
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE

VEGFC as a prognostic cytokine biomarker linking lymph node metastasis to immune suppression in breast cancer
1 Department of Nursing, Jenteh Junior College of Medicine, Nursing and Management, Miaoli, Taiwan
2 Research Assistant Center, Show Chwan Memorial Hospital, Changhua, Taiwan
3 Department of Surgery, Show Chwan Memorial Hospital, Changhua, Taiwan
4 Department of Post-Baccalaureate Medicine, College of Medicine, National Chung Hsing University, Taichung, Taiwan
* Corresponding Authors: Yu-Chieh Tsai. Email: ; Hung-Yu Lin. Email:
European Cytokine Network 2026, 37(2), 121-135. https://doi.org/10.32604/ecn.2026.079012
Received 12 January 2026; Accepted 11 April 2026; Issue published 30 June 2026
 View Full Text
View Full Text Download PDF
Download PDFAbstract
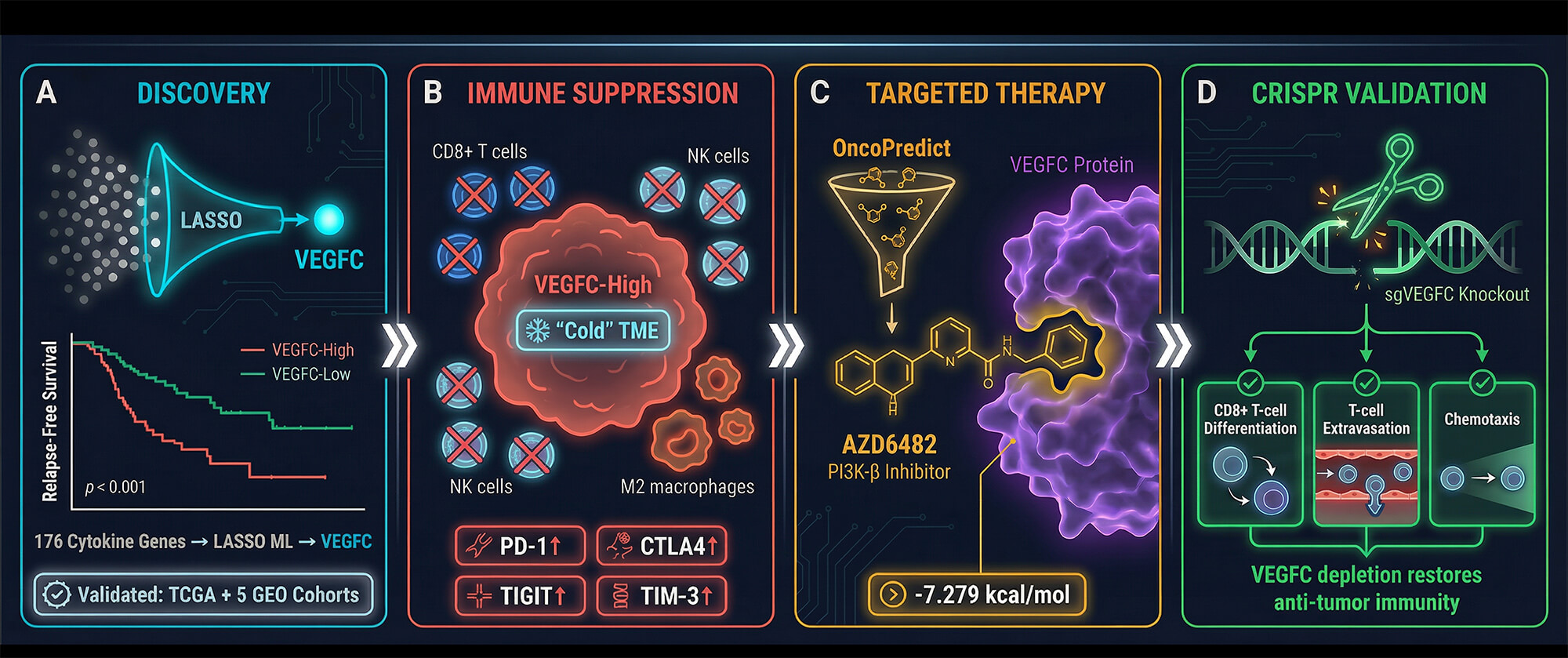
Backgrounds: Lymph node metastasis is a critical determinant of breast cancer prognosis, yet the specific microenvironmental cytokines driving this process remain elusive. This study aims to identify key prognostic cytokines linking nodal metastasis to tumor microenvironment (TME) remodeling and to evaluate their clinical utility. Methods: A predefined panel of 176 microenvironmental genes was evaluated using differential expression analysis and the Least Absolute Shrinkage and Selection Operator (LASSO) algorithm on the TCGA-BRCA cohort to identify optimal predictors of nodal metastasis. Prognostic value was assessed via Kaplan-Meier, subgroup, and multivariate Cox regression analyses, and validated across five independent GEO datasets. Immune infiltration and therapeutic potential were evaluated using Gene Set Enrichment Analysis (GSEA), CIBERSORT deconvolution, pharmacogenomic screening, and molecular docking simulations. Results: Vascular Endothelial Growth Factor C (VEGFC) emerged as the top biomarker significantly upregulated in node-positive tumors. Multivariate analysis confirmed that VEGFC acts as a robust, independent predictor of poor relapse-free survival (RFS). Notably, subgroup analyses revealed this prognostic penalty is exceptionally pronounced in hormone receptor-positive Luminal A and B subtypes. Mechanistically, elevated VEGFC is strongly associated with an immunosuppressive “cold” TME, characterized by reduced cytotoxic CD8+ T-cell infiltration and enriched M2-macrophage polarization across multiple algorithms. Furthermore, pharmacogenomic analyses and docking simulations indicated potential sensitivity to PI3K/AKT inhibitors in VEGFC-high tumors. Conclusions: VEGFC is a robust, independent precision biomarker specifically indicating poor prognosis in Luminal breast cancers. It is significantly associated with an immune-excluded TME, suggesting that targeting the VEGFC axis may offer a novel strategy to reverse immune evasion.Graphic Abstract
Keywords
Supplementary Material
Supplementary Material FileBreast cancer remains the most commonly diagnosed malignancy among women worldwide, representing a critical global health challenge with substantial morbidity and mortality. In 2022, an estimated 2.3 million new cases and 670,000 deaths were reported globally, establishing breast cancer as the leading cause of cancer-related mortality in women [1–4].
Current clinical management of breast cancer relies heavily on established biomarkers, including estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2), which guide treatment decisions and predict therapeutic responses [5–7]. While the molecular classification of breast cancer into luminal A, luminal B, HER2-enriched, and basal-like subtypes has refined prognostication, the accurate prediction of metastatic progression remains a significant clinical challenge across all subtypes. Standard biomarkers primarily reflect cellular growth signaling and hormonal sensitivity rather than the intrinsic capability for dissemination. Consequently, identifying novel molecular drivers that specifically govern lymph node metastasis is essential to improve risk stratification and develop targeted interventions for patients with aggressive disease phenotypes [8,9].
Lymph node metastasis represents one of the most powerful prognostic factors in breast cancer, with the number and extent of nodal involvement strongly correlating with disease-free survival and overall survival [10]. The mechanisms underlying lymphatic dissemination are complex and incompletely understood, involving intricate interactions between tumor cells and the surrounding tumor microenvironment (TME) [11–13]. Understanding the molecular drivers of lymph node metastasis is essential for identifying patients at the highest risk of disease progression and for developing targeted interventions to interrupt metastatic cascades.
Within the tumor microenvironment (TME), cytokines serve as critical orchestrators of cellular communication, regulating immune responses, angiogenesis, and tumor progression [14–17]. These soluble mediators can exert both pro- and anti-tumorigenic effects depending on the cellular context and the broader cytokine milieu. Pro-inflammatory cytokines, such as interleukin-6 (IL-6), tumor necrosis factor-alpha (TNF-α), and IL-1β, promote tumor cell proliferation, invasion, and metastasis through the activation of signaling pathways, including Nuclear Factor kappa-light-chain-enhancer of activated B cells (NF-κB) and Signal Transducer and Activator of Transcription 3 (STAT3) [18–20]. Conversely, immunosuppressive cytokines, such as IL-10 and transforming growth factor-beta (TGF-β), facilitate immune evasion by recruiting regulatory T cells and myeloid-derived suppressor cells, thereby dampening anti-tumor immunity [21–25]. Furthermore, chemokines (including members of the CC chemokine ligand (CCL) and CXC chemokine ligand (CXCL) families) regulate the trafficking of immune cells into the TME and can either promote or suppress tumor immunity depending on the recruited cell populations [26–29]. Among the diverse array of cytokines implicated in cancer progression, vascular endothelial growth factor C (VEGFC) has emerged as a key regulator of lymphangiogenesis and lymphatic metastasis [30–33]. VEGFC promotes the formation of lymphatic vessels within and around tumors, providing conduits for cancer cell dissemination to regional lymph nodes. Beyond its canonical role in lymphangiogenesis, emerging evidence suggests that VEGFC may exert immunomodulatory functions within the TME, potentially influencing the recruitment and activity of immune effector cells [30,34,35]. However, the precise mechanisms by which VEGFC shapes the immune landscape in breast cancer remain incompletely characterized, warranting further investigation.
The advent of artificial intelligence (AI) and machine learning (ML) algorithms has revolutionized biomedical research, enabling the extraction of meaningful patterns from high-dimensional datasets and facilitating the discovery of novel biomarkers with improved diagnostic and prognostic accuracy [36–39]. In oncology, AI-powered approaches have demonstrated remarkable success in multiple domains, including early cancer detection from medical imaging, the prediction of treatment responses, the identification of molecular subtypes, and personalized therapy selection [40–43]. Machine learning techniques, particularly supervised learning algorithms such as support vector machines, random forests, and penalized regression methods, have proven highly effective in feature selection and biomarker discovery from transcriptomic, proteomic, and multi-omics datasets [44–46]. Among these methods, the Least Absolute Shrinkage and Selection Operator (LASSO) has gained widespread adoption for high-dimensional data analysis due to its ability to simultaneously perform variable selection and regularization, thereby identifying the most informative features while preventing overfitting [47–49]. LASSO regression employs L1 penalization to shrink regression coefficients, effectively setting less important features to zero and retaining only those with the strongest predictive power [50]. This approach has been successfully applied to identify prognostic gene signatures [48,49,51], predict treatment outcomes, and stratify patient risk in multiple cancer types, including breast, lung, colorectal, and hepatocellular carcinomas [52–54]. The integration of LASSO with other machine learning classifiers has further enhanced the accuracy and robustness of biomarker identification, as demonstrated in recent studies of cancer recurrence prediction and immune profiling [55–57]. Despite these advances, the application of machine learning to systematically screen comprehensive cytokine expression profiles for prognostic biomarkers associated with lymph node metastasis in breast cancer remains underexplored.
In this study, we employed a machine learning-driven approach to address this critical knowledge gap. Utilizing LASSO penalized regression on comprehensive transcriptomic data from The Cancer Genome Atlas Breast Invasive Carcinoma (TCGA-BRCA) cohort, we systematically screened a predefined panel of cytokine-related genes to identify robust molecular predictors of lymph node metastasis. Our objectives were: (1) to identify key cytokine biomarkers associated with nodal involvement using the LASSO algorithms; (2) to validate the prognostic significance of the identified biomarkers across multiple independent patient cohorts; (3) to characterize the immunological landscape and the mechanistic role of the lead biomarker in shaping the tumor immune microenvironment; and (4) to identify potential therapeutic agents targeting the discovered biomarker pathway using pharmacogenomic profiling and molecular docking approaches. Through this integrative computational biology framework combining machine learning feature selection, multi-cohort validation, transcriptomic profiling, immune landscape analysis, and drug sensitivity prediction, we aimed to establish a novel cytokine-based biomarker for lymph node metastasis in breast cancer and to elucidate its functional role in modulating anti-tumor immunity, ultimately informing precision medicine strategies for high-risk patients.
2.1 Data Acquisition and Processing
RNA-sequencing (RNA-seq) expression data and corresponding clinical information for the Breast Invasive Carcinoma (TCGA-BRCA) cohort were obtained from The Cancer Genome Atlas (TCGA) database. The dataset was filtered to include patients with complete clinical data regarding nodal status, age, and survival outcomes. For external validation of survival analyses, five independent breast cancer microarray datasets (GSE45255, GSE17705, GSE25065, GSE25055, and GSE20711) were retrieved from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/). The detailed patient selection process and the specific inclusion/exclusion criteria for the GEO datasets are illustrated in a CONSORT-style flow diagram (Supplementary Fig. S1).
2.2 Machine Learning-Driven Feature Selection
To identify robust prognostic cytokines associated with lymph node metastasis, we employed the Least Absolute Shrinkage and Selection Operator (LASSO) machine learning algorithm using the glmnet package (version 4.1-8) in R (version 4.5.3). The initial input for the LASSO model was a predefined, comprehensive panel of 176 genes encoding major cytokines, chemokines, and critical growth factors (including the IL, IFN, TNF, CCL, CXCL, VEGF, FGF, and TGF families). This gene list was systematically curated based on the internationally recognized KEGG ‘Cytokine-cytokine receptor interaction’ pathway (hsa04060) and the ImmPort immunology database, aiming to capture the core soluble mediators that orchestrate the tumor microenvironment. To maintain an unbiased feature selection process, we did not perform pre-filtering based on differential expression; rather, the entire panel of 176 features was subjected to LASSO regularization. LASSO was specifically selected over other machine learning classifiers (such as Random Forests or Support Vector Machines) due to its L1 regularization property. This L1 penalty effectively shrinks the regression coefficients of less informative or highly correlated features to exactly zero, yielding a highly sparse and interpretable model that is uniquely suited for clinical biomarker screening. To strictly prevent overfitting and ensure model robustness, the optimal penalty parameter (λ) was determined via rigorous 10-fold cross-validation, minimizing the binomial deviance. A binomial logistic regression model was constructed to differentiate between node-negative (N0) and node-positive (non-N0) statuses. The optimal λ was determined via 10-fold cross-validation to minimize binomial deviance. Feature selection results and coefficient profiles were visualized using the ggplot2 package (version 4.0.2).
2.3 Clinical Association, Diagnostic and Survival Analysis
The diagnostic performance of VEGFC in distinguishing tumor tissue from normal breast tissue was evaluated using Receiver Operating Characteristic (ROC) curve analysis, with the Area Under the Curve (AUC) calculated to quantify accuracy. Associations between VEGFC expression and clinical characteristics, including nodal stage (N0 vs. N1/N2/N3) and age stratification (<40 vs. >60 years), were analyzed. These analyses and visualizations were performed using the TCGAplot R package. Relapse-free survival (RFS) analysis was performed to evaluate the prognostic value of VEGFC. Patients were stratified into high- and low-expression groups based on optimal cut-offs. Subgroup survival analyses were additionally conducted to evaluate the prognostic impact across distinct molecular subtypes (e.g., Luminal A, Luminal B, HER2-enriched, and Basal-like). Kaplan-Meier survival curves were generated, and statistical significance was assessed using the log-rank test. To ascertain the independent prognostic value of VEGFC, multivariate Cox proportional hazards regression analysis was performed, adjusting for relevant clinicopathological factors. All survival analyses were conducted using the survival and survminer packages (version 0.5.2) in R.
2.4 Immune Microenvironment and Pan-Cancer Analysis
To characterize the immunological landscape, Gene Set Enrichment Analysis (GSEA) was utilized to identify immune pathways downregulated in VEGFC-high tumors. To validate these pathway-level findings at the cellular level, comprehensive immune deconvolution analysis was performed using the TIMER3.0 platform. The continuous Spearman correlation between VEGFC expression and the absolute abundance of specific immune cell infiltrates (including CD8+ T cells and M2 macrophages) was quantified across multiple algorithms, specifically incorporating CIBERSORT and CIBERSORT-ABS. For pan-cancer analysis, systematic correlations between VEGFC expression and key immune signatures—including immune checkpoints (e.g., PDCD1, CTLA4), chemokine receptors, and chemokines—were evaluated across diverse tumor types in the TCGA database (https://portal.gdc.cancer.gov/). Heatmaps illustrating these correlations were generated using the TCGAplot R package (v8.0.0), identifying distinct immune modulation patterns in breast cancer compared to other malignancies.
Targeted therapeutic candidates were identified using the OncoPredict R package (v1.2), which predicts in vivo drug sensitivity (IC50) based on gene expression profiles. We performed a correlation analysis between predicted IC50 values and VEGFC expression across the TCGA-BRCA cohort and independent validation datasets (GSE7390, GSE21653, and GSE88770). A negative correlation indicated that higher VEGFC expression was associated with lower IC50 values, signifying increased sensitivity to the candidate drug.
2.6 Molecular Docking Simulation
To explore the structural interaction between the identified therapeutic agent (AZD6482) and the VEGFC protein, molecular docking simulations were performed. The 3D structure of VEGFC was prepared, and PyRx was utilized for virtual screening to score and rank binding affinities across potential binding pockets. AutoDock Vina was employed to calculate binding energies and simulate the docking pose. The resulting protein-ligand complex and binding pocket topology were visualized in 3D using PyMOL. Detailed 2D interaction maps, including hydrogen bonds and hydrophobic contacts, were generated using LigPlot+.
2.7 Functional Validation via CRISPR-Cas9 Screening
The biological function of VEGFC was validated using the Q-omics AI platform (https://qomics.ai/), which integrates data from CRISPR-Cas9 knockout screens. We analyzed the functional impact of high-efficacy sgVEGFC treatment on cellular pathways. Gene Ontology (GO) enrichment analysis was performed to identify biological processes, specifically those related to immune cell recruitment and function (e.g., T-cell differentiation and extravasation), that were significantly suppressed following VEGFC depletion.
All statistical comparisons were performed using R software (version 4.5.3). Differences between groups were assessed using the Wilcoxon rank-sum test or Student’s t-test, as appropriate. Correlations were evaluated using Pearson or Spearman correlation coefficients. A p-value of <0.05 was considered statistically significant.
3.1 Machine Learning-Driven Feature Selection Identifies VEGFC as a Robust Prognostic Predictor in Breast Cancer
To establish a biological foundation for our biomarker discovery pipeline, we first conducted a differential expression analysis of the comprehensive 176-gene cytokine panel between node-negative (N0) and node-positive (non-N0) primary breast cancer tissues. As illustrated in the volcano plot (Supplementary Fig. S2), multiple immune mediators exhibited significant transcriptional alterations during nodal metastasis, with VEGFC demonstrating prominent upregulation in the non-N0 cohort. Building upon this biological context, we next sought to pinpoint the most robust molecular determinants with the strongest predictive capability. We applied the Least Absolute Shrinkage and Selection Operator (LASSO) machine learning algorithm to the TCGA-BRCA RNA-seq dataset. This approach allowed us to screen high-dimensional genomic data to isolate variables with the strongest predictive capability for nodal status (N0 vs. non-N0). The optimal penalty parameter (λ) was determined via 10-fold cross-validation, minimizing binomial deviance (figure 1A). Based on the resultant coefficient profiles (figure 1B), we identified a signature of candidate genes, among which VEGFC emerged as the most significant feature associated with nodal involvement (figure 1C).

Figure 1: Identification of VEGFC as a prognostic biomarker in breast cancer using machine learning and multi-cohort validation. (A–C) Feature selection for lymph node metastasis prediction using the Least Absolute Shrinkage and Selection Operator (LASSO) machine learning algorithm. (A) Cross-validation plot showing binomial deviance vs. log(λ) to identify the optimal parameter. (B) LASSO coefficient profiles of candidate genes. (C) Distribution of coefficient values identifying VEGFC as the most significant predictor for distinguishing nodal status (N0 vs. non-N0). (D) Receiver operating characteristic (ROC) curve analysis demonstrating the diagnostic accuracy of VEGFC in differentiating breast tumor tissue from normal tissue (AUC = 0.773). (E) Correlation between VEGFC expression and nodal stage; expression is significantly elevated in advanced nodal stages (N2 and N3) compared to N0. (F) Association between VEGFC levels and patient age, showing significantly higher expression in younger patients (<40 years) compared to the older group (>60 years). (G–L) Kaplan-Meier curves comparing relapse-free survival (RFS) between VEGFC-high and VEGFC-low groups in the TCGA-BRCA cohort (G) and five independent validation datasets: GSE45255 (H), GSE17705 (I), GSE25065 (J), GSE25055 (K), and GSE20711 (L). VEGFC: Vascular Endothelial Growth Factor C; TCGA-BRCA: The Cancer Genome Atlas—Breast Invasive Carcinoma; ns: not significant; AUC: Area Under the Curve; *p < 0.05, **p < 0.01.
We subsequently evaluated the clinical relevance and diagnostic potential of VEGFC in the context of disease progression by assessing its discriminative capacity for nodal status. Receiver operating characteristic (ROC) curve analysis for distinguishing node-negative (N0) from node-positive (N+) cases yielded an area under the curve (AUC) of 0.623 (95% CI: 0.590–0.657) (figure 1D). Furthermore, VEGFC expression demonstrated good accuracy in distinguishing breast tumor tissue from adjacent normal tissue (AUC = 0.773; Supplementary Fig. S3). While a single cytokine transcript exhibits modest diagnostic discriminative ability for a highly complex, multifactorial process like lymph node metastasis, consistent with our LASSO prediction, VEGFC expression was significantly upregulated in patients with advanced nodal disease (N2 and N3) compared to node-negative (N0) patients (figure 1E). Furthermore, analysis of clinical demographics revealed an inverse correlation with age; younger patients (<40 years) exhibited significantly higher VEGFC levels compared to the older cohort (>60 years) (figure 1F), suggesting a potential role for this cytokine in early-onset aggressive disease.
To confirm the prognostic value of VEGFC across diverse patient populations, we performed a comprehensive survival analysis using the TCGA-BRCA cohort and five independent validation datasets (GSE45255, GSE17705, GSE25065, GSE25055, and GSE20711). Kaplan-Meier survival curves consistently demonstrated that patients with high VEGFC expression experienced significantly shorter RFS compared to those with low expression (figure 1G–L). To rigorously validate the independent prognostic value of VEGFC and address potential confounding clinical factors, we subsequently performed a multivariate Cox proportional hazards regression analysis. The model was adjusted for critical clinicopathological covariates, including patient age, tumor stage, estrogen receptor (ER) status, and HER2 status. Consistent with our univariate findings, the multivariate analysis confirmed that VEGFC expression serves as a robust, independent predictor of RFS. Specifically, patients with low VEGFC expression exhibited a significantly reduced risk of relapse compared to the high-expression reference cohort (Hazard Ratio [HR] = 0.67, 95% Confidence Interval [CI]: 0.46–0.98, p = 0.039; Supplementary Fig. S4). These findings substantiate the clinical relevance of VEGFC, demonstrating that its prognostic utility remains highly significant even after stratifying for traditional biomarkers and disease stage. Given the well-established biological heterogeneity of breast cancer, we performed subgroup Kaplan-Meier analyses to investigate whether the prognostic significance of VEGFC varies across distinct molecular subtypes (Supplementary Fig. S5). Strikingly, the prognostic penalty of VEGFC was highly pronounced in hormone receptor-positive tumors. Elevated VEGFC expression was a highly significant predictor of poor RFS in both the Luminal A (p = 0.0077) and Luminal B (p = 0.00062) sub-cohorts, which collectively represent the majority of breast cancer cases. In contrast, the prognostic divergence did not reach statistical significance in the HER2-enriched (p = 0.24) and Triple-Negative Breast Cancer (TNBC) (p = 0.86) subgroups. This suggests that while robust epithelial-derived oncogenic drivers may dominate survival outcomes in highly aggressive intrinsic subtypes, VEGFC serves as a remarkably potent and precise microenvironmental prognostic biomarker specifically for Luminal breast cancer.
3.2 Elevated VEGFC Expression Potentially Contributes to an Immunosuppressive Tumor Microenvironment by Dampening Innate and Adaptive Immune Responses
To elucidate the biological mechanisms underlying the poor prognosis associated with high VEGFC levels, we performed a comprehensive transcriptomic analysis using the TCGA-BRCA cohort. Following patient stratification into high- and low-expression groups (figure 2A, B), we utilized Gene Set Enrichment Analysis (GSEA) to characterize the immunological landscape.

Figure 2: Elevated VEGFC expression is associated with distinct immune suppression signatures in the breast cancer tumor microenvironment. (A) Schematic workflow of the computational analysis pipeline. RNA-seq data from the TCGA-BRCA cohort were utilized to profile VEGFC expression, stratify patients into high and low expression groups based on a median cutoff, and perform Gene Set Enrichment Analysis (GSEA) to evaluate immunogenic pathway activity. (B) Patient stratification and differential gene expression. The top panel displays the ranked distribution of VEGFC expression across the cohort, with the dashed line marking the median separation between low (blue) and high (red) groups. The bottom heatmap visualizes the expression profiles of the top differentially expressed genes associated with VEGFC status. (C) Summary bubble plot of GSEA results identifying significantly downregulated immune pathways in the VEGFC-high group. The y-axis lists specific gene sets related to immune function (including NK cell, T cell, Dendritic cell (DC), and B cell responses), while the x-axis represents the negative enrichment score, indicating suppression. Bubble size corresponds to the number of genes in the set, and color intensity represents the False Discovery Rate (FDR) q-value. (D) Immune deconvolution analysis via the TIMER3.0 platform, incorporating the CIBERSORT algorithm. The bar chart illustrates the Spearman correlation (Rho) between VEGFC expression and the infiltration levels of various immune cell populations in breast cancer. VEGFC: Vascular Endothelial Growth Factor C; TCGA-BRCA: The Cancer Genome Atlas Breast Invasive Carcinoma; NK: Natural Killer; CD8: Cluster of Differentiation 8.
Our analysis revealed a striking negative correlation between VEGFC expression and key immune effector pathways. Specifically, patients with high VEGFC exhibited significant downregulation of gene signatures governing both innate and adaptive immunity (figure 2C). Notably, pathways associated with Natural Killer (NK) cell function (e.g., “NK Cell Influenza Response”) and T-cell mediated cytotoxicity (e.g., “CD8 T Cell Response”, “PBMC Zostavax T Cell Response”) were markedly suppressed in the high-expression group. Furthermore, we observed a concomitant reduction in signatures related to Dendritic Cell (DC) maturation and B-cell activity. To explicitly validate these GSEA pathway-level findings at the cellular level, we performed comprehensive immune deconvolution analysis on the TCGA-BRCA cohort using the TIMER3.0 platform. This allowed us to quantify the correlation between VEGFC expression and the absolute abundance of specific immune cell infiltrates using multiple algorithms, including CIBERSORT. As depicted in figure 2D, the deconvolution analysis provided striking cellular validation of our transcriptomic models. VEGFC expression demonstrated a significant negative correlation with the infiltration of cytotoxic CD8+ T cells, NK cells and M1-polarized macrophages. Conversely, VEGFC levels were strongly positively correlated with the recruitment of immunosuppressive myeloid populations, particularly M2-polarized macrophages. These cellular-level quantifications solidly substantiate our GSEA claims, confirming that VEGFC potentially contributes to an immunosuppressive ‘cold’ TME not merely by downregulating immune pathways, but by physically altering the immune cell composition toward an exhausted and excluded phenotype. These findings indicate that VEGFC overexpression does not merely correlate with tumor aggressiveness but actively contributes to shaping an immunosuppressive (“cold”) tumor microenvironment, potentially by impairing the recruitment and activation of cytotoxic lymphocytes and antigen-presenting cells.
3.3 Pan-Cancer Analysis Reveals VEGFC as a Broad Modulator of the Tumor Immune Microenvironment, Distinctly Promoting Immune Exhaustion in Breast Cancer
To determine whether the immunosuppressive role of VEGFC is unique to breast cancer or a pan-cancer phenomenon, we performed a systematic correlation analysis across diverse cancer types in the TCGA database. This analysis evaluated the relationship between VEGFC expression and key immunological signatures, including immune checkpoints, chemokine receptors, and chemokines.
In the analysis of immune checkpoints (figure 3A), we observed a striking heterogeneity across tumor types. While some cancers, such as Thymoma (THYM) and Testicular Germ Cell Tumors (TGCT), exhibited significant negative correlations (blue) between VEGFC and checkpoint markers, breast cancer (BRCA) displayed a consistent and significant positive correlation (red). Specifically, in BRCA, elevated VEGFC levels were strongly associated with the upregulation of critical exhaustion markers, including Programmed Cell Death 1 (PDCD1/PD-1), Cytotoxic T-Lymphocyte Associated Protein 4 (CTLA4), CD274 (Programmed Cell Death 1 Ligand 1; PD-L1), T-cell Immunoreceptor with Ig and ITIM domains (TIGIT), and Hepatitis A Virus Cellular Receptor 2 (HAVCR2/TIM-3). This positive correlation suggests that while immune cells may be present in the VEGFC-high tumor microenvironment, they are likely in a dysfunctional or exhausted state, aligning with the poor prognosis observed in figure 1.

Figure 3: Pan-cancer landscape of VEGFC-associated immunological signatures. Systematic correlation analysis evaluating the relationship between VEGFC expression and immune-related genes across pan-cancer types in the TCGA database. (A) Heatmap displaying the correlation between VEGFC expression and key immune checkpoint genes (including PDCD1, CTLA4, LAG3, and TIGIT). (B) Correlation profile of VEGFC with chemokine receptors (e.g., CCR and CXCR family members). (C) Association analysis between VEGFC levels and chemokines (e.g., CCL and CXCL family members). The color gradient indicates the strength of the correlation (Red: positive correlation; Blue: negative correlation), and asterisks denote statistical significance (*p < 0.05; **p < 0.01). VEGFC: Vascular Endothelial Growth Factor C; PDCD1: Programmed Cell Death Protein 1; CTLA4: Cytotoxic T-Lymphocyte–Associated Protein 4; LAG3: Lymphocyte-Activation Gene 3; TIGIT: T-cell Immunoreceptor with Ig and ITIM Domains; CCR: C-C Motif Chemokine Receptor; CXCR: C-X-C Motif Chemokine Receptor.
Furthermore, the analysis of chemokine receptors (figure 3B) and chemokines (figure 3C) revealed that VEGFC expression in BRCA is significantly linked to a specific inflammatory milieu. Unlike diverse patterns seen in other malignancies like Kidney Chromophobe (KICH) or Brain Lower Grade Glioma (LGG), BRCA showed significant positive correlations with myeloid- and regulatory T cell-associated chemoattractants, such as C-C Motif Chemokine Ligand 2 (CCL2), C-C Motif Chemokine Receptor 2 (CCR2), C-C Motif Chemokine Receptor 5 (CCR5), and C-X-C Motif Chemokine Ligand 12 (CXCL12). The co-expression of these recruitment signals alongside exhaustion markers supports a model where VEGFC drives the infiltration of immunosuppressive populations (e.g., Myeloid-Derived Suppressor Cells [MDSCs] or Tregs) rather than cytotoxic effectors, thereby reinforcing the “cold” immune phenotype identified in our GSEA analysis.
3.4 Pharmacogenomic Profiling Identifies AZD6482 as a Potent Therapeutic Agent Targeting VEGFC-Overexpressing Tumors
Given the prognostic significance of VEGFC in breast cancer, we sought to identify targeted therapies that could selectively inhibit tumors with high VEGFC expression. We utilized the OncoPredict algorithm to screen for drug sensitivity across the TCGA-BRCA cohort. This analysis revealed that the PI3K-β inhibitor AZD6482 exhibited a significant negative correlation, across multiple datasets, between its predicted IC50 values and VEGFC expression (figure 4A). Specifically, tumors with elevated VEGFC levels showed lower IC50 values, indicating heightened sensitivity to AZD6482. This therapeutic vulnerability was robustly validated across three independent breast cancer datasets (GSE7390, GSE21653, and GSE88770), where the inverse relationship between VEGFC abundance and drug resistance was consistently observed (figure 4B–E).

Figure 4: Identification of AZD6482 as a targeted therapeutic and functional validation of VEGFC via CRISPR-Cas9 screening. (A) Pharmacogenomic profiling using the OncoPredict algorithm to identify drug candidates. The heatmap illustrates the correlation between drug sensitivity (predicted IC50) and VEGFC expression across multiple breast cancer datasets. Blue tiles represent negative correlations (indicating that higher VEGFC expression predicts higher drug sensitivity). The PI3K-β inhibitor AZD6482 is highlighted as a top candidate. (B–E) Correlation analysis between VEGFC expression and AZD6482 IC50 values. A significant negative correlation confirms that VEGFC-high tumors are more sensitive to AZD6482 in the TCGA-BRCA cohort (B) and three independent validation datasets: GSE7390 (C), GSE21653 (D), and GSE88770 (E). (F) Molecular docking simulation of AZD6482 binding to the VEGFC protein. The upper panel visualizes five potential binding pockets on the tertiary structure of VEGFC. The lower panel displays the surface topology and calculated binding energies (kcal/mol) for each pocket, identifying Pocket 1 as the optimal binding site with the lowest energy (−7.279 kcal/mol). (G) Two-dimensional interaction LigPlot+ map of the AZD6482-VEGFC complex within Pocket 1. Key molecular interactions include hydrophobic contacts with residues such as Phe109 and Tyr77, and a critical hydrogen bond (3.05 Å) formed with Ile279. (H) Functional annotation analysis using the Q-omics CRISPR-Cas9 screening platform. The scatter plot identifies 56 Gene Ontology (GO) biological functions (blue dots) that are significantly suppressed following high-efficacy sgVEGFC knockout. (I–K) Box plots validating the functional impact of VEGFC depletion. High-efficacy sgVEGFC treatment resulted in the significant downregulation of (I) CD8+ T-cell differentiation (p = 0.028), (J) T-cell extravasation (p = 0.012), and (K) Regulation of chemotaxis (p = 0.015), further confirming the role of VEGFC in modulating immune cell behavior. VEGFC: Vascular Endothelial Growth Factor C; PI3K-β: Phosphoinositide 3-Kinase Beta; TCGA-BRCA: The Cancer Genome Atlas Breast Invasive Carcinoma; CD8: Cluster of Differentiation 8.
To characterize the structural basis of this interaction, we performed molecular docking simulations between AZD6482 and the VEGFC protein. Among five potential binding sites, Pocket 1 was identified as the optimal binding domain, exhibiting the lowest binding energy (−7.279 kcal/mol) (figure 4F). Analysis of the protein-ligand complex revealed a stable interaction profile, stabilized by hydrophobic contacts with residues Phe109 and Tyr77, and a critical hydrogen bond (3.05 Å) formed with Ile279 (figure 4G). These structural data support the potential of AZD6482 to directly interact with and inhibit VEGFC signaling.
Finally, to elucidate the functional impact of VEGFC modulation, we leveraged the Q-omics AI platform to analyze data from CRISPR-Cas9 knockout screens. We examined the biological consequences of high-efficacy sgVEGFC treatment and found that depletion of VEGFC led to the significant downregulation of 56 specific biological functions out of 7063 screened Gene Ontology (GO) terms (figure 4H). Notably, the suppression of VEGFC resulted in significant reductions in pathways critical for immune cell recruitment and function, including CD8+ T-cell differentiation (p = 0.028), T-cell extravasation (p = 0.012), and the regulation of chemotaxis (p = 0.015) (figure 4I–K). These findings underscore the multifaceted role of VEGFC in modulating the tumor immune microenvironment and suggest that its therapeutic targeting could disrupt these essential oncogenic and immunomodulatory axes.
Cytokines function as critical messengers within the tumor microenvironment (TME), orchestrating complex interactions between neoplastic cells and the immune system that can either restrain or fuel malignancy. In this study, we performed a systematic, machine learning-driven screen of a comprehensive cytokine panel—encompassing interleukins, interferons, tumor necrosis factors, chemokines, and growth factors—to identify key determinants of metastatic progression in breast cancer. From this extensive candidate list, the Least Absolute Shrinkage and Selection Operator (LASSO) algorithm identified VEGFC as the most significant prognostic cytokine associated with lymph node metastasis. Our multi-cohort validation consistently demonstrated that elevated VEGFC expression is not only predictive of advanced nodal stage (N2/N3) and early-onset disease (age <40) but also serves as a robust biomarker for poor relapse-free survival. Beyond its canonical role in lymphangiogenesis, our transcriptomic and pan-cancer analyses revealed that VEGFC actively shapes an immunosuppressive “cold” TME by downregulating critical cytotoxic effectors, including CD8+ T cells and NK cells, while correlating with immune exhaustion markers (figure 5). Furthermore, we validated the therapeutic actionability of this axis, identifying the PI3K-β inhibitor AZD6482 as a potent targeted agent and confirming via CRISPR screening that VEGFC depletion restores essential immune recruitment pathways such as T-cell extravasation. Collectively, these findings position VEGFC as a master cytokine regulator of immune exclusion and a high-priority therapeutic target in aggressive breast carcinoma. Figure 5 summarizes the integrative framework of our study, illustrating how machine learning–based feature selection identified VEGFC as a prognostic cytokine, validated across multiple cohorts, and mechanistically linked to immune suppression and therapeutic vulnerability in breast cancer.

Figure 5: Integrative framework: From machine learning discovery of VEGFC to its role in metastasis-associated immune suppression and targeted therapy. (A) Discovery & Prognosis: Workflow utilized to identify VEGFC from a comprehensive panel of cytokines using TCGA RNA-seq data and LASSO machine learning. High VEGFC expression is clinically linked to lymph node metastasis (N2/N3), early onset (<40 years), and poor relapse-free survival (RFS). (B) Mechanism: Proposed model of VEGFC-mediated immune suppression. Tumor-derived VEGFC inhibits T-cell extravasation and downregulates NK cell cytotoxicity while promoting T-cell exhaustion (upregulation of PD-1, CTLA4, TIGIT), driving a “cold” tumor microenvironment. (C) Therapeutic Targeting: Pharmacogenomic screening (OncoPredict) and molecular docking identify the PI3K-β inhibitor AZD6482 as a targeted agent that binds preferentially to VEGFC (Pocket 1). (D) Functional Validation: CRISPR-Cas9 mediated depletion of VEGFC (sgVEGFC) functionally rescues anti-tumor immunity by restoring CD8+ T-cell differentiation and T-cell extravasation pathways. VEGFC: Vascular Endothelial Growth Factor C; TCGA: The Cancer Genome Atlas; LASSO: Least Absolute Shrinkage and Selection Operator; NK cell: Natural Killer Cell; PD-1: Programmed Cell Death Protein 1; CTLA4: Cytotoxic T-Lymphocyte–Associated Protein 4; TIGIT: T-cell Immunoreceptor with Ig and ITIM Domains; TIM-3: T-cell Immunoglobulin and Mucin-domain containing-3; PI3K-β: Phosphoinositide 3-Kinase Beta; CRISPR-Cas9: Clustered Regularly Interspaced Short Palindromic Repeats—CRISPR-associated protein 9; CD8: Cluster of Differentiation 8; sgVEGFC: single guide RNA targeting VEGFC.
Our finding that VEGFC expression is strongly associated with lymph node metastasis and poor prognosis in breast cancer aligns with and substantially extends previous clinical and preclinical investigations. A meta-analysis by Liang et al. demonstrated that high VEGFC expression was significantly associated with lymph node metastasis (OR = 3.28) and reduced overall survival in breast cancer patients [58], while Mohammed et al. reported that VEGFC was an independent prognostic factor in a 10-year follow-up study of 177 breast cancer cases (HR = 2.85; 95% CI: 1.02–8.02; p = 0.047) [59]. Similarly, Zhang et al. found that elevated VEGFC correlated with both shorter disease-free survival and overall survival, particularly in non-Asian populations [60]. These observations are consistent with our multi-cohort validation, demonstrating that VEGFC-high patients exhibited significantly shorter relapse-free survival across the TCGA-BRCA cohort and five independent validation datasets. Our study advances this body of literature by employing an unbiased machine learning approach—LASSO regression—to systematically screen a comprehensive cytokine panel, thereby establishing VEGFC as the most statistically robust predictor of nodal involvement among numerous candidate biomarkers.
The mechanistic role of VEGFC in promoting lymphangiogenesis and facilitating lymphatic metastasis has been extensively documented in experimental models. Seminal work by Mandriota et al. demonstrated that transgenic VEGFC overexpression in pancreatic β-cell tumors induced peritumoral and intratumoral lymphangiogenesis, with tumor cell masses frequently observed within lymphatic vessels and metastatic spread to pancreatic lymph nodes [61]. Hirakawa et al. further showed that VEGFC-induced lymphangiogenesis in sentinel lymph nodes promoted metastatic dissemination beyond regional nodes to distant organs including lungs [62]. Cao et al. revealed a collaborative interplay between FGF-2 and VEGFC, wherein dual expression resulted in disorganized peritumoral lymphatics, frequent intralymphatic tumor cell emboli, and widespread pulmonary and lymph node metastases [63]. These findings establish VEGFC as a critical driver of lymphatic metastatic cascades and are in line with our clinical observation that elevated VEGFC expression correlates with advanced nodal stages (N2/N3) and aggressive disease phenotypes.
Importantly, our study provides novel insights into the immunomodulatory functions of VEGFC beyond its canonical role in lymphangiogenesis. While previous studies have predominantly focused on VEGFC’s canonical effects on vascular and lymphatic biology, a paradigm shift in recent literature highlights its active, direct role in myeloid-cell reprogramming and immune evasion. In the contemporary immunological context, VEGFC is increasingly recognized as a key architect of the immunosuppressive tumor microenvironment. For instance, Banerjee et al. (2023) recently elucidated that VEGF-C-expressing tumor-associated macrophages (TAMs) critically rewire the metastatic fate of breast cancer cells, establishing a localized immunosuppressive niche that facilitates dissemination [34]. Similarly, Zhang et al. demonstrated that VEGFR signaling on myeloid cells directly dictates immune suppression, including the upregulation of PD-L1; importantly, disrupting this axis successfully restored CD8+ T-cell infiltration and activation [64]. Furthermore, recent investigations have shown that tumor-derived VEGFC signaling actively recruits myeloid-derived suppressor cells (MDSCs) and polarizes macrophages toward a pro-tumoral M2 phenotype, thereby dampening NK cell responses and adaptive immunity. Mechanistically, VEGFR-3-expressing TAMs are highly chemotactic toward VEGF-C, and VEGFC-dependent activation of VEGFR-3 in colorectal TAMs has been shown to weaken antitumor adaptive immunity and promote cancer immune escape, effects that were abolished upon VEGFR-3 inhibition [65]. Moreover, VEGF-C/VEGFR-3 signaling drives macrophage polarization toward an immunosuppressive phenotype through STAT6-dependent transcriptional reprogramming, while VEGFR-3+ TAMs simultaneously upregulate VEGF-C/D expression within the peritumoral stroma, creating a self-reinforcing lymphangiogenic and immunosuppressive loop [65]. Consistent with these findings, pharmacological inhibition of VEGFR3/FLT4 with the selective oral inhibitor fruquintinib markedly reduced the infiltration of immunosuppressive myeloid cell populations, including TAMs and CD11b+ precursors, in murine breast (4T1, E0771) and colorectal (MC38, CT26) tumor models, while simultaneously increasing CD4+ and CD8+ T-cell proportions and shifting macrophage-derived lymphatic endothelial cell progenitors (M-LECPs) toward pro-inflammatory phenotypes [66]. Notably, higher VEGFR3/FLT4 expression in metastatic tumors correlated with poorer patient survival, underscoring the clinical relevance of VEGFC/VEGFR3-mediated myeloid immunosuppression as a therapeutic target [66]. In line with this, our GSEA analysis revealed that VEGFC-high tumors exhibit profound downregulation of NK cell cytotoxicity, CD8+ T-cell responses, dendritic cell maturation, and B-cell activity, collectively indicative of a “cold” immunosuppressive microenvironment. Furthermore, our pan-cancer correlation analysis uniquely demonstrated that VEGFC expression in breast cancer—unlike in several other tumor types—positively correlates with immune checkpoint exhaustion markers including PDCD1 (PD-1), CTLA4, TIGIT, and HAVCR2 (TIM-3). This finding aligns with the work of Tietscher et al., who showed that breast tumors with exhausted T cells exhibit high co-expression of PD-1, CTLA-4, LAG-3, TIM-3, and TIGIT [67]. Our data demonstrate that elevated VEGFC not only correlates with checkpoint exhaustion markers but also with specific chemokine signatures (CCL2, CCR2, CCR5, CXCL12) associated with recruitment of immunosuppressive myeloid and regulatory T cell populations. Collectively, these findings position VEGFC as a dual regulator of lymphangiogenesis and immune exclusion, suggesting that its pro-tumoral effects are mediated through both facilitation of metastatic dissemination and suppression of anti-tumor immunity.
Our observation that elevated VEGFC expression is significantly associated with younger age (<40 years) is consistent with epidemiologic studies demonstrating that young-onset breast cancer presents with more aggressive clinicopathologic features. Fabiano et al. reported that young breast cancer patients (≤40 years) exhibited higher frequencies of HER2 positivity, triple-negative phenotype, high histologic grade, and lymph node metastasis compared to older patients [68]. Similarly, Svanøe et al. found that age <40 was significantly associated with hormone receptor negativity, HER2 positivity, lymph-node metastasis, higher proliferation (Ki-67), and shorter survival [69]. Lian et al. demonstrated that younger women (≤40 years) with luminal A subtype breast cancer had significantly worse disease-free survival and distant metastasis-free survival after adjusting for other prognostic factors (HR = 2.06; 95% CI: 1.15–3.69) [70]. These observations support our finding that VEGFC overexpression in younger patients may contribute to the aggressive phenotype and poor prognosis characteristic of early-onset breast cancer. The biological mechanisms underlying this age-related association remain incompletely understood but may involve differences in hormonal milieu, immune function, or tumor microenvironment composition between younger and older patients.
The application of machine learning algorithms, particularly LASSO regression, for prognostic biomarker discovery in breast cancer has gained considerable traction in recent years. Fang et al. developed a LASSO-Cox regression-based prognostic risk model that effectively stratified breast cancer patients into high-risk and low-risk groups, with high-risk patients exhibiting increased tumor proliferation, immune evasion, and reduced immune cell infiltration [55]. Our study builds upon these methodological advances by applying LASSO to a focused cytokine panel for the specific purpose of predicting lymph node metastasis status (N0 vs. non-N0), identifying VEGFC as the single most significant predictor. The advantage of this targeted approach is that it reduces dimensionality while maintaining biological interpretability, enabling clinicians to potentially stratify patients based on a single, measurable biomarker rather than complex multi-gene signatures.
The therapeutic targeting of VEGFC and its downstream signaling pathways represents a promising but underexplored avenue for intervention in metastatic breast cancer. While considerable attention has been directed toward VEGF-A inhibition (e.g., bevacizumab), direct VEGFC blockade has received less clinical development. Our pharmacogenomic profiling identified the PI3K-β inhibitor AZD6482 as a candidate therapeutic agent that exhibits heightened sensitivity in VEGFC-high tumors, a finding supported by preclinical evidence. Zhu et al. demonstrated that PI3K/Akt and MAPK/ERK1/2 signaling pathways are critically involved in IGF-1-induced VEGFC upregulation and lymphatic metastasis in MDA-MB-231 breast cancer cells, and that the Akt inhibitor LY294002 completely blocked IGF-1-induced VEGFC expression [71]. Similarly, Korhonen et al. revealed that lymphangiogenesis requires Ang2/Tie/PI3K signaling and that deletion of the PI3K catalytic p110α subunit or small-molecule inhibition of PI3K decreased VEGF-C-induced lymphangiogenesis [72]. Wang et al. noted that AZD6482 (KIN-193), a PI3Kβ-selective inhibitor derived from TGX221, has been developed for the treatment of solid tumors and that PI3Kβ inhibitors selectively inhibit the growth of PTEN-deficient tumors [73]. The PI3K pathway is one of the most frequently altered pathways in breast cancer, and PI3K inhibitors have shown clinical promise in combination with endocrine therapy [74]. Our molecular docking simulations further confirmed a stable binding interaction between AZD6482 and VEGFC (binding energy = −7.279 kcal/mol), suggesting a potential direct mechanism of action. However, the clinical translation of PI3K inhibitors has been challenged by toxicity and limited single-agent efficacy, highlighting the need for patient selection based on predictive biomarkers such as VEGFC expression.
The functional validation of VEGFC using CRISPR-Cas9 knockout technology represents a critical strength of our study and aligns with emerging applications of genome editing in cancer biology. Hou et al. integrated genome-wide CRISPR immune screens with transcriptomic data to identify immune resistance regulators and demonstrated their potential as therapeutic targets to improve immunotherapy efficacy [75]. In the context of VEGF signaling, CRISPR-mediated knockout studies have yielded valuable mechanistic insights. For instance, knockout of VEGFR2/KDR in squamous thyroid cancer cells resulted in dramatically decreased colony formation and invasion abilities (30% and 60% reduction, respectively) [76], while lentiviral delivery of CRISPR/Cas9 targeting VEGFA in retinal pigment epithelium cells achieved up to 84% indel formation and 78% reduction in secreted VEGFA protein [77]. Notably, the suppression of VEGFC resulted in significant reductions in pathways critical for immune cell recruitment and function, including CD8+ T-cell differentiation (p = 0.028), T-cell extravasation (p = 0.012), and regulation of chemotaxis (p = 0.015) (figure 4I–K). At first glance, this acute depletion effect appears to contradict our clinical TCGA findings, where high VEGFC correlates with an immunosuppressive ‘cold’ TME. However, we propose that this paradox highlights a highly complex, context- and dose-dependent role of VEGFC in tumor immunology. Physiologically, basal VEGFC signaling is indispensable for maintaining the structural integrity of lymphatic/vascular endothelia and basal chemokine gradients, which are prerequisites for normal T-cell extravasation. Consequently, absolute CRISPR-mediated ablation of VEGFC effectively dismantles this baseline trafficking infrastructure, leading to the transcriptomic downregulation of T-cell recruitment. Conversely, in the chronic tumor microenvironment, pathological VEGFC hyper-secretion actively reshapes the stroma into a tolerogenic barrier, preferentially recruiting immunosuppressive M2-macrophages and actively trapping or exhausting cytotoxic T cells. Thus, both the absolute loss and the pathological excess of VEGFC severely compromise anti-tumor T-cell immunity via distinct spatial and transcriptomic mechanisms, underscoring the nuanced dual-role of the VEGFC-lymphatic axis.
Several limitations exist in this study. This study relies primarily on retrospective transcriptomic data subject to batch effects inherent in TCGA and GEO databases. Consequently, while our computational deconvolution and GSEA analyses highlight a strong association between VEGFC and a ‘cold’ tumor microenvironment, these in silico data do not establish direct mechanistic causation. Future in vitro co-culture assays and in vivo functional experiments—such as utilizing VEGFC overexpression or targeted knockout murine models—are absolutely warranted to explicitly validate its direct immunomodulatory effects. Furthermore, mRNA expression levels were used as proxies for protein abundance, which may not correlate perfectly due to post-translational regulation. Pharmacogenomic predictions and molecular docking simulations lack experimental validation through in vitro binding assays or cell-based drug sensitivity studies. CRISPR-Cas9 functional insights derive from computational screening rather than direct experimental interrogation in breast cancer models. Finally, potential overfitting of the LASSO model requires prospective validation in independent cohorts, and the generalizability of our findings to ethnically diverse populations and breast cancer subtypes remains uncertain, limiting clinical translation.
In conclusion, while our multi-layered computational approach combining machine learning, transcriptomic profiling, pharmacogenomics, and functional annotation provides compelling evidence for VEGFC as a master regulator of lymph node metastasis and immune suppression in breast cancer, these findings should be interpreted as hypothesis-generating discoveries requiring extensive experimental validation, protein-level confirmation, functional studies in physiologically relevant model systems, and prospective clinical validation before translating into clinical practice.
Acknowledgement: The authors extend their sincere appreciation to the Research Assistant Center, Show Chwan Memorial Hospital, Taiwan, for providing essential technical support throughout this study. During the preparation of this work, the authors utilized Gemini 3.0 strictly for language editing, grammar correction, and enhancing the overall readability of the manuscript. After using this tool, the authors comprehensively reviewed and edited the content as needed and took full responsibility for the content of the publication. The authors explicitly declare that the AI tool did not participate in any aspect of study design, data analysis, chart generation, data interpretation, or scientific reasoning. All scientific deductions and intellectual content are entirely the original work of the human authors.
Funding Statement: This research was funded by the National Science and Technology Council, Taiwan (NSTC 112-2314-B-442-003, NSTC 113-2314-B-442-002, NSTC 114-2314-B-442-001-MY3 [recipient: Hung-Yu Lin]), and by the Show Chwan Memorial Hospital (SRD-113041 [recipient: Yu-Chieh Tsai]).
Author Contributions: The authors confirm contribution to the paper as follows: Conceptualization, Hung-Yu Lin and Hsing-Ju Wu; methodology, Hung-Yu Lin; software, Hsing-Ju Wu; validation, Yu-Chieh Tsai; formal analysis, Hsing-Ju Wu; investigation, Hsing-Ju Wu; resources, Yu-Chieh Tsai; data curation, Hung-Yu Lin; writing—original draft preparation, Hung-Yu Lin and Hsing-Ju Wu; writing—review and editing, Yu-Chieh Tsai; visualization, Hung-Yu Lin; supervision, Yu-Chieh Tsai; project administration, Hsing-Ju Wu; funding acquisition, Yu-Chieh Tsai. All authors reviewed and approved the final version of the manuscript.
Availability of Data and Materials: All original genomic and clinical data used in this study are publicly available from the TCGA (https://portal.gdc.cancer.gov/) and GEO databases (https://www.ncbi.nlm.nih.gov/geo/) under the accession numbers GSE45255, GSE17705, GSE25065, GSE25055, and GSE20711. The complete R scripts and custom codes utilized for data processing, machine learning feature selection, and statistical analyses have been deposited in a public repository and can be freely accessed at: https://github.com/HYLin-501/VEGFC-Breast-Cancer-Analysis. The data that support the findings of this study are available from the Corresponding Author, upon reasonable request.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare no conflicts of interest.
Supplementary Materials: The supplementary material is available online at https://www.techscience.com/doi/10.32604/ecn.2026.079012/s1.
References
1. Katsura C, Ogunmwonyi I, Kankam HK, Saha S. Breast cancer: presentation, investigation and management. Br J Hosp Med 2022;83(2):1–7. doi:10.12968/hmed.2021.0459. [Google Scholar] [PubMed] [CrossRef]
2. Giaquinto AN, Sung H, Newman LA et al. Breast cancer statistics 2024. CA Cancer J Clin 2024;74(6):477–95. doi:10.3322/caac.21863. [Google Scholar] [PubMed] [CrossRef]
3. Arnold M, Morgan E, Rumgay H et al. Current and future burden of breast cancer: global statistics for 2020 and 2040. Breast 2022;66(8):15–23. doi:10.1016/j.breast.2022.08.010. [Google Scholar] [PubMed] [CrossRef]
4. Kim J, Harper A, McCormack V et al. Global patterns and trends in breast cancer incidence and mortality across 185 countries. Nat Med 2025;31(4):1154–62. doi:10.1038/s41591-025-03502-3. [Google Scholar] [PubMed] [CrossRef]
5. Calhoun BC, Collins LC. Predictive markers in breast cancer: an update on ER and HER2 testing and reporting. Semin Diagn Pathol 2015;32(5):362–9. doi:10.1053/j.semdp.2015.02.011. [Google Scholar] [PubMed] [CrossRef]
6. Yousuf A, Khan NU. Targeting MDM2-p53 interaction for breast cancer therapy. Oncol Res 2025;33(4):851–61. doi:10.32604/or.2025.058956. [Google Scholar] [PubMed] [CrossRef]
7. Zhang F, Lin M, Jiang Y, Zhou F. The role of IL-33 in immunotherapy for breast cancer: targets and signalling pathways. Eur Cytokine Netw 2025;36(1):1–5. doi:10.1684/ecn.2025.0500. [Google Scholar] [PubMed] [CrossRef]
8. Lin HY, Chu PY. Mitochondrial calcium uniporter as biomarker and therapeutic target for breast cancer: prognostication, immune microenvironment, epigenetic regulation and precision medicine. J Adv Res 2025;70:445–61. doi:10.1016/j.jare.2024.04.015. [Google Scholar] [PubMed] [CrossRef]
9. Lin HY, Wu HJ, Chu PY. Multi-omics and experimental analysis unveil theragnostic value and immunological roles of inner membrane mitochondrial protein (IMMT) in breast cancer. J Transl Med 2023;21(1):189. doi:10.1186/s12967-023-04035-4. [Google Scholar] [PubMed] [CrossRef]
10. Andersson Y, Frisell J, Sylvan M, de Boniface J, Bergkvist L. Breast cancer survival in relation to the metastatic tumor burden in axillary lymph nodes. J Clin Oncol 2010;28(17):2868–73. doi:10.1200/JCO.2009.24.5001. [Google Scholar] [PubMed] [CrossRef]
11. de Visser KE, Joyce JA. The evolving tumor microenvironment: from cancer initiation to metastatic outgrowth. Cancer Cell 2023;41(3):374–403. doi:10.1016/j.ccell.2023.02.016. [Google Scholar] [PubMed] [CrossRef]
12. Mahjabeen F, Habbani SF, Mohammed SI. Molecular mechanisms of lymphatic metastasis in breast cancer: an updated review. Cancers 2025;17(13):2134. doi:10.3390/cancers17132134. [Google Scholar] [PubMed] [CrossRef]
13. Tang C, Wang P, Li X et al. Lymph node status have a prognostic impact in breast cancer patients with distant metastasis. PLoS One 2017;12(8):e0182953. doi:10.1371/journal.pone.0182953. [Google Scholar] [PubMed] [CrossRef]
14. Kałużna S, Świerczewska M, Ciesiółka S, Partyka M, Nowicki Mł, Wojtowicz K. The role of exosomes as a key factor of cytostatic resistance in cancer: mechanisms of action, potential biomarkers, and possible exosome-based therapies. Oncol Res 2025;34(1):2. doi:10.32604/or.2025.070356. [Google Scholar] [PubMed] [CrossRef]
15. Lin HY, Wu HJ, Chu PY. Igniting the tumor: targeting mitochondrial stress to prime breast cancer for immunotherapy. Eur Cytokine Netw 2025;36(3):24–37. doi:10.1684/ecn.2025.0504. [Google Scholar] [PubMed] [CrossRef]
16. Liu J, Gao F, Wang D, Zhou R, Huang C. Effects and mechanisms of exosomes in microenvironment angiogenesis in breast cancer: an updated review. Oncol Res 2025;33(6):1323–34. doi:10.32604/or.2024.059113. [Google Scholar] [PubMed] [CrossRef]
17. Lu C, Zhang H, Bhawal UK et al. Mast cells in the solid tumor microenvironment: multiple roles and targeted therapeutic potential. Oncol Res 2025;33(12):3657–78. doi:10.32604/or.2025.069703. [Google Scholar] [PubMed] [CrossRef]
18. Mao H, Zhao X, Sun SC. NF-κB in inflammation and cancer. Cell Mol Immunol 2025;22(8):811–39. doi:10.1038/s41423-025-01310-w. [Google Scholar] [CrossRef]
19. Pavitra E, Kancharla J, Gupta VK et al. The role of NF-κB in breast cancer initiation, growth, metastasis, and resistance to chemotherapy. Biomed Pharmacother 2023;163(3):114822. doi:10.1016/j.biopha.2023.114822. [Google Scholar] [CrossRef]
20. Zhou J, Ottewell PD. The role of IL-1B in breast cancer bone metastasis. J Bone Oncol 2024;46(8):100608. doi:10.1016/j.jbo.2024.100608. [Google Scholar] [PubMed] [CrossRef]
21. Cao P, Sun Z, Zhang F et al. TGF-β enhances immunosuppression of myeloid-derived suppressor cells to induce transplant immune tolerance through affecting arg-1 expression. Front Immunol 2022;13:919674. doi:10.3389/fimmu.2022.919674. [Google Scholar] [CrossRef]
22. Davis RJ, Van Waes C, Allen CT. Overcoming barriers to effective immunotherapy: MDSCs, TAMs, and Tregs as mediators of the immunosuppressive microenvironment in head and neck cancer. Oral Oncol 2016;58:59–70. doi:10.1016/j.oraloncology.2016.05.002. [Google Scholar] [PubMed] [CrossRef]
23. Li K, Shi H, Zhang B et al. Myeloid-derived suppressor cells as immunosuppressive regulators and therapeutic targets in cancer. Signal Transduct Target Ther 2021;6(1):362. doi:10.1038/s41392-021-00670-9. [Google Scholar] [PubMed] [CrossRef]
24. Lindau D, Gielen P, Kroesen M, Wesseling P, Adema GJ. The immunosuppressive tumour network: myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology 2013;138(2):105–15. doi:10.1111/imm.12036. [Google Scholar] [PubMed] [CrossRef]
25. Li Z, Yue C, Hou S et al. MALAT1 promotes epithelial-mesenchymal transition of pancreatic cancer cells through the miR-141-5p-TGF-ß-TGFBR1/TGFBR2 axis. Eur Cytokine Netw 2024;35(3):28–37. doi:10.1684/ecn.2024.0495. [Google Scholar] [CrossRef]
26. Jou E. Type 1 and type 2 cytokine-mediated immune orchestration in the tumour microenvironment and their therapeutic potential. Explor Target Antitumor Ther 2023;4(3):474–97. doi:10.37349/etat.2023.00146. [Google Scholar] [PubMed] [CrossRef]
27. Jung H, Paust S. Chemokines in the tumor microenvironment: implications for lung cancer and immunotherapy. Front Immunol 2024;15:1443366. doi:10.3389/fimmu.2024.1443366. [Google Scholar] [PubMed] [CrossRef]
28. Kohli K, Pillarisetty VG, Kim TS. Key chemokines direct migration of immune cells in solid tumors. Cancer Gene Ther 2022;29(1):10–21. doi:10.1038/s41417-021-00303-x. [Google Scholar] [PubMed] [CrossRef]
29. Nagarsheth N, Wicha MS, Zou W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat Rev Immunol 2017;17(9):559–72. doi:10.1038/nri.2017.49. [Google Scholar] [PubMed] [CrossRef]
30. Chen JY, Lai YS, Chu PY, Chan SH, Wang LH, Hung WC. Cancer-derived VEGF-C increases chemokine production in lymphatic endothelial cells to promote CXCR2-dependent cancer invasion and MDSC recruitment. Cancers 2019;11(8):1120. doi:10.3390/cancers11081120. [Google Scholar] [PubMed] [CrossRef]
31. Chen Z, Varney ML, Backora MW et al. Down-regulation of vascular endothelial cell growth factor-C expression using small interfering RNA vectors in mammary tumors inhibits tumor lymphangiogenesis and spontaneous metastasis and enhances survival. Cancer Res 2005;65(19):9004–11. doi:10.1158/0008-5472.CAN-05-0885. [Google Scholar] [PubMed] [CrossRef]
32. Li J, Yan Z, Ma J et al. ZKSCAN5 activates VEGFC expression by recruiting SETD7 to promote the lymphangiogenesis, tumour growth, and metastasis of breast cancer. Front Oncol 2022;12:875033. doi:10.3389/fonc.2022.875033. [Google Scholar] [PubMed] [CrossRef]
33. Skobe M, Hawighorst T, Jackson DG et al. Induction of tumor lymphangiogenesis by VEGF-C promotes breast cancer metastasis. Nat Med 2001;7(2):192–8. doi:10.1038/84643. [Google Scholar] [PubMed] [CrossRef]
34. Banerjee K, Kerzel T, Bekkhus T et al. VEGF-C-expressing TAMs rewire the metastatic fate of breast cancer cells. Cell Rep 2023;42(12):113507. doi:10.1016/j.celrep.2023.113507. [Google Scholar] [PubMed] [CrossRef]
35. Long J, Hu Z, Xue H et al. Vascular endothelial growth factor (VEGF) impairs the motility and immune function of human mature dendritic cells through the VEGF receptor 2-RhoA-cofilin1 pathway. Cancer Sci 2019;110(8):2357–67. doi:10.1111/cas.14091. [Google Scholar] [PubMed] [CrossRef]
36. Alum EU. AI-driven biomarker discovery: enhancing precision in cancer diagnosis and prognosis. Discov Oncol 2025;16(1):313. doi:10.1007/s12672-025-02064-7. [Google Scholar] [CrossRef]
37. Huang J, Xiang Y, Gan S et al. Application of artificial intelligence in medical imaging for tumor diagnosis and treatment: a comprehensive approach. Discov Oncol 2025;16(1):1625. doi:10.1007/s12672-025-03307-3. [Google Scholar] [PubMed] [CrossRef]
38. Jiang Z, Zhang H, Gao Y, Sun Y. Multi-omics strategies for biomarker discovery and application in personalized oncology. Mol Biomed 2025;6(1):115. doi:10.1186/s43556-025-00340-0. [Google Scholar] [PubMed] [CrossRef]
39. You Y, Lai X, Pan Y et al. Artificial intelligence in cancer target identification and drug discovery. Signal Transduct Target Ther 2022;7(1):156. doi:10.1038/s41392-022-00994-0. [Google Scholar] [PubMed] [CrossRef]
40. Ahmad I, Alqurashi F. Early cancer detection using deep learning and medical imaging: a survey. Crit Rev Oncol Hematol 2024;204(1):104528. doi:10.1016/j.critrevonc.2024.104528. [Google Scholar] [PubMed] [CrossRef]
41. Li Z, Li Y, Xiang J et al. AI-enabled virtual spatial proteomics from histopathology for interpretable biomarker discovery in lung cancer. Nat Med 2026;32(1):231–44. doi:10.1038/s41591-025-04060-4. [Google Scholar] [PubMed] [CrossRef]
42. Lu L, Dercle L, Zhao B, Schwartz LH. Deep learning for the prediction of early on-treatment response in metastatic colorectal cancer from serial medical imaging. Nat Commun 2021;12(1):6654. doi:10.1038/s41467-021-26990-6. [Google Scholar] [PubMed] [CrossRef]
43. Prelaj A, Miskovic V, Zanitti M et al. Artificial intelligence for predictive biomarker discovery in immuno-oncology: a systematic review. Ann Oncol 2024;35(1):29–65. doi:10.1016/j.annonc.2023.10.125. [Google Scholar] [PubMed] [CrossRef]
44. He Y, Ma J, Wang A et al. A support vector machine and a random forest classifier indicates a 15-miRNA set related to osteosarcoma recurrence. Onco Targets Ther 2018;11:253–69. doi:10.2147/OTT.S148394. [Google Scholar] [PubMed] [CrossRef]
45. Hussen BM, Abdullah SR, Hidayat HJ, Samsami M, Taheri M. Integrating AI and RNA biomarkers in cancer: advances in diagnostics and targeted therapies. Cell Commun Signal 2025;23(1):430. doi:10.1186/s12964-025-02434-2. [Google Scholar] [PubMed] [CrossRef]
46. Siemers FM, Bajorath J. Differences in learning characteristics between support vector machine and random forest models for compound classification revealed by Shapley value analysis. Sci Rep 2023;13(1):5983. doi:10.1038/s41598-023-33215-x. [Google Scholar] [PubMed] [CrossRef]
47. Liu XY, Wu SB, Zeng WQ, Yuan ZJ, Xu HB. LogSum + L2 penalized logistic regression model for biomarker selection and cancer classification. Sci Rep 2020;10(1):22125. doi:10.1038/s41598-020-79028-0. [Google Scholar] [CrossRef]
48. Qin JN, Dai WB, Zhang WH et al. Identification of optimal biomarkers associated with distant metastasis in breast cancer using Boruta and Lasso machine learning algorithms. BMC Cancer 2025;25(1):1311. doi:10.1186/s12885-025-14664-1. [Google Scholar] [PubMed] [CrossRef]
49. Tian Y, Chen LE, Jiang Y. LASSO-based screening for potential prognostic biomarkers associated with glioblastoma. Front Oncol 2023;12:1057383. doi:10.3389/fonc.2022.1057383. [Google Scholar] [PubMed] [CrossRef]
50. Wu TT, Chen YF, Hastie T, Sobel E, Lange K. Genome-wide association analysis by lasso penalized logistic regression. Bioinformatics 2009;25(6):714–21. doi:10.1093/bioinformatics/btp041. [Google Scholar] [PubMed] [CrossRef]
51. Zhou N, Zhou M, Ding N, Li Q, Ren G. An 11-gene signature risk-prediction model based on prognosis-related miRNAs and their target genes in lung adenocarcinoma. Front Oncol 2021;11:726742. doi:10.3389/fonc.2021.726742. [Google Scholar] [PubMed] [CrossRef]
52. Al Mehedi Hasan M, Maniruzzaman M, Huang J, Shin J. Statistical and machine learning based platform-independent key genes identification for hepatocellular carcinoma. PLoS One 2025;20(2):e0318215. doi:10.1371/journal.pone.0318215. [Google Scholar] [PubMed] [CrossRef]
53. Xun D, Li X, Huang L, Zhao Y, Chen J, Qi X. Machine learning-based analysis identifies a 13-gene prognostic signature to improve the clinical outcomes of colorectal cancer. J Gastrointest Oncol 2024;15(5):2100–16. doi:10.21037/jgo-24-325. [Google Scholar] [PubMed] [CrossRef]
54. Yu S, Wang C, Ouyang J et al. Identification of candidate biomarkers correlated with the pathogenesis of breast cancer patients. Sci Rep 2025;15(1):8770. doi:10.1038/s41598-025-93208-w. [Google Scholar] [PubMed] [CrossRef]
55. Fang Y, Zheng R, Xiao Y, Zhang Q, Liu J, Wu J. Machine learning-based diagnostic and prognostic models for breast cancer: a new frontier on the clinical application of natural killer cell-related gene signatures in precision medicine. Front Immunol 2025;16:1581982. doi:10.3389/fimmu.2025.1581982. [Google Scholar] [PubMed] [CrossRef]
56. Shen Y, Goparaju C, Yang Y et al. Recurrence prediction of lung adenocarcinoma using an immune gene expression and clinical data trained and validated support vector machine classifier. Transl Lung Cancer Res 2023;12(10):2055–67. doi:10.21037/tlcr-23-473. [Google Scholar] [PubMed] [CrossRef]
57. Wang X, Guan J, Feng L et al. A machine learning-based immune response signature to facilitate prognosis prediction in patients with endometrial cancer. Sci Rep 2024;14(1):30801. doi:10.1038/s41598-024-81040-7. [Google Scholar] [PubMed] [CrossRef]
58. Liang B, Li Y. Prognostic significance of VEGF-C expression in patients with breast cancer: a meta-analysis. Iran J Public Health 2014;43(2):128–35. doi:10.1007/s13277-013-1211-3. [Google Scholar] [CrossRef]
59. Mohammed RAA, Green A, El-Shikh S, Paish EC, Ellis IO, Martin SG. Prognostic significance of vascular endothelial cell growth factors-A, -C and-D in breast cancer and their relationship with angio- and lymphangiogenesis. Br J Cancer 2007;96(7):1092–100. doi:10.1038/sj.bjc.6603678. [Google Scholar] [PubMed] [CrossRef]
60. Zhang Z, Luo G, Tang H, Cheng C, Wang P. Prognostic significance of high VEGF-C expression for patients with breast cancer: an update meta analysis. PLoS One 2016;11(11):e0165725. doi:10.1371/journal.pone.0165725. [Google Scholar] [PubMed] [CrossRef]
61. Mandriota SJ, Jussila L, Jeltsch M et al. Vascular endothelial growth factor-C-mediated lymphangiogenesis promotes tumour metastasis. EMBO J 2001;20(4):672–82. doi:10.1093/emboj/20.4.672. [Google Scholar] [PubMed] [CrossRef]
62. Hirakawa S, Brown LF, Kodama S, Paavonen K, Alitalo K, Detmar M. VEGF-C-induced lymphangiogenesis in sentinel lymph nodes promotes tumor metastasis to distant sites. Blood 2007;109(3):1010–7. doi:10.1182/blood-2006-05-021758. [Google Scholar] [PubMed] [CrossRef]
63. Cao R, Ji H, Feng N et al. Collaborative interplay between FGF-2 and VEGF-C promotes lymphangiogenesis and metastasis. Proc Natl Acad Sci U S A 2012;109(39):15894–9. doi:10.1073/pnas.1208324109. [Google Scholar] [PubMed] [CrossRef]
64. Zhang Y, Huang H, Coleman M et al. VEGFR2 activity on myeloid cells mediates immune suppression in the tumor microenvironment. JCI Insight 2021;6(23):e150735. doi:10.1172/jci.insight.150735. [Google Scholar] [PubMed] [CrossRef]
65. Kannan S, Rutkowski JM. VEGFR-3 signaling in macrophages: friend or foe in disease? Front Immunol 2024;15:1349500. doi:10.3389/fimmu.2024.1349500. [Google Scholar] [PubMed] [CrossRef]
66. Suárez L, Martínez-Azcona M, Serrano-Mendioroz I, Fernández-Rubio L, Rodríguez-Ruiz ME, Rouzaut A. Fruquintinib saddles tumor immune tolerance by curbing pro-tumoral immature myeloid cell populations. Front Immunol 2025;16:1699980. doi:10.3389/fimmu.2025.1699980. [Google Scholar] [PubMed] [CrossRef]
67. Tietscher S, Wagner J, Anzeneder T et al. A comprehensive single-cell map of T cell exhaustion-associated immune environments in human breast cancer. Nat Commun 2023;14(1):98. doi:10.1038/s41467-022-35238-w. [Google Scholar] [PubMed] [CrossRef]
68. Fabiano V, Mandó P, Rizzo M et al. Breast cancer in young women presents with more aggressive pathologic characteristics: retrospective analysis from an Argentine national database. JCO Glob Oncol 2020;6:639–46. doi:10.1200/JGO.19.00228. [Google Scholar] [PubMed] [CrossRef]
69. Svanøe AA, Humlevik ROC, Knutsvik G et al. Age-related phenotypes in breast cancer: a population-based study. Int J Cancer 2024;154(11):2014–24. doi:10.1002/ijc.34863. [Google Scholar] [PubMed] [CrossRef]
70. Lian W, Fu F, Lin Y et al. The impact of young age for prognosis by subtype in women with early breast cancer. Sci Rep 2017;7(1):11625. doi:10.1038/s41598-017-10414-x. [Google Scholar] [PubMed] [CrossRef]
71. Zhu C, Qi X, Chen Y, Sun B, Dai Y, Gu Y. PI3K/Akt and MAPK/ERK1/2 signaling pathways are involved in IGF-1-induced VEGF-C upregulation in breast cancer. J Cancer Res Clin Oncol 2011;137(11):1587–94. doi:10.1007/s00432-011-1049-2. [Google Scholar] [PubMed] [CrossRef]
72. Korhonen EA, Murtomäki A, Jha SK et al. Lymphangiogenesis requires Ang2/Tie/PI3K signaling for VEGFR3 cell-surface expression. J Clin Investig 2022;132(15):e155478. doi:10.1172/JCI155478. [Google Scholar] [PubMed] [CrossRef]
73. Wang X, Ding J, Meng LH. PI3K isoform-selective inhibitors: next-generation targeted cancer therapies. Acta Pharmacol Sin 2015;36(10):1170–6. doi:10.1038/aps.2015.71. [Google Scholar] [PubMed] [CrossRef]
74. Juric D, Janku F, Rodón J et al. Alpelisib plus fulvestrant in PIK3CA-altered and PIK3CA-wild-type estrogen receptor-positive advanced breast cancer: a phase 1b clinical trial. JAMA Oncol 2019;5(2):e184475. doi:10.1001/jamaoncol.2018.4475. [Google Scholar] [PubMed] [CrossRef]
75. Hou J, Wang Y, Shi L et al. Integrating genome-wide CRISPR immune screen with multi-omic clinical data reveals distinct classes of tumor intrinsic immune regulators. J Immunother Cancer 2021;9(2):e001819. doi:10.1136/jitc-2020-001819. [Google Scholar] [PubMed] [CrossRef]
76. Tsai ML, Lee CH, Huang LC et al. CRISPR-mediated knockout of VEGFR2/KDR inhibits cell growth in a squamous thyroid cancer cell line. FEBS Open Bio 2022;12(5):993–1005. doi:10.1002/2211-5463.13399. [Google Scholar] [PubMed] [CrossRef]
77. Holmgaard A, Askou AL, Benckendorff JNE et al. In vivo knockout of the vegfa gene by lentiviral delivery of CRISPR/Cas9 in mouse retinal pigment epithelium cells. Mol Ther Nucleic Acids 2017;9:89–99. doi:10.1016/j.omtn.2017.08.016. [Google Scholar] [CrossRef]
Cite This Article
 Copyright © 2026 The Author(s). Published by Tech Science Press.
Copyright © 2026 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools