
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE

Promotion of Growth of True Pulmonary Arteries as a First Step in Staged Repair of Pulmonary Atresia, Ventricular Septal Defect and Major Aorto-Pulmonary Collateral Arteries
1 Congenital Cardiothoracic Surgeon, Department of Cardiothoracic Surgery, Erasmus MC, Rotterdam, 3015 CE, The Netherlands
2 Pediatric Cardiology, Department of Pediatric Cardiology, Erasmus MC Sophia Childrens Hospital, Rotterdam, 3015 GJ, The Netherlands
3 Research Coordinatior, Department of Cardiothoracic Surgery, Erasmus MC, Rotterdam, 3015 CE, The Netherlands
* Corresponding Author: Pieter C. van de Woestijne. Email:
Congenital Heart Disease 2024, 19(6), 593-601. https://doi.org/10.32604/chd.2025.060607
Received 06 November 2024; Accepted 20 January 2025; Issue published 27 January 2025
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Background: Construction of a central shunt by connection of hypoplastic true pulmonary arteries to the ascending aorta (AO) can be performed as a first step in staged repair of pulmonary atresia with ventricular septal defect and major aorto-pulmonary collateral arteries (PA-VSD-MAPCAs) intended to promote growth and development of the central pulmonary arteries. Methods: To determine early and intermediate-term growth of true pulmonary arteries after their connection to the AO as a first step in staged repair of PA-VSD-MAPCAs, we reviewed all angiographic studies and CT imaging of patients, treated in our tertiary referral center in the last 26 years (1991 until 2017) for PA-VSD-MAPCAs with such a shunt. Results: A total of 13 patients (6 male) underwent direct end-to-side connection of the true pulmonary artery with the AO.). The absolute median size of both PAs showed statistically significant absolute growth over time. The absolute LPA growth was estimated to be 0.013 mm/day (p < 0.001). The absolute RPA growth was estimated to be 0.010 mm/day (p = 0.001). In total 9/13 (69%) reached total repair and 4 were considered not suitable for final repair. Conclusions: Creation of a central shunt by connection of true pulmonary arteries to the AO promotes their growth in patients with PA-VSD-MAPCAs, particular the LPA, although not reaching normal size. Patients with pulmonary atresia, and diminutive pulmonary arteries are a high-risk group, with a lower complete repair rate compared to those with better sized PAs. Alternative techniques or interventions should be explored in these patients to augment the size of the pulmonary arteries.Graphic Abstract
Keywords
In patients with pulmonary atresia with ventricular septal defect and major aorto-pulmonary collateral arteries (PA-VSD-MAPCAs), natural history studies showed very limited survival. Therefore several different strategies were described to achieve a normal biventricular situation with correction of the anomalies [1]. Successful complete repair in these patients depends on an adequate size of the central pulmonary arterial system [2–6]. The patients with diminutive pulmonary arteries may benefit from rehabilitation of those true pulmonary arteries (tPAs) as a first step in surgical management (Fig. 1) [2,6]. Tiny tPAs are typically connected to a normal pulmonary vascular bed with possibly potential for full growth [3,4]. In this regard, earlier surgical approaches included initial palliative surgical procedures to increase pulmonary blood flow and stimulate growth of the tPAS.
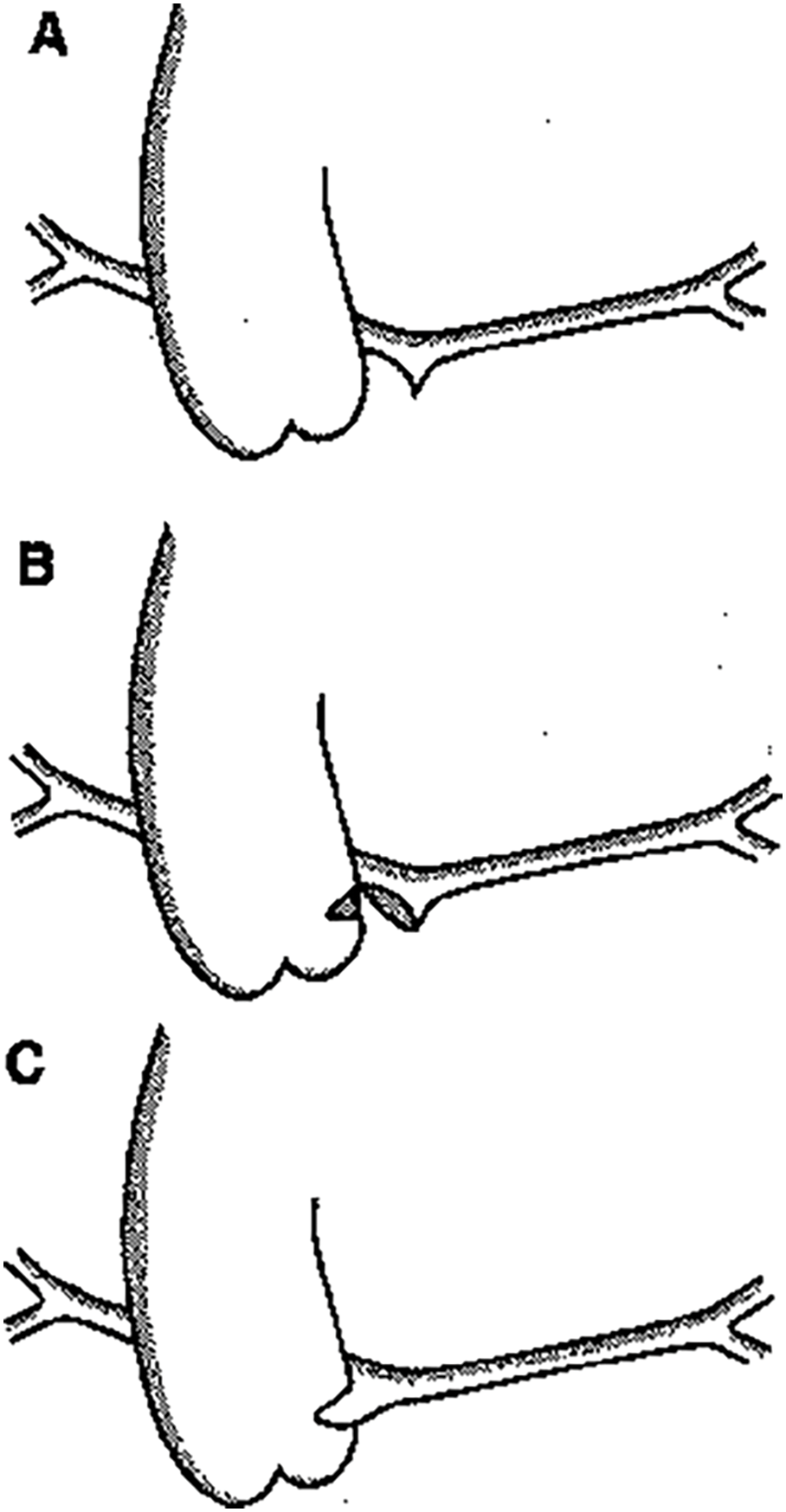
Figure 1: (A). The hypoplastic pulmonary trunk is detached from the heart as proximal as possible. (B). The pulmonary trunk is incised on the right lateral side. As posterior as possible a side-biting clamp is placed on the left dorsal side of the ascending aorta. A transverse incision is made in the aorta. (C). A direct end-to-side anastomosis between pulmonary trunk and ascending aorta is made with 7-0 Prolene®
Several surgical methods to connect the tPAs with the aorta have been described [5]. It was shown that direct end-to-side anastomosis of the main PA in the lateral wall of the aorta, also termed the Melbourne shunt [7] led to better growth of the true pulmonary arteries and to less thrombotic occlusion than connection of the tPAs with the aorta using a Gore-Tex central shunt [8,9].
In addition to complexity of the cardiac defect, co-morbidity due to concomitant genetic disorders [10–12] is associated with a high mortality as well, both in untreated [13] as well as in surgically treated patients [8,14].
To evaluate the growth of the tPAs in PA-VSD-MAPCAs after the Melbourne shunt, we reviewed all patients with diminutive PAs treated with this shunt in the Erasmus MC in Rotterdam, a tertiary referral center. The relation between genetic abnormalities on growth to the tPA and mortality was studied as this was described in other studies as a risk factor for unfavorable outcome [12,15].
All patients with PA-VSD-MAPCAs with diminuitive tPA’s were treated with construction of a central shunt (all Melbourne shunts) from January 1991 to December 2017. Thirteen patients were included in this study. Six of these patients were described before [16]. All patients were followed in the Erasmus MC Sophia Childrens Hospital, The Netherlands. The local Medical Ethical Committee approved this study with no need for informed consent (MEC 12-477).
Through median sternotomy the aorta and pulmonary artery were approached. The diminutive main pulmonary trunk and left and right PAs were mobilized. After heparinization the main pulmonary artery was transected as proximal as possible and the cardiac end was oversewn. The right lateral side of the pulmonary artery mainstem was incised into the origin of the right pulmonary artery. A side-biting clamp was positioned on the left lateral side of the ascending aorta and a lateral transverse incision is made. A direct anastomosis between pulmonary artery and aorta was made with 7.0 polypropylene. After declamping the aorta, antegrade flow in the pulmonary arterial system was checked by inspection and by perioperative TEE. Heparin was not antagonized.
Demographic and medical data were collected retrospectively from medical records. Follow up was complete. Angiographic studies and CT imaging (in 4 patients) were used to measure the size of right (RPA) and left pulmonary artery (LPA) just proximal to the origin of the first branch, see Fig. 2 [17]. Since 2015 (the last four patients in this cohort), we routinely applied CT imaging as first diagnostic tool instead of angiography in these patients, often in the newborn period. Measurements were done prior to the Melbourne shunt and prior to further unifocalization. The measurements at the two time-points were related to body size by calculating maximum pulmonary artery branch diameter in mm/body surface area in m2. Also z-scores were calculated before and after shunt surgery.
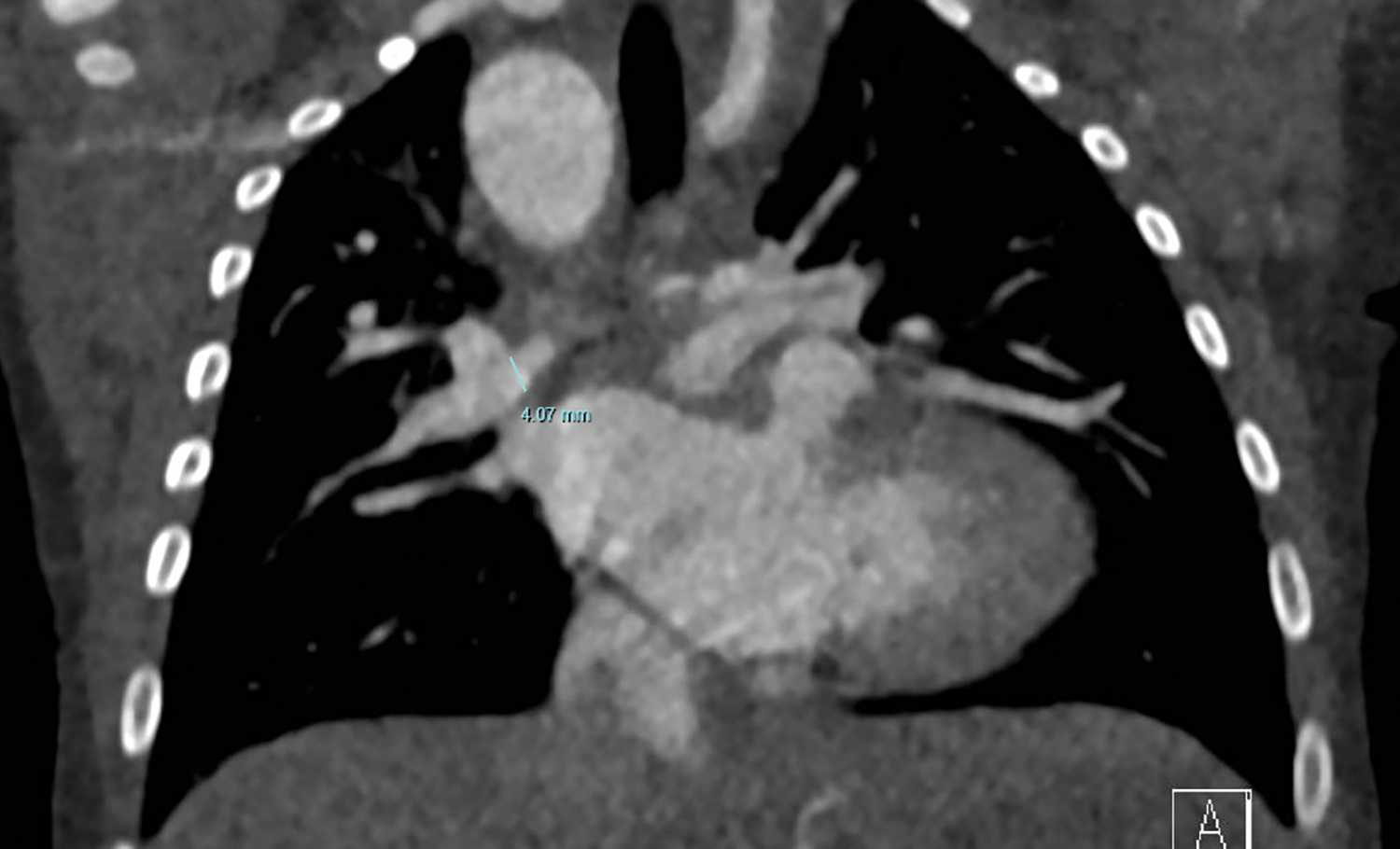
Figure 2: CT image with measurement of RPA
Continuous data were presented as median and range. Comparisons of continuous data were done with Student t-test (Gaussian) or Wilcoxon rank-sum (non-Gaussian). Categorical data are presented as counts with percentage. Comparisons were done with the chi-squared test or Fisher exact test, as appropriate. Pulmonary artery growth was modeled using a linear mixed-effect model, with random intercept and slope over time for patients, and time and other risk factors (if applicable) as fixed effects. Normality of residuals was assed using qqplots. Complete case analyses were utilized. A p-value of <0.05 was considered statistically significant.
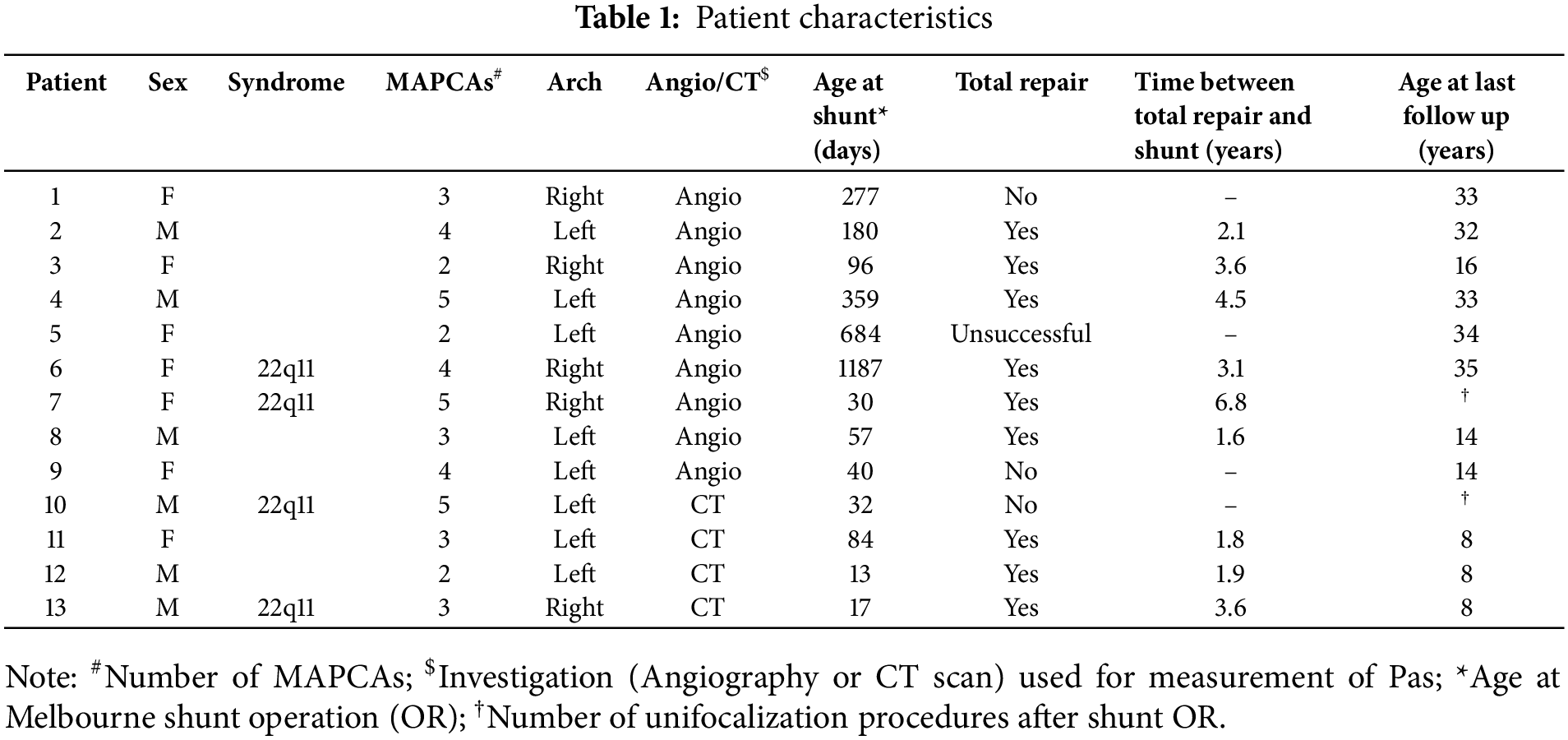
Thirteen patients were included in the study. Table 1 provides individual details of included patients. Four patients (31%) had associated genetic defects, being 22q11 deletion in all. There were 6 boys and 7 girls with a median weight at first study of 3.3 kg. Almost half of the patients (5/13) had a right sided aortic arch. The median age at first evaluation, prior to surgery, was 27 days (range 2–1126). The median age at time of the Melbourne shunt was 84 days (range 17–1187 days). Median time difference between the two measurements of the pulmonary arteries was 235 days (range 39–593 days). Patients with 22q11 had smaller BSA-indexed RPA (median: 1.45 vs. 2.8, p = 0.019) and LPA, but the latter did not reach statistical significance (median: 1.6 vs. 2.8, p = 0.07).
There was no early mortality after the shunt procedures.
4.1 Pulmonary Artery Absolute Growth
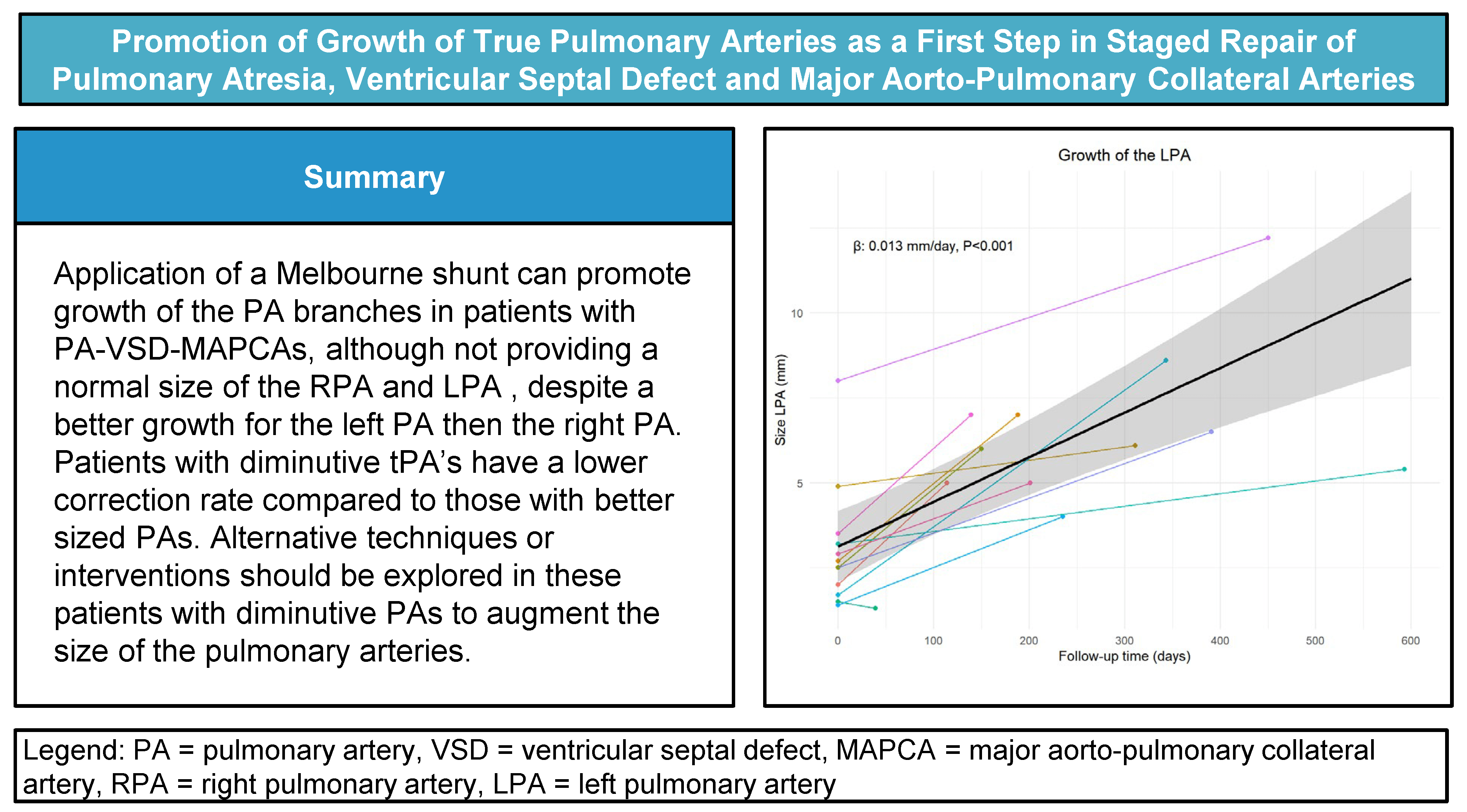
For the analyses of the pulmonary artery growth, in one patient with an absent left pulmonary artery was excluded from LPA analyses. Table 2 shows detailed PA measurements of all patients. Fig. 3A,B presents the individual growth and average absolute growth trajectories of the LPA (A) and the RPA (B). The absolute median size of both PAs showed statistically significant absolute growth over time. The absolute LPA growth was estimated to be 0.013 mm/day (CI: 0.006 to 0.016, p < 0.001), with a median LPA start diameter of 2.6 mm (range 1.4–8 mm) and 6.1 mm (range 1.3–12.2) at last measurement (Fig. 3A). The absolute RPA growth was estimated to be 0.010 mm/day (CI: 0.005 to 0.015, p = 0.001), with a median RPA start diameter of 2.5 mm (range 1.4–6 mm) and 4.4 mm (range 1.3–10.1) at last measurement (Fig. 3B). Absolute growth did not differ significantly between patients with and without 22q11 syndrome in both LPA (no syndrome: 0.015 mm/day vs. 22q11: 0.013 mm/day, Pinteraction = 0.80) and RPA (no syndrome: 0.014 mm/day vs. 22q11: 0.006 mm/day, Pinteraction = 0.14).
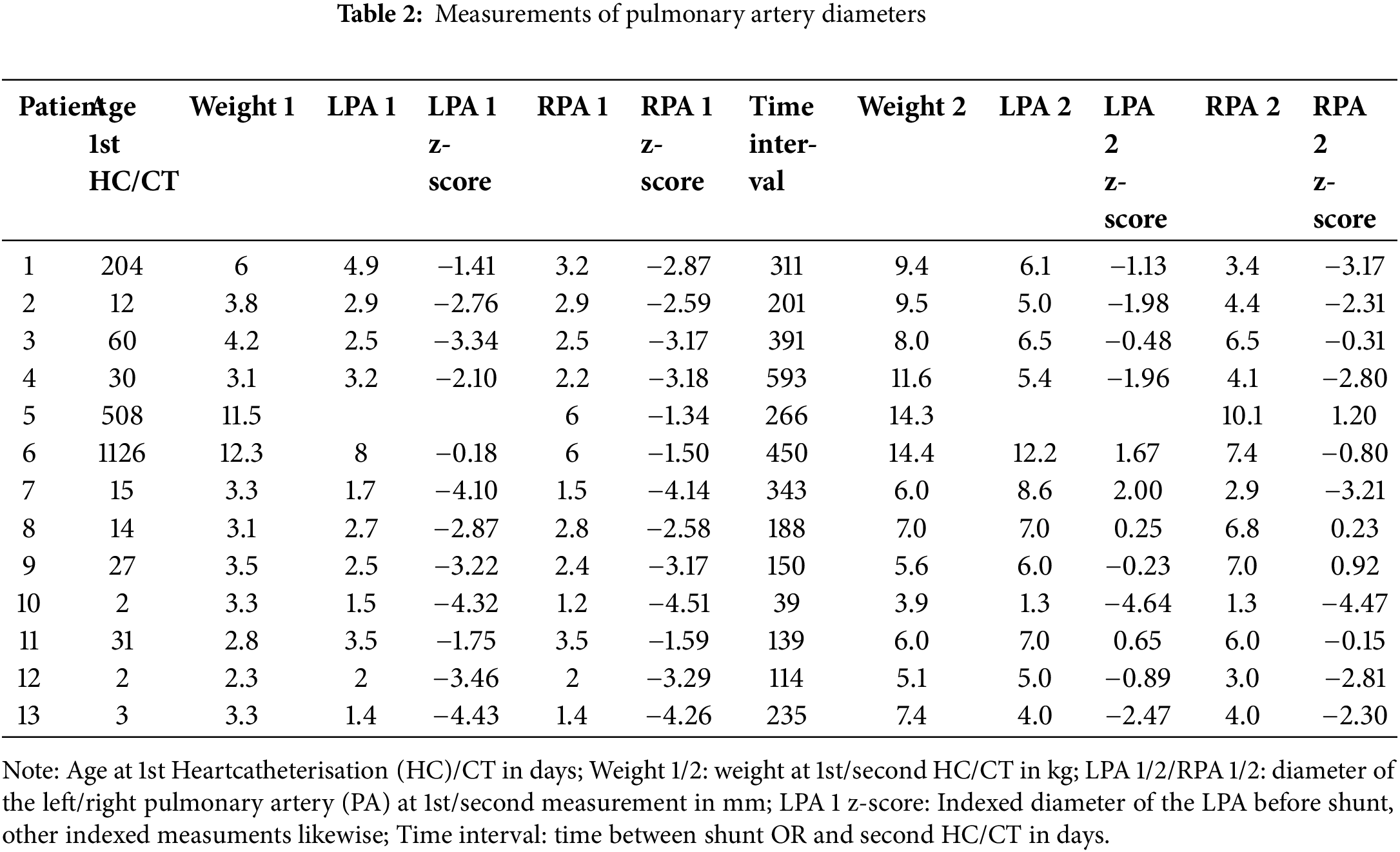
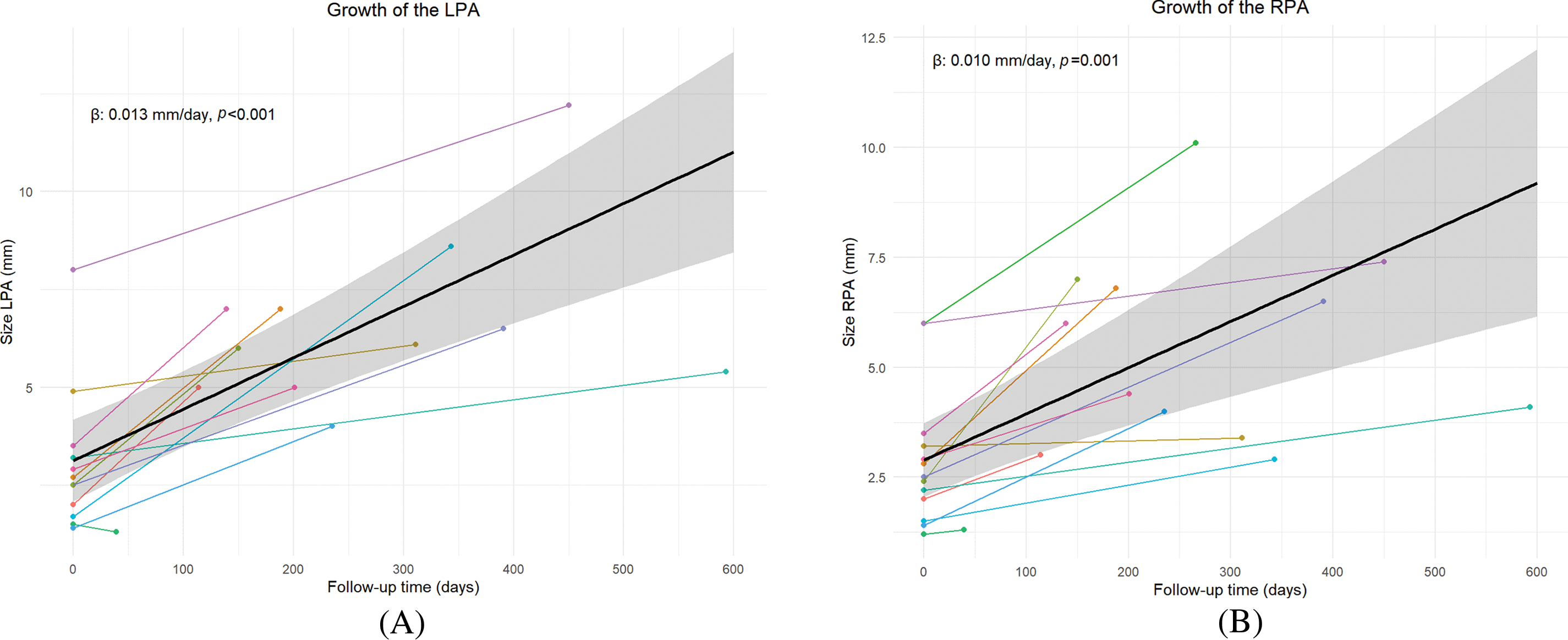
Figure 3: Growth of the LPA (A) and RPA (B) as estimated by the linear mixed model (black line). Colored lines depict individual patient trajectories
4.2 Pulmonary Artery BSA-Indexed Growth
Related to body size there was still growth of LPA estimated as 0.017 mm/m2/day (95% CI: (0.003 to 0.032), p = 0.024, with a median at last measurement of 16.9 mm/m2 (range 5.4–24.6 mm/m2), compared to 12.2 mm/m2 (range 6.7–17.5 mm/m2). For the RPA no significant indexed growth was observed with estimated log-transformed growth of 0.001 mm/m2/day (95% CI: −0.0003 to 0.001), p = 0.20, with at last measurement a median of 10.8 mm/m2 (range 5.4–21.9 mm/m2), compared to 10.7 mm/m2 (range 5.7–17.5 mm/m2) (Fig. S1A,B). Related to body surface area, median growth after Melbourne shunt was 28% and 1% for the LPA and RPA, respectively.
4.3 Pulmonary Artery Z-Score Growth
Compared to normal z-scores for LPA and RPA indexed to body size [17], most of the PAs (7/12 for LPA and 9/13 for RPA) did not reach normal size at last measurement (Table 2). Yet, both the z-scores of the LPA (0.009 z-score/day CI: 0.004 to 0.014, p = 0.002) and RPA (0.007 z-score/day CI: 0.002 to 0.011, p = 0.008) increased significantly over time (Fig. S2A,B).
There was no early mortality. Two patients, both with 22q11 deletion died during follow up. One patient developed a major cerebral bleeding the day after correction. The other patient died at age 2.5 years, being not suitable for correction based on his diminutive pulmonary vascular bed and pulmonary hypertension. He died from respiratory failure during a severe pneumonia.
4.5 Follow Up and Complete Repair
During follow up complete records of all patients were available. All patients received (bilateral) unifocalization and subsequent evaluation. During the study period 4 were deemed unrepairable, one of them had 22q11 deletion. These patients occurred throughout the study period. Nine patients (9/13, 69%) have undergone complete repair with implantation of a right ventricular to pulmonary artery (RV-PA) conduit including closure of the VSD.
Depending on the source of pulmonary circulation, PA-VSD- has a wide range of clinical presentations and treatment strategies. In the subgroup of patients with PA-VSD-MAPCAs and diminutive PAs, augmentation of pulmonary blood flow to promote pulmonary artery growth can lead to complete repair in 42%–60% [14,18,19]. In our small series 69% of the patients reached corrective surgery. This is comparable with the study of Mumtaz et al. in which the correction rate after Melbourne shunt was 63% [5].
The concept of creating antegrade flow in the native, even diminutive, true pulmonary arteries was first described in the 1990s [4]. By creating an end-to side connection between the ascending aorta and the diminutive tPAs, rehabilitation of these arteries, with distribution to most of the pulmonary segments, can develop adequately sized Pas [14]. The avoidance of a tube prosthesis in these small patients can have a potential benefit in promoting outgrowth of the tiny PAs without risk of distortion and no need for anticoagulation [9]. In this study, promotion of growth of tPAs was established in the majority of patients, particularly for the LPA, but growth did not result in a normal pulmonary artery size for either RPA or LPA. A study from Simsek et al. where tube prosthesis were used they reported adequate blood flow and growth in all but two patients and thrombosis in one patient [20].
The syndromic group, with less pronounced PA growth and complex collateral anatomy may have contributed to the higher incidence of not reaching complete repair. This underdevelopment and inability of adequate growth of PAs has been previously described by Mastromoro et al. [15]. and observational studies have shown more complex MAPCA anatomy in 22q11 patients compared with non-syndromic patients in the PA-VSD-MAPCA group [12]. The effect of underdeveloped bronchopulmonary arterial supply as an important determinant of mortality, achievement of definitive repair, and post-repair reoperation was previously described by Amark et al. [21].
In this study a significant difference in absolute PA growth could be demonstrated in the period between the preoperative imaging of the Melbourne shunt and the second evaluation before unifocalization. Although promotion of tPA growth (appearing better for the left PA than for the right PA) was demonstrated in this patient group, the rate of successful repair is lower compared to our previously published full cohort of patients of the spectrum of PA/VSD/MAPCAs, including those with better sized pulmonary arteries [22]. The difference between outgrowth of the left and right PA is still unclear but one aspect could be outgrowth of the aorta with compression on the right PA. Because of the small size of our series no (negative) relation between 22q11.2-deletion and the surgical outcome could be demonstrated. In the four patients who not achieved final correction we could speculate that a lack of sufficient antegrade blood flow may have contributed to this but in this study, we were not able to demonstrate this relationship.
Given the current high rate of prenatal diagnosis of congenital heart defects (CHDs), including of PA-VSD-MAPCAs, prenatal genetic diagnosis can improve prenatal counseling in cases with this severe CHD [23].
Application of a Melbourne shunt can promote growth of the PA branches in patients with PA-VSD-MAPCAs, although not providing a normal size of the RPA and LPA, despite a better growth for the left PA then the right PA. The correction rate in our series was 69%, in comparison with other series, patients with diminutive tPA’s have a lower correction rate compared to those with better sized PAs. Alternative techniques or interventions should be explored in these patients with diminutive Pas to augment the size of the pulmonary arteries.
Acknowledgement: Not applicable.
Funding Statement: The authors received no specific funding for this study.
Author Contributions: The authors confirm contribution to the paper as follows: Conceptualization, Pieter C van de Woestijne and Ad JJC Bogers; methodology, Pieter C van de Woestijne, Ad JJC Bogers and Kevin M Veen; software, Pieter C van de Woestijne and Kevin M Veen; validation, Pieter C van de Woestijne, Ad JJC Bogers, Kevin M Veen and Ingrid M van Beynum; formal analysis, Pieter C van de Woestijne and Kevin M Veen; investigation, Pieter C van de Woestijne, Kevin M Veen and Ingrid M van Beynum; resources, Pieter C van de Woestijne and Ingrid M van Beynum; data curation, Pieter C van de Woestijne and Ingrid M van Beynum; writing—original draft preparation, Pieter C van de Woestijne; writing—review and editing, Ad JJC Bogers, Ingrid M van Beynum and Kevin M Veen; visualization, Pieter C van de Woestijne and Kevin M Veen; supervision, Ad JJC Bogers and Ingrid M van Beynum; project administration Pieter C van de Woestijne; funding acquisition, NA. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: The authors confirm that the data supporting the findings of this study are available within the article and its Supplementary Materials.
Ethics Approval: All patients with PA-VSD-MAPCAs with diminuitive tPA’s were treated with construction of a central shunt (all Melbourne shunts) from January 1991 to December 2017. Thirteen patients were included in this study. Six of these patients were described before [16]. All patients were followed in the Erasmus MC Sophia Childrens Hospital, The Netherlands. The local Medical Ethical Committee approved this study with no need for informed consent (MEC 12-477).
Conflicts of Interest: The authors declare no conflict of interests to report regarding the present study.
Supplementary Materials: The supplementary material is available online at https://doi.org/10.32604/chd.2025.060607.
References
1. Ikai A. Surgical strategies for pulmonary atresia with ventricular septal defect associated with major aortopulmonary collateral arteries. Gen Thorac Cardiovasc Surg. 2018;66(7):390–7. doi:10.1007/s11748-018-0948-4. [Google Scholar] [PubMed] [CrossRef]
2. Chetaille P, Fraisse A, Ghez O, Kreitmann B, Voisin M, Aubert F, et al. Rehabilitation of hypoplastic pulmonary arteries and anatomic correction of pulmonary atresia with interventricular communication. Rehabilitation des arteres pulmonaires hypoplasiques et correction anatomique des atresies pulmonaires avec communication interventriculaire. Arch Mal Coeur Vaiss. 2001;94(5):446–51. doi:10.1016/s1010-7940(01)00855-7. [Google Scholar] [PubMed] [CrossRef]
3. Malhotra SP, Hanley FL. Surgical management of pulmonary atresia with ventricular septal defect and major aortopulmonary collaterals: a protocol-based approach. Semin Thorac Cardiovasc Surg Pediatr Card Surg Annu. 2009:145–51. doi:10.1053/j.pcsu.2009.01.017. [Google Scholar] [PubMed] [CrossRef]
4. Rome JJ, Mayer JE, Castaneda AR, Lock JE. Tetralogy of Fallot with pulmonary atresia. Rehabilitation of diminutive pulmonary arteries. Circulation. 1993;88(4):1691–8. doi:10.1161/01.cir.88.4.1691. [Google Scholar] [PubMed] [CrossRef]
5. Mumtaz MA, Rosenthal G, Qureshi A, Prieto L, Preminger T, Lorber R, et al. Melbourne shunt promotes growth of diminutive central pulmonary arteries in patients with pulmonary atresia, ventricular septal defect, and systemic-to-pulmonary collateral arteries. Ann Thorac Surg. 2008;85(6):2079–83; discussion 83–4. doi:10.1016/j.athoracsur.2008.01.098. [Google Scholar] [PubMed] [CrossRef]
6. Barozzi L, Brizard CP, Galati JC, Konstantinov IE, Bohuta L, d’Udekem Y. Side-to-side aorto-GoreTex central shunt warrants central shunt patency and pulmonary arteries growth. Ann Thorac Surg. 2011;92(4):1476–82. doi:10.1016/j.athoracsur.2011.05.105. [Google Scholar] [PubMed] [CrossRef]
7. Watterson KG, Wilkinson JL, Karl TR, Mee RB. Very small pulmonary arteries: central end-to-side shunt. Ann Thorac Surg. 1991;52(5):1132–7. doi:10.1016/0003-4975(91)91294-6. [Google Scholar] [PubMed] [CrossRef]
8. Fouilloux V, Bonello B, Kammache I, Fraisse A, Mace L, Kreitmann B. Management of patients with pulmonary atresia, ventricular septal defect, hypoplastic pulmonary arteries and major aorto-pulmonary collaterals: focus on the strategy of rehabilitation of the native pulmonary arteries. Arch Cardiovasc Dis. 2012;105(12):666–75. doi:10.1016/j.acvd.2012.08.003. [Google Scholar] [PubMed] [CrossRef]
9. Metras D, Chetaille P, Kreitmann B, Fraisse A, Ghez O, Riberi A. Pulmonary atresia with ventricular septal defect, extremely hypoplastic pulmonary arteries, major aorto-pulmonary collaterals. Eur J Cardiothorac Surg. 2001;20(3):590–6. doi:10.1016/S1010-7940(01)00855-7. [Google Scholar] [PubMed] [CrossRef]
10. Carotti A, Digilio MC, Piacentini G, Saffirio C, Di Donato RM, Marino B. Cardiac defects and results of cardiac surgery in 22q11.2 deletion syndrome. Dev Disabil Res Rev. 2008;14(1):35–42. doi:10.1002/ddrr.6. [Google Scholar] [PubMed] [CrossRef]
11. Chessa M, Butera G, Bonhoeffer P, Iserin L, Kachaner J, Lyonnet S, et al. Relation of genotype 22q11 deletion to phenotype of pulmonary vessels in tetralogy of Fallot and pulmonary atresia-ventricular septal defect. Heart. 1998;79(2):186–90. doi:10.1136/hrt.79.2.186. [Google Scholar] [PubMed] [CrossRef]
12. Mahle WT, Crisalli J, Coleman K, Campbell RM, Tam VK, Vincent RN, et al. Deletion of chromosome 22q11.2 and outcome in patients with pulmonary atresia and ventricular septal defect. Ann Thorac Surg. 2003;76(2):567–71. doi:10.1016/S0003-4975(03)00516-2. [Google Scholar] [PubMed] [CrossRef]
13. Bull K, Somerville J, Ty E, Spiegelhalter D. Presentation and attrition in complex pulmonary atresia. J Am Coll Cardiol. 1995;25(2):491–9. doi:10.1016/0735-1097(94)00364-V. [Google Scholar] [PubMed] [CrossRef]
14. Dragulescu A, Kammache I, Fouilloux V, Amedro P, Metras D, Kreitmann B, et al. Long-term results of pulmonary artery rehabilitation in patients with pulmonary atresia, ventricular septal defect, pulmonary artery hypoplasia, and major aortopulmonary collaterals. J Thorac Cardiovasc Surg. 2011;142(6):1374–80. doi:10.1016/j.jtcvs.2011.05.010. [Google Scholar] [PubMed] [CrossRef]
15. Mastromoro G, Calcagni G, Versacci P, Putotto C, Chinali M, Lambiase C, et al. Left pulmonary artery in 22q11.2 deletion syndrome. Echocardiographic evaluation in patients without cardiac defects and role of Tbx1 in mice. PLoS One. 2019;14(4):e0211170. doi:10.1371/journal.pone.0211170. [Google Scholar] [PubMed] [CrossRef]
16. de Jong PL, van de Woestijne PC, Witsenburg M, Bogers A. The effect of a central aortopulmonary shunt in staged repair of pulmonary atresia, ventricular septal defect and systemic-pulmonary collateral arteries. CVE. 1999;4(1):27–9. [Google Scholar]
17. Sievers HH, Onnasch DG, Lange PE, Bernhard A, Heintzen PH. Dimensions of the great arteries, semilunar valve roots, and right ventricular outflow tract during growth: normative angiocardiographic data. Pediatr Cardiol. 1983;4(3):189–96. doi:10.1007/BF02242254. [Google Scholar] [PubMed] [CrossRef]
18. Wang X, Lu Z, Li S, Yan J, Yang K, Wang Q. Systemic to pulmonary artery versus right ventricular to pulmonary artery shunts for patients with pulmonary atresia, ventricular septal defect, and hypoplastic pulmonary arteries. J Card Surg. 2015;30(11):840–5. doi:10.1111/jocs.12634. [Google Scholar] [PubMed] [CrossRef]
19. Zhang Y, Hua Z, Yang K, Zhang H, Yan J, Wang X, et al. Outcomes of the rehabilitative procedure for patients with pulmonary atresia, ventricular septal defect and hypoplastic pulmonary arteries beyond the infant period. Eur J Cardiothorac Surg. 2014;46(2):297–303. doi:10.1093/ejcts/ezt622. [Google Scholar] [PubMed] [CrossRef]
20. Simsek B, Ozyuksel A, Saygi M, Demiroluk S, Basaran M. Revisiting the central aortopulmonary shunt procedure. Turk Gogus Kalp Damar Cerrahisi Derg. 2023;31(2):207–14. doi:10.5606/tgkdc.dergisi. [Google Scholar] [CrossRef]
21. Amark KM, Karamlou T, O’Carroll A, MacDonald C, Freedom RM, Yoo SJ, et al. Independent factors associated with mortality, reintervention, and achievement of complete repair in children with pulmonary atresia with ventricular septal defect. J Am Coll Cardiol. 2006;47(7):1448–56. doi:10.1016/j.jacc.2005.10.068. [Google Scholar] [PubMed] [CrossRef]
22. van de Woestijne P, Mokhles M, van Beynum I, de Jong P, Wilschut J, Bogers A. Staged correction of pulmonary atresia, ventricular septal defect, and collateral arteries. J Card Surg. 2022;37(4):960–6. doi:10.1111/jocs.16299. [Google Scholar] [PubMed] [CrossRef]
23. van Nisselrooij AEL, Lugthart MA, Clur SA, Linskens IH, Pajkrt E, Rammeloo LA, et al. The prevalence of genetic diagnoses in fetuses with severe congenital heart defects. Genet Med. 2020;22(7):1206–14. doi:10.1038/s41436-020-0791-8. [Google Scholar] [PubMed] [CrossRef]
Cite This Article
 Copyright © 2024 The Author(s). Published by Tech Science Press.
Copyright © 2024 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools