH, Eid MM, El-Kenway EMEl. Advances and challenges in feature selection methods: a comprehensive review. J Artif Intellig Metaheuris. 2024;7(1):67–77. doi:10.54216/jaim.070105. [Google Scholar] [CrossRef]
11. Wang A, Liu H, Yang J, Chen G. Ensemble feature selection for stable biomarker identification and cancer classification from microarray expression data. Comput Biol Med. 2022;142(33):105208–18. doi:10.1016/j.compbiomed.2021.105208. [Google Scholar] [PubMed] [CrossRef]
12. Mazlan AU, Sahabudin NA, Remli MA, Ismail NSN, Mohamad MS, Nies HW, et al. A review on recent progress in machine learning and deep learning methods for cancer classification on gene expression data. Processes. 2021 Aug;9(8):1–12. doi:10.3390/pr9081466. [Google Scholar] [CrossRef]
13. Khanna M, Singh LK, Shrivastava K, Singh R. An enhanced and efficient approach for feature selection for chronic human disease prediction: a breast cancer study. Heliyon. 2024;10(5):26799–820. doi:10.1016/j.heliyon.2024.e26799. [Google Scholar] [PubMed] [CrossRef]
14. Yaqoob A, Musheer Aziz R, verma NK. Applications and techniques of machine learning in cancer classification: a systematic review. Human-Centric Intell Syst. 2023;3(4):588–615. doi:10.1007/s44230-023-00041-3. [Google Scholar] [CrossRef]
15. Rundo L, Militello C, Vitabile S, Russo G, Sala E, Gilardi MC. A survey on nature-inspired medical image analysis: a step further in biomedical data integration. Fundam Inform. 2019;171(1–4):345–65. doi:10.3233/FI-2020-1887. [Google Scholar] [CrossRef]
16. Alrefai N, Ibrahim O. Optimized feature selection method using particle swarm intelligence with ensemble learning for cancer classification based on microarray datasets. Neural Comput Appl. 2022;34(16):13513–528. doi:10.1007/s00521-022-07147-y. [Google Scholar] [CrossRef]
17. Quazi S. Artificial intelligence and machine learning in precision and genomic medicine. Med Oncol. 2022;39(8):1–18. doi:10.1007/s12032-022-01711-1. [Google Scholar] [PubMed] [CrossRef]
18. Gad AG. Particle swarm optimization algorithm and its applications: a systematic review. Arch Comput Methods Eng. 2022 Aug;29(5):2531–61. doi:10.1007/s11831-021-09694-4. [Google Scholar] [CrossRef]
19. Bommert A, Welchowski T, Schmid M, Rahnenführer J. Benchmark of filter methods for feature selection in high-dimensional gene expression survival data. Brief Bioinform. 2022;23(1):1–13. doi:10.1093/bib/bbab354. [Google Scholar] [PubMed] [CrossRef]
20. Abasabadi S, Nematzadeh H, Motameni H, Akbari E. Hybrid feature selection based on SLI and genetic algorithm for microarray datasets. J Supercomput. 2022;78(18):19725–53. doi:10.1007/s11227-022-04650-w. [Google Scholar] [PubMed] [CrossRef]
21. Kafrawy PEl, Fathi H, Qaraad M, Kelany AK, Chen X. An efficient SVM-based feature selection model for cancer classification using high-dimensional microarray data. IEEE Access. 2021;9:155353–69. doi:10.1109/ACCESS.2021.3123090. [Google Scholar] [CrossRef]
22. Han F, Yang C, Wu YQ, Zhu JS, Ling QH, Song YQ. A gene selection method for microarray data based on binary PSO encoding gene-to-class sensitivity information. IEEE/ACM Trans Comput Biol Bioinform. 2017;14(1):85–96. doi:10.1109/TCBB.2015.2465906. [Google Scholar] [PubMed] [CrossRef]
23. Jin C, Jin SW. Gene selection approach based on improved swarm intelligent optimisation algorithm for tumour classification. IET Syst Biol. 2016;10(3):107–15. doi:10.1049/iet-syb.2015.0064. [Google Scholar] [PubMed] [CrossRef]
24. Saraf TOQ, Fuad N, Taujuddin NSAM. Framework of meta-heuristic variable length searching for feature selection in high-dimensional data. Computers. 2023;12(1):1–13. doi:10.3390/computers12010007. [Google Scholar] [CrossRef]
25. Sayed S, Nassef M, Badr A, Farag I. A Nested Genetic Algorithm for feature selection in high-dimensional cancer Microarray datasets. Expert Syst Appl. 2019;121(1):233–43. doi:10.1016/j.eswa.2018.12.022. [Google Scholar] [CrossRef]
26. Tran B, Xue B, Zhang M. Variable-length particle swarm optimization for feature selection on high-dimensional classification. IEEE Trans Evol Comput. 2019;23(3):473–87. doi:10.1109/tevc.2018.2869405. [Google Scholar] [CrossRef]
27. Budiman H, Wang SLi, Morsidi F, Foo Ng T, Chin Neoh S. Self-adaptive ensemble-based differential evolution with enhanced population sizing. In: 2020 2nd International Conference on Cybernetics and Intelligent System, ICORIS 2020. IEEE; 2020. doi:10.1109/ICORIS50180.2020.9320767. [Google Scholar] [CrossRef]
28. Alharbi F, Vakanski A. Machine learning methods for cancer classification using gene expression data: a review. Bioengineering. 2023;10(173):1–26. doi:10.3390/bioengineering10020173. [Google Scholar] [PubMed] [CrossRef]
29. Wang SL, Morsidi F, Ng TF, Budiman H, Neoh SC. Insights into the effects of control parameters and mutation strategy on self-adaptive ensemble-based differential evolution. Inf Sci. 2020;514(3):203–33. doi:10.1016/j.ins.2019.11.046. [Google Scholar] [CrossRef]
30. Houssein EH, Gad AG, Hussain K, Suganthan PN. Major advances in particle swarm optimization: theory, analysis, and application. Swarm Evol Comput. 2021;63:100868–84. doi:10.1016/j.swevo.2021.100868. [Google Scholar] [CrossRef]
31. Almugren N, Alshamlan H. A survey on hybrid feature selection methods in microarray gene expression data for cancer classification. IEEE Access. 2019;7:78533–48. doi:10.1109/ACCESS.2019.2922987. [Google Scholar] [CrossRef]
32. Rani RR, Ramyachitra D. Microarray cancer gene feature selection using spider monkey optimization algorithm and cancer classification using SVM. Procedia Comput Sci. 2018;143(6):108–16. doi:10.1016/j.procs.2018.10.358. [Google Scholar] [CrossRef]
33. Agarwalla P, Mukhopadhyay S. Bi-stage hierarchical selection of pathway genes for cancer progression using a swarm based computational approach. Appl Soft Comput J. 2018 Jan;62:230–50. doi:10.1016/j.asoc.2017.10.024. [Google Scholar] [CrossRef]
34. Wang A, An N, Chen G, Liu L, Alterovitz G. Subtype dependent biomarker identification and tumor classification from gene expression profiles. Knowl Based Syst. 2018 Apr;146(5439):104–17. doi:10.1016/j.knosys.2018.01.025. [Google Scholar] [CrossRef]
35. Qu C, Zhang L, Li J, Deng F, Tang Y, Zeng X. Improving feature selection performance for classification of gene expression data using Harris Hawks optimizer with variable neighborhood learning. Brief Bioinform. 2021;22(5):1–15. doi:10.1093/bib/bbab097. [Google Scholar] [PubMed] [CrossRef]
36. Teng L, Feng Z, Fang X, Teng S, Wang H, Kang P. Unsupervised feature selection with adaptive residual preserving. Neurocomputing. 2019 Nov;367(1):259–72. doi:10.1016/j.neucom.2019.05.097. [Google Scholar] [CrossRef]
37. Ram PK, Kuila P. Dynamic scaling factor based differential evolution with multi-layer perceptron for gene selection from pathway information of microarray data. Multimed Tools Appl. 2023;82(9):13453–78. doi:10.1007/s11042-022-13964-z. [Google Scholar] [CrossRef]
38. Jaddi NS, Saniee Abadeh M. Gene selection of non-small cell lung cancer data for adjuvant chemotherapy decision using cell separation algorithm. Appl Intell. 2020 Nov;50(11):3822–36. doi:10.1007/s10489-020-01740-1. [Google Scholar] [CrossRef]
39. Hashmi A, Ali W, Abulfaraj A, Binzagr F, Alkayal E. Enhancing cancerous gene selection and classification for high-dimensional microarray data using a novel hybrid filter and differential evolutionary feature selection. Cancers. 2024 Dec;16(23):3913. doi:10.3390/cancers16233913. [Google Scholar] [PubMed] [CrossRef]
40. Książek W, Gandor M, Pławiak P. Comparison of various approaches to combine logistic regression with genetic algorithms in survival prediction of hepatocellular carcinoma. Comput Biol Med. 2021 Jul;134:104431. doi:10.1016/j.compbiomed.2021.104431. [Google Scholar] [PubMed] [CrossRef]
41. Cleofas-Sánchez L, Sánchez JS, García V. Gene selection and disease prediction from gene expression data using a two-stage hetero-associative memory. Prog Artif Intell. 2019 Apr;8(1):63–71. doi:10.1007/s13748-018-0148-6. [Google Scholar] [CrossRef]
42. Al-Shammary D, Albukhnefis AL, Alsaeedi AH, Al-Asfoor M. Extended particle swarm optimization for feature selection of high-dimensional biomedical data. Concurr Comput. 2022;34(10):1–12. doi:10.1002/cpe.6776. [Google Scholar] [CrossRef]
43. Gangavarapu T, Patil N. A novel filter-wrapper hybrid greedy ensemble approach optimized using the genetic algorithm to reduce the dimensionality of high-dimensional biomedical datasets. Appl Soft Comput J. 2019;81(1):105538–58. doi:10.1016/j.asoc.2019.105538. [Google Scholar] [CrossRef]
44. Venkataramana L, Jacob SG, Ramadoss R. A Parallel Multilevel Feature Selection algorithm for improved cancer classification. J Parallel Distrib Comput. 2020 Apr;138(5):78–98. doi:10.1016/j.jpdc.2019.12.015. [Google Scholar] [CrossRef]
45. Jain I, Jain VK, Jain R. Correlation feature selection based improved-Binary Particle Swarm Optimization for gene selection and cancer classification. Appl Soft Comput. 2018 Jan;62(16):203–15. doi:10.1016/j.asoc.2017.09.038. [Google Scholar] [CrossRef]
46. Mabu AM, Prasad R, Yadav R. Optimised feature selection and entropy-based graph classification of gene expression data. Int J Med Eng Inform. 2020;12(4):354–74. doi:10.1504/ijmei.2020.108239. [Google Scholar] [PubMed] [CrossRef]
47. Mabu AM, Prasad R, Yadav R. Gene expression dataset classification using artificial neural network and clustering-based feature selection. Int J Swarm Intell Res. 2020 Jan;11(1):65–86. doi:10.4018/IJSIR.2020010104. [Google Scholar] [CrossRef]
48. Xie S, Zhang Y, Lv D, Chen X, Lu J, Liu J. A new improved maximal relevance and minimal redundancy method based on feature subset. J Supercomput. 2023;79(3):3157–80. doi:10.1007/s11227-022-04763-2. [Google Scholar] [PubMed] [CrossRef]
49. Alomari OA, Khader AT, Betar MA, Awadalllah MA. A novel gene selection method using modified MRMR and hybrid Bat Inspired with Hill Climbing Algorithm. Appl Intell. 2020;48(1):4429–47. doi:10.1007/s10489-018-1207-1. [Google Scholar] [CrossRef]
50. Koul N, Manvi SS. Framework for classification of cancer gene expression data using Bayesian hyper-parameter optimization. Med Biol Eng Comput. 2021;59(11):2353–71. doi:10.1007/s11517-021-02442-7. [Google Scholar] [PubMed] [CrossRef]
51. Mahto R, Ahmed SU, ur Rahman R, Aziz RM, Roy P, Mallik S. A novel and innovative cancer classification framework through a consecutive utilization of hybrid feature selection. BMC Bioinformatics. 2023 Dec;24(1):479–505. doi:10.1186/s12859-023-05605-5. [Google Scholar] [PubMed] [CrossRef]
52. Sun L, Zhang X, Xu J, Wang W, Liu R. A gene selection approach based on the fisher linear discriminant and the neighborhood rough set. Bioengineered. 2018;9(1):144–51. doi:10.1080/21655979.2017.1403678. [Google Scholar] [PubMed] [CrossRef]
53. Dhrif H, Kubat M, Giraldo LGS, Wuchty S. A stable hybrid method for feature subset selection using particle swarm optimization with local search. In: GECCO 2019—Proceedings of the 2019 Genetic and Evolutionary Computation Conference. Association for Computing Machinery, Inc. ACM; 2019 Jul. p. 13–21. doi:10.1145/3321707.3321816. [Google Scholar] [CrossRef]
54. Houssein EH, Abdelminaam DS, Hassan HN, Al-Sayed MM, Nabil E. A hybrid barnacles mating optimizer algorithm with support vector machines for gene selection of microarray cancer classification. IEEE Access. 2021;9:64895–905. doi:10.1109/ACCESS.2021.3075942. [Google Scholar] [CrossRef]
55. Uzma, Halim Z. An ensemble filter-based heuristic approach for cancerous gene expression classification. Knowl Based Syst. 2021 Oct;234(2):107560–82. doi:10.1016/j.knosys.2021.107560. [Google Scholar] [CrossRef]
56. Lai CM. Multi-objective simplified swarm optimization with weighting scheme for gene selection. Appl Soft Comput J. 2018 Apr;65:58–68. doi:10.1016/j.asoc.2017.12.049. [Google Scholar] [CrossRef]
57. Prabhakar SK, Lee SW. Transformation based tri-level feature selection approach using wavelets and swarm computing for prostate cancer classification. IEEE Access. 2020 Jun;8(2):127462–76. doi:10.1109/ACCESS.2020.3006197. [Google Scholar] [CrossRef]
58. Andjelkovic J, Ljubic B, Hai AA, Stanojevic M, Pavlovski M. Sequential machine learning in prediction of common cancers. Inform Med Unlocked. 2022 Nov;30:100928–50. doi:10.1016/j.imu.2022.100928. [Google Scholar] [CrossRef]
59. Ray SS, Misra S. Genetic algorithm for assigning weights to gene expressions using functional annotations. Comput Biol Med. 2019;104:149–62. doi:10.1016/j.compbiomed.2018.11.011. [Google Scholar] [PubMed] [CrossRef]
60. Peng C, Wu X, Yuan W, Zhang X, Zhang Y, Li Y, et al. Multilayer recursive feature elimination based on an embedded genetic algorithm for cancer classification. IEEE/ACM Trans Comput Biol Bioinform. 2021;18(2):621–32. doi:10.1109/TCBB.2019.2921961. [Google Scholar] [PubMed] [CrossRef]
61. Almugren N, Alshamlan H. FF-SVM: new firefly-based gene selection algorithm for microarray cancer classification. In: 2019 IEEE Conference on Computational Intelligence in Bioinformatics and Computational Biology, CIBCB 2019; 2019; Siena, Italy. doi:10.1109/CIBCB.2019.8791236. [Google Scholar] [CrossRef]
62. Tsanousa A, Ntoufa S, Papakonstantinou N, Stamatopoulos K, Angelis L. Study of gene expressions’ correlation structures in subgroups of Chronic Lymphocytic Leukemia Patients. J Biomed Inform. 2019;95(2):103211–26. doi:10.1016/j.jbi.2019.103211. [Google Scholar] [PubMed] [CrossRef]
63. Sun L, Zhang XY, Qian YH, Xu JC, Zhang SG, Tian Y. Joint neighborhood entropy-based gene selection method with fisher score for tumor classification. Appl Intell. 2019 Apr;49(4):1245–59. doi:10.1007/s10489-018-1320-1. [Google Scholar] [CrossRef]
64. Breitenbach T, Rasbach L, Liang C, Jahnke P. A principal feature analysis. J Comput Sci. 2022;58:101502–22. doi:10.1016/j.jocs.2021.101502. [Google Scholar] [CrossRef]
65. Huang M, Ye X, Imakura A, Sakurai T. Sequential reinforcement active feature learning for gene signature identification in renal cell carcinoma. J Biomed Inform. 2022 Apr;128:104049–61. doi:10.1016/j.jbi.2022.104049. [Google Scholar] [PubMed] [CrossRef]
66. Li J, Cheng K, Wang S, Morstatter F, Trevino RP. Feature selection: a data perspective. ACM Comput Surv. 2017;50(6):1–24. doi:10.1145/3136625. [Google Scholar] [CrossRef]
67. Wang A, Liu H, Chen G. Chaotic harmony search based multi-objective feature selection for classification of gene expression profiles. In: 2021 IEEE 9th International Conference on Bioinformatics and Computational Biology, ICBCB 2021. Taiyuan, China: Institute of Electrical and Electronics Engineers Inc.; 2021 May. p. 107–12. doi:10.1109/ICBCB52223.2021.9459222. [Google Scholar] [CrossRef]
68. Hussain SM. Multi-metaheuristic feature selection model for high-dimensional biomedical data. In: First International Conference on Mathematical Modeling and Computational Science (ICMMCS). Singapore: Springer; 2020. p. 309–21. [Google Scholar]
69. Nematzadeh H, García-Nieto J, Navas-Delgado I, Aldana-Montes JF. Automatic frequency-based feature selection using discrete weighted evolution strategy. Appl Soft Comput. 2022;130(1):109699–711. doi:10.1016/j.asoc.2022.109699. [Google Scholar] [CrossRef]
70. Zhang QB, Ding LX. A new crossover mechanism for genetic algorithms with variable-length chromosomes for path optimization problems. Expert Syst Appl. 2016;60(10):183–9. doi:10.1016/j.eswa.2016.04.005. [Google Scholar] [CrossRef]
71. Hammami M, Bechikh S, Hung CC, Ben Said L. A Multi-objective hybrid filter-wrapper evolutionary approach for feature selection. Memet Comput. 2019 Jun;11(2):193–208. doi:10.1007/s12293-018-0269-2. [Google Scholar] [CrossRef]
72. Baliarsingh SK, Vipsita S, Dash B. A new optimal gene selection approach for cancer classification using enhanced Jaya-based forest optimization algorithm. Neural Comput Appl. 2020 Jun;32(12):8599–616. doi:10.1007/s00521-019-04355-x. [Google Scholar] [CrossRef]
73. Dash R. An adaptive harmony search approach for gene selection and classification of high dimensional medical data. J King Saud Univ-Comput Inf Sci. 2021;33(2):195–207. doi:10.1016/j.jksuci.2018.02.013. [Google Scholar] [CrossRef]
74. Panda M. Elephant search optimization combined with deep neural network for microarray data analysis. J King Saud Univ-Comput Inform Sci. 2020 Oct;32(8):940–8. doi:10.1016/j.jksuci.2017.12.002. [Google Scholar] [CrossRef]
75. Cumbo F, Cappelli E, Weitschek E. A brain-inspired hyperdimensional computing approach for classifying massive DNA methylation data of cancer. Algorithms. 2020;13(9):233. doi:10.3390/A13090233. [Google Scholar] [CrossRef]
76. Canatalay PJ, Ucan ON. A Bidirectional LSTM-RNN and GRU method to exon prediction using splice-site mapping. Appl Sci. 2022 May;12(9):4390. doi:10.3390/app12094390. [Google Scholar] [CrossRef]
77. Macías-García L, Martínez-Ballesteros M, Luna-Romera JM, García-Heredia JM, García-Gutiérrez J, Riquelme-Santos JC. Autoencoded DNA methylation data to predict breast cancer recurrence: machine learning models and gene-weight significance. Artif Intell Med. 2020;110:101976–92. doi:10.1016/j.artmed.2020.101976. [Google Scholar] [PubMed] [CrossRef]
78. Duan M, Wang Y, Qiao Y, Wang Y, Pan X, Hu Z. Pan-cancer identification of the relationship of metabolism-related differentially expressed transcription regulation with non-differentially expressed target genes via a gated recurrent unit network. Comput Biol Med. 2022;148(2):105883–98. doi:10.1016/j.compbiomed.2022.105883. [Google Scholar] [PubMed] [CrossRef]
79. Kaushik M, Joshi RC, Kushwah AS, Gupta MK, Banerjee M. Cytokine gene variants and socio-demographic characteristics as predictors of cervical cancer: a machine learning approach. Comput Biol Med. 2021;134:104559–79. doi:10.1016/j.compbiomed.2021.104559. [Google Scholar] [PubMed] [CrossRef]
80. Castillo-Secilla D, Gálvez JM, Carrillo-Perez F. KnowSeq R-Bioc package: the automatic smart gene expression tool for retrieving relevant biological knowledge. Comput Biol Med. 2021;133(1):1–12. doi:10.1016/j.compbiomed.2021.104387. [Google Scholar] [PubMed] [CrossRef]
81. Bruno P, Calimeri F, Kitanidis AS, De Momi E. Data reduction and data visualization for automatic diagnosis using gene expression and clinical data. Artif Intell Med. 2020;107:101884–902. doi:10.1016/j.artmed.2020.101884. [Google Scholar] [PubMed] [CrossRef]
82. Sun D, Li A, Tang B, Wang M. Integrating genomic data and pathological images to effectively predict breast cancer clinical outcome. Comput Methods Programs Biomed. 2018 Jul;161:45–53. doi:10.1016/j.cmpb.2018.04.008. [Google Scholar] [PubMed] [CrossRef]
83. Ramadhan ML. Breast and colon cancer classification from gene expression profiles using data mining techniques. Symmetry. 2020;12(408):1–15. doi:10.3390/sym12030408. [Google Scholar] [CrossRef]
84. Dabba A, Tari A, Meftali S. A novel grey wolf optimization algorithm based on geometric transformations for gene selection and cancer classification. J Supercomput. 2024;80(4):4808–40. doi:10.1007/s11227-023-05643-z. [Google Scholar] [CrossRef]
85. Kaur H, Bhalla S, Raghava GPS. Classification of early and late stage liver hepatocellular carcinoma patients from their genomics and epigenomics profiles. PLoS One. 2019 Sep;14(9):1–12. doi:10.1371/journal.pone.0221476. [Google Scholar] [PubMed] [CrossRef]
86. Li H, Zheng X, Gao J, Leung KS. Whole transcriptome analysis reveals non-coding RNA’s competing endogenous gene pairs as novel form of motifs in serous ovarian cancer. Comput Biol Med. 2022;148:105881–902. doi:10.1016/j.compbiomed.2022.105881. [Google Scholar] [PubMed] [CrossRef]
87. Hu Y, Dingerdissen H, Gupta S, Kahsay R, Shanker V. Identification of key differentially expressed MicroRNAs in cancer patients through pan-cancer analysis. Comput Biol Med. 2018;103:183–97. doi:10.1016/j.compbiomed.2018.10.021. [Google Scholar] [PubMed] [CrossRef]
88. Hong CF, Chen YC, Chen WC, Tu KC, Tsai MH. Construction of diagnosis system and gene regulatory networks based on microarray analysis. J Biomed Inform. 2018 May;81:61–73. doi:10.1016/j.jbi.2018.03.008. [Google Scholar] [PubMed] [CrossRef]
89. Shao W, Wang T, Sun L, Dong T, Han Z. Multi-task multi-modal learning for joint diagnosis and prognosis of human cancers. Med Image Anal. 2020;65(1):101795. doi:10.1016/j.media.2020.101795. [Google Scholar] [PubMed] [CrossRef]
90. Zhang Z, Sun C, Liu ZP. Discovering biomarkers of hepatocellular carcinoma from single-cell RNA sequencing data by cooperative games on gene regulatory network. J Comput Sci. 2022;65:101881. doi:10.1016/j.jocs.2022.101881. [Google Scholar] [CrossRef]
91. Lawrence MO. An efficient feature selection and classification system for microarray cancer data using genetic algorithm and deep belief networks. Multimed Tools Appl. 2025;84(8):4393–434. doi:10.1007/s11042-024-18802-y. [Google Scholar] [CrossRef]
92. Feng G. Feature selection algorithm based on optimized genetic algorithm and the application in high-dimensional data processing. PLoS One. 2024 May;19(2):e0303088. doi:10.1371/journal.pone.0303088. [Google Scholar] [PubMed] [CrossRef]
93. Qian J. Multisensor detection design via a weighted scheme with AUC and information theory. IEEE Trans Instrum Meas. 2023;73(1):1–10. doi:10.1109/tim.2023.3341111. [Google Scholar] [CrossRef]
94. Dey A. Identification of biomarkers for arsenicosis employing multiple kernel learning embedded multiobjective swarm intelligence. IEEE Trans Nanobioscience. 2022;22(2):383–92. doi:10.1109/tnb.2022.3194091. [Google Scholar] [PubMed] [CrossRef]
95. Dixit S, Kumar A, Srinivasan K. A current review of machine learning and deep learning models in oral cancer diagnosis: recent technologies, open challenges, and future research directions. Diagnostics. 2023;13(7):1353. doi:10.3390/diagnostics13071353. [Google Scholar] [PubMed] [CrossRef]
96. Sahu B. Hybrid grasshopper optimization algorithm with simulated annealing for feature selection using high dimensional dataset. In: 2023 1st International Conference on Advanced Innovations in Smart Cities (ICAISC). Jeddah, Saudi Arabia: IEEE; 2023. p. 1–6. [Google Scholar]
97. Amiriebrahimabadi M. A comprehensive survey of feature selection techniques based on whale optimization algorithm. Multimed Tools Appl. 2024;83(16):47775–846. doi:10.1007/s11042-023-17329-y. [Google Scholar] [CrossRef]
98. Sharma A, Rani R. C-HMOSHSSA: gene selection for cancer classification using multi-objective meta-heuristic and machine learning methods. Comput Methods Programs Biomed. 2019 Sep;178:219–35. doi:10.1016/j.cmpb.2019.06.029. [Google Scholar] [PubMed] [CrossRef]
99. Wang J. Establishing a machine learning model for predicting nutritional risk through facial feature recognition. Front Nutr. 2023;10(1):1219193–202. doi:10.3389/fnut.2023.1219193. [Google Scholar] [PubMed] [CrossRef]
100. Bales B. GA-SFS: a two-stage hybrid algorithm for feature selection in biomedical data. In: 2025 IEEE 15th Annual Computing and Communication Workshop and Conference (CCWC). IEEE; 2025. p. 00421–6. [Google Scholar]
101. Balamurugan P, Bharathy AMV, Marimuthu K, Niranchana R. Type-specific classification of bronchogenic carcinomas using bi-layer mutated particle swarm optimisation. Int J Comput Aided Eng Technol. 2020;13(3):360–70. doi:10.1504/IJCAET.2020.109520. [Google Scholar] [CrossRef]
102. Elreedy D. A theoretical distribution analysis of synthetic minority oversampling technique (SMOTE) for imbalanced learning. Mach Learn. 2024;113(7):4903–23. doi:10.1007/s10994-022-06296-4. [Google Scholar] [CrossRef]
103. Zhang H, Zhang R, Nie F, Li X. An efficient framework for unsupervised feature selection. Neurocomputing. 2019;366(6):194–207. doi:10.1016/j.neucom.2019.07.020. [Google Scholar] [CrossRef]
104. Digra M. Enhancing remote sensing image analysis: optimization of a hybrid deep network through HHO algorithm. Multimed Tools Appl. 2015 Jan;1(1):1–21. doi:10.1007/s11042-024-20499-y. [Google Scholar] [CrossRef]
105. Eissa N, Khairuddin U, Yusof R, Madani A. A metaheuristic technique for cluster-based feature selection of DNA methylation data for cancer. Comput Mat Continua. 2023;74(2):2817–38. doi:10.32604/cmc.2023.033632. [Google Scholar] [CrossRef]
106. Raufi AG. Advances in liquid biopsy technology and implications for pancreatic cancer. Int J Mol Sci. 2023;24(4):4238–63. doi:10.3390/ijms24044238. [Google Scholar] [PubMed] [CrossRef]
107. Akter S. Recent advances in ovarian cancer: therapeutic strategies, potential biomarkers, and technological improvements. Cells. 2022;11(4):650–62. doi:10.3390/cells11040650. [Google Scholar] [PubMed] [CrossRef]
108. Wang Y. Gene selection in a gene decision space with application to gene expression data classification. J Intell Fuzzy Syst. 2023;45(3):5021–44. doi:10.3233/JIFS-231569. [Google Scholar] [CrossRef]
109. He R, Lu J, Feng J, Lu Z, Shen K, Xu K. Advancing immunotherapy for melanoma: the critical role of single-cell analysis in identifying predictive biomarkers. Front Immunol. 2024;15(1):1–9. doi:10.3389/fimmu.2024.1435187. [Google Scholar] [PubMed] [CrossRef]
110. Aladdin AM, Rashid TA. A new lagrangian problem crossover—A systematic review and meta-analysis of crossover standards. Systems. 2023 Mar;11(3):144. doi:10.3390/systems11030144. [Google Scholar] [CrossRef]
111. Nouri-Moghaddam B. A novel bio-inspired hybrid multi-filter wrapper gene selection method with ensemble classifier for microarray data. Neural Comput Appl. 2023;35(16):11531–61. doi:10.1007/s00521-021-06459-9. [Google Scholar] [PubMed] [CrossRef]
112. Wang B, Sun Y, Xue B, Zhang M. A hybrid differential evolution approach to designing deep convolutional neural networks for image classification. Lect Notes Comput Sci. 2018;11320(1):237–50. doi:10.1007/978-3-030-03991-2_24. [Google Scholar] [CrossRef]
113. Ryerkerk M, Averill R, Deb K, Goodman E. A survey of evolutionary algorithms using metameric representations. Genet Program Evolvable Mach. 2019;20(4):441–78. doi:10.1007/s10710-019-09356-2. [Google Scholar] [CrossRef]
114. Wang B, Sun Y, Xue B, Zhang M. Evolving deep convolutional neural networks by variable-length particle swarm optimization for image classification. In: 2018 IEEE Congress on Evolutionary Computation (CEC); 2018; Rio de Janeiro, Brazil. p. 1–8. doi:10.1109/CEC.2018.8477735. [Google Scholar] [CrossRef]
115. Lee M. The geometry of feature space in deep learning models: a holistic perspective and comprehensive review. Mathematics. 2023 May;11(10):2375. doi:10.3390/math11102375. [Google Scholar] [CrossRef]
116. Celli F, Cumbo F, Weitschek E. Classification of large DNA methylation datasets for identifying cancer drivers. Big Data Res. 2018 Sep;13(3):21–8. doi:10.1016/j.bdr.2018.02.005. [Google Scholar] [CrossRef]
117. Tang H, Yu X, Liu R, Zeng T. Vec2image: an explainable artificial intelligence model for the feature representation and classification of high-dimensional biological data by vector-to-image conversion. Brief Bioinform. 2022 Mar;23(2):1–17. doi:10.1093/bib/bbab584. [Google Scholar] [PubMed] [CrossRef]
118. Matsuda M, Futamura Y, Ye X, Sakurai T. Distortion-free PCA on sample space for highly variable gene detection from single-cell RNA-seq data. Front Comput Sci. 2023;17(1):1–10. doi:10.1007/s11704-022-1172-z. [Google Scholar] [CrossRef]
119. Kumari R, Singh J, Gosain A. B-HPD: bagging-based hybrid approach for the early diagnosis of Parkinson’s disease. Intell Decis Technol. 2024;18(2):1385–401. doi:10.3233/idt-230331. [Google Scholar] [CrossRef]
120. Schwalbe G, Finzel B. A comprehensive taxonomy for explainable artificial intelligence: a systematic survey of surveys on methods and concepts. Data Min Knowl Discov. 2024;38(5):3043–101. doi:10.1007/s10618-022-00867-8. [Google Scholar] [CrossRef]
121. Mohammed B. A review on explainable artificial intelligence methods, applications, and challenges. Indones J Electr Eng Informatics. 2023;11(4):1007–24. doi:10.52549/ijeei.v11i4.5151. [Google Scholar] [CrossRef]
122. Brahim Belhaouari S. Bird’s Eye View feature selection for high-dimensional data. Sci Rep. 2023;13(1):1–21. doi:10.1038/s41598-023-39790-3. [Google Scholar] [PubMed] [CrossRef]
123. Pham TH, Raahemi B. Bio-inspired feature selection algorithms with their applications: a systematic literature review. IEEE Access. 2023;11:43733–58. doi:10.1109/access.2023.3272556. [Google Scholar] [CrossRef]
124. Haque MN, Sharmin S, Ali AA, Sajib AA, Shoyaib M. Use of relevancy and complementary information for discriminatory gene selection from high-dimensional gene expression data. PLoS One. 2021 Oct;16(10):e0230164. doi:10.1371/journal.pone.0230164. [Google Scholar] [PubMed] [CrossRef]
125. Das A, Neelima N, Deepa K, Ozer T. Gene selection based cancer classification with adaptive optimization using deep learning architecture. IEEE Access. 2024;12(5):62234–55. doi:10.1109/ACCESS.2024.3392633. [Google Scholar] [CrossRef]
126. Ahmed M, Sulaiman MH, Mohamad AJ. Improved barnacle mating optimizer-based least square support vector machine to predict COVID-19 confirmed cases with total vaccination. Cybern Inf Technol. 2023 Mar;23(1):125–40. doi:10.2478/cait-2023-0007. [Google Scholar] [CrossRef]
127. Janardhanaprabhu S, Malathi V. Brain tumor detection using depth-first search tree segmentation. J Med Syst. 2019 Aug;43(8):254–66. doi:10.1007/s10916-019-1366-6. [Google Scholar] [PubMed] [CrossRef]
128. Jeremiah I. Integration of specific local search methods in metaheuristic algorithms for optimizing the feature selection process: a survey authors. SLU J Sci Technol. 2022 Aug;4(1&2):34–48. doi:10.56471/slujst.v4i.267. [Google Scholar] [CrossRef]
129. Prat A, Guarneri V, As Pascual T, Bras O-Maristany F, Sanfeliu E, Par L. Development and validation of the new HER2DX assay for predicting pathological response and survival outcome in early-stage HER2-positive breast cancer. eBioMedicine. 2022;75(1):103801–15. doi:10.1016/j.ebiom.2021.103801. [Google Scholar] [PubMed] [CrossRef]
130. Xie W, Wang L, Yu K, Shi T, Li W. Improved multi-layer binary firefly algorithm for optimizing feature selection and classification of microarray data. Biomed Signal Process Control. 2023;79(1):104080–92. doi:10.1016/j.bspc.2022.104080. [Google Scholar] [CrossRef]
131. Mandal AK, Nadim MD, Saha H, Sultana T, Hossain MD, Huh EN. Feature subset selection for high-dimensional, low sampling size data classification using ensemble feature selection with a wrapper-based search. IEEE Access. 2024 Mar;12:62341–57. doi:10.1109/ACCESS.2024.3390684. [Google Scholar] [CrossRef]
132. Duo H, Li Y, Lan Y, Tao J, Yang Q, Xiao Y. Systematic evaluation with practical guidelines for single-cell and spatially resolved transcriptomics data simulation under multiple scenarios. Genome Biol. 2024 Dec;25(1):1–29. doi:10.1186/s13059-024-03290-y. [Google Scholar] [PubMed] [CrossRef]
133. Wang Y, Chen YG, Ahn KW, Lin CW. A realistic FastQ-based framework FastQ-Design for ScRNA-seq study design issues. Commun Biol. 2025 Dec;8(1):1–17. doi:10.1038/s42003-025-07938-8. [Google Scholar] [PubMed] [CrossRef]
134. Mauro SDi, Scamporrino A, Filippello A, Pino ADi, Scicali R. Clinical and molecular biomarkers for diagnosis and staging of NAFLD. Int J Mol Sci. 2021 Nov;22(21):11905. doi:10.3390/ijms222111905. [Google Scholar] [PubMed] [CrossRef]
135. Alsaeedi AH, Al-Mahmood HHR, Alnaseri ZF, Aziz MR, Al-Shammary D, Ibaida A. Fractal feature selection model for enhancing high-dimensional biological problems. BMC Bioinformatics. 2024 Dec;25(1):12–35. doi:10.1186/s12859-023-05619-z. [Google Scholar] [PubMed] [CrossRef]
136. Ng GYL, Tan SC, Ong CS. On the use of QDE-SVM for gene feature selection and cell type classification from scRNA-seq data. PLoS One. 2023 Oct;18:1–14. doi:10.1371/journal.pone.0292961. [Google Scholar] [PubMed] [CrossRef]
137. Lupat R, Perera R, Loi S, Li J. Moanna: multi-omics autoencoder-based neural network algorithm for predicting breast cancer subtypes. IEEE Access. 2023;11(1):10912–24. doi:10.1109/ACCESS.2023.3240515. [Google Scholar] [CrossRef]
138. Nematzadeh H, García-Nieto J, Aldana-Montes JF, Navas-Delgado I. Pattern recognition frequency-based feature selection with multi-objective discrete evolution strategy for high-dimensional medical datasets. Expert Syst Appl. 2024 Sep;249(4):123521–36. doi:10.1016/j.eswa.2024.123521. [Google Scholar] [CrossRef]
139. Sun L. Mixed measure-based feature selection using the Fisher score and neighborhood rough sets. Appl Intell. 2022;52(15):17264–88. doi:10.1007/s10489-021-03142-3. [Google Scholar] [CrossRef]
140. Mansur A, Vrionis A, Charles JP, Hancel K, Panagides JC. The role of artificial intelligence in the detection and implementation of biomarkers for hepatocellular carcinoma: outlook and opportunities. Cancers. 2023 Jun;15(11):2928. doi:10.3390/cancers15112928. [Google Scholar] [PubMed] [CrossRef]
141. Wang B, van der Kloet F, Kes MBMJ, Luirink J, Hamoen LW. Improving gene set enrichment analysis (GSEA) by using regulation directionality. Microbiol Spectr. 2024 Mar;12(3):1–12. doi:10.1128/spectrum.03456-23. [Google Scholar] [PubMed] [CrossRef]
142. Inoue J, Inazawa J. Cancer-associated miRNAs and their therapeutic potential. J Hum Genet. 2021;66(9):937–45. doi:10.1038/s10038-021-00938-6. [Google Scholar] [PubMed] [CrossRef]
143. Mohamed TIA, Ezugwu AE, Fonou-Dombeu JV, Mohammed M, Greeff J. A novel feature selection algorithm for identifying hub genes in lung cancer. Sci Rep. 2023 Dec;13(1):1–19. doi:10.1038/s41598-023-48953-1. [Google Scholar] [PubMed] [CrossRef]
144. Aljohani A. Optimizing patient stratification in healthcare: a comparative analysis of clustering algorithms for EHR Data. Int J Comput Intell Syst. 2024 Dec;17(1):1–23. doi:10.1007/s44196-024-00568-8. [Google Scholar] [CrossRef]
145. Morid MA, Sheng ORL, Dunbar J. Time series prediction using deep learning methods in healthcare. ACM Trans Manag Inf Syst. 2023 Jan;14(1):1–29. doi:10.1145/3531326. [Google Scholar] [CrossRef]
146. Daisy A, Porkodi R. Classification of human cancer diseases gene expression profiles using genetic algorithm by integrating protein protein interactions along with gene expression profiles. In: Proceedings of the 2018 International Conference on Current Trends towards Converging Technologies, ICCTCT 2018. Coimbatore, India: IEEE; 2018. p. 1–7. doi:10.1109/ICCTCT.2018.8550878. [Google Scholar] [CrossRef]
147. Ghafori S, Gharehchopogh FS. Advances in spotted hyena optimizer: a comprehensive survey. Arch Comput Methods Eng. 2022 May;29(3):1569–90. doi:10.1007/s11831-021-09624-4. [Google Scholar] [CrossRef]
148. Mostafa G, Mahmoud H, Abd El-Hafeez T, ElAraby ME. Feature reduction for hepatocellular carcinoma prediction using machine learning algorithms. J Big Data. 2024 Dec;11(88):1–27. doi:10.1186/s40537-024-00944-3. [Google Scholar] [CrossRef]
149. Kanya Kumari L, Naga Jagadesh B. An adaptive teaching learning based optimization technique for feature selection to classify mammogram medical images in breast cancer detection. Int J Syst Assur Eng Management. 2024;15(1):35–48. doi:10.1007/s13198-021-01598-7. [Google Scholar] [CrossRef]
150. Patel JJ, Hadia SK. Two-stage feature selection method created for 20 neurons artificial neural networks for automatic breast cancer detection. Trends Sci. 2023 Feb;20(2):1–28. doi:10.48048/tis.2023.4027. [Google Scholar] [CrossRef]
151. Zhang H. Large-scale clustering with structured optimal bipartite graph. IEEE Trans Pattern Anal Mach Intell. 2023;45(8):9950–63. doi:10.1109/tpami.2023.3277532. [Google Scholar] [PubMed] [CrossRef]
152. Al-Obeidat F, Rocha Á, Akram M, Razzaq S, Maqbool F. Maqbool, (CDRGI)—Cancer detection through relevant genes identification. Neural Comput Appl. 2022 Jun;34(11):8447–54. doi:10.1007/s00521-021-05739-8. [Google Scholar] [CrossRef]
153. Mohamed TIA, Ezugwu AE, Fonou-Dombeu JV, Ikotun AM, Mohammed M. A bio-inspired convolution neural network architecture for automatic breast cancer detection and classification using RNA-Seq gene expression data. Sci Rep. 2023 Dec;13(1):14644–63. doi:10.1038/s41598-023-41731-z. [Google Scholar] [PubMed] [CrossRef]
154. Minyilu Y, Abebe M, Meshesha M. Applying multimodal data fusion based on deep learning methods for the diagnoses of neglected tropical diseases: a systematic review. medRxiv. 2024 Jan;2(1):1–12. doi:10.1101/2024.01.07.24300957. [Google Scholar] [CrossRef]
155. Yassi M, Chatterjee A, Parry M. Application of deep learning in cancer epigenetics through DNA methylation analysis. Brief Bioinform. 2023 Nov;24(6):1–22. doi:10.1093/bib/bbad411. [Google Scholar] [PubMed] [CrossRef]
156. Hosseinalipour A, Gharehchopogh FS, Masdari M, Khademi A. Toward text psychology analysis using social spider optimization algorithm. Concurr Comput. 2021 Sep;33(17):1–23. doi:10.1002/cpe.6325. [Google Scholar] [CrossRef]
157. Bérchez-Moreno F, Durán-Rosal AM, Hervás Martínez C, Gutiérrez PA, Fernández JC. A memetic dynamic coral reef optimisation algorithm for simultaneous training, design, and optimisation of artificial neural networks. Sci Rep. 2024 Dec;14(1):1–22. doi:10.1038/s41598-024-57654-2. [Google Scholar] [PubMed] [CrossRef]
158. Chen Y, Deng X, Li Y, Han Y, Peng Y. Comprehensive molecular classification predicted microenvironment profiles and therapy response for HCC. Hepatology. 2024 Sep;80(3):536–51. doi:10.1097/HEP.0000000000000869. [Google Scholar] [PubMed] [CrossRef]
159. Armstrong G, Martino C, Rahman G, Gonzalez A, Vázquez-Baeza Y. Uniform manifold approximation and projection (UMAP) reveals composite patterns and resolves visualization artifacts in microbiome data. mSystems. 2021 Oct;6(5):1–6. doi:10.1128/msystems.00691-21. [Google Scholar] [PubMed] [CrossRef]
160. Driessen DAJJ, Zámecnik P, Dijkema T, Pegge SAH, van Engen-van Grunsven ACH, Takes RP, et al. High-accuracy nodal staging of head and neck cancer with USPIO-enhanced MRI: a new reading algorithm based on node-to-node matched histopathology. Invest Radiol. 2022 Dec;57(12):810–8. doi:10.1097/RLI.0000000000000902. [Google Scholar] [PubMed] [CrossRef]
161. Yang Y, Sun H, Zhang Y, Zhang T, Gong J, Wei Y. Dimensionality reduction by UMAP reinforces sample heterogeneity analysis in bulk transcriptomic data. Cell Rep. 2021 Jul;36(4):1–26. doi:10.1016/j.celrep.2021.109442. [Google Scholar] [PubMed] [CrossRef]
162. Venkatesan VK, Kuppusamy Murugesan KR, Chandrasekaran KA, Thyluru Ramakrishna M, Khan SB, Almusharraf A, et al. Cancer diagnosis through contour visualization of gene expression leveraging deep learning techniques. Diagnostics. 2023 Nov;13(22):3452. doi:10.3390/diagnostics13223452. [Google Scholar] [PubMed] [CrossRef]
163. Gao S, Wang P, Feng Y, Xie X, Duan M, Fan Y. RIFS2D: a two-dimensional version of a randomly restarted incremental feature selection algorithm with an application for detecting low-ranked biomarkers. Comput Biol Med. 2021;133:104405–15. doi:10.1016/j.compbiomed.2021.104405. [Google Scholar] [PubMed] [CrossRef]
164. Freitas D, Lopes LG, Morgado-Dias F. Particle Swarm Optimisation: a historical review up to the current developments. Entropy. 2020 Mar;22(3):362. doi:10.3390/E22030362. [Google Scholar] [PubMed] [CrossRef]
165. Makrodimitris S, Reinders MJT, van Ham RCHJ. Metric learning on expression data for gene function prediction. Bioinformatics. 2020 Feb;36(4):1182–90. doi:10.1093/bioinformatics/btz731. [Google Scholar] [PubMed] [CrossRef]
166. Xu X, Li J, Zhu Z, Zhao L, Wang H. A comprehensive review on synergy of multi-modal data and AI technologies in medical diagnosis. Bioengineering. 2024;11(3):219. doi:10.3390/bioengineering11030219. [Google Scholar] [PubMed] [CrossRef]
167. Aburass S, Dorgham O, Shaqsi JA. A hybrid machine learning model for classifying gene mutations in cancer using LSTM. Syst Soft Comput. 2024 Dec;6:200110–20. doi:10.1016/j.sasc.2024.200110. [Google Scholar] [CrossRef]
168. Horton R, Lucassen A. Ethical considerations in research with genomic data. New Bioethics. 2023;29(1):37–51. doi:10.1080/20502877.2022.2060590. [Google Scholar] [PubMed] [CrossRef]
169. Fernandes S, Abbas S, Saini JR, Andrew andrewj CJ. An anonymization-based privacy-preserving data collection protocol for digital health data. Front Public Health. 2023;10(1):1–13. doi:10.3389/fpubh.2023.1125011. [Google Scholar] [PubMed] [CrossRef]
170. Sardor Shukhratovich M. De-identification and anonymisation: legal and technical approaches article info Tsul legal report. Business Law Family Law Int Priv Law TSUL Legal Rep. 2024;5(1):2181–96. doi:10.51788/tsul.lr.5.1./TCYN1311. [Google Scholar] [CrossRef]
171. Tawfik SM, Elhosseiny AA, Galal AA, William MB, Qansuwa E, Elbaz RM, et al. Health inequity in genomic personalized medicine in underrepresented populations: a look at the current evidence. Funct Integr Genomics. 2023;23(1):54. doi:10.1007/s10142-023-00979-4. [Google Scholar] [PubMed] [CrossRef]
172. Aldrighetti CM, Niemierko A, Van Allen E, Willers H, Kamran SC. Racial and ethnic disparities among participants in precision oncology clinical studies. JAMA Netw Open. 2021 Nov;4(11):E2133205. doi:10.1001/jamanetworkopen.2021.33205. [Google Scholar] [PubMed] [CrossRef]
173. Roper K, Abdel-Rehim A, Hubbard S, Carpenter M, Rzhetsky A, Soldatova L. Testing the reproducibility and robustness of the cancer biology literature by robot. J R Soc Interface. 2022;19(189):1–13. doi:10.1098/rsif.2021.0821. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2025 The Author(s). Published by Tech Science Press.
Copyright © 2025 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools