
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE
The transcriptome analysis of cleft lip/palate-related PTCH1 variants in GMSM-K cells show carcinogenic potential
1 Department of Orthodontics, Peking University School and Hospital of Stomatology, Beijing, China
2 Central Laboratory, Peking University School and Hospital of Stomatology, Beijing, China
3 Biomedical Engineering Department, Peking University, Beijing, China
* Corresponding Authors: Feng Chen, ; Xiaoshuai Huang,
(This article belongs to the Special Issue: Recent Advancement in Cancer Molecular Signaling)
BIOCELL 2023, 47(1), 205-214. https://doi.org/10.32604/biocell.2022.022572
Received 16 March 2022; Accepted 24 May 2022; Issue published 26 September 2022
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Cancer progression involves the sonic hedgehog (SHH) pathway, in which the receptor PTCH1 actives the downstream pathways. Dysfunction of PTCH1 can lead to nevoid basal cell carcinoma Syndrome (NBCCs) including neoplastic disease and congenital disorder. To evaluate the relationship between PTCH1 and cancer, we applied the CRISPR/Cas9 system to knock out PTCH1 in oral nontumorous epithelial cells (GMSM-K). Then we screened six PTCH1 variants associated with cleft lip/palate (CL/P), one of the congenital disorders in NBCCs, and generated PTCH1 variant and wild-type recombinant PTCH1−/− GMSM-K cell lines. Transcriptome sequencing was conducted in these cell lines. The results revealed that differentially expressed genes (DEGs) in PTCH1−/− GMSM-K were enriched in extracellular compartments, contributing epithelial diseases by pathway enrichment analysis. RT-PCR confirmed that KRT34, KRT81, KRT86, PDGFB, and WNT10B genes, associated with extracellular compartments were highly expressed in PTCH1−/− . The Kyoto Encyclopedia of Genes and Genomes analysis also suggested that DEGs are closely related to focal adhesion, transcriptional misregulation, and proteoglycans in breast and gastric cancers. Comparative analysis of samples revealed that the CL/P-associated PTCH1 variants A443G and V908G are potentially carcinogenic. These findings provide new insights into the carcinogenic potential of PTCH1 dysfunction.Keywords
PTCH1, a receptor in the sonic hedgehog (SHH) pathway, contains two large extracellular domains. The degradation of PTCH1 activates smoothened (SMO), a seven-transmembrane protein whose activation stimulates glioma-associated oncogene (Gli) transcription factors (GLI 1, 2, 3), which upregulates the SHH pathway (Carballo et al., 2018). The SHH pathway controls the formation of bones, limbs, and the central nervous system. In mature tissues, SHH signaling pathways regulate the proliferation and differentiation of various stem cells. In many tumors, abnormalities of SHH signaling pathways have been found. For example, nevoid basal cell carcinoma syndrome (NBCCs)-associated skin basal cell carcinoma (BCC) and medulloblastoma have the abnormal expression of SHH signal pathway related genes (Athar et al., 2014; Northcott et al., 2019). Abnormal expression of the SHH pathway is also related to gastrointestinal carcinogenesis and the changes in the tumor microenvironment (Zhang et al., 2021).
Dysfunction of PTCH1 may cause NBCCs, including basal cell carcinoma and developmental disorders. Neoplastic diseases include BCC, odontogenic keratocyst, ovarian fibroma, and medulloblastoma (Jiang and Hui, 2008). The common malformations of palmar or plantar pits in NBCCs are an important diagnostic criterion. Developmental disorders include rib abnormalities, macrocephaly and cleft lip/palate (CL/P). Genome-wide association studies and case reports have linked PTCH1 to CL/P (Zhao et al., 2018), that PTCH1 can serve as a bridge between CL/P and cancerous disease. CL/P increased the risk of several types of cancer in a multi-family comparative study, but other studies have found conflicting results (Bille et al., 2005). Although these studies indicate that CL/P may be linked to cancer, they did not evaluate genetic factors. Therefore, we investigated the mechanisms underlying the relationship between CL/P and cancer using PTCH1.
PTCH1 contains 12 transmembrane domains, 2–6 of which are sterol-sensing domains linked to lipid transportation. In addition, PTCH1 contains two extracellular domains and one intracellular domain (Gong et al., 2018). CL/P-associated variants are present in almost all domains, and a minority (40%) are missense variants (Zhong et al., 2021). Other types of variants such as frameshift and nonsense variants can cause early termination of PTCH1, indicating the same PTCH1 dysfunction as the dysfunction of PTCH1 due to the CRISPR/Cas9 system. Therefore, we focused on missense variants of PTCH1 to study whether it leads to the functional defects of PTCH1 and may have carcinogenic potential. Conditional loss of PTCH1 in animal models has shown that both the FGF pathway and WNT signaling are involved in CL/P (Kurosaka et al., 2014). The connection between SHH and WNT pathways also plays an important role in the formation of BCC (Noubissi et al., 2018). Therefore, understanding the oncogenic potential of CL/P-related PTCH1 variants is needed to uncover the downstream cellular and molecular changes caused by PTCH1 deficiency. Because most malignant tumors caused by PTCH1 deficiency are epithelial, we selected GMSM-K, an oral epithelial cell line, for further study.
In this study, we used the CRISPR/Cas9 system to generate PTCH1-knockout GMSM-K cells (PTCH1−/−). We selected six CL/P-associated PTCH1 variants (P295S, A392V, A443G, T728M, V908G, and T1052M) and overexpressed variant and wild-type PTCH1 in PTCH1−/− cells to generate stably transfected cells. A transcriptome analysis was performed to assess changes in PTCH1-knockout GMSM-K cells and the carcinogenic potential of PTCH1 variants.
GMSM-K cells were a gift from Profs. Gilchrist and Cornell (Gilchrist et al., 2000) and were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum (Gibco), 100 U/mL penicillin, and 100 μg/mL streptomycin at 37°C in a 5% CO2 atmosphere. The culture medium was refreshed every other day. 293T cells were cultured under the same conditions.
The LentiCRISPR V2 vector was a gift from Dr. Ting Han (Lv et al., 2020). The guide RNA sequence (AAATGTACGAGCACTTCAAG) for PTCH1 knockout was from Broad GPP (Broad Institute). We synthesized the gRNA positive and negative strands and annealed them to form sticky ends. The annealed product was ligated into the linear vector, which was digested by BsmBI (NEB; R0580). The full-length coding sequence of PTCH1, provided by Dr. Tiejun Li (Yu et al., 2014), was cloned into the lenti-EFS-P2A-BSD vector. Variants of PTCH1 were generated using a mutagenesis kit (Transgen; FM111) following the manufacturer’s instructions.
GMSM-K PTCH1−/− cell line construction
The gRNA-carried vector was co-transfected with pSPAX2 and pMD2.G in 293T cells in a ratio of 5:3:2. Lentivirus in the supernatant was collected 72 h after transfection and added to the GMSM-K cells. Three days later, stably transfected cells were selected by puromycin (1 μg/mL). The cells were transferred to a 96-well plate (<1 cell/well). Single-cell clones were collected and subjected to Western blotting to identify PTCH1−/− cells. PTCH1−/− cells were verified by TA cloning. A pair of primers (gRNA-F: 5′-CCCGGTGTATCTGTT-CAAGCTATCTGCTCC-3′, gRNA-R: 5′-GAAGAATTGCATAACCAGCGAGTCTGCACG-3′) were used to PCR target splicing site; then the PCR product was cloned into the pTOPO-TA Cloning vector (Mei5bio, MF019). The vector was transferred into the competent cell (Trans5α, CD201) and inoculated onto an ampicillin agar plate, where it was grown and then picked up five clones for sequencing.
PTCH1 wild-type/variants overexpressed cell conduction in PTCH1−/− cells
PTCH1 wild-type and variant lentiviral vectors were generated as described above. PTCH1−/− cells were infected with lentivirus and selected using 15 μg/mL blasticidin (Yeasen; 60218ES10).
Cells were washed three times with PBS and lysed in 1% SDS lysis buffer (20 mM HEPES, 10 mM NaCl, 2 mM CaCl2; pH 8.0) containing protease inhibitor cocktail (CWBIO; CW2200), phosphatase inhibitor cocktail (CWBIO, CW2383), and 50 U/mL benzonase (Yeasen; 20156ES25). The BCA Protein Assay Kit (CWBIO; CW0014S) was used to measure protein concentrations. Samples were separated by 10% SDS-PAGE and transferred to 0.45 μm PVDF membranes. Membranes were incubated with anti-PTCH1 rabbit antibodies (1:1000; ABclonal; A0826). An anti-GAPDH (CST; 5174) rabbit antibody was used as the protein loading control followed by HRP-linked secondary antibodies. Samples were visualized using a gel imager.
RNA isolation, reverse transcription and quantitative reverse transcription PCR (RT-PCR)
Total cellular RNA was extracted using the RNApure Cell Kit (CWBIO; CW0560) following the manufacturer’s protocols. Next, 1 μg of RNA was retrotranscribed to cDNA using ReverTra Ace qPCR RT Master Mix (Toyobo; FSQ-201). The mRNA levels of target genes were determined by Real-Time PCR on an ABI 7500 Real-Time PCR system (Thermo Fisher Scientific, USA) using SYBR-Green Quantitative PCR Mix (Mei5Bio; MF797) following the manufacturer’s instructions. The GAPDH gene was used as a control in the experiment. The sequences of the primers synthesized by Ruibiotech (Beijing, China) can be found in Suppl. Table S1.
Cell proliferation, apoptosis, and cell cycle
GMSM-K cells were seeded into 96-well plates at 5 × 103/well. Cell proliferation was measured using the Cell Counting Kit-8 (CCK-8; Beyotime; C0039) assay according to the manufacturer’s instructions.
GMSM-K cells were seeded in six-well plates at 1 × 105/well and incubated for 48 h. Apoptosis and cell cycle were assayed using an Annexin V-FITC Apoptosis Detection Kit (Dojindo; AD10) and Cell Cycle Analysis Kit (Dojindo; C543) according to the manufacturer’s instructions. Apoptosis and cell cycle were analyzed using a FACSCalibur flow cytometer (BD).
Total RNA was purified using oligo(dT)-attached magnetic beads and subjected to mRNA enrichment. Purified mRNA was fragmented and reverse transcribed and validated for quality control to generate the sequencing library. The library was sequenced on the BGISEQ-500 platform (Beijing Genomics Institute [BGI], Shenzhen, China). Sequencing data were provided by the BGI. Analysis of differentially expressed genes (DEGs) was performed using DESeq2 (v. 1.4.5) with a Q-value of ≤0.05. A heatmap of gene expression was generated using pheatmap (v. 1.0.8). Gene ontology (GO) (http://www.geneontology.org/) and Encyclopedia of Genes and Genomes (KEGG) (https://www.kegg.jp/) enrichment analyses of DEGs were performed using Phyper (https://en.wikipedia.org/wiki/Hypergeometric_distribution) based on the hypergeometric test. The String Database and Cytoscape v. 3.7.1 were used to construct a protein–protein interaction network and screen out the top 10 hub genes according to the score. The significance levels of terms and pathways were corrected according to a Q-value of ≤0.05.
All data were presented as means ± sem. Statistical analysis between two groups was performed using unpaired Student’s t-test to determine the p values.
We previously reported 25 variants of PTCH1 associated with CL/P, most of which were frameshift, nonsense, splicing, and in-frame deletion variants. All these variants may cause early termination of PTCH1, so we focused on the 10 missense variants (Zhong et al., 2021). D436N and S827G were excluded because they are predicted to be benign. Three variants (P295S, A392V, and T728M) were shown to have unequivocal pedigree evidence (Mansilla et al., 2006; Ming et al., 2002; Zhao et al., 2018), while T728M was detected in two pedigrees and was likely pathogenic, A392V showed strong linkage with inherited CL/P in our previous study. V908G was lethal, and the carrier died in utero (Ribeiro et al., 2006). Given that the domains of PTCH1 have different functions, we selected P295S and A392V in the first extracellular domain and V908G in the second extracellular domain. Both extracellular domains are involved in SHH binding. T728M is present in the intracellular loop, which is reportedly associated with the cell cycle; A443G is present in the sterol-sensing domain and may affect lipid transportation; T1052M causes severe craniofacial dysplasia. Two variants were found in NSCL/P (P295S and A392V), while the other four were found in SCL/P (Table 1).
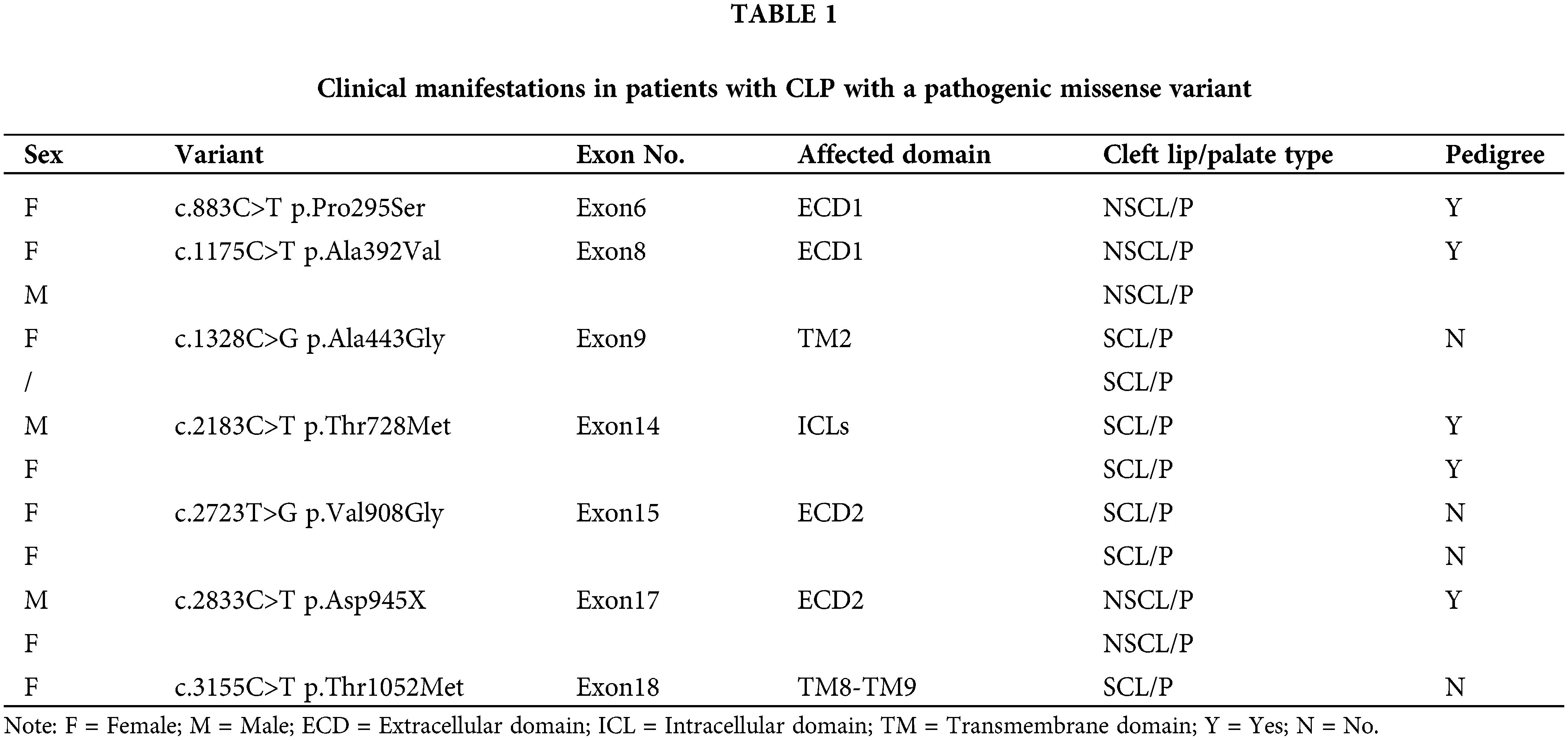
PTCH1−/− cell line construction in GMSM-K using the CRISPR/Cas9 system
We used the CRISPR/Cas9 system to knockout PTCH1 (Fig. 1A). Next, the TA cloning assay (n = 5) showed deletion of 12 bases and insertion of 1 base at the target site in both alleles (Fig. 1B), causing a frameshift downstream and a stop codon at residue 426. The western blotting assay showed that the target size band where PTCH1 was lost in PTCH1−/− GMSM-K cells (Fig. 1C). The mRNA of PTCH1 degraded and the expression of Gli1 was upregulated threefold (Fig. 1D), revealing the deletion of PTCH1. These results suggested that we could generate a PTCH1−/− cell line of GMSM-K.
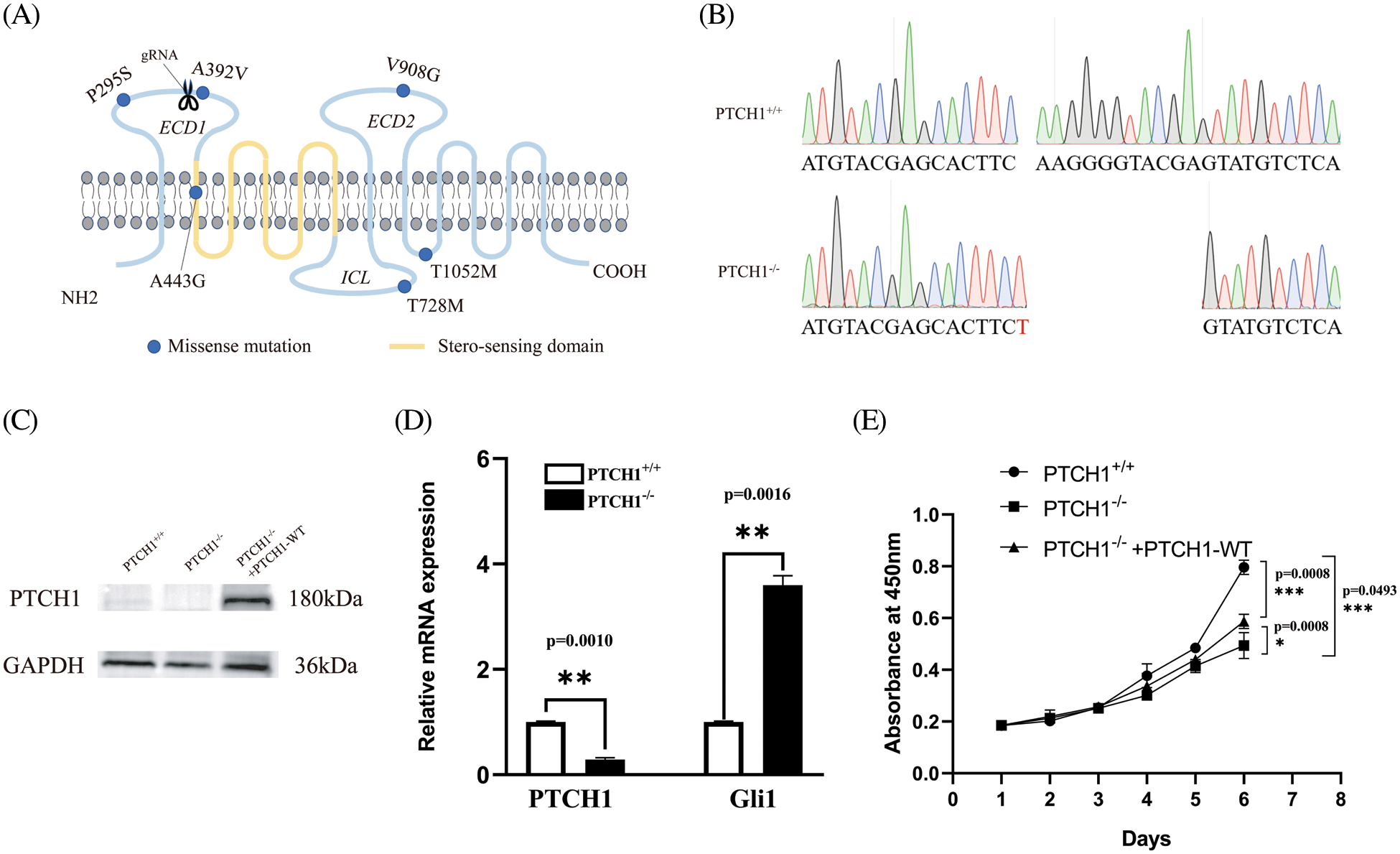
Figure 1: PTCH1−/− cell line construction in GMSM-K using the CRISPR/Cas9 system. (A) All six PTCH1 variants are shown in blue dots (missense variant). The scissors represent the position of guide RNA designed for knocking out PTCH1 using the CRISPR/cas9 system. ECD: extracellular domain; ICL: intracellular loop. (B) DNA sequencing reveals deletion of 12 bases deletes and insertion of 1 base in both alleles (n = 5). (C) Western blot verified knocking out of PTCH1 and PTCH1 overexpressed in PTCH1−/− cells with PTCH1-WT (PTCH1−/− +PTCH1-WT). (D) Relative mRNA level of PTCH1 and Gli1. (E) Proliferation assay of the three cell lines (GMSM-K, PTCH1−/−, PTCH1−/−+PTCH1-WT); *p < 0.05, **p < 0.01 and ***p < 0.001 vs. control.
PTCH1−/− inhibits GMSM-K cell proliferation by modulating the cell cycle
The cell proliferation assay using the CCK-8 method showed that the proliferation of PTCH1−/− cells was significantly reduced compared to PTCH1+/+ GMSM-K cells (Fig. 1E). Both the apoptosis and cell cycle assays showed that the apoptosis rate was similar in the two groups (Figs. 2A–2C). The cell cycle assay showed that the S-phase rate in PTCH1+/+ cells was higher than in PTCH1−/− cells, indicating more DNA synthesis in PTCH1+/+ cells (Figs. 2D–2F). Therefore, the slowing of the PTCH1−/− cell cycle led to a decrease in cell proliferation.
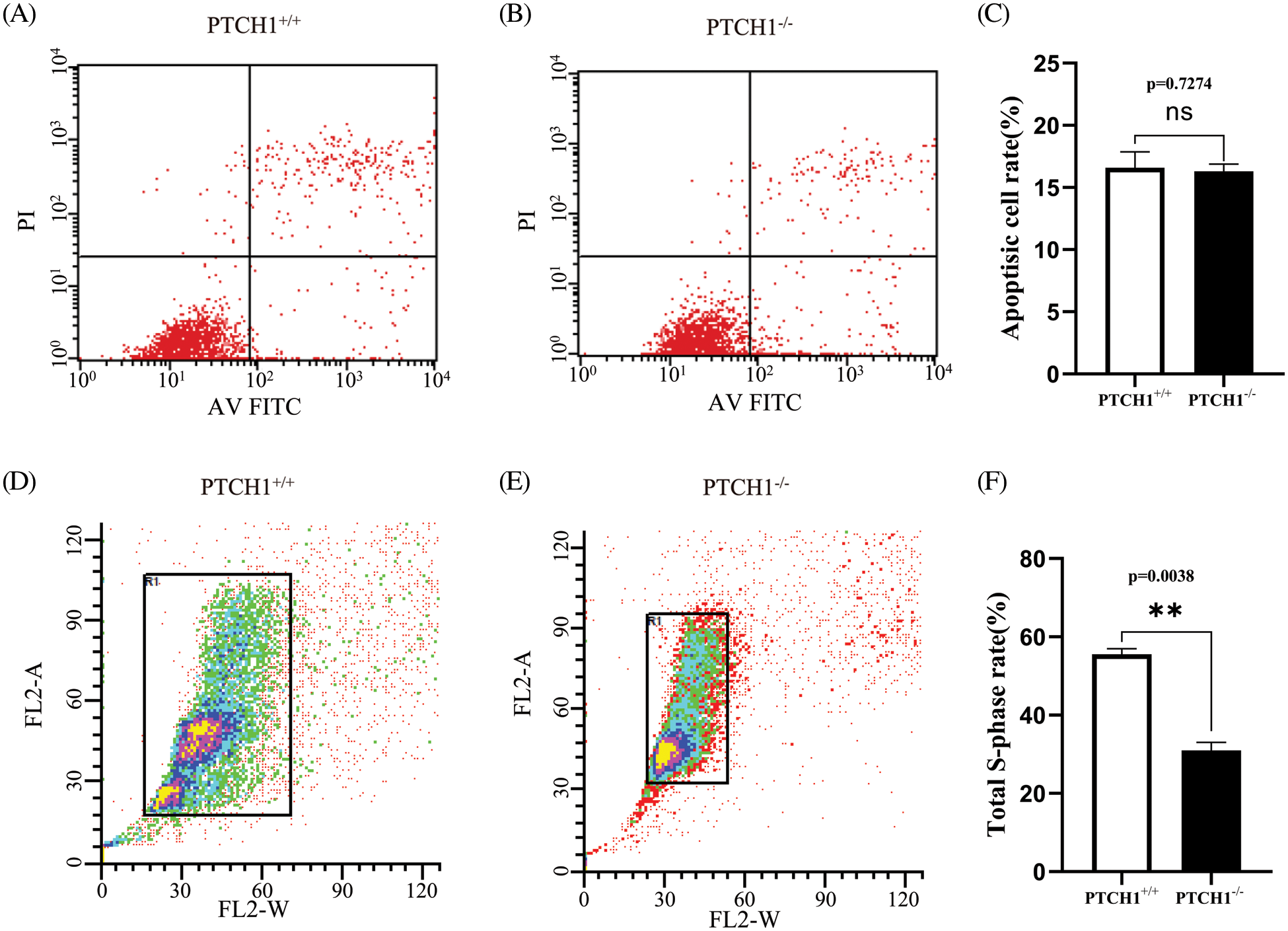
Figure 2: PTCH1−/− inhibits GMSM-K cell proliferation by modulating the cell cycle. (A–B) Apoptosis flow cytometry chart of PTCH1+/+ (A) and PTCH1−/− (B). Annexin V-FITC and PI staining. (C) Total apoptosis ratio of PTCH1+/+ and PTCH1−/−. (D–E) Cell cycle flow cytometry chart of PTCH1+/+ and PTCH1−/−. (F) Total S-phase cell ratio of PTCH1+/+ and PTCH1−/−; ns, no significance, **p < 0.01 vs. control.
DEGs in PTCH1−/− cells were enriched in extracellular matrix components
There were 624 DEGs between PTCH1−/− and PTCH1+/+ GMSM-K cells, while 405 genes were down-regulated and 219 genes were up-regulated (Fig. 3A). In addition, a GO enrichment analysis showed that 135 DEGs were associated with extracellular matrix-related systems, including the extracellular space, the extracellular matrix, the extracellular region, and the collagen-containing extracellular matrix (Fig. 3B). The 135 involved genes included: BGLAP, BGN, COL8A1, COL13A1, FN1, COL5A2, and LAMA4. These 135 DEGs were linked to several epithelial diseases in the KEGG disease enrichment analysis, including pachyonychia congenita, bullous congenital ichthyosiform erythroderma, palmoplantar keratoderma, and white sponge nevus (Fig. 3C). This coincides with the epithelial manifestations of NBCCs. KRT34, KRT81, and KRT86 are linked to epithelial diseases, and RT-PCR showed that their expression levels were increased, respectively, in PTCH1−/− GMSM-K cells compared to PTCH1+/+ cells (Fig. 3D). Platelet-derived growth factor (PDGF) and WNT/β-catenin systems may contribute to the development of basal cell carcinoma (Litvinov et al., 2021; Sellheyer, 2011). Both pathways are extracellular matrix components. In this study, we checked the gene expression of PDGF and WNT/β-catenin systems between PTCH1+/+ and PTCH1−/− GMSM-K cells and observed highly expressed PDGFB and WNT10B in PTCH1−/− cells (Figs. 3E and 3F).
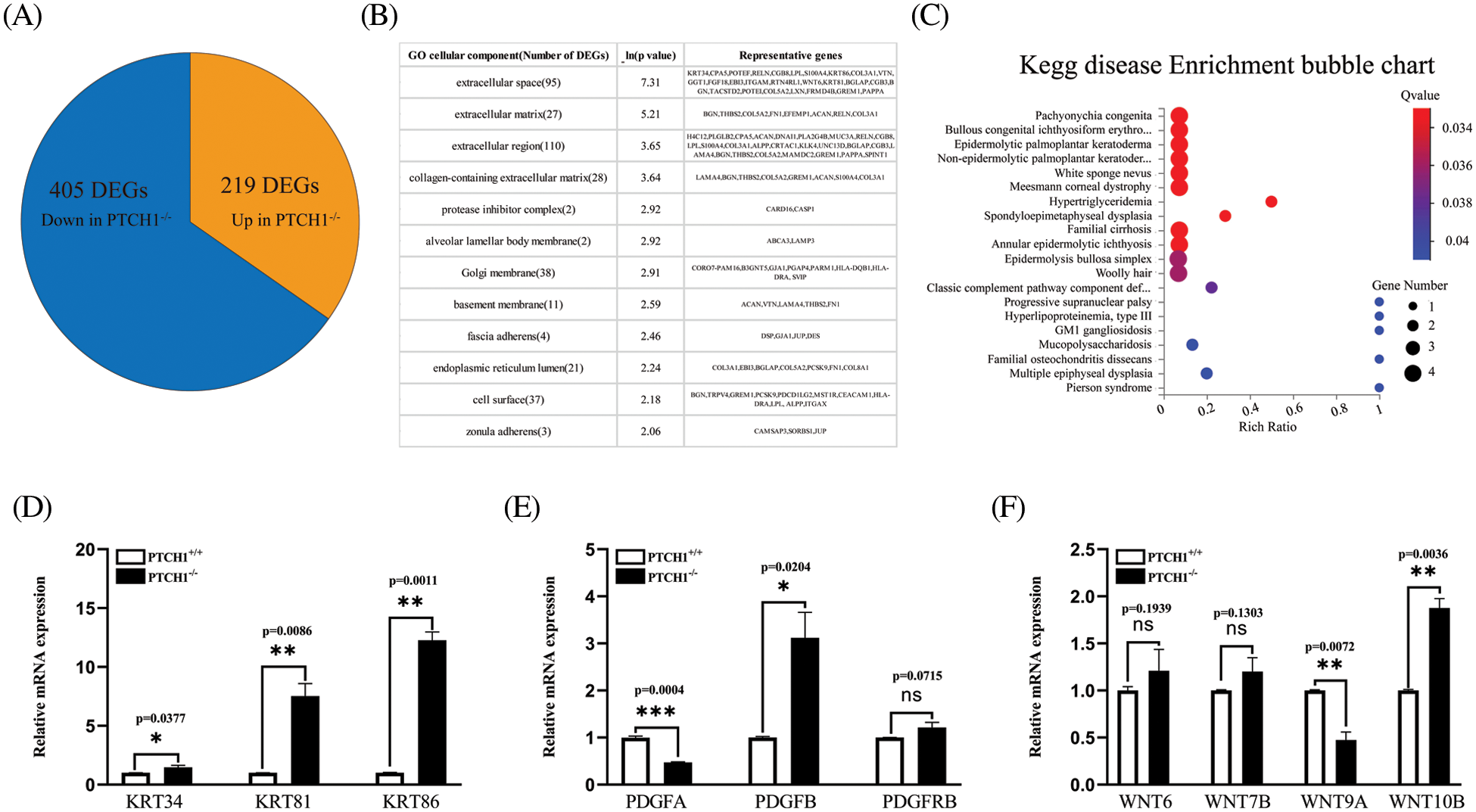
Figure 3: Differentially expressed genes (DEGs) in PTCH1−/− cells were enriched in extracellular matrix components. (A) Up- and down-regulated DEGs numbers in the PTCH1-/- cell line compared to the PTCH1+/+GMSM-K cells. (B) DEGs enriched in extracellular compartments in gene ontology cellular enrichment analysis. (C) Disease enrichment of DEGs in extracellular compartments shows epithelial disease accumulation. (D) Relative mRNA expression of KRT34, KRT81, and KRT86. (E) Relative mRNA expression of platelet-derived growth factor (PDGF)A, PDGFB, and PDGFRB. (F) Relative mRNA expression of WNT6, WNT7B, WNT9A, and WNT10B; *p < 0.05, **p < 0.01, ***p < 0.001 vs. control; ns, no significance.
The oncogenic pathways of PTCH1 dysfunction and the oncogenic possibility of PTCH1 variants
Most DEGs were enriched in human diseases and organismal systems. We selected 285 cancer-related genes in KEGG pathway annotations associated with malignant tumors (Fig. 4A). The KEGG network analysis of these oncogenic genes showed that pathways involving cancer had 27 nodes. Moreover, pathways in cancers are closely related to focal adhesion, transcriptional misregulation and proteoglycans in cancer. Some of these oncogenes are related to breast cancer and gastric cancer (Fig. 4B).
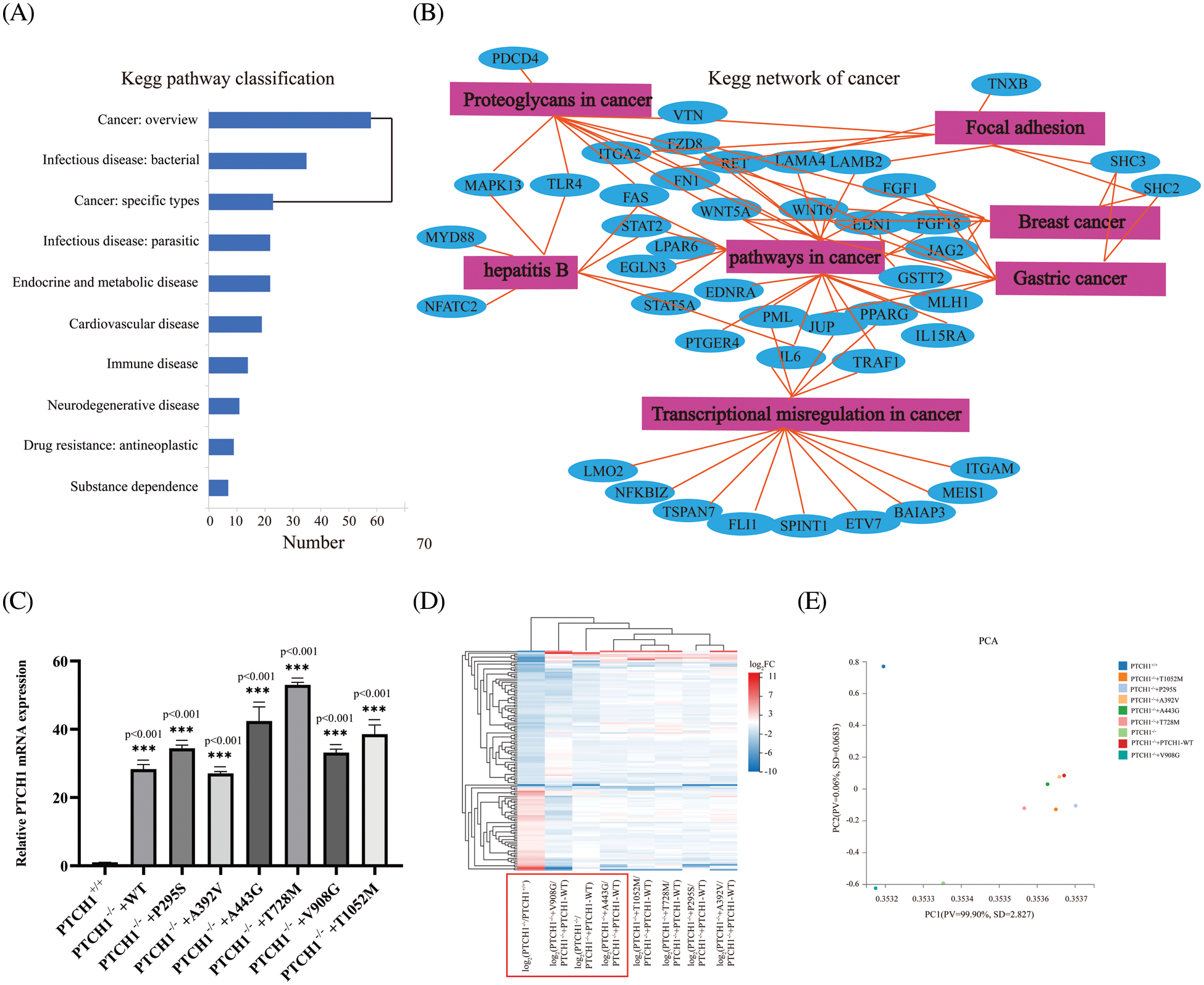
Figure 4: Roles of PTCH1 variants in oncogenesis. (A) Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway annotation of differentially expressed genes (DEGs). (B) KEGG network of cancer-associated genes. (C) Relative mRNA expression of PTCH1 in GMSM-K PTCH1+/+ cells and PTCH1 wildtype and variants recombinant cell lines. (D) Clustering heat map of extracellular compartments DEGs in PTCH1 nine cell lines. (E) Principal component analysis (PCA) of extracellular compartments DEGs in nine cell lines of PTCH1. ***p < 0.001 vs. control.
We overexpressed the PTCH1 wild-type and variants to generate PTCH1−/−+PTCH1-WT, PTCH1−/−+P295S, PTCH1−/−+A392V, PTCH1−/−+A443G, PTCH1−/−+T728M, PTCH1−/−+V908G, and PTCH1−/−+T1052M cells (Fig. 4C). We conducted transcriptome analysis of the PTCH1-overexpressing cell lines. First, a clustering heat map analysis of 8 GMSM-K cell lines was performed based on extracellular-compartment DEGs. PTCH1−/−+V908G/PTCH1−/−+PTCH1-WT and PTCH1−/−+A443G/PTCH1−/−+PTCH1-WT were closer to PTCH1−/−/PTCH1−/−+PTCH1-WT cells, suggesting that V908G and A443G are deleterious (Fig. 4D). A principal components analysis showed that, unlike the other variants, V908G exhibited a greater propensity to be loss-of-function in the SHH signaling pathway. PTCH1−/−+A392V was more resembled to PTCH1−/−+PTCH1-WT than other variants, showing a likely benign trait (Fig. 4E). Therefore, V908G and A443G are more inclined to oncogenesis from the perspective of extracellular compartments.
Potential pathogenicity of PTCH1 variants A443G and V908G
To explore the effect of PTCH1 variants on the SHH signaling pathway, mRNA levels of Gli1 in GMSM-K cells that stably expressed wildtype and mutant PTCH1 were detected by RT-PCR. The level of Gli1 in PTCH1−/−+A443G and PTCH1−/−+V908G was higher than in other recombinant cell lines, which means A443G and V908G are loss-of-function mutations (Fig. 5A). Expression levels of PDGFB, WNT10B, KRT34, KRT81, and KRT86 increased in PTCH1−/−+A443G and PTCH1−/−+V908G compared to other recombinant cell lines (Figs. 5B–5F). The transcriptome data showed that DEGs of PTCH1−/−+A443G enriched in intermediate filament containing KRT34 (Fig. 5G). The DEGs of PTCH1−/−+V908G enriched in the extracellular compartments, similar to the expression of PTCH1−/− (Fig. 5H). These results suggest that PTCH1 variants A443G and V908G were likely pathogenic in terms of carcinogenesis.
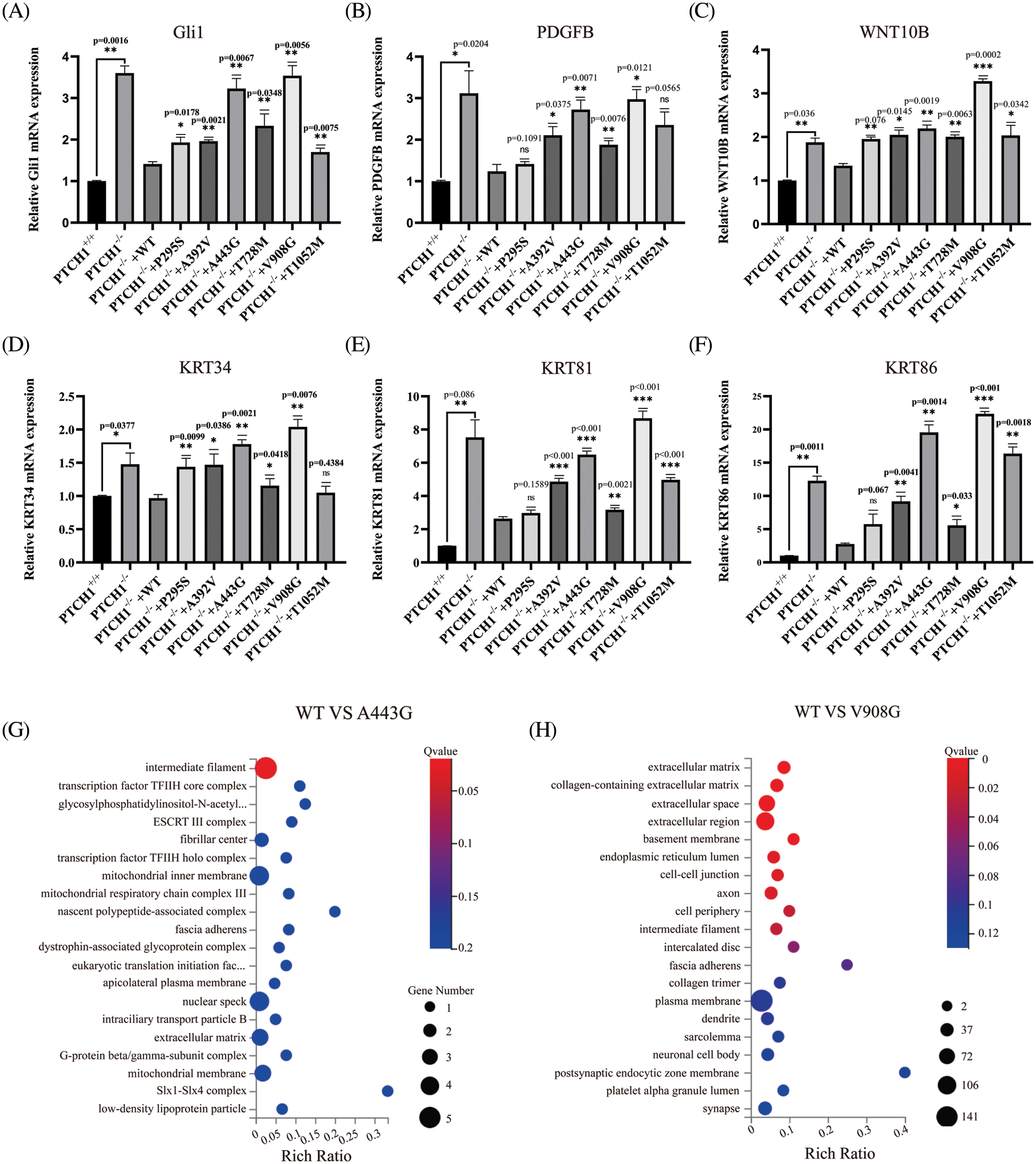
Figure 5: Possible pathogenicity of PTCH1 variants A443G and V908G. Relative mRNA expression of Gli1 (A), platelet-derived growth factor (PDGF)B (B), WNT10B (C), KRT34 (D), KRT81 (E), and KRT86 (F). (G) DEGs of A443G enrich in intermediate filament in GO cellular enrichment analysis. (H) DEGs of V908G enrich in extracellular compartments in GO cellular enrichment analysis. *p < 0.05, **p < 0.01, ***p < 0.001 vs. control; ns, no significance.
In the SHH signaling pathway, PTCH1 acts as the receptor and is involved in the progression of cancers. Dysfunction of PTCH1 may cause basal cell carcinoma and developmental disorders, including rib abnormalities, macrocephaly, and cleft lip/palate (CL/P). There have been several studies about the relationship between PTCH1 and CL/P, while the carcinogenesis of CL/P-related PTCH1 variants is still unclear. In this study, we used CRISPR/Cas9 system combined with transcriptome analysis and found the likely pathogenicity of PTCH1 variants A443G and V908G in terms of carcinogenesis. We also speculated that the abnormal expression of PDGFB, WNT10B, KRT34, KRT81, and KRT86 may contribute to the carcinogenic process. Our findings implicated that alterations in the extracellular compartments may play an important role in the carcinogenesis of PTCH1-related diseases.
In this study, the CRISPR/Cas9 system was used to delete PTCH1 in GMSM-K cells. GMSM-K is a human polyclonal oral epithelial cell line that is a proper model for studying CL/P and was used to assess carcinogenicity because cancers caused by PTCH1 dysfunction are of epithelial origin (Gilchrist et al., 2000). Oral leukoplakia and fibrosis are precancerous lesions, and the GMSM-K cell line enables a mechanistic analysis of tumorigenesis because it is also a non-tumor cell line, thus allowing us to monitor the precancerous process. Traditional methods to knock-down PTCH1 may not work well in this scenario. The activated SHH pathway can increase the expression of PTCH1, while the downregulation of PTCH1 can contribute to SHH pathway activation, forming a negative feedback loop (Ribes and Briscoe, 2009). The knockout of PTCH1 in GMSM-K cells at the genetic level may be the only effective method, although a cell proliferation assay showed that the knockout of PTCH1 inhibited GMSM-K cell proliferation. We also noticed that the overexpression of PTCH1 only slightly relieved the PTCH1−/− cells. This phenomenon coincided with previous reports, which indicated that the overexpression of PTCH1 can attenuate the proliferation of other cells (Liu et al., 2018). One biochemical analysis revealed the binding of PTCH1 to the M-phase promoting factor (MPF) through its intracellular domain, thus affecting MPF’s subcellular localization. Eventually, the cell cycle was affected by the overexpression of PTCH1 (Barnes et al., 2001).
CL/P has been reported to be linked to carcinogenesis, but there are conflicts (Bille et al., 2005; Dietz et al., 2012; Metzis et al., 2013; Steinwachs et al., 2000). A nationwide study in Denmark of 8783 cancer patients investigated whether congenital malformations contribute to cancer and showed a higher risk only in CL/P among the malformations investigated (odds ratio for all cancer = 1.8) (Zhu et al., 2002). In a large study involving 2349 cases, CL/P was linked to the risk of leukemia (Nishi et al., 2000). Therefore, CL/P may be associated with an elevated risk of cancer. Leukemia and colorectal cancer are reportedly associated with CL/P (p < 0.01) (Menezes et al., 2009). However, these studies did not evaluate genotype or the mechanism analysis. Our GO cellular component analysis showed that changes in the extracellular matrix (ECM) may contribute to carcinogenesis, which is consistent with the result of another study (Athar et al., 2006). Dysfunction of PDGF contributes to the development of BCC; it is a bridge between the epithelium and the ECM and is involved in skin and hair development (Andrae et al., 2008). In PTCH1−/− cells, the expression of PDGFB was observed to increase up to threefold, whereas the expression of PDGFA was lower. Therefore, the SHH pathway may affect PDGF in epithelial cells. However, mesenchymal cells also contribute to the ECM. Therefore, we plan to investigate the epithelial–ECM interaction in the future. We observed the association of KRT81 and KRT86 with epithelial diseases. KRT81 is a type II hair keratin reportedly overexpressed in and related to the invasiveness of breast cancer cells (Nanashima et al., 2017). The expression of KRT81 is affected by microRNAs and linked to non-small-cell lung cancer and non-Hodgkin’s lymphoma (Robles and Ryan, 2016). Therefore, PTCH1−/− cells may increase the expression of KRT81 via downstream microRNAs. KRT86 is associated with monilethrix, characterized by fragile hair (de Cruz et al., 2012). KRT81 and KRT86 are highly expressed in PTCH1−/− cells and may contribute to epithelial diseases. Also, the WNT/β-catenin system contributes to carcinogenesis by responding to oncogenic SHH signaling in the skin (Yang et al., 2008). Canonical WNT/β-catenin controls skin self-renewal, lineage determination, and terminal differentiation (Wend et al., 2012). Among the WNT proteins expressed in GMSM-K cells, only WNT10B was highly expressed in PTCH1−/− cells. WNT10B expression is linked to fibrosis in systemic sclerosis, which is associated with heightened cancer risk (Fragoulis et al., 2020). Whether these ECM changes contribute to BCC development warrants further studies involving animal models.
Our current data reveal that the malfunction of PTCH1 resulting in abnormal ECM, while the DEGs indicate several cancers. However, all these conclusions need verification by animal and immunohistochemical experiments in the future. The involvement of KRT81/86, PDGF, and WNT/β-catenin is hypothetical and requires further studies. Nevertheless, our work provides insights into the pathogenetic role of PTCH1 and offers some explanations of the relation between CL/P and carcinoma.
Our results suggest that dysfunction of PTCH1 may contribute to carcinogenesis, and PDGF, KRT81/86, and WNT/β-catenin may be involved in the process. PTCH1−/− cancer-associated DEGs are linked to several types of cancer, including gastric cancer and breast cancer. Among the PTCH1 variants, V908G and A443G were predicted to be likely pathogenic. Our results thus provide mechanistic insight into the carcinogenicity of PTCH1.
Acknowledgement: We would like to thank Dr. Ting Han and Hong Zou for their kind support throughout this research. We appreciate the kind support from the High-performance Computing Platform of Peking University.
Author Contribution: Feng Chen and Xiaoshuai Huang designed the experiments. Mingzhao Li carried out the experiments and wrote the paper. Mingzhao Li, Wenbin Huang, and Shiying Zhang analyzed the transcriptome data. Qian Zhang and Nan Jiang participated in the interpretation of the experimental results and the revision of the manuscript.
Availability of Data and Materials: The data of transcriptome used in this article were uploaded to NCBI, BioProject No. PRJNA789749.
Funding Statement: This work was supported by the Natural Science Foundation of China (Nos. 81870747, 82170916), Clinical Medicine Plus X–Young Scholars Project (PKU2021LCXQ003), and the Fundamental Research Funds for the Central Universities (BMU2021YJ001).
Conflicts of Interest: The authors declare that they have no conflicts of interest regarding the present study.
References
Andrae J, Gallini R, Betsholtz C (2008). Role of platelet-derived growth factors in physiology and medicine. Genes & Development 22: 1276–1312. DOI 10.1101/gad.1653708. [Google Scholar] [CrossRef]
Athar M, Li C, Kim AL, Spiegelman VS, Bickers DR (2014). Sonic Hedgehog signaling in basal cell nevus syndrome. Cancer Research 74: 4967–4975. DOI 10.1158/0008-5472.CAN-14-1666. [Google Scholar] [CrossRef]
Athar M, Tang X, Lee JL, Kopelovich L, Kim AL (2006). Hedgehog signalling in skin development and cancer. Experimental Dermatology 15: 667–677. DOI 10.1111/j.1600-0625.2006.00473.x. [Google Scholar] [CrossRef]
Barnes EA, Kong M, Ollendorff V, Donoghue DJ (2001). Patched1 interacts with cyclin B1 to regulate cell cycle progression. The Embo Journal 20: 2214–2223. DOI 10.1093/emboj/20.9.2214. [Google Scholar] [CrossRef]
Bille C, Winther JF, Bautz A, Murray JC, Olsen J, Christensen K (2005). Cancer risk in persons with oral cleft--a population-based study of 8,093 cases. American Journal Epidemiology 161: 1047–1055. DOI 10.1093/aje/kwi132. [Google Scholar] [CrossRef]
Carballo GB, Honorato JR, de Lopes GPF, Spohr T (2018). A highlight on Sonic hedgehog pathway. Cell Communication and Signaling 16: 11. DOI 10.1186/s12964-018-0220-7. [Google Scholar] [CrossRef]
de Cruz R, Horev L, Green J, Babay S, Sladden M, Zlotogorski A, Sinclair R (2012). A novel monilethrix mutation in coil 2A of KRT86 causing autosomal dominant monilethrix with incomplete penetrance. The British Journal of Dermatology 166: 20–26. DOI 10.1111/j.1365-2133.2012.10861.x. [Google Scholar] [CrossRef]
Dietz A, Pedersen DA, Jacobsen R, Wehby GL, Murray JC, Christensen K (2012). Risk of breast cancer in families with cleft lip and palate. Annals of Epidemiology 22: 37–42. DOI 10.1016/j.annepidem.2011.09.003. [Google Scholar] [CrossRef]
Fragoulis GE, Daoussis D, Pagkopoulou E, Garyfallos A, Kitas GD, Dimitroulas T (2020). Cancer risk in systemic sclerosis: Identifying risk and managing high-risk patients. Expert Review of Clinical Immunology 16: 1105–1113. DOI 10.1080/1744666X.2021.1847641. [Google Scholar] [CrossRef]
Gilchrist EP, Moyer MP, Shillitoe EJ, Clare N, Murrah VA (2000). Establishment of a human polyclonal oral epithelial cell line. Oral Surgery, Oral Medicine, Oral Pathology, Oral Radiology, Endodontics 90: 340–347. DOI 10.1067/moe.2000.107360. [Google Scholar] [CrossRef]
Gong X, Qian H, Cao P, Zhao X, Zhou Q, Lei J, Yan N (2018). Structural basis for the recognition of Sonic Hedgehog by human Patched1. Science 361: 2584–2586. DOI 10.1126/science.aas8935. [Google Scholar] [CrossRef]
Jiang J, Hui CC (2008). Hedgehog signaling in development and cancer. Developmental Cell 15: 801–812. DOI 10.1016/j.devcel.2008.11.010. [Google Scholar] [CrossRef]
Kurosaka H, Iulianella A, Williams T, Trainor PA (2014). Disrupting hedgehog and WNT signaling interactions promotes cleft lip pathogenesis. The Journal of Clinical Investigation 124: 1660–1671. DOI 10.1172/JCI72688. [Google Scholar] [CrossRef]
Litvinov IV, Xie P, Gunn S, Sasseville D, Lefrançois P (2021). The transcriptional landscape analysis of basal cell carcinomas reveals novel signalling pathways and actionable targets. Life Science Alliance 4: 7–0651. DOI 10.26508/lsa.202000651. [Google Scholar] [CrossRef]
Liu J, Jiang J, Hui X, Wang W, Fang D, Ding L (2018). Mir-758-5p suppresses glioblastoma proliferation, migration and invasion by targeting ZBTB20. Cellular Physiology Biochemistry 48: 2074–2083. DOI 10.1159/000492545. [Google Scholar] [CrossRef]
Lv L, Chen P, Cao L, Li Y, Zeng Z et al.(2020). Discovery of a molecular glue promoting CDK12-DDB1 interaction to trigger cyclin K degradation. eLife 9. DOI 10.7554/eLife.59994. [Google Scholar] [CrossRef]
Mansilla MA, Cooper ME, Goldstein T, Castilla EE, Lopez Camelo JS, Marazita ML, Murray JC (2006). Contributions of PTCH gene variants to isolated cleft lip and palate. The Cleft Palate-Craniofacial Journal 43: 21–29. DOI 10.1597/04-169r.1. [Google Scholar] [CrossRef]
Menezes R, Marazita ML, Goldstein McHenry T, Cooper ME, Bardi K, Brandon C, Letra A, Martin RA, Vieira AR (2009). AXIS inhibition protein 2, orofacial clefts and a family history of cancer. Journal of American Dental Association 140: 80–84. DOI 10.14219/jada.archive.2009.0022. [Google Scholar] [CrossRef]
Metzis V, Courtney AD, Kerr MC, Ferguson C, Rondón Galeano MC, Parton RG, Wainwright BJ, Wicking C (2013). Patched1 is required in neural crest cells for the prevention of orofacial clefts. Human Molecular Genetics 22: 5026–5035. DOI 10.1093/hmg/ddt353. [Google Scholar] [CrossRef]
Ming JE, Kaupas ME, Roessler E, Brunner HG, Golabi M, Tekin M, Stratton RF, Sujansky E, Bale SJ, Muenke M (2002). Mutations in PATCHED-1, the receptor for SONIC HEDGEHOG, are associated with holoprosencephaly. Human Genetics 110: 297–301. DOI 10.1007/s00439-002-0695-5. [Google Scholar] [CrossRef]
Nanashima N, Horie K, Yamada T, Shimizu T, Tsuchida S (2017). Hair keratin KRT81 is expressed in normal and breast cancer cells and contributes to their invasiveness. Oncology Reports 37: 2964–2970. DOI 10.3892/or.2017.5564. [Google Scholar] [CrossRef]
Nishi M, Miyake H, Takeda T, Hatae Y (2000). Congenital malformations and childhood cancer. Medical and Pediatric Oncology 34: 250–254. DOI 10.1002/(sici)1096-911x(200004)34:4<250::aid-mpo3>3.0.co;2-w. [Google Scholar] [CrossRef]
Northcott PA, Robinson GW, Kratz CP, Mabbott DJ, Pomeroy SL et al. (2019). Medulloblastoma. Nature Reviews. Disease Primers 5: 11. DOI 10.1038/s41572-019-0063-6. [Google Scholar] [CrossRef]
Noubissi FK, Yedjou CG, Spiegelman VS, Tchounwou PB (2018). Cross-talk between Wnt and Hh signaling pathways in the pathology of basal cell carcinoma. International Journal of Environmental Research Public Health 15: 7–1442. DOI 10.3390/ijerph15071442. [Google Scholar] [CrossRef]
Ribeiro LA, Murray JC, Richieri-Costa A(2006). PTCH mutations in four Brazilian patients with holoprosencephaly and in one with holoprosencephaly-like features and normal MRI. American Journal of Medical Genetics A 140: 2584–2586. DOI 10.1002/ajmg.a.31369. [Google Scholar] [CrossRef]
Ribes V, Briscoe J (2009). Establishing and interpreting graded Sonic Hedgehog signaling during vertebrate neural tube patterning: The role of negative feedback. Cold Spring Harbor Perspectives in Biology 1: a002014. DOI 10.1101/cshperspect.a002014. [Google Scholar] [CrossRef]
Robles AI, Ryan BM (2016). KRT81 miR-SNP rs3660 is associated with risk and survival of NSCLC. Annals of Oncology 27: 360–361. DOI 10.1093/annonc/mdv552. [Google Scholar] [CrossRef]
Sellheyer K (2011). Basal cell carcinoma: Cell of origin, cancer stem cell hypothesis and stem cell markers. The British Journal of Dermatology 164: 696–711. DOI 10.1111/j.1365-2133.2010.10158.x. [Google Scholar] [CrossRef]
Steinwachs EF, Amos C, Johnston D, Mulliken J, Stal S, Hecht JT (2000). Nonsyndromic cleft lip and palate is not associated with cancer or other birth defects. American Journal of Medical Genetics 90: 17–24. DOI 10.1002/(ISSN)1096-8628. [Google Scholar] [CrossRef]
Wend P, Wend K, Krum SA, Miranda-Carboni GA (2012). The role of WNT10B in physiology and disease. Acta Physiologica 204: 34–51. DOI 10.1111/j.1748-1716.2011.02296.x. [Google Scholar] [CrossRef]
Yang SH, Andl T, Grachtchouk V, Wang A, Liu J et al. (2008). Pathological responses to oncogenic Hedgehog signaling in skin are dependent on canonical Wnt/beta3-catenin signaling. Nature Genetics 40: 1130–1135. DOI 10.1038/ng.192. [Google Scholar] [CrossRef]
Yu FY, Hong YY, QuJF, Chen F, Li TJ (2014). The large intracellular loop of ptch1 mediates the non-canonical Hedgehog pathway through cyclin B1 in nevoid basal cell carcinoma syndrome. International Journal of Molecular Medicine 34: 507–512. DOI 10.3892/ijmm.2014.1783. [Google Scholar] [CrossRef]
Zhang J, Fan J, Zeng X, Nie M, Luan J, Wang Y, Ju D, Yin K (2021). Hedgehog signaling in gastrointestinal carcinogenesis and the gastrointestinal tumor microenvironment. Acta Pharmaceutica Sinica B 11: 609–620. DOI 10.1016/j.apsb.2020.10.022. [Google Scholar] [CrossRef]
Zhao H, Zhong W, Leng C, Zhang J, Zhang M et al. (2018). A novel PTCH1 mutation underlies nonsyndromic cleft lip and/or palate in a Han Chinese family. Oral Diseases 24: 1318–1325. DOI 10.1111/odi.12915. [Google Scholar] [CrossRef]
Zhong W, Zhao H, Huang W, Zhang M, Zhang Q et al. (2021). Identification of rare PTCH1 nonsense variant causing orofacial cleft in a Chinese family and an up-to-date genotype-phenotype analysis. Genes & Diseases 8: 689–697. DOI 10.1016/j.gendis.2019.12.010. [Google Scholar] [CrossRef]
Zhu JL, Basso O, Hasle H, Winther JF, Olsen JH, Olsen J (2002). Do parents of children with congenital malformations have a higher cancer risk? A nationwide study in Denmark. British Journal of Cancer 87: 524–528. DOI 10.1038/sj.bjc.6600488. [Google Scholar] [CrossRef]
Appendix

Cite This Article
 Copyright © 2023 The Author(s). Published by Tech Science Press.
Copyright © 2023 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools