
 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
REVIEW

Multiscale Numerical Simulation of Dynamic Damage and Fracture in Metallic Materials: A Review
1 College of Mechanical Engineering, Chongqing University of Technology, Banan, China
2 School of Environment and Resources, Southwest University of Science and Technology, Mianyang, China
3 State Key Laboratory of Precision Welding and Joining of Materials and Structures, Harbin Institute of Technology, Harbin, China
4 Magnesium Research Center, Kumamoto University, Kumamoto, Japan
5 Faculty of Advanced Science and Technology, Kumamoto University, Kumamoto, Japan
* Corresponding Authors: Lusheng Wang. Email: ; Shaojie Gu. Email:
Computers, Materials & Continua 2026, 87(3), 1 https://doi.org/10.32604/cmc.2026.077091
Received 02 December 2025; Accepted 26 February 2026; Issue published 09 April 2026
 View Full Text
View Full Text Download PDF
Download PDFAbstract
This paper provides a comprehensive review of recent advances in multi-scale modeling for simulating dynamic damage and fracture in metallic materials, a critical area due to the widespread application of metals and their susceptibility to complex failure in engineering practice. The paper first outlines the mechanisms of damage evolution and crack propagation across different spatial and temporal scales. It then introduces commonly used simulation approaches spanning micro- to macro-scales for studying damage and fracture in metals, analyzing the evolution of mechanical properties from defect initiation to ultimate failure, and elucidating the underlying damage mechanisms at different scales. Finally, the review summarizes multi-scale coupling strategies and mechanisms, as well as the integration of machine learning (ML) into multi-scale frameworks. These advanced approaches are recognized as key tools for improving predictive accuracy and computational efficiency, facilitating the scalability of multi-scale damage modeling for metallic materials in large-scale engineering applications and digital twin platforms. This review aims to provide a theoretical foundation for future research toward more reliable, efficient, and predictive multi-scale modeling of metallic materials.Keywords
Metallic materials, due to their outstanding mechanical properties, high strength, and corrosion resistance, find extensive applications in aerospace, automotive manufacturing, naval engineering, energy systems, and infrastructure construction [1]. However, during service, they are frequently exposed to complex environments characterized by high temperatures, high pressures, corrosive media, and high-velocity impacts. These harsh conditions lead to the development of various microstructural features, including point defects, dislocation accumulation, nanoscale void formation, and microcrack initiation. Such changes can progressively result in plastic deformation, fatigue damage, and ultimately, fracture failure. This process displays a pronounced multiscale nature, ranging from atomic-scale lattice slip to macroscopic crack propagation [2–4]. Therefore, it is crucial to elucidate the key factors and governing mechanisms that underlie damage evolution in metallic materials to enhance our fundamental understanding of their dynamic failure behavior.
Under external loading, the structural characteristics of metallic materials evolve, and their fracture and damage behaviors exhibit pronounced scale effects [5–7]. This damage process spans from the nanoscale to the macroscale and involves multiple temporal and spatial hierarchies, making it a highly complex multiscale problem [8,9], as illustrated in Fig. 1. At the microscopic scale, the formation of point defects and vacancies provides the initial conditions for subsequent structural evolution. As the scale increases, these defects gradually evolve into dislocations, voids, and cracks, which grow through mutual interactions. Ultimately, cracks propagate rapidly at the macroscopic scale, leading to material failure [10–14]. Macroscopic experiments provide validation for microscopic mechanisms, while microscopic models offer theoretical support for predicting macroscopic performance. Therefore, conducting multiscale simulations of metallic materials—from microscopic defects to macroscopic cracks—and enabling the transfer of physical quantities across scales is essential for improving the reliability of damage and fracture predictions.
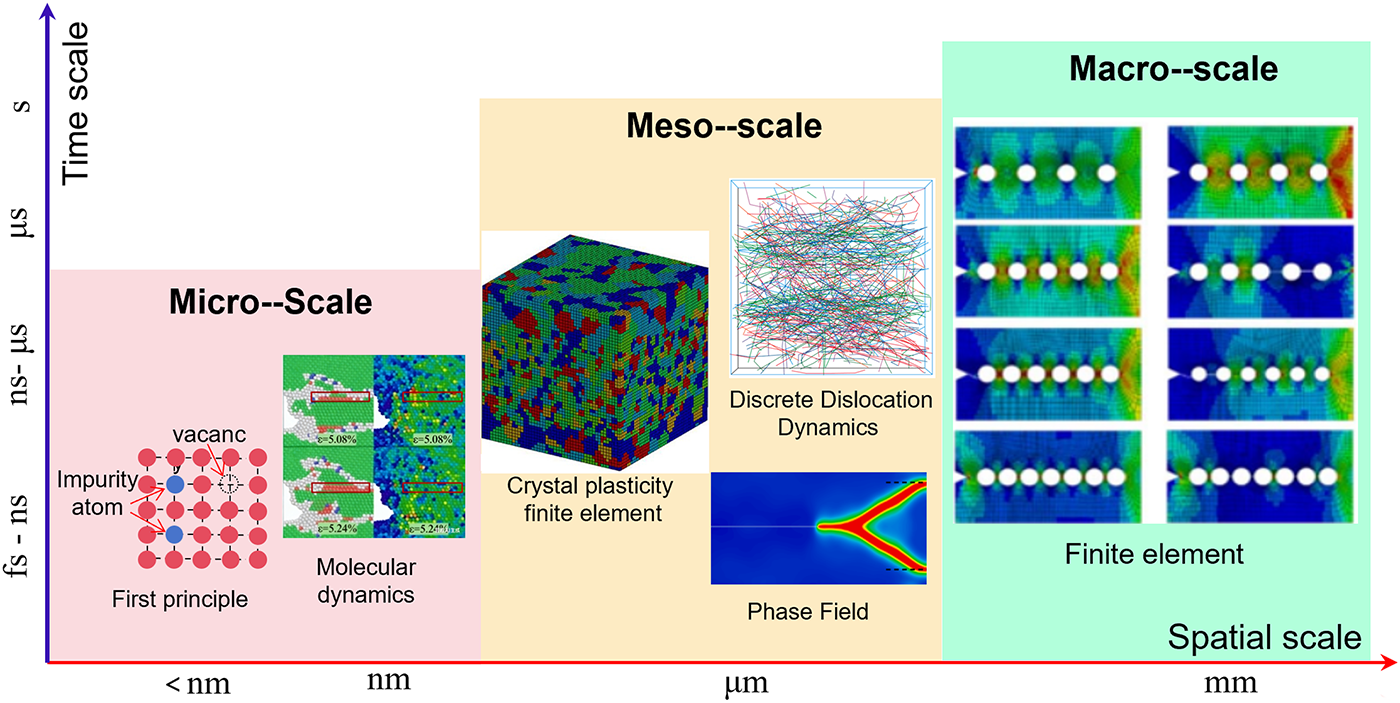
Figure 1: Representative deformation and damage features across different spatial and temporal scales. Insets are adapted from Refs. [15–19].
In recent years, with the advancement of computational capabilities, multiscale numerical simulations have been widely applied to study dynamic damage and fracture in metals. Fracture in metallic materials is not a single-scale phenomenon; it involves a progressive evolution of microscopic defects into macroscopic crack propagation, accompanied by complex changes in mechanical behavior. While single-scale simulations can partially verify the feasibility of parameter transfer, they are limited in capturing the dynamic coupling between microscopic defects and macroscopic stress fields during crack evolution. As shown in Fig. 2, multiscale simulation methods are generally divided into microscopic, mesoscopic, and macroscopic scales. At the microscopic scale, first-principles calculations provide key physical parameters such as elastic constants, defect formation energies, and interfacial binding energies, while molecular dynamics (MD) describes atomic motion and the nucleation and evolution of dislocations, nanopores, and cracks. At the mesoscopic scale, crystal plasticity finite element methods (CPFEM) and phase-field approaches simulate phase transformations and the evolution of micron-scale voids and cracks. At the macroscopic scale, finite element methods (FEM) analyze fracture patterns and performance of materials and structures. Because single-scale methods cannot fully capture the coupled multiscale and multi-physics nature of damage evolution, research in multiscale fracture mechanics is gradually shifting from single-scale analyses toward multiscale coupled modeling, offering new approaches for predicting material failure and assessing service life.
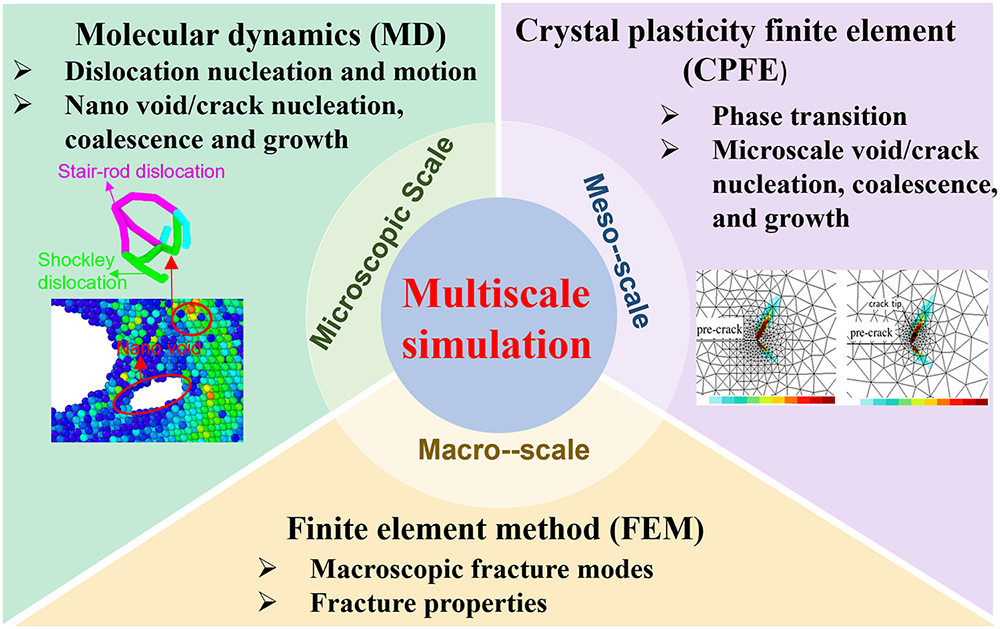
Figure 2: Multiscale simulation methods and their corresponding application domains.
Existing review studies primarily focus on multiscale theoretical frameworks or examine specific aspects of metallic material failure, such as damage evolution, fatigue failure, or macroscopic fracture modeling. However, comprehensive reviews addressing damage evolution and failure mechanisms of metallic materials across different scales—including under multiscale coupling conditions—are still limited. To address this gap, the present work provides a systematic review of the damage and fracture behaviors of metallic materials from a multiscale perspective. By examining mechanisms that operate across various spatial and temporal scales, this review aims to offer methodological guidance for elucidating failure mechanisms and pathways for improving lifetime prediction and structural optimization. Wu et al. [20] reviewed the mechanical behavior and life prediction of titanium alloys under very-high-cycle fatigue, analyzing crack initiation and propagation while clarifying the influence of micro-defects on fatigue life. Wang et al. [21] summarized approaches that integrate experiments with multiscale models to reveal damage evolution and enhance the accuracy of fatigue-life predictions. Karantza and Manolakos [22] investigated the formation, evolution, and fracture mechanisms of adiabatic shear bands in metals and alloys, discussing the roles of microstructure and thermal softening, the behaviors of different materials, and emphasizing the need for multiscale-coupled models to uncover the relationships between thermal softening, damage evolution, and macroscopic mechanical response, thereby providing a deeper understanding of dynamic failure mechanisms. Cheng et al. [23] reviewed damage mechanisms and multiscale modeling methods in composite materials, highlighting the importance of multiscale models in linking microstructural characteristics with macroscopic performance and identifying the integration of artificial intelligence and reduced-order modeling as a promising future direction.
This review systematically examines the multiscale simulation methods utilized to investigate the dynamic damage and fracture processes of metallic materials. The main content is organized into five sections. Section 2 introduces commonly used microscale simulation approaches and summarizes the corresponding damage mechanisms of metals at the atomic and nanoscale. Section 3 presents mesoscale modeling methods and outlines recent advancements in understanding damage evolution and fracture behavior at the micrometer scale. Section 4 discusses macroscale simulation techniques employed to evaluate deformation, damage progression, and fracture failure from a continuum perspective. Section 5 focuses on multiscale modeling strategies, with particular emphasis on the integration of multiscale methods with ML. Finally, the paper offers a comprehensive summary of current multiscale numerical simulation research and highlights potential directions for future development.
2 Numerical Simulation Methods at the Microscale
The evolution of defects in metallic materials at the microscale is challenging to observe directly using experimental techniques, thereby complicating the study of these defects. To elucidate the mechanisms of formation and evolution of microscale defects, researchers typically utilize numerical simulation methods, such as MD and first-principles FP calculations. These methods facilitate a detailed analysis of atomic interactions, defect formation processes, and crack nucleation behaviors, offering insights into the changes in the mechanical properties of metals. Moreover, integrating experimental results with computational simulations not only validates the accuracy of the models but also provides theoretical guidance for understanding macroscopic mechanical behavior.
2.1 First-Principles Calculation
Given that many properties of solid materials are inherently governed by electronic behavior, analyzing them at the electronic scale is crucial. FP calculations based on density functional theory (DFT) facilitate efficient atomistic-level simulations of essential electronic structure features, including band structures, density of states, and charge-density distributions [24]. The basic workflow of first-principles calculations is illustrated in Fig. 3, which can be divided into six stages from bottom to top:
I. Model construction and parameter selection: The computational model is established by defining the lattice parameters, atomic occupations, and crystallographic symmetry, providing the foundational structural description for subsequent simulations.
II. Geometry optimization: The lattice constants and atomic positions are optimized by appropriately selecting the k-point mesh density and convergence criteria, thereby relieving internal stresses and obtaining a stable equilibrium structure.
III. Electronic structure calculations: The electronic wave functions, total energy, charge density, and structural stability of the system are calculated to characterize its electronic properties.
IV. Result analysis and validation: The band structure, optical properties, elastic properties, and magnetic behavior of the material are analyzed to validate the simulation results and assess the predicted physical properties.
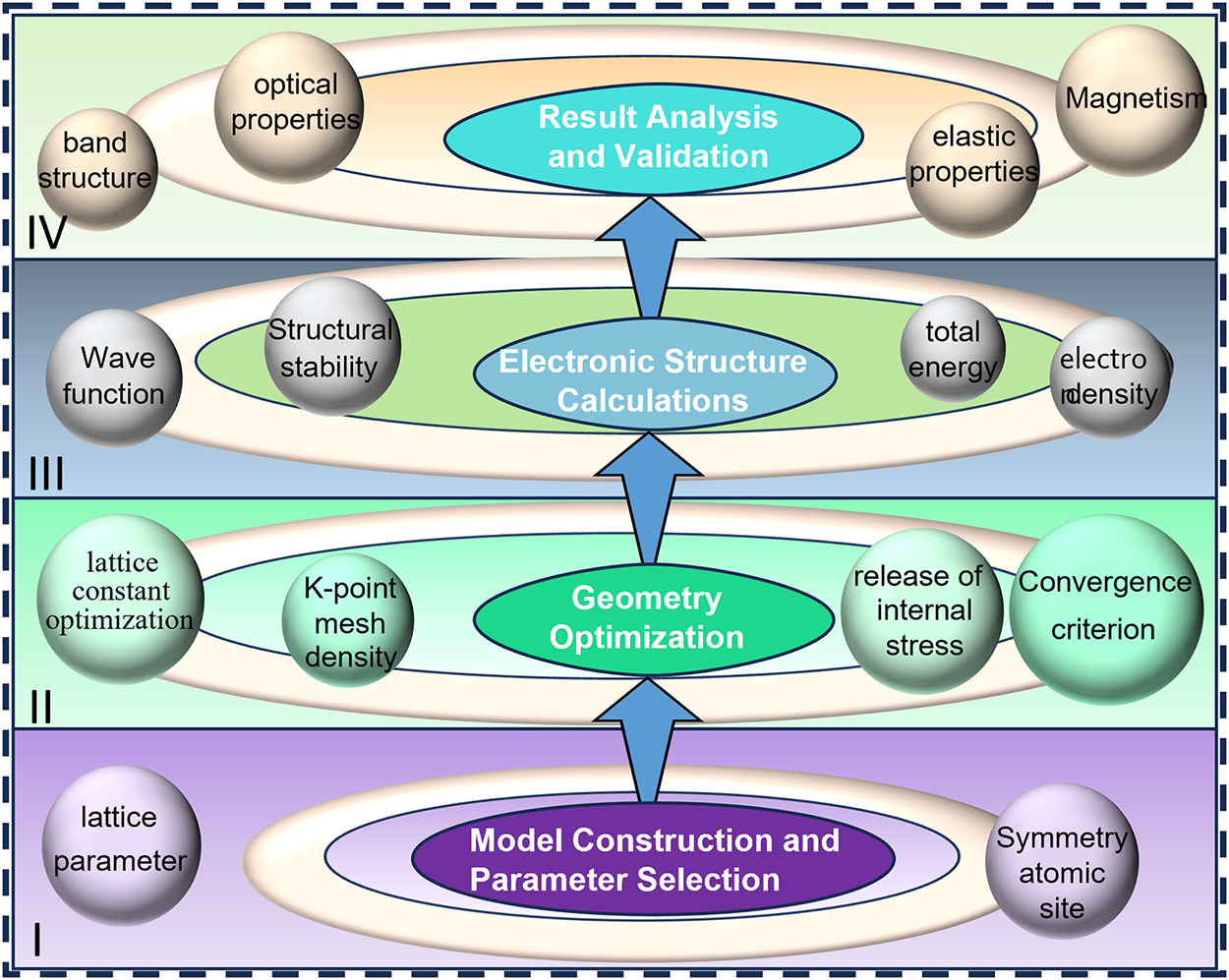
Figure 3: Flowchart illustrating the workflow of first-principles calculations.
First-principles calculations provide interatomic interaction potentials for molecular dynamics simulations, offering a robust theoretical foundation for studying material behavior at electronic and atomic scales. Furthermore, they supply essential physical parameters that facilitate the transfer of electronic-scale information to mesoscopic and macroscopic levels, thereby supporting parameter transfer and multiscale modeling.
Point defects are prevalent forms of microscopic damage in materials. They can arise intrinsically, such as from vacancies or self-interstitial atoms, or extrinsically through the incorporation of dopant atoms, and they significantly influence the mechanical properties of materials. These defects induce lattice distortions, alter interatomic bonding strengths, and affect the local mechanical stability of atoms, thereby modifying the overall mechanical behavior of the material [25]. Interstitial atoms in metals and hydrogen storage alloys exhibit specific site preferences; by modifying local bonding environments and electronic structure distributions, they can impact structural stability as well as mechanical properties such as strength and plasticity [26,27]. Mosquera-Lois et al. [28] combined first-principles calculations with ML to accurately identify stable defect configurations and elucidate how point defect-induced structural changes influence system energy and electronic structure. Dopant atoms, which form substitutional point defects by replacing host atoms in the lattice, can modulate local atomic occupancy and bonding characteristics, thereby affecting damage mechanisms and crack propagation behavior, ultimately enhancing the mechanical performance and damage tolerance of alloys [29,30]. In summary, point defects influence material strength, plasticity, and mechanical stability by altering lattice distortion, atomic bonding, and electronic structure. The integration of ML with first-principles calculations provides a novel and efficient approach for studying point defects. Fig. 4 illustrates the validation of ML based analyses in point defect studies, offering an effective pathway for investigating the impact of defects on material damage. Collectively, these studies demonstrate that point defects, as microscopic imperfections, profoundly affect mechanical properties, and the combination of artificial intelligence with first-principles methods enables rapid and accurate identification of stable defect configurations, thereby accelerating defect research.
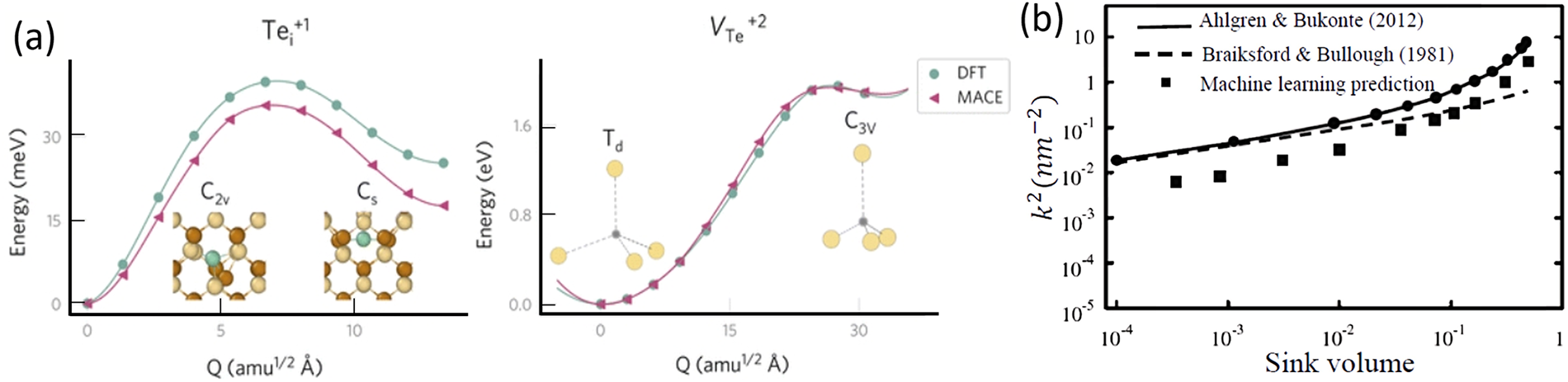
Figure 4: Validation of machine-learning approaches in point-defect studies: (a) comparison between defect-formation energies obtained from DFT and MACE calculations; (b) comparison of yield-strength predictions from machine-learning models and traditional approaches. Insets adapted from Refs. [31,32].
It is noteworthy that, in addition to point defects, interfaces—being a critical microstructural feature in metallic materials—play a key role in determining material properties. Due to discontinuities in atomic arrangement and variations in chemical composition, interfaces are prone to stress concentration, lattice distortion, and local energy fluctuations, which in turn influence dislocation motion, crack propagation, and phase transformation behavior. In nanostructured metals, interfaces possess unique structural characteristics and can serve as effective defect sinks, promoting the recombination of vacancies and interstitial atoms [33]. Moreover, the termination mode, stacking structure, and elemental segregation at interfaces affect interfacial bonding strength and fracture location, thereby governing whether cracks propagate along the interface or into the matrix [34–37]. First-principles and ML studies have further demonstrated that calculating interfacial segregation and binding energies can identify alloying elements that enhance interfacial cohesion and suppress undesirable interfacial reactions [38]. Such elements can optimize interfacial electronic structure and bonding, improving material toughness, reducing the formation of brittle phases, and effectively delaying damage and crack propagation. Taken together, interfacial fracture dominates the strength and toughness of metal composites and represents a critical stage in the damage evolution process, providing essential theoretical guidance for understanding interfacial failure mechanisms and designing toughened composites.
First-principles calculations play a pivotal role in multiscale modeling. As a method independent of empirical interatomic potentials, these calculations enable the determination of key physical parameters, including elastic constants, stacking-fault energies, twin-boundary energies, diffusion barriers, and interfacial bonding energies. Moreover, by capturing fundamental electronic-level interactions, they provide mechanistic insights into the origins of macroscopic mechanical behavior, thereby offering a robust theoretical foundation for materials design and property optimization. Ultimately, first-principles calculations serve not only as an essential tool for understanding the intrinsic properties of metallic materials but also as a critical driving force in advancing multiscale simulation frameworks.
2.2 Molecular Dynamics Simulation
In experimental studies, material damage and fracture are typically characterized at the macroscopic scale, while the fundamental mechanisms underlying these processes occur at the microscopic level. MD simulations facilitate the observation of defect formation, irradiation damage, crack propagation, and other phenomena at the atomic scale, thereby providing deeper insights into material damage and fracture behavior [39–41]. MD is a microscopic simulation method grounded in classical mechanics, specifically Newton’s equations of motion, to describe particle systems. The typical computational workflow, illustrated in Fig. 5, encompasses model parameter loading, simulation initialization, energy minimization, equilibration, and result output. In recent years, advancements in high-performance computing and the development of diverse interatomic potentials have propelled MD into a pivotal paradigm for multiscale scientific research, leading to its widespread application in studies of phase transformations, mechanical behavior, microstructural evolution, and irradiation damage in metallic materials.
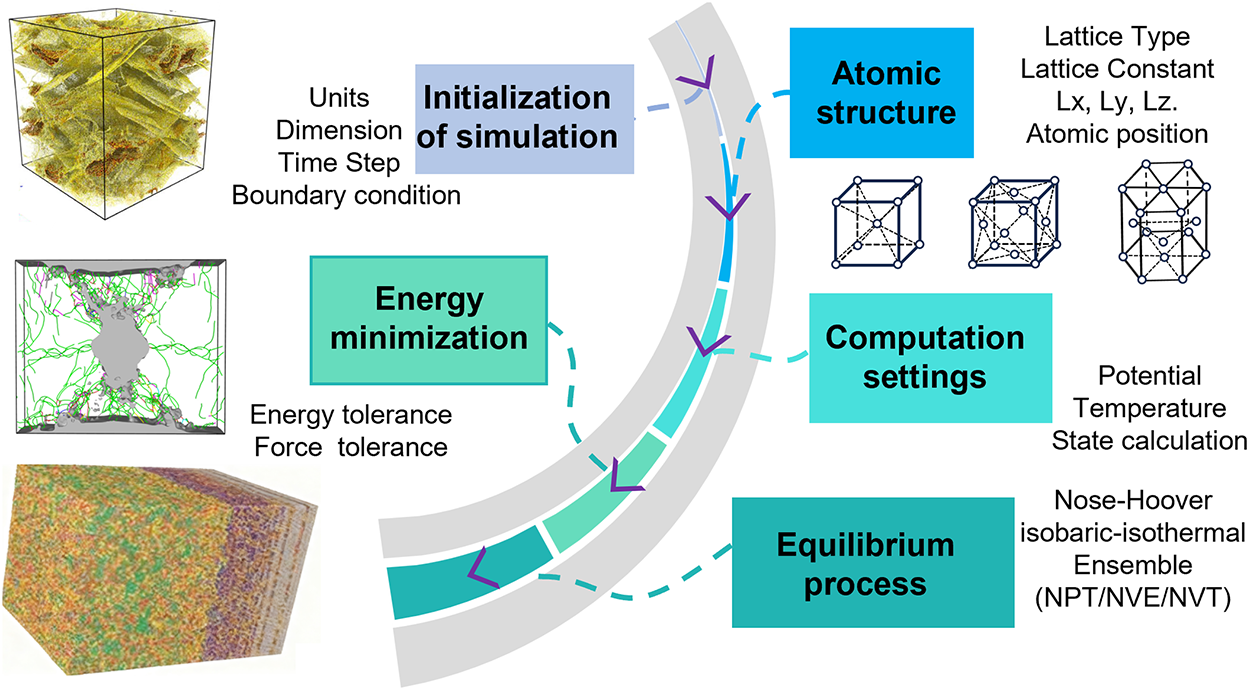
Figure 5: Workflow of molecular dynamics simulations. Insets adapted from Refs. [42–44].
Nano-defects, including nanoscale cracks and voids, represent the primary microscopic features governing material damage and fracture. Under applied external loads, voids gradually grow and coalesce, leading to the progressive accumulation of internal damage and significant local density reductions, with microscopic damage eventually developing into macroscopic fracture. As representative microscopic defects, nanoscale voids and cracks markedly influence the yield strength, crack nucleation, and propagation behavior of metallic materials through stress concentration and local structural alterations. Their evolution is closely associated with crack size, loading conditions, and anisotropic deformation mechanisms, thereby accelerating material damage and failure [45–49]. At the atomic scale, these defects dominate the mechanisms of damage evolution and failure, providing crucial theoretical insights for understanding material degradation and fatigue behavior under complex service conditions.
Microstructural features play a critical role in governing damage evolution as well as crack initiation and propagation in metallic materials. Liu et al. [50] investigated the influence of microstructure on fatigue crack growth in steels and observed that materials with more homogeneous microstructures exhibit lower crack growth rates, while the fatigue crack growth threshold decreases with increasing grain size. Yin et al. [15] demonstrated that the refinement and uniform dispersion of the γ’phase in nickel-based superalloys increase phase-boundary density, promote dislocation nucleation and emission, and consequently alleviate crack-tip stress concentration, thereby suppressing crack propagation. Xue et al. [51] further reported that crack growth behavior in the B2 phase of Ti2AlNb alloys strongly depends on crystallographic orientation, leading to ductile, brittle, or mixed-mode fracture. Their results indicate that crack-tip stress concentration can induce a BCC→HCP phase transformation and the formation of stacking faults, enhancing plastic deformation and improving toughness. Collectively, these studies highlight that microstructural characteristics critically influence mechanical performance by modulating crack propagation pathways and activating distinct plastic deformation mechanisms.
Irradiation damage serves as a significant source of damage in metallic materials, leading to the generation of numerous defects whose accumulation alters the microstructure and substantially degrades mechanical performance. Zhou et al. [52] employed MD simulations to demonstrate that helium bubbles influence the dynamic fracture behavior of aluminum; their expansion and coalescence are governed by dislocation activity, resulting in a reduction of dynamic tensile strength, with the weakening effect intensifying as the bubble volume fraction increases. Liu et al. [53] reported that helium bubbles preferentially agglomerate along the basal plane in α-Zr, with their size dictating variations in strength and ductility. These bubbles facilitate crack propagation along the basal plane and contribute to brittle fracture, thereby affecting the deformation and failure mechanisms of hexagonal close-packed (HCP) metals. Overall, irradiation-induced defects, particularly helium bubbles, reshape the microstructural landscape and modulate dislocation motion and crack-growth pathways. Their size, distribution, and evolution critically determine their influence on the deformation behavior, mechanical properties, and fracture response of irradiated metals.
In summary, the damage and fracture behavior of materials is governed by multiple micro-scale factors. Microdefects, such as nanovoids and nano-cracks, promote crack initiation and propagation through stress concentration, while microstructural features dictate deformation and fracture modes. Irradiation-induced defects, including helium bubbles, further alter the microstructure and degrade mechanical performance. Table 1 compares the characteristics of two representative microscale simulation methods: first-principles calculations and molecular dynamics. First-principles methods facilitate the analysis of defect formation at the electronic scale. however, they are computationally intensive and often lack direct experimental validation. Conversely, MD can elucidate phase transformations and dislocation mechanisms at the atomic scale, although its accuracy is heavily dependent on the selected interatomic potential. In recent years, ML has emerged as a powerful tool in materials science. Fig. 6 illustrates the integration of molecular dynamics with ML, which offers new pathways for predicting and elucidating the formation and evolution of microdefects in materials.
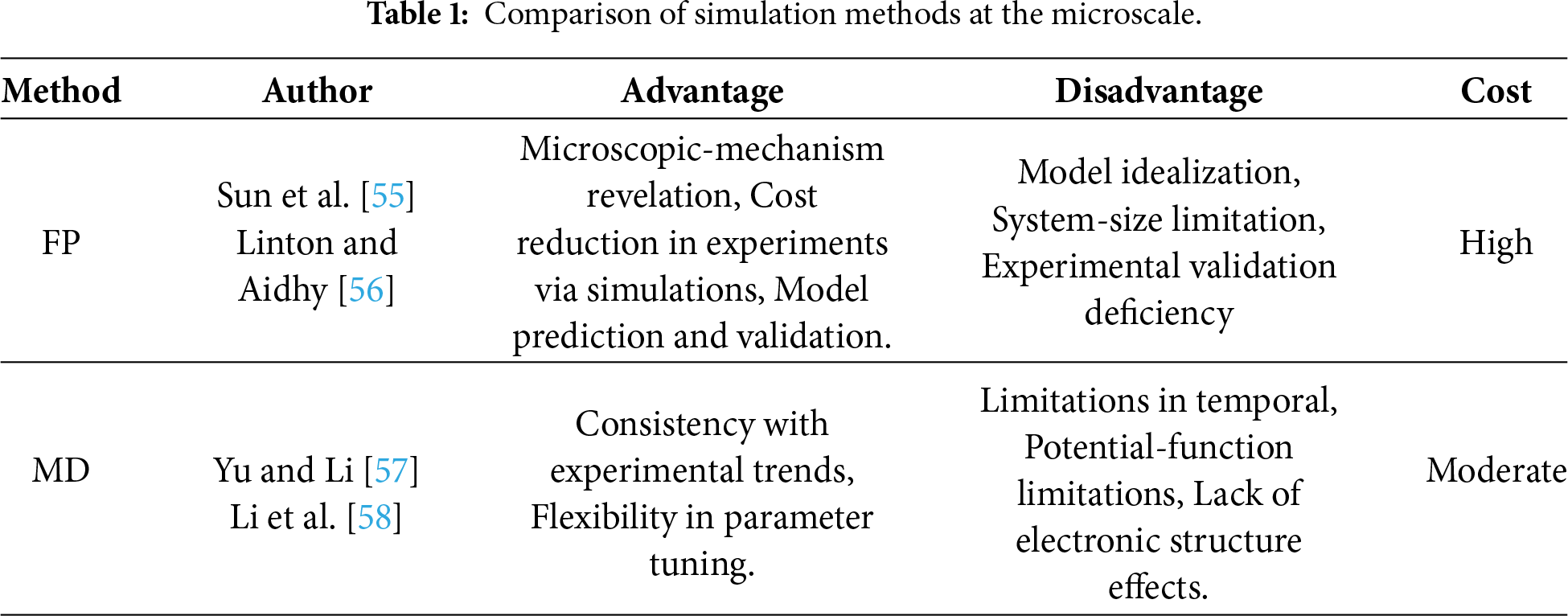
Figure 6: The workflow for molecular dynamics simulations alongside machine-learning-based predictions of the mechanical properties of high-entropy alloys. (a) The procedural steps involved in the molecular dynamics simulation. (b) The prediction of dislocation-related quantities utilizing both direct training methods and pretrained machine-learning models. The inset images have been adapted from reference [54].
Microscopic-scale numerical simulations have made significant progress in revealing the mechanisms of material damage and fracture, but several common challenges remain:
(1) High computational cost: First-principles calculations can accurately describe point defects, interfaces, and atomic bonding at the electronic level, but they are computationally expensive and limited in time and spatial scales.
(2) Limited system size and timescale for MD: MD can handle larger systems but struggles to capture long-term and large-scale damage evolution relevant to experiments.
(3) Difficulty in mapping to macroscopic behavior: While microscopic simulations can reveal the effects of point defects, nanopores, and cracks on crack propagation, effectively transferring this atomic-scale information to mesoscopic and macroscopic models remains a key challenge.
(4) Strong dependency on potentials: MD results are highly sensitive to the choice of interatomic potentials, and different potentials may lead to different predictions of defect evolution and mechanical response.
The future research directions of microscopic-scale simulations mainly focus on the following aspects:
(1) Cross-scale parameter transfer and model coupling: Incorporating physical parameters obtained from first-principles calculations into MD and higher-scale models to achieve a consistent description of damage mechanisms across scales.
(2) Integration of ML with numerical simulations: Using ML to accelerate defect configuration exploration and potential construction, significantly improving computational efficiency while maintaining accuracy.
(3) Damage studies under complex environments: Strengthening investigations of defect evolution and fracture behavior under coupled conditions such as irradiation and hydrogen exposure, providing support for critical applications in nuclear and aerospace materials.
3 Numerical Simulation Methods at the Mesoscopic Scale
During fatigue crack fracture, mesoscopic defects, such as micro-cracks and micro-voids continuously nucleate and accumulate in the vicinity of a macroscopic crack tip. The interaction between these initial defects and the propagating macro-crack constitutes a critical mechanism governing crack growth. These mesoscopic defects can induce material softening and local stress concentration, thereby altering the stress state around the crack tip [59]. To investigate their effects, various mesoscopic-scale numerical simulation methods have been developed. Discrete dislocation dynamics (DDD) facilitate the characterization of dislocation generation and motion; crystal plasticity finite element (CPFE) capture the influence of grain morphology and slip activity on deformation; and phase-field (PF) models enable the simulation of crack initiation and propagation. Collectively, these approaches provide robust tools for elucidating damage and fracture mechanisms from a mesoscopic perspective.
3.1 Discrete Dislocation Dynamics Simulation
Damage and fracture in metallic materials are highly complex processes rooted in the evolution of micro-scale defects, including lattice imperfections, dislocation activity, void coalescence, and the initiation and propagation of nanoscale cracks. Dislocations play a central role in plastic deformation, and their interactions with other defects largely determine a material’s strength and ductility. DDD grounded in dislocation elasticity theory, neglects atomistic details while explicitly resolving the nucleation, interaction, and motion of individual dislocations [60]. This approach enables DDD to capture the essential features of crystalline plasticity at the mesoscale. As a computational method that operates between atomistic and continuum scales, DDD directly simulates plastic deformation through dislocation evolution, thereby providing detailed insights into the mechanisms and kinetics of dislocation-mediated plasticity. Fig. 7 illustrates the primary workflow of a DDD simulation:
I. Dislocation initialization, during which dislocation lines are discretized into nodes and segments.
II. Force calculation on dislocation segments and nodes based on linear elasticity and dislocation theory.
III. Dislocation motion, where node velocities are determined using mobility laws, followed by time integration to update nodal positions.
IV. Result post-processing, which includes the evaluation of plastic strain generated by dislocation motion, stress–strain response, and the evolution of the dislocation network.
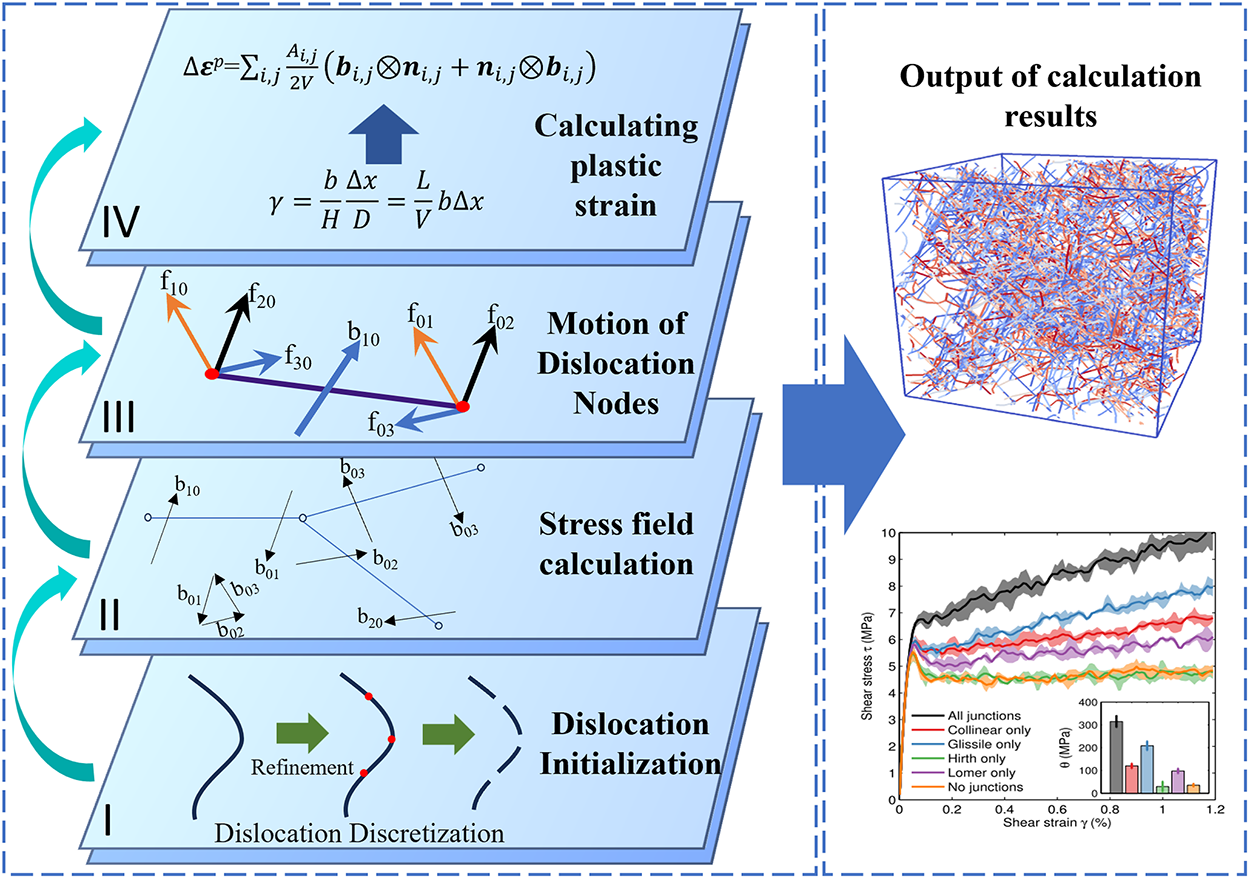
Figure 7: Workflow of a DDD simulation. Insets adapted from Refs. [61].
This section reviews recent advances in applying DDD to investigate the role of dislocations in the dynamic damage and fracture of metallic materials, highlighting the underlying mechanisms revealed through mesoscale simulations.
Advanced experimental characterization techniques are essential for revealing crack initiation and the evolution of surface morphology. However, microscale experiments typically capture only the post-deformation configuration of dislocations and microstructures, making it challenging to observe dislocation evolution and its interactions with other defects in a real-time, comprehensive, and in-situ manner. With the rapid development of computational technologies, numerical simulation approaches have become increasingly powerful. Among these, DDD offers distinct advantages in modeling dislocation microstructure evolution and damage accumulation, providing a crucial link between microscopic mechanisms and macroscopic fracture behavior. DDD enables the simulation of key processes such as dislocation density evolution, slip-system interactions, and cross-slip, all of which govern the yielding and strain-hardening behavior of single crystals. The simulation results can accurately capture the macroscopic mechanical response, even under high strain-rate loading conditions [62]. Li et al. [63] examined the compression behavior of Mg single crystals and found that strain hardening is primarily governed by immobile dislocations and dislocation loops generated by basal-plane slip transfer. These defects increase the dislocation density and create obstacles to dislocation motion, thereby controlling the hardening response. As temperature increases, thermally activated processes are enhanced, leading to a further increase in the hardening rate. Collectively, these studies demonstrate that DDD provides a systematic framework for elucidating how dislocation density evolution, slip-system interactions, and cross-slip govern yielding, strain hardening, and fatigue damage evolution in single crystals. They underscore the critical role of microscopic dislocation behavior in shaping macroscopic mechanical responses.
Compared to single-crystal materials, polycrystalline materials exhibit more complex mechanical responses and damage evolution due to their intricate microstructures. Grain boundaries serve not only as significant obstacles and regulators of dislocation motion but also interact with defects such as dislocations, vacancies, and micro voids, influencing the mechanisms of crack nucleation and propagation. In heterogeneous polycrystalline materials, for instance, micro void growth is governed by local dislocation density and dislocation accumulation: small voids grow slowly due to limited dislocation sources, while larger voids more readily propagate into nanoscale cracks [64,65]. Furthermore, in gradient nanocrystalline materials, the distribution of residual stresses affects dislocation nucleation and motion, thereby modifying yield strength and flow stress. The combined effects of gradient structures and non-uniform residual stresses alter dislocation–grain boundary interactions and dislocation density distributions, ultimately influencing damage evolution and crack propagation [66]. Tak et al. [67], through DDD simulations, elucidated the relationships among steady-state creep rate, stress, temperature, and grain size, demonstrating that dislocation pinning at grain boundaries and obstacles affects dislocation mobility, thereby impacting creep behavior and damage evolution. Fig. 8 illustrates the mechanical performance and deformation mechanisms of gradient nanocrystalline aluminum alloys studied via DDD, showing that dislocation accumulation at interfaces generates back stresses that induce synergistic strengthening, resulting in a substantial enhancement of material strength.
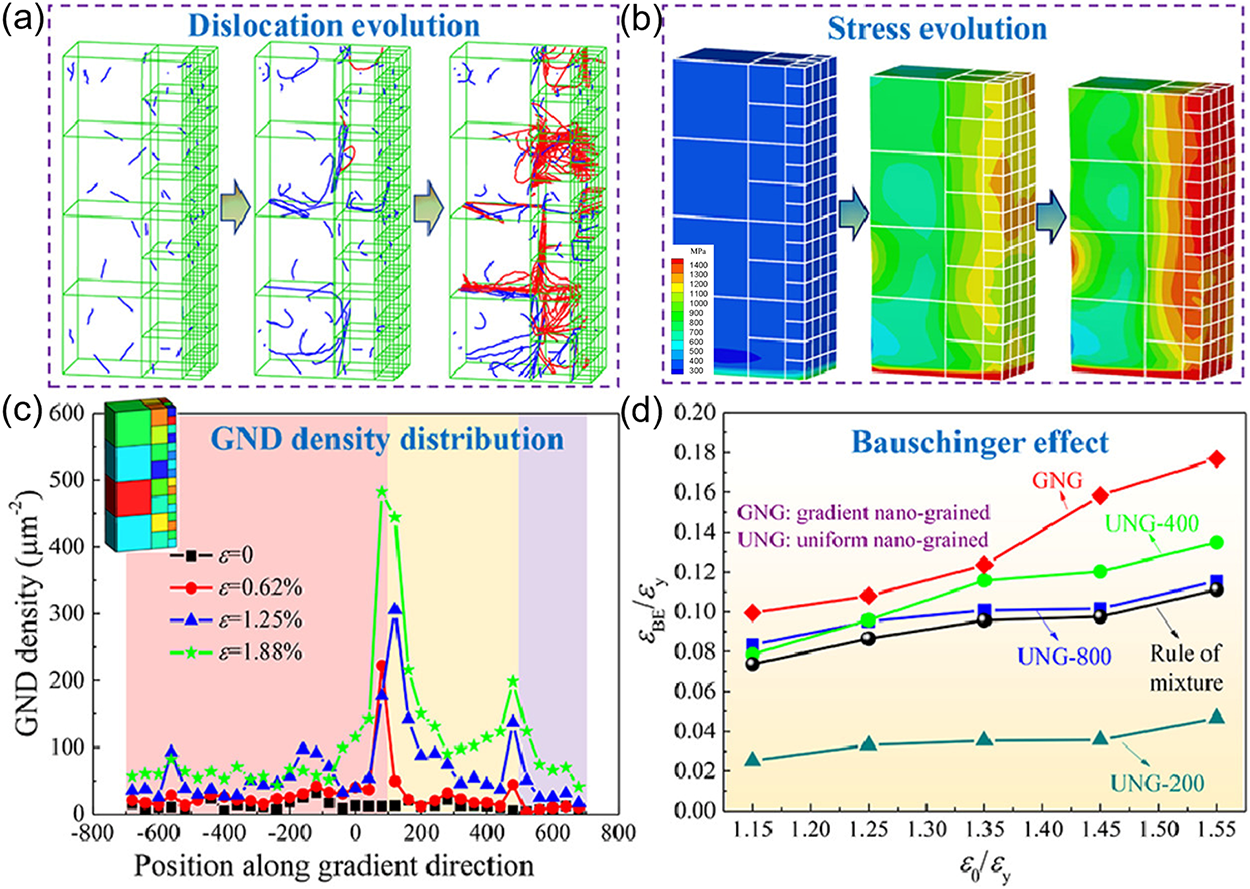
Figure 8: Microstructural evolution and microscopic mechanical responses of GNG and UNG samples with different grain sizes under monotonic compression. (a) Dislocation evolution, (b) stress evolution, (c) geometrically necessary dislocation (GND) density distribution, and (d) spatial variation of GND density along the gradient direction [68].
These studies systematically elucidate how dislocation density evolution, slip system interactions, and cross-slip in single-crystal materials microscopically regulate yield strength, strain hardening, and fatigue damage evolution, highlighting the critical influence of microscopic dislocation behavior on macroscopic mechanical responses. Overall, DDD demonstrates a unique advantage in revealing the complex mechanical behaviors of polycrystalline materials, including crack propagation, creep, and damage evolution.
In polycrystalline materials, the grain structure and dislocation activity are the primary factors governing mechanical behavior. Under irradiation, dislocation loops and other microstructural perturbations are generated, and the accumulation and interaction of these defects significantly alter the material’s mechanical response and damage evolution. Traditional experiments face challenges in directly resolving the dynamic interactions between microscopic dislocations and irradiation-induced defects; however, DDD simulations can effectively reveal the underlying mechanisms of these processes. Irradiation-induced defects, such as dislocations, vacancy clusters, and nanoscale voids, interact with dislocations to modify plastic deformation behavior, enhancing hardening, regulating local plasticity, and mitigating damage accumulation, while also influencing crack nucleation and propagation. Furthermore, material composition and defect characteristics modulate yield strength and strain localization [69–72]. DDD studies have elucidated the microscopic mechanisms by which irradiation defects control plasticity, hardening, and damage evolution, clarifying their role in governing crack nucleation and propagation.
DDD simulations have elucidated the microscale mechanisms through which dislocation behaviors regulate the mechanical properties and damage evolution of single crystals, polycrystals, and irradiated materials. In single crystals, the evolution of dislocations and interactions among slip systems govern yield strength and the initiation of microcracks. In polycrystalline materials, grain boundaries, residual stresses, and the distribution of micropores alter dislocation motion and crack propagation, thereby influencing damage accumulation. Following irradiation, the formation of dislocation loops, clusters, and nanovoids affects strain localization and cross-slip processes, modifying hardening behavior, retarding damage progression, and ultimately influencing crack-initiation paths. Overall, the DDD method facilitates microscale investigations of dislocation-defect interactions and establishes a quantitative link between microscopic mechanisms and macroscopic mechanical responses. It provides a robust theoretical basis for interpreting damage evolution and crack propagation while offering distinct advantages for materials design and performance prediction.
3.2 Crystal Plasticity Finite Element Method
With the deepening study of the micromechanical behavior and fracture mechanisms of metallic materials, it has become evident that damage at the micron scale plays a critical role in macroscopic failure. Consequently, the crystal plasticity finite element method (CPFEM) has emerged as a pivotal tool for investigating damage evolution at this scale. DDD effectively simulates the microscopic deformation of crystals under external loads, offering valuable insights into dislocation motion, slip, and the interactions that govern plastic deformation [73,74]. Through numerical simulation and analysis, CPFEM elucidates the fundamental behaviors and underlying mechanisms of crystal plasticity. By integrating crystal orientation and slip system constitutive relationships into the finite element framework, CPFEM accurately captures plastic deformation and local damage evolution under applied loads. As a result, it serves as a robust numerical approach for analyzing material mechanical performance and crack propagation. Fig. 9 illustrates the comprehensive numerical procedure for implementing CPFEM in the commercial finite element software ABAQUS through the user-defined subroutine UMAT [75].
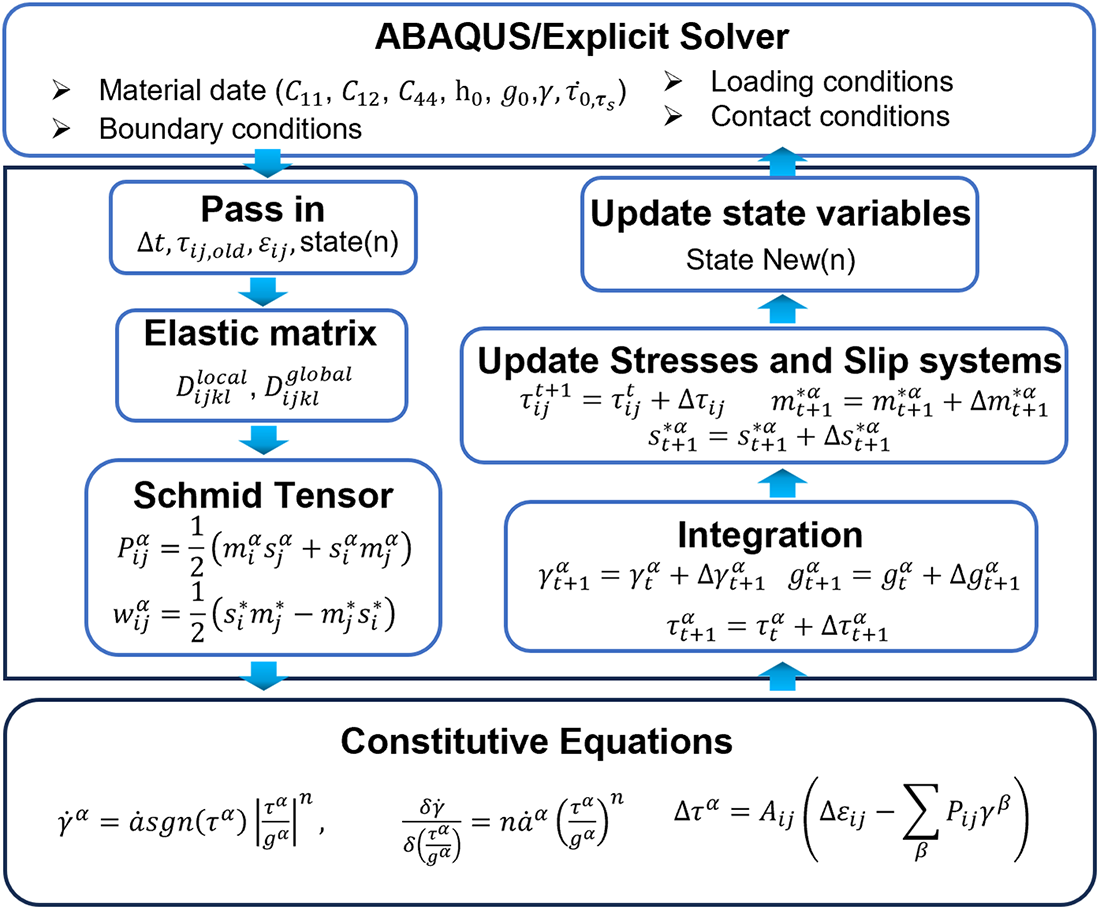
Figure 9: Implementation procedure of the crystal plasticity finite element method in ABAQUS [76].
In studies of crystalline materials, the CPFEM is widely employed to analyze the mechanical response and fracture behavior of single crystals at the mesoscale, thereby elucidating their underlying deformation mechanisms and fracture characteristics. Wu and Sun [77] demonstrated how microscale slip activity, grain orientation, and strain localization jointly influence the macroscopic mechanical performance of crystalline metals based on CPFEM. By further integrating the cohesive zone model, they successfully captured damage evolution and crack-initiation paths under multiple interacting mechanisms, providing an effective framework for predicting fracture behavior under complex loading conditions. Wang et al. [78] developed a CPFEM model that incorporates slip-system activation and dislocation evolution to investigate nanoscale scratching of single-crystal copper, revealing a strong orientation dependence of stress fields, plasticity, and surface damage. In the case of fretting fatigue in nickel-based single crystals, crystal orientation governs slip-system distribution and slip morphology, thereby shaping both surface and subsurface stress–strain fields; cracks typically propagate along the primary slip band, and the equivalent plastic strain serves as a reliable indicator for crack initiation [79,80]. Overall, CPFEM effectively correlates crystal orientation, slip behavior, and stress–strain evolution, enabling accurate characterization of damage development and crack nucleation under multi-mechanism coupling. This makes CPFEM a robust mesoscale modeling tool for predicting fracture behavior in both single- and polycrystalline materials subjected to complex loading conditions.
The macroscopic mechanical behavior of polycrystalline materials is governed by the coupled effects of microstructural evolution and external loading. By integrating crystal plasticity with other computational approaches, it is possible to capture the spatiotemporal evolution of features such as grain structures, grain boundaries, and voids, thereby quantitatively explaining the heterogeneity of stress–strain fields and the mechanisms of damage evolution. For instance, CPFE simulations of void growth in FCC polycrystals have demonstrated that microstructural heterogeneity suppresses void expansion, indicating that traditional homogeneous isotropic models tend to overestimate void growth rates [81]. Moreover, grain size, shape, orientation, and grain boundary characteristics collectively influence stress distribution, damage initiation, and crack propagation in metallic materials, while dynamic recrystallization, multi-oriented fine-grain structures, and grain boundary effects modulate dislocation nucleation and local strain distribution, significantly affecting overall mechanical performance and failure behavior [82–84]. In summary, these studies, centered on CPFE and combined with molecular dynamics, statistical damage models, and particle swarm optimization, achieve cross-scale characterization from grain boundaries and grains to voids and macroscopic responses under consistent physical constraints. This integrated approach not only captures microstructural evolution and damage initiation but also elucidates macroscopic phenomena and validates underlying mechanisms, providing valuable guidance for material strengthening design and crack propagation studies.
In recent years, the advancement of computational methods and intelligent algorithms has led to an increasing integration of CPFEM with optimization techniques and ML. These approaches not only enhance computational efficiency and predictive accuracy but also provide deeper insights into plastic deformation, structural evolution, and damage formation in polycrystalline materials. Hu et al. [85] coupled CPFEM with particle swarm optimization to develop a multiscale predictive framework that quantitatively characterizes texture evolution and heterogeneous deformation during the compression of polycrystalline copper. They found that spatial variations in grain rotation angles are significant contributors to local heterogeneous deformation and damage initiation. Mao et al. [86] employed a deep learning-driven CPFEM model, which improved stress-strain computation efficiency by 29.3 times. Dorward et al. [87] combined Gaussian processes with Sobol global sensitivity analysis, revealing significant interactions among parameters and identifying certain geometric constants as primary influencers of material yield and hardening behavior. Keshavarz et al. [88] proposed a framework that integrates physics-informed neural networks with CPFEM, incorporating equilibrium equations and constitutive relations as physical constraints to achieve high-fidelity predictions of stress-strain responses, plastic deformation gradients, and dislocation density evolution under large deformation. Although these methods do not directly simulate damage and crack propagation, the combination of CPFEM with other computational approaches effectively and accurately captures localized plasticity, heterogeneous deformation, and the influence of critical parameters in polycrystalline materials, thus providing a robust foundation for subsequent damage and fracture studies.
CPFEM provides a robust connection between microscopic deformation mechanisms and macroscopic material responses, enabling the evaluation of how crystal orientation, grain boundaries, and texture evolution influence stress-strain behavior and crack initiation. When integrated with other computational techniques, CPFEM can accurately reproduce experimentally observed crack initiation sites and propagation paths, thereby enhancing the validation of microscale mechanisms. Furthermore, the incorporation of artificial intelligence approaches, such as deep learning surrogates and physics-informed neural networks, significantly reduces computational costs and improves the generalizability of the models.
The PF is a mesoscale simulation approach particularly well-suited for modeling the evolution of microstructures within materials. Unlike macroscopic models that treat the material as a continuum or microscopic models that focus on localized atomic interactions, the PF method provides a continuous representation capable of accurately capturing the formation and evolution of grains, phase boundaries, and cracks. Since damage and fracture in metallic materials are closely linked to microstructural evolution, the PF method offers an effective means to elucidate the underlying mechanisms governing deformation, damage accumulation, and crack propagation from a mesoscale perspective. The PF method has a broad scope of applicability, encompassing single-grain growth, crack initiation and propagation, phase transformations, solute redistribution, and compositional reconfiguration. This review primarily highlights the use of the PF method to simulate the mechanical degradation of metallic materials during hydrogen embrittlement and further summarizes recent advances in PF–CPFEM coupled frameworks for analyzing deformation and damage evolution.
In recent years, researchers have employed phase-field methods to develop coupled models that address hydrogen diffusion and mechanical behavior, with a particular emphasis on reduced fracture toughness, crack interactions, and the synergistic effects of fatigue and diffusion. These studies have elucidated the mechanisms underlying damage evolution and crack propagation in metals subjected to hydrogen embrittlement. By integrating hydrogen diffusion with mechanical or fatigue behavior within a phase-field fracture framework, researchers can elucidate how hydrogen accumulation results in local degradation of mechanical properties, reduced fracture toughness, and the nucleation and growth of cracks [89–92]. Overall, phase-field approaches not only enhance our understanding of crack initiation, propagation, and fracture toughness degradation under hydrogen embrittlement but also improve the accuracy and computational efficiency of crack growth predictions.
The integration of crystal plasticity finite element (CPFE) and phase-field (PF) methods has emerged as a prominent research focus, as it facilitates the simultaneous consideration of stress evolution and microstructural changes at the continuum scale. This methodology enables a systematic analysis of twin formation mechanisms, interface migration, and interactions with dislocations and cracks. Fig. 10 illustrates the CPFE–PF coupling strategy: the CPFE method resolves the elastic and plastic deformation gradients at material points under prescribed boundary conditions, while the PF model explicitly simulates twin evolution, governed by a non-conserved phase-field equation that drives the minimization of the system’s total energy. Liu et al. [93] employed the CPFE–PF approach to investigate the effects of twins and dislocations on the plastic deformation of hexagonal metals, revealing that twin boundary migration coupled with dislocations significantly alters local stress distributions. However, the driving force for twin boundary migration must be adjusted to eliminate self-stresses induced by boundary defects to avoid biased predictions. To address this issue, Jiang et al. [94] proposed a stress-correction scheme to mitigate self-stress effects, while Mo et al. [95] developed a double-interface model utilizing CPFE–PF, accurately capturing the evolution dynamics of twin fronts and side interfaces, including key characteristics such as interface energy, driving force, and interface velocity. Overall, the CPFE–PF coupling method allows for the concurrent description of mechanical responses and twin microstructural evolution at the continuum scale, providing a robust framework for elucidating twin formation mechanisms, interface migration, and interactions with dislocations, while offering more precise insights into the influence of twinning on the macroscopic mechanical behavior of metals.
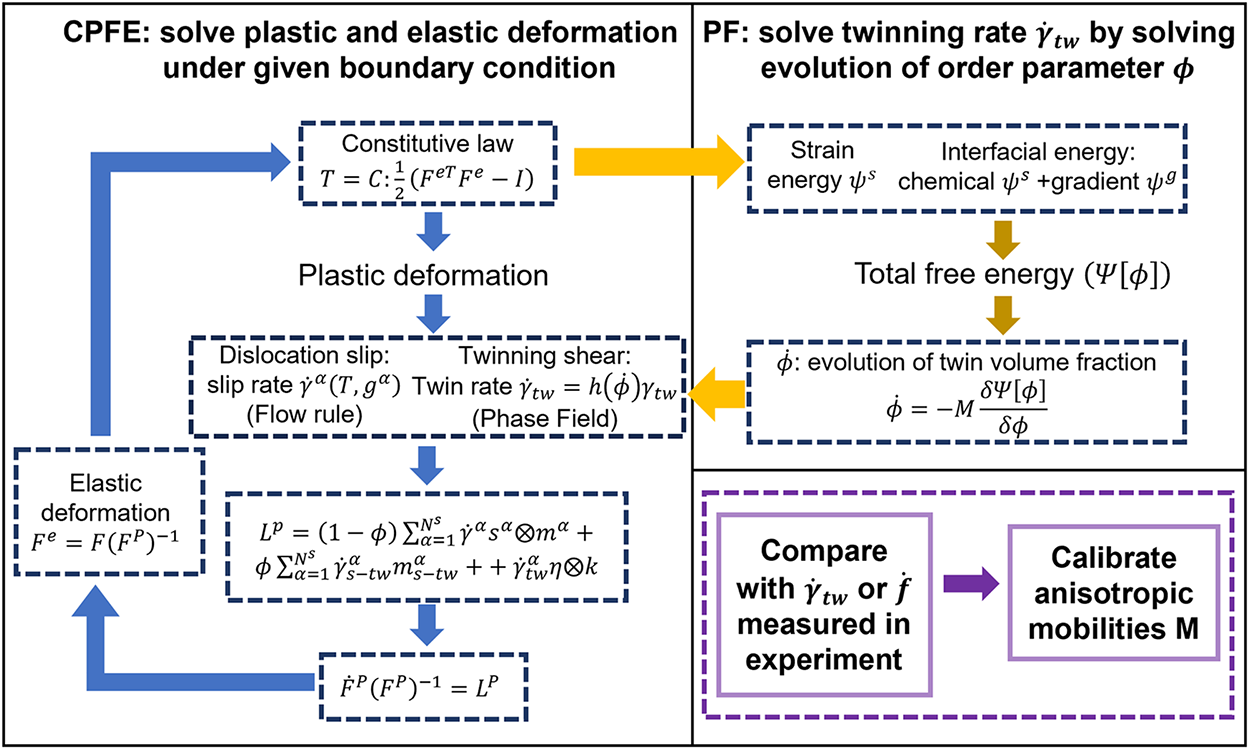
Figure 10: Coupling strategy between the crystal plasticity finite element method and the phase-field approach.
The phase-field (PF) method is a powerful tool for investigating damage and fracture in metallic materials at the mesoscopic scale. By explicitly modeling the interaction between hydrogen diffusion and mechanical fields, the PF method facilitates the description of crack initiation, propagation, interaction, and the coupled effects of fatigue and diffusion. Furthermore, the integration of crystal plasticity finite element methods with the phase-field approach (CPFE–PF) allows for the simultaneous consideration of plastic deformation and interfacial evolution. Recent advancements in the PF method present several notable advantages: (1) it eliminates the need for explicit tracking of interface positions; (2) it accurately captures material heterogeneity, defect characteristics, and anisotropy; (3) it does not require predefined microstructural morphologies. Nevertheless, several limitations persist: (1) the computational cost remains relatively high; (2) numerical predictions are often challenging to validate experimentally; (3) the PF framework encompasses numerous material and model parameters, many of which are difficult to determine accurately.
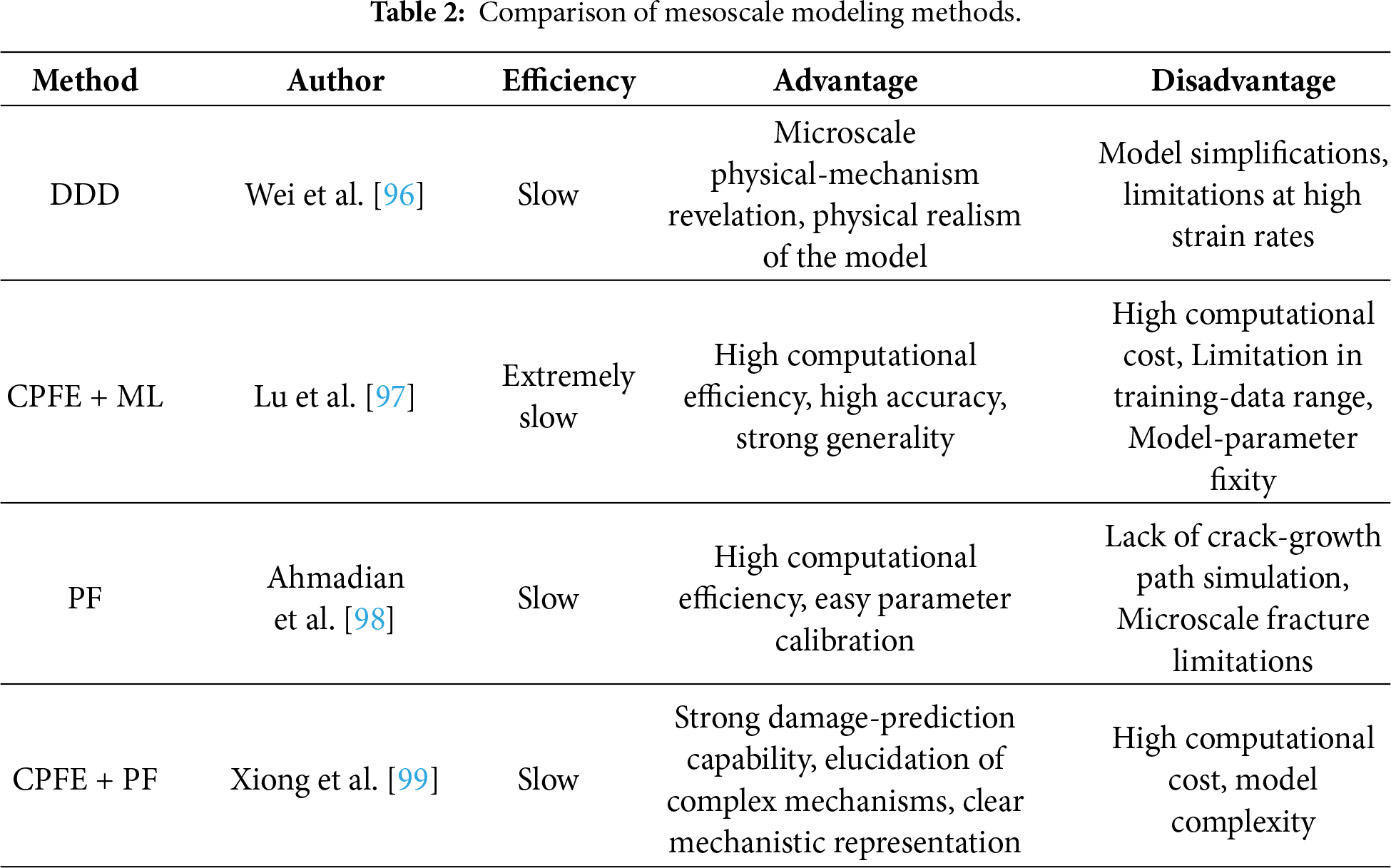
At the mesoscale, the mechanical behavior of materials is significantly influenced by dislocation evolution, grain morphology, and phase-boundary characteristics. The previous sections have reviewed various mesoscale modeling approaches applied to the study of damage and fracture in metallic materials. Under macroscopic loading, subcritical cracks frequently form within crystalline materials, and these micro-defects can substantially alter their mechanical responses. Therefore, characterizing such defects at the mesoscale is essential to enhance the safety and reliability of materials in engineering applications. Table 2 summarizes the primary mesoscale research methods and representative studies, offering a comparative overview of the capabilities of different approaches in elucidating damage evolution and crack behavior.
Mesoscopic scale numerical simulations play an important role in revealing the transitional mechanisms of material damage and crack propagation, serving as a critical bridge between microscopic defect evolution and macroscopic mechanical response. The advantages of mesoscopic-scale simulations include:
(1) Revealing the link between microstructure evolution and macroscopic mechanical behavior.
(2) Studying processes that are difficult to observe in experiments, such as dislocation shielding at crack tips, irradiation defect–dislocation interactions, and hydrogen diffusion–fracture coupling.
(3) Ease of integration with other numerical methods and multi-physics models, providing good potential for multiscale extension.
However, mesoscopic-scale simulations still have several limitations:
(1) High computational cost, which makes direct application to large-scale or long-term engineering problems challenging.
(2) Strong dependence on initial conditions, boundary conditions, and model assumptions, which can affect the stability of results.
(3) Difficulties in quantitative validation against experiments, particularly in crack nucleation and early-stage damage. Future research should focus on developing efficient coupling frameworks and data-driven approaches to improve computational efficiency and predictive capability, thereby providing theoretical guidance for engineering applications.
4 Numerical Simulation Approaches at the Macroscale
Macroscale fracture research represents a crucial intersection between fundamental material theory and engineering applications, with its primary value in establishing quantitative links between constitutive behavior and structural performance. By elucidating failure mechanisms under external loading, this research direction not only provides a scientific basis for correlating microstructural features—such as grain boundaries, dislocations, and second-phase particles—with macroscopic properties, including toughness, strength, and fatigue life, but also offers essential theoretical support for damage-tolerant design, service life prediction, and safety assessment of engineering structures.
The FEM is a fundamental approach for investigating material damage and fracture mechanics. By discretizing a continuous medium into a finite number of elements and constructing interpolation functions for each element, FEM formulates the governing equations for nodal displacements and stress fields across the solution domain. Fracture analysis using FEM is generally conducted through three primary techniques: the Virtual Crack Closure Technique (VCCT), the J-integral method, and the Extended Finite Element Method (XFEM). Table 3 presents a comparison of various macroscale simulation methods along with their application characteristics. Each method possesses unique advantages: the FLJC method is effective for analyzing two-dimensional multiple interacting cracks; VCCT is computationally efficient but constrained to predefined crack paths; and XFEM offers significant versatility, avoiding the need for remeshing, though its implementation is more complex and may still be affected by mesh quality.
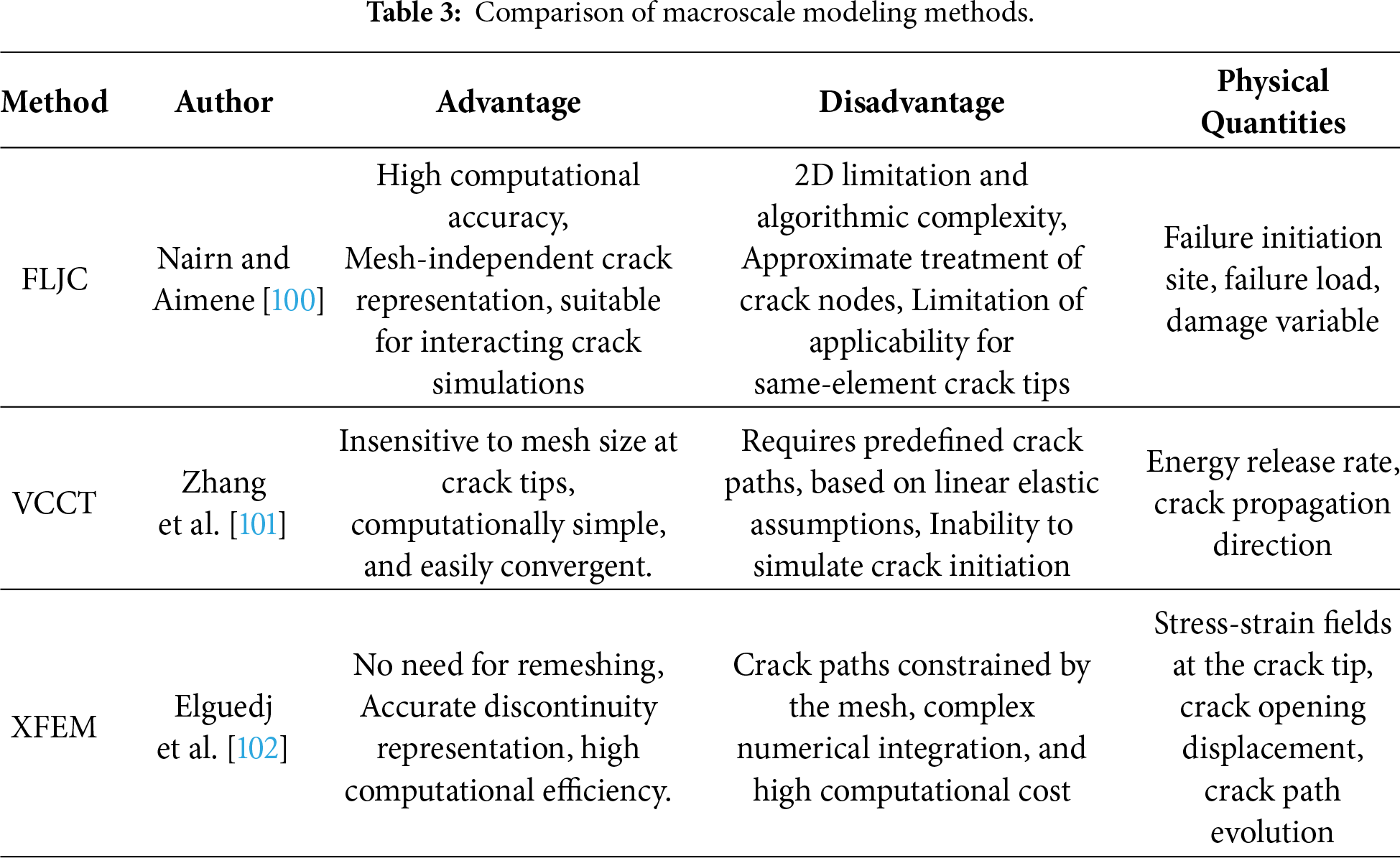
The J-integral-based numerical framework effectively characterizes the crack-tip energy release rate and R-curve evolution under elastoplastic conditions through contour integration, accurately capturing the crack driving force via a mesh-independent treatment. By integrating damage constitutive models with plasticity mechanisms and incorporating an elastoplastic fracture framework based on J/ΔJ integrals, it precisely describes crack initiation and propagation, thereby enhancing the reliability of crack growth predictions and lifetime assessments in complex metallic materials [103,104]. Fig. 11 illustrates the load–crack opening displacement and J–R curves for HfNbTaTiZr SENB specimens tested at room temperature. Importantly, the improved TA-CCT method, coupled with the XFEM–VCCT framework, facilitates efficient and stable simulations of arbitrary crack growth paths, thereby improving the accuracy of crack-path and fracture-parameter predictions under cyclic loading in complex structures [105,106]. Furthermore, XFEM-based multiscale coupling approaches can elucidate the microscale mechanisms of crack nucleation and propagation in polycrystalline metals, detailing the influence of dislocation motion, transformation-induced stresses, and crystal structure on crack paths, propagation behavior, and ductile-to-brittle transitions [107–110].
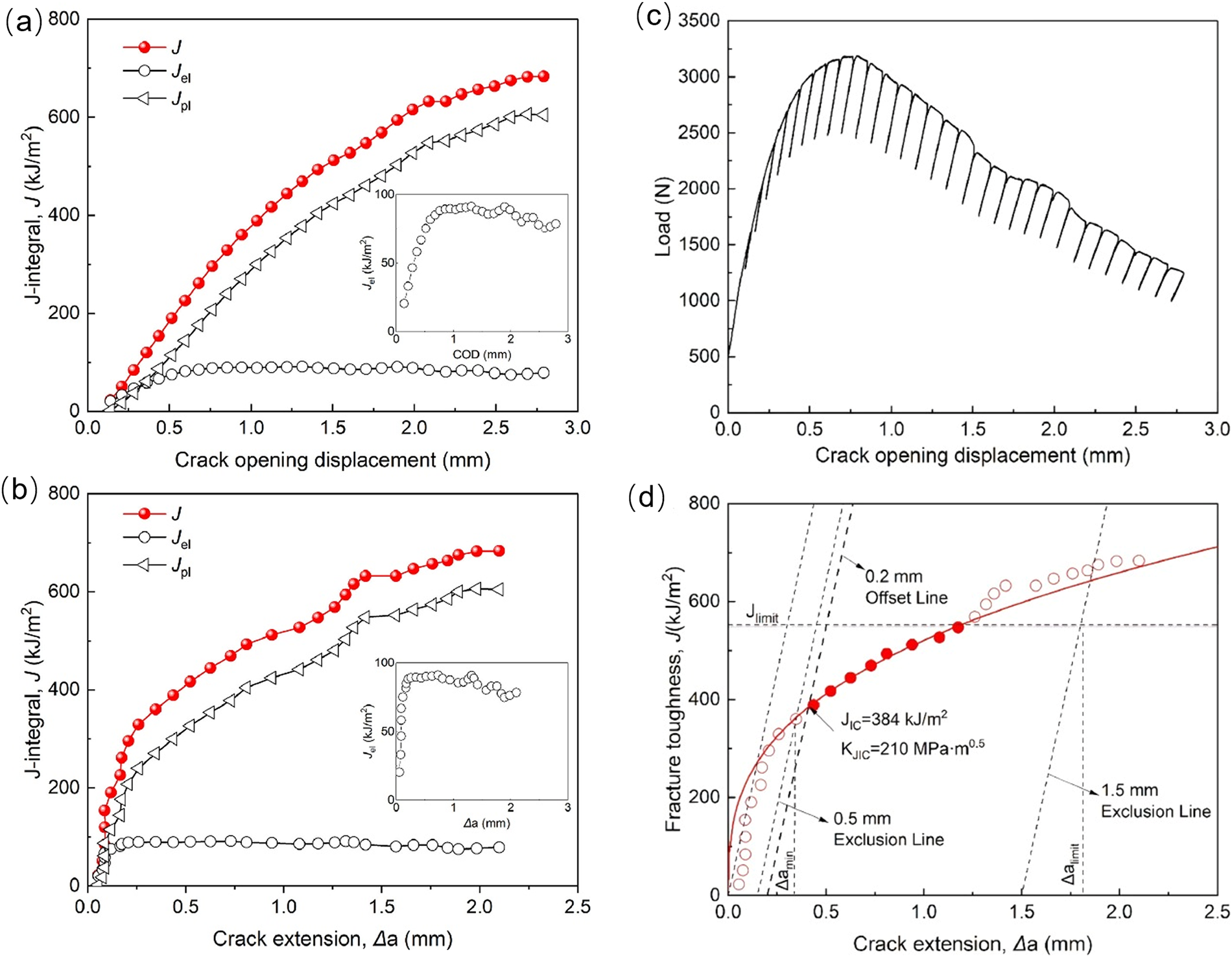
Figure 11: Finite Element Model of the CT Specimen and Evaluation of the Cyclic J-Integral (ΔJ) (a) crack opening displacement (b) crack extension. (c) Load-crack opening displacement curve and (d) J-R curve for HfNbTaTiZr SENB specimen tested at room temperature [103].
Fatigue life prediction has long faced the dilemma between “microscopic mechanism fidelity” and “macroscopic computational efficiency.” Professor Shibanuma’s team has recently established a complete multiscale modeling framework that resolves this conflict. The core innovation of their approach lies in the introduction of a physics-driven bridging strategy. Unlike traditional methods that rely on experimental determination of Paris law parameters, this strategy uses “data-driven parameter identification,” where the Paris law parameters required for the macroscopic phase are directly obtained by inverting crack growth rate data generated by the microscopic model in the “transition zone.” This approach not only ensures physical consistency and high accuracy in cross-scale coupling but has also reportedly achieved more than a 90% improvement in computational efficiency. Beyond efficiency and accuracy, this framework has also demonstrated outstanding coupling precision when applied to non-homogeneous materials [111–114].
In summary, numerical frameworks based on the J-integral, TA-CCT, and XFEM, when integrated with damage constitutive laws, plasticity mechanisms, or crystal plasticity, provide high-precision tools for capturing crack driving forces, growth paths, and multiple nucleation modes under elastoplastic and cyclic loading, thus offering reliable methodologies for analyzing damage evolution, fracture behavior, and lifetime prediction in complex metallic materials and components. Numerical simulation frameworks based on the J-integral, TA-CCT, and XFEM, when coupled with damage constitutive models, plasticity mechanisms, and crystal plasticity, accurately characterize crack-tip energy release rates, crack growth paths, and multiple crack nucleation modes under elastoplastic and cyclic loading. This integration provides a reliable computational tool for analyzing damage evolution, fracture behavior, and lifetime prediction in complex metallic structures.
Macroscopic scale fracture simulations establish a connection between material constitutive behavior and structural performance, serving as an important tool for engineering damage-limit design and lifetime prediction. However, several challenges remain:
(1) Reliance on empirical or semi-empirical constitutive models: Most macroscopic models incorporate microscopic damage mechanisms to a limited extent.
(2) High number of model parameters: Some parameters need to be determined through specific experiments, which limits the general applicability of the models.
Future research directions for macroscopic-scale numerical simulations include:
(1) Enhanced coupling with mesoscopic models: Integrating macroscopic fracture models with mesoscopic-scale approaches such as crystal plasticity and dislocation dynamics to achieve a more realistic multiscale description.
(2) Integration of data-driven and ML methods: Reducing the cost of parameter calibration while improving computational efficiency.
5 Multiscale Coupled Modeling Approaches
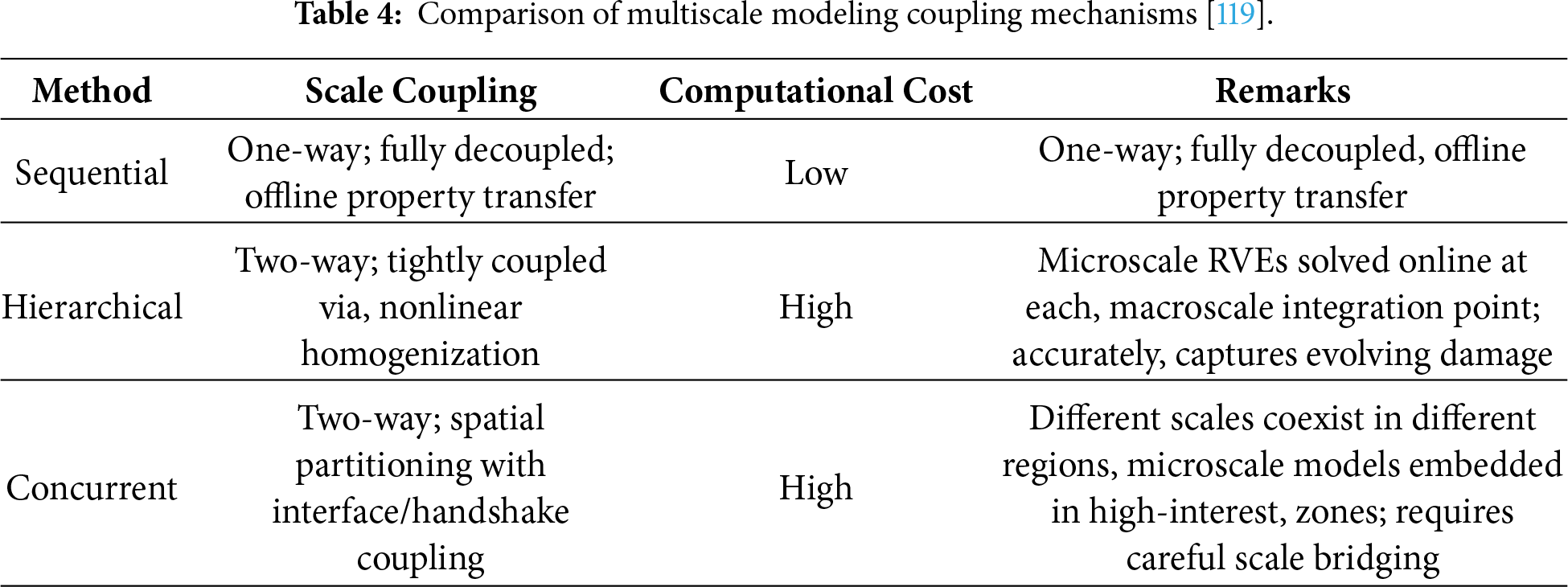
To elucidate the intrinsic nature of scale effects in material fracture, traditional fracture-prediction approaches based on empirical relations or macroscopic constitutive models are insufficient to accurately capture the damage and failure behaviors governed by multiple interacting mechanisms across disparate length scales. The fracture process fundamentally originates from atomic-scale bond breaking and defect evolution, with its progression spanning atomic, mesoscale, and macroscopic levels. Consequently, establishing effective information transfer and a unified mechanistic description across these scales has become a central challenge in multiscale fracture mechanics. To address this issue, researchers have developed a hierarchical modeling framework that bridges atomistic insights with macroscopic performance predictions, encompassing molecular dynamics simulations, continuum-scale finite element analysis, and macro–micro coupled as well as intelligent modeling techniques. Based on their implementation and coupling strategies, multiscale approaches can be broadly categorized into three types: sequential, hierarchical, and concurrent methods. Table 4 summarizes the advantages and limitations of each method in terms of scale bridging capability, computational cost, accuracy, and their effectiveness in capturing damage mechanisms in composite materials [115–118].
Multiscale simulations are widely used to analyze the physical and mechanical properties of materials; however, challenges remain in computational efficiency and the accuracy of parameter transfer across scales. Recent reviews have summarized strategies that integrate ML with FEM to enhance both the efficiency and precision of material simulations. By learning input–output mappings from training data, ML has been increasingly combined with multiscale modeling for parameter inversion and mechanical performance analysis in materials science. Fig. 12 illustrates the coupling framework between ML and multiscale simulations.
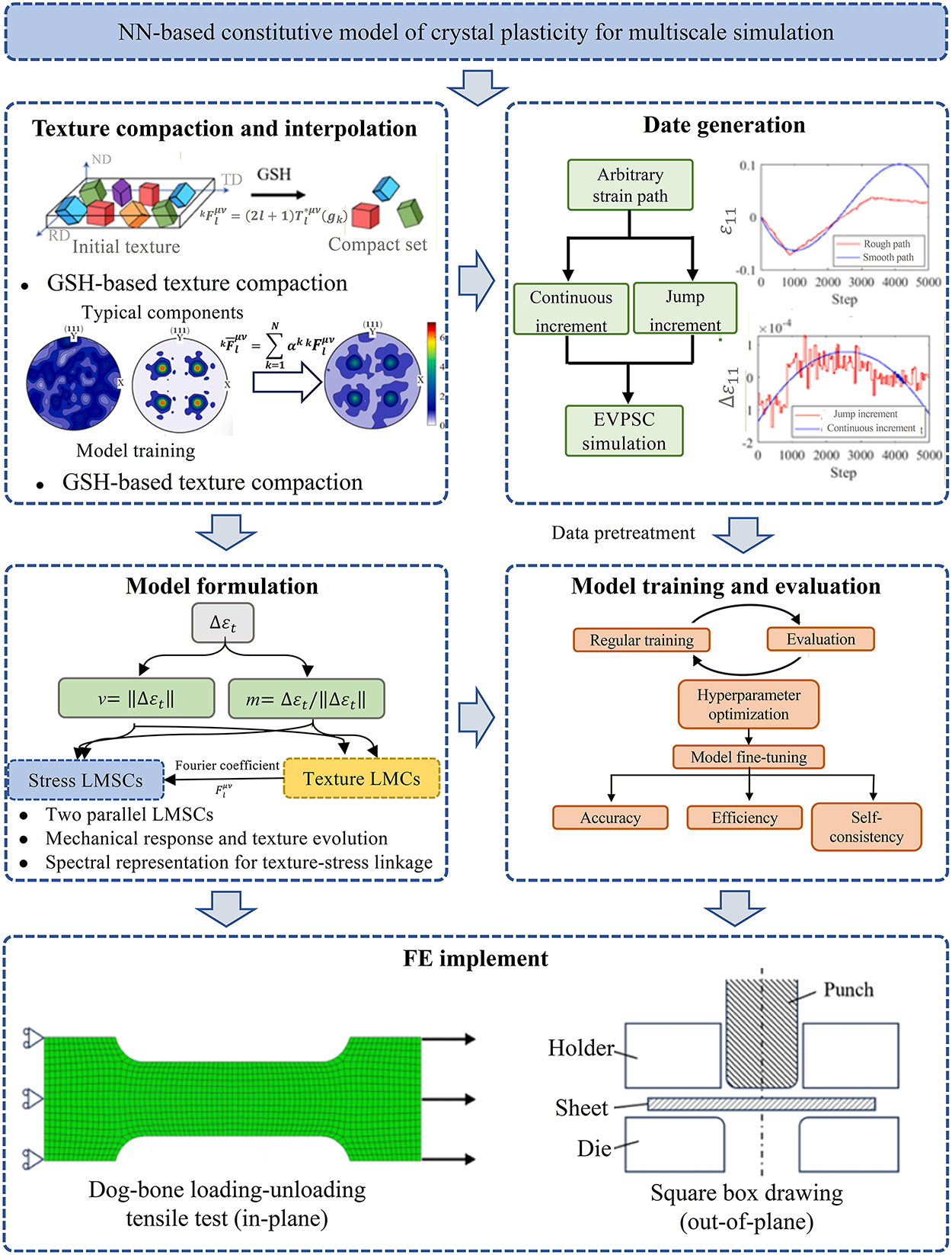
Figure 12: Workflow of a machine-learning-based hierarchical modeling framework [120].
In multiscale simulation studies, modeling approaches based on the cohesive zone model (CZM) provide a critical bridge connecting atomic-scale dislocation evolution mechanisms with macroscopic crack propagation behavior. By introducing traction–separation (T–S) relationships, CZM can describe the nonlinear separation process in the fracture region within a continuum framework and enable cross-scale mapping from atomic-scale energy release to macroscopic fracture toughness. Atomic-scale cohesive models have been constructed for FCC single crystals (Cu and Ni) to directly extract T–S relations. For Cu, the T–S response agrees with an exponential cohesive law, whereas Ni exhibits significant nonlinear deviations in the softening regime, revealing the influence of microscopic heterogeneity on macroscopic fracture parameters and providing atomic-scale guidance for cross-scale cohesive model parameterization and macroscopic crack growth prediction [121]. Recent studies employing multiscale coupling methods, such as molecular dynamics and crystal plasticity finite element simulations, have elucidated the roles of crack propagation, phase transformations, twinning, and microstructural defects in controlling ductile-to-brittle transitions and damage evolution from atomic-scale interface evolution and dislocation motion to macroscopic mechanical behavior [122–124]. CZM-based multiscale approaches, by extracting atomic-scale T–S curves, enable the mapping of microscopic mechanisms—such as dislocation activity, interface separation, and phase transformation—onto macroscopic crack propagation behavior, providing a quantitative bridge from atomic to continuum scales for fracture toughness prediction, damage evolution analysis, and cross-scale model parameterization.
Intelligent modeling that integrates ML with physical information has significantly enhanced the capabilities of multiscale mapping. Consequently, numerous studies have concentrated on developing cross-scale fracture models, beginning from the atomic scale. These models capture microscopic physical features, thereby providing a robust theoretical foundation for predicting failure and assessing the lifetime of complex material systems. Wang et al. [125] proposed a method that combines multiscale simulations with ML to predict the dynamic fracture toughness of nickel-based alloys, optimizing network architectures and sample sizes while evaluating the performance of various models. Additionally, several studies have integrated crystal plasticity, FFT solvers, Voronoi-based polycrystal modeling, and ML to construct high-fidelity multiscale. macro–micro coupling frameworks. These frameworks enable accurate mapping from microscopic slip, lamellar interactions, and microstructural evolution to macroscopic stress–strain responses, quantitatively revealing how microstructural heterogeneity regulates damage, crack nucleation, and cyclic hardening [126,127]. With the deep integration of high-performance computing and artificial intelligence, research in multiscale fracture mechanics is transitioning from static parameter transfer to dynamic mechanism coupling, thereby opening new avenues for failure prediction and lifetime assessment in complex material systems.
Within a closed-loop multiscale framework that bridges microscopic and macroscopic behavior, molecular dynamics simulations are employed to accurately capture defect formation, dislocation motion, and microcrack initiation within crystals. This approach reveals the fundamental mechanisms of stress concentration and crack propagation at the atomic scale. The results from the microscopic simulations are then mapped onto continuum finite element models to reproduce and predict the material’s mechanical response at the macroscopic level. By comparing model predictions with experimental measurements and calibrating microscopic parameters based on feedback, a fully integrated optimization is achieved from atomic structure to macroscopic performance. This methodology not only enables quantitative characterization of fracture behavior across various length scales and loading conditions but also provides actionable theoretical guidance for the design of high-performance materials, facilitating a shift in materials development from an ‘experience-driven’ to a ‘mechanism-driven’ paradigm.
Multiscale methods have demonstrated strong capability in simulating damage and fracture in metallic materials, and their applicability has been further expanded to practical engineering problems by incorporating diverse physical effects and damage mechanisms. As one of the critical issues governing material failure, damage and fracture can now be analyzed across scales—from atomic interactions to the structural response at the macro level—through multiscale modeling frameworks. This work reviews recent advances in the application of multiscale models to damage and fracture in materials. Various multiscale approaches have been continuously refined to address increasingly complex engineering scenarios, including fracture behavior, defect initiation, plastic deformation, and multiple failure criteria. Existing multiscale models have been widely employed to capture complex fracture phenomena such as crack branching, nucleation, and coalescence. Building on the literature surveyed, further developments have been made at each scale to better characterize the energy partitioning, defect initiation, crack nucleation, propagation, and ultimate failure in metallic materials.
Despite significant progress, challenges persist in simulation efficiency and the accuracy of parameter transfer across scales. In high-fidelity multiscale models of damage evolution and fracture—especially those designed to capture complex multi-physics phenomena—the computational cost can be prohibitive. As a result, adaptive strategies and reduced-order models (ROMs) have emerged as indispensable tools, providing substantial potential to decrease computational expenses while preserving high accuracy. ROMs simplify intricate physical processes by reducing the number of degrees of freedom or by utilizing surrogate models trained on high-fidelity simulation data. In the context of multiscale damage modeling, ROMs effectively capture essential damage mechanisms at both micro- and macro-scales, facilitating faster and more scalable simulations for large-scale applications.
Integrating artificial intelligence and ML into multiscale modeling frameworks has emerged as an effective strategy for addressing the high computational costs and limited efficiency associated with traditional simulations of damage and fracture in metallic materials. A typical approach involves conducting high-fidelity simulations at a single scale and constructing a database of the resulting physical parameters. Based on this database, a ML model is trained to learn the implicit mapping between features across different scales. Once the model is trained, new input data can be rapidly processed to predict the corresponding parameters at other scales. This inference step is highly parallelizable and computationally efficient, significantly alleviating the costs and time requirements of conventional multiscale computations. Future research should focus on developing adaptive multiscale coupling architectures. Real-time multiscale simulation systems based on digital twin technologies are emerging as a frontier direction, enabling dynamic calibration of cross-scale model parameters using experimental observations. Such advancements provide new tools for analyzing damage evolution in intelligent materials and open new possibilities for failure prediction and lifetime assessment in complex material systems.
Acknowledgement: The authors gratefully acknowledge the financial and infrastructural support provided by the National Natural Science Foundation of China, the Chongqing Science and Technology Committee, Foundation of National Key Laboratory of Computational Physics and the Chongqing Municipal Education Commission. We also thank laboratory staff and technical support personnel for assistance with data acquisition and computing resources.
Funding Statement: This review was funded by the National Natural Science Foundation of China, grant number 52405341; Foundation of National Key Laboratory of Computational Physics, grant number 6142A05QN24012; Chongqing Science and Technology Committee, grant number CSTB2023NSCQ-MSX0363; The Science and Technology Research Program of Chongqing Municipal Education Commission, grant number KJQN202301117.
Author Contributions: Conceptualization: Lusheng Wang and Bin Gao; methodology: Bin Gao; validation: Xinyu Jiang; formal analysis: Lusheng Wang, Bin Gao and Yanhong Peng; investigation: Lusheng Wang and Shaojie Gu; resources: Lusheng Wang and Jun Ding; writing—original draft preparation: Bin Gao; writing—review and editing: Lusheng Wang, Bin Gao, Yanhong Peng, Xin Yang, Hongzhou Yan and Shaojie Gu; supervision: Lusheng Wang; project administration: Lusheng Wang; funding acquisition: Lusheng Wang. All authors reviewed and approved the final version of the manuscript.
Availability of Data and Materials: Not applicable.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare no conflict of interest.
References
1. Ritchie RO. The conflicts between strength and toughness. Nat Mater. 2011;10(11):817–22. doi:10.1038/nmat3115. [Google Scholar] [PubMed] [CrossRef]
2. Schuh CA, Hufnagel TC, Ramamurty U. Mechanical behavior of amorphous alloys. Acta Mater. 2007;55(12):4067–109. doi:10.1016/j.actamat.2007.01.052. [Google Scholar] [CrossRef]
3. Lejaeghere K, Bihlmayer G, Björkman T, Blaha P, Blügel S, Blum V, et al. Reproducibility in density functional theory calculations of solids. Sci. 2016;351(6280):aad3000. doi:10.1126/science.aad3000. [Google Scholar] [PubMed] [CrossRef]
4. Fan T, Ruan Z, Nie B, Liao Y, Huang B, Xu Z, et al. First-principles investigation of point defects on the thermal conductivity and mechanical properties of aluminum at room temperature. Coatings. 2023;13(8):1357. doi:10.3390/coatings13081357. [Google Scholar] [CrossRef]
5. Chen J, Cao T, Xu W, Wu J, Hu Y, Cao L, et al. Analysis of thin-wall effect mechanism based on stress rupture properties and fracture characteristics of DD10 Ni-based single-crystal alloy. Mater Charact. 2024;218:114494. doi:10.1016/j.matchar.2024.114494. [Google Scholar] [CrossRef]
6. Li F, Wen Z, Wu Z, Pei H, Yue Z. Fatigue life assessment of nickel-based single crystals considering equivalent initial flaw size model and anisotropy. Int J Fatigue. 2022;160(19):106886. doi:10.1016/j.ijfatigue.2022.106886. [Google Scholar] [CrossRef]
7. Abraham FF, Broughton JQ, Bernstein N, Kaxiras E. Spanning the continuum to quantum length scales in a dynamic simulation of brittle fracture. Europhys Lett. 1998;44(6):783–7. doi:10.1209/epl/i1998-00536-9. [Google Scholar] [CrossRef]
8. Tang H, Huang H, Liu C, Liu Z, Yan W. Multi-Scale modelling of structure-property relationship in additively manufactured metallic materials. Int J Mech Sci. 2021;194:106185. doi:10.1016/j.ijmecsci.2020.106185. [Google Scholar] [CrossRef]
9. Wu WP, Yao ZZ. Molecular dynamics simulation of stress distribution and microstructure evolution ahead of a growing crack in single crystal nickel. Theor Appl Fract Mech. 2012;62:67–75. doi:10.1016/j.tafmec.2013.01.008. [Google Scholar] [CrossRef]
10. Petersson CLM, Fredriksson A, Melin S, Ahadi A, Hansson P. A molecular dynamics study on the influence of vacancies and interstitial helium on mechanical properties of tungsten. J Nucl Mater. 2023;580(5):154378. doi:10.1016/j.jnucmat.2023.154378. [Google Scholar] [CrossRef]
11. Qian C, Qiu Y, He Z, Mu W, Tang Y, Wang H, et al. Effects of interstitial carbon atoms on texture structure and mechanical properties of FeMnCoCr alloys. PLoS One. 2020;15(12):e0242322. doi:10.1371/journal.pone.0242322. [Google Scholar] [PubMed] [CrossRef]
12. Sun SP, Hu YF, Li XP, Chen Y, Wang HJ, Yu Y, et al. The effect of point defect on mechanical properties of MoSi2. Int J Mod Phys B. 2017;31(16–19):1744081. doi:10.1142/s0217979217440817. [Google Scholar] [CrossRef]
13. Li J, Dong X, Xie H, Xu C, Liu J, Zhang J. Process-induced evolution of prismatic dislocation loop and its effect on mechanical properties. Mater Today Commun. 2022;31:103754. doi:10.1016/j.mtcomm.2022.103754. [Google Scholar] [CrossRef]
14. Könke C. Coupling of micro- and macro-damage models for the simulation of damage evolution in ductile materials. Comput Struct. 1997;64(1–4):643–53. doi:10.1016/S0045-7949(96)00157-5. [Google Scholar] [CrossRef]
15. Yin Y, Lu S, Wang L, Song K, Huang X, Ding J, et al. Atomic-scale insights into the strength and plasticity enhancement of Ni-based superalloys with refinement dispersion of precipitates. Chem Phys Lett. 2025;861(1):141837. doi:10.1016/j.cplett.2024.141837. [Google Scholar] [CrossRef]
16. Li YL, Kohar CP, Mishra RK, Inal K. A new crystal plasticity constitutive model for simulating precipitation-hardenable aluminum alloys. Int J Plast. 2020;132:102759. doi:10.1016/j.ijplas.2020.102759. [Google Scholar] [CrossRef]
17. Amouzou-Adoun YA, Jebahi M, Fivel M, Forest S, Lecomte JS, Schuman C, et al. On elastic gaps in strain gradient plasticity: 3D discrete dislocation dynamics investigation. Acta Mater. 2023;252(8):118920. doi:10.1016/j.actamat.2023.118920. [Google Scholar] [CrossRef]
18. Liu Y, Feng Y, Wu D, Chen X, Gao W. Virtual modelling integrated phase field method for dynamic fracture analysis. Int J Mech Sci. 2023;252(105008):108372. doi:10.1016/j.ijmecsci.2023.108372. [Google Scholar] [CrossRef]
19. Ma LH, Song ZZ, Zhang K, Wang L, Zhou W, Zhang Q. Simulation of crack growth for metal plates with holes based on cohesive zone model. Mater Res Express. 2022;9(8):086512. doi:10.1088/2053-1591/ac8394. [Google Scholar] [CrossRef]
20. Wu Y, He W, Ma H, Nie X, Liang X, Pan J, et al. Titanium alloy materials with very high cycle fatigue: a review. Materials. 2024;17(12):2987. doi:10.3390/ma17122987. [Google Scholar] [PubMed] [CrossRef]
21. Wang Y, Wang W, Zhang B, Li CQ. A review on mixed mode fracture of metals. Eng Fract Mech. 2020;235(6):107126. doi:10.1016/j.engfracmech.2020.107126. [Google Scholar] [CrossRef]
22. Karantza KD, Manolakos DE. A review on the adiabatic shear banding mechanism in metals and alloys considering microstructural characteristics, morphology and fracture. Metals. 2023;13(12):1988. doi:10.3390/met13121988. [Google Scholar] [CrossRef]
23. Cheng ZQ, Liu H, Tan W. Advanced computational modelling of composite materials. Eng Fract Mech. 2024;305(2):110120. doi:10.1016/j.engfracmech.2024.110120. [Google Scholar] [CrossRef]
24. Kohn W. Nobel lecture: electronic structure of matter—wave functions and density functionals. Rev Mod Phys. 1999;71(5):1253–66. doi:10.1103/revmodphys.71.1253. [Google Scholar] [CrossRef]
25. Hui J, Zhang X, Liu T, Liu W, Wang B. First-principles calculation of twin boundary energy and strength/embrittlement in hexagonal close-packed titanium. Mater Des. 2022;213(3):110331. doi:10.1016/j.matdes.2021.110331. [Google Scholar] [CrossRef]
26. Chen Y, Xue Z, Zhang S, Liu Y, Zhang X. First principles calculations of the influence of nitrogen content on the mechanical properties of α-Ti. Mater Chem Phys. 2020;248:122891. doi:10.1016/j.matchemphys.2020.122891. [Google Scholar] [CrossRef]
27. Yang Y, Wang L. Hydrogen solution in tetrahedral or octahedral interstitial sites in zirconium-cobalt hydrogen storage alloy: a first-principles study. J Alloys Compd. 2021;859(25):157881. doi:10.1016/j.jallcom.2020.157881. [Google Scholar] [CrossRef]
28. Mosquera-Lois I, Kavanagh SR, Ganose AM, Walsh A. Machine-learning structural reconstructions for accelerated point defect calculations. npj Comput Mater. 2024;10(1):121. doi:10.1038/s41524-024-01303-9. [Google Scholar] [CrossRef]
29. Yang Y, Wang L. Firs-principles investigation of ZrCo and its hydrides. Int J Hydrogen Energy. 2022;47(58):24398–405. doi:10.1016/j.ijhydene.2022.05.210. [Google Scholar] [CrossRef]
30. Li J, Huang Y, Zhang X, Zhang L. Influence of Mg doping on the structure and mechanical properties of Al2Cu precipitated phase by first-principles calculations. Mater. 2023;17(1):93. doi:10.3390/ma17010093. [Google Scholar] [PubMed] [CrossRef]
31. Mosquera-Lois I, Klarbring J, Walsh A. Point defect formation at finite temperatures with machine learning force fields. Chem Sci. 2025;16(20):8878–88. doi:10.1039/d4sc08582e/v2/response1. [Google Scholar] [CrossRef]
32. Yang K, Zhu Y. A general scheme for point defect sink strength calculation and related machine-learning-based expressions. Int J Plast. 2024;172(7):103855. doi:10.1016/j.ijplas.2023.103855. [Google Scholar] [CrossRef]
33. Gao F, Chen D, Hu W, Weber WJ. Energy dissipation and defect generation in nanocrystalline silicon carbide. Phys Rev B. 2010;81(18):184101. doi:10.1103/physrevb.81.184101. [Google Scholar] [CrossRef]
34. Gao N, Zhong Z, Liu L, Lu X, Fan T, Zhou X. First-principles study on the interfacial stability, mechanical behavior and failure mechanism of Al4C3(0 0 0 1)/Al (1 1 1) interfaces. Surf Interfaces. 2025;62:106277. doi:10.1016/j.surfin.2025.106277. [Google Scholar] [CrossRef]
35. Qiu C, Su Y, Chen B, Yang J, Li Z, Ouyang Q, et al. First-principles investigation of interfacial stability, mechanical behavior and failure mechanism of β-SiC(1 1 1)/Al(1 1 1) interfaces. Comput Mater Sci. 2020;175:109608. doi:10.1016/j.commatsci.2020.109608. [Google Scholar] [CrossRef]
36. Gao Y, Liu X, Wei L, Zhang X, Chen M. The segregation behavior of elements at the Ti/TiFe coherent interface: first-principles calculation. Surf Interfaces. 2022;34(2):102321. doi:10.1016/j.surfin.2022.102321. [Google Scholar] [CrossRef]
37. Zhang X, Zhang L, Zhang Z, Huang X. Effect of solute atoms segregation on Al grain boundary energy and mechanical properties by first-principles study. Mech Mater. 2023;185(23):104775. doi:10.1016/j.mechmat.2023.104775. [Google Scholar] [CrossRef]
38. Du X, Qu N, Zhang X, Chen J, Cui P, Huang J, et al. Accelerated first-principles calculations based on machine learning for interfacial modification element screening of SiCp/Al composites. Mater. 2024;17(6):1322. doi:10.3390/ma17061322. [Google Scholar] [PubMed] [CrossRef]
39. Doan T, Le-Quang H, To QD. Coupled molecular dynamics and micromechanics study of planar elastic properties of graphene with void defects. Mech Mater. 2020;147:103450. doi:10.1016/j.mechmat.2020.103450. [Google Scholar] [CrossRef]
40. Huang X, Yi J, Ding J, Song K, Lu S, Liu H, et al. Radiation damage behavior and mechanism in RAFM steel: orientation effect. Vacuum. 2022;205:111445. doi:10.1016/j.vacuum.2022.111445. [Google Scholar] [CrossRef]
41. Zhang Y, Jiang S, Zhu X, Zhao Y. Mechanisms of crack propagation in nanoscale single crystal, bicrystal and tricrystal nickels based on molecular dynamics simulation. Results Phys. 2017;7(1):1722–33. doi:10.1016/j.rinp.2017.04.039. [Google Scholar] [CrossRef]
42. Pogorelko VV, Mayer AE. Dynamic tensile fracture of iron: molecular dynamics simulations and micromechanical model based on dislocation plasticity. Int J Plast. 2023;167(3):103678. doi:10.1016/j.ijplas.2023.103678. [Google Scholar] [CrossRef]
43. Gao T, Song H, Wang B, Gao Y, Liu Y, Xie Q, et al. Molecular dynamics simulations of tensile response for FeNiCrCoCu high-entropy alloy with voids. Int J Mech Sci. 2023;237(4):107800. doi:10.1016/j.ijmecsci.2022.107800. [Google Scholar] [CrossRef]
44. Zhu Y, Gong Q, Yi M. Molecular dynamics investigation of shock-induced deformation behavior and failure mechanism in metallic materials. Arch Comput Meth Eng. 2024;31(4):2317–44. doi:10.1007/s11831-023-10045-8. [Google Scholar] [CrossRef]
45. Sui H, Yu L, Liu W, Chen L, Duan H. Three dimensional dislocation-loop emission criterion for void growth of ductile metals. Int J Plast. 2020;131:102746. doi:10.1016/j.ijplas.2020.102746. [Google Scholar] [CrossRef]
46. He Y, Zhou G, Liu YX, Wang H, Xu DS, Yang R. Atomistic simulation of microvoid formation and its influence on crack nucleation in hexagonal titanium. Acta Phys Sin. 2018;67(5):27–33. (In Chinese). doi:10.7498/aps.67.20171670. [Google Scholar] [CrossRef]
47. Wang B, Han YH, Zhu DM, Liu TY, Wang R, Ma XY. Effect of micro-defects on mechanical properties and plastic deformation characteristics of Ti2AlNb diffusion bonded layers. J Plast Eng. 2020;27(11):202–9. (In Chinese). [Google Scholar]
48. Hong YS. Further exploration on characteristic region of crack initiation for very-high-cycle fatigue. Chin J Theor Appl Mech. 2022;54(8):2101–18. (In Chinese). doi:10.1016/j.ijfatigue.2015.11.029. [Google Scholar] [CrossRef]
49. Li N, Ding N, Zhou J, Liu L, Zaïri F, Yang Y. Mechanical properties and deformation behavior of the magnesium crystal with nano-cracks. Modelling Simul Mater Sci Eng. 2021;29(3):035006. doi:10.1088/1361-651x/abde8c. [Google Scholar] [CrossRef]
50. Liu F, Lin X, Yang H, Wen X, Li Q, Liu F, et al. Effect of microstructure on the fatigue crack growth behavior of laser solid formed 300M steel. Mater Sci Eng A. 2017;695:258–64. doi:10.1016/j.msea.2017.04.001. [Google Scholar] [CrossRef]
51. Xue K, Zhang Y, Meng M, Wang L, Li P. Fracture behavior of B2 phase matrix of Ti2AlNb-based alloy with microcracks of different orientations. Eng Fract Mech. 2023;279(4):109050. doi:10.1016/j.engfracmech.2023.109050. [Google Scholar] [CrossRef]
52. Zhou T, Zhao F, Zhou H, Zhang F, Wang P. Atomistic simulation and continuum modeling of the dynamic tensile fracture and damage evolution of solid single crystalline Al with He bubble. Int J Mech Sci. 2022;234(5):107681. doi:10.1016/j.ijmecsci.2022.107681. [Google Scholar] [CrossRef]
53. Liu SM, Li SH, Han WZ. Effect of ordered helium bubbles on deformation and fracture behavior of α-Zr. J Mater Sci Technol. 2019;35(7):1466–72. doi:10.1016/j.jmst.2019.03.015. [Google Scholar] [CrossRef]
54. Nguyen HG, Young SJ, Le TD, Chatzinotas S, Fang TH. Deformation mechanisms of AlCoCrCuFeNi: a molecular dynamics and machine learning approach. Mater Today Nano. 2025;31(7):100662. doi:10.1016/j.mtnano.2025.100662. [Google Scholar] [CrossRef]
55. Sun H, Lin Y, Song P, Yu W, Lai S, Wang G, et al. Insight into the strengthening and fracture mechanism of Au/Ni3Al interface via first-principles calculations. Surf Interfaces. 2025;72(1):106977. doi:10.1016/j.surfin.2025.106977. [Google Scholar] [CrossRef]
56. Linton N, Aidhy DS. Mechanistic understanding of vacancy formation energies in FCC concentrated alloys from DFT calculations. Acta Mater. 2025;289(15):120874. doi:10.1016/j.actamat.2025.120874. [Google Scholar] [CrossRef]
57. Yu Y, Li Q. Molecular dynamics study on crack propagation of Fe and Fe-Ni: temperature, crystallographic orientation and elemental segregation. Mater Des. 2025;260(11):115104. doi:10.1016/j.matdes.2025.115104. [Google Scholar] [CrossRef]
58. Li L, Chang L, Zhao J, Zhao J. Molecular dynamics simulation of fatigue crack propagation behavior at different twin boundaries in metallic titanium. Mater Today Commun. 2025;49:113813. doi:10.1016/j.mtcomm.2025.113813. [Google Scholar] [CrossRef]
59. Tamuzs VP, Petrova VE. On macrocrack-microdefect interaction. Int Appl Mech. 2002;38(10):1157–77. doi:10.1023/A:1022250111016. [Google Scholar] [CrossRef]
60. Wang Z, Ghoniem N, Swaminarayan S, LeSar R. A parallel algorithm for 3D dislocation dynamics. J Comput Phys. 2006;219(2):608–21. doi:10.1016/j.jcp.2006.04.005. [Google Scholar] [CrossRef]
61. Sills RB, Bertin N, Aghaei A, Cai W. Dislocation networks and the microstructural origin of strain hardening. Phys Rev Lett. 2018;121(8):085501. doi:10.1103/PhysRevLett.121.085501. [Google Scholar] [PubMed] [CrossRef]
62. Akhondzadeh S, Kang M, Sills RB, Ramesh KT, Cai W. Direct comparison between experiments and dislocation dynamics simulations of high rate deformation of single crystal copper. Acta Mater. 2023;250(855):118851. doi:10.1016/j.actamat.2023.118851. [Google Scholar] [CrossRef]
63. Li M, Tian X, Jiang W, Wang Q, Fan H. Mechanism of strain hardening of magnesium single-crystals: discrete dislocation dynamics simulations. J Mech Phys Solids. 2023;173:105238. doi:10.1016/j.jmps.2023.105238. [Google Scholar] [CrossRef]
64. Liang S, Zhu Y, Huang M, Li Z. Studying dislocation-induced shielding effect on the crack-tip in polycrystal by discrete dislocation dynamics. Int J Solids Struct. 2019;156–157:148–62. doi:10.1016/j.ijsolstr.2018.08.012. [Google Scholar] [CrossRef]
65. Liu J, Yuan S, Li Z, Huang M, Zhao L, Zhu Y. Size-dependent microvoid growth in heterogeneous polycrystals. Int J Plast. 2022;158:103410. doi:10.1016/j.ijplas.2022.103410. [Google Scholar] [CrossRef]
66. Lu S, Ao N, Kan Q, Wu S, Kang G, Zhang X. Effect of residual stress in gradient-grained metals: dislocation dynamics simulations. Int J Mech Sci. 2023;256:108518. doi:10.1016/j.ijmecsci.2023.108518. [Google Scholar] [CrossRef]
67. Tak TN, Prakash A, Keralavarma SM, Samajdar I, Guruprasad PJ. A discrete dislocation dynamics model of creep in polycrystals. J Mech Phys Solids. 2023;179:105385. doi:10.1016/j.jmps.2023.105385. [Google Scholar] [CrossRef]
68. Lu S, Zhao J, Huang M, Li Z, Kang G, Zhang X. Multiscale discrete dislocation dynamics study of gradient nano-grained materials. Int J Plast. 2022;156:103356. doi:10.1016/j.ijplas.2022.103356. [Google Scholar] [CrossRef]
69. Chen Y, Wang S, Feng H, Li W, Liu B, Li J, et al. Irradiation hardening behavior of high entropy alloys using random field theory informed discrete dislocation dynamics simulation. Int J Plast. 2023;162(9):103497. doi:10.1016/j.ijplas.2022.103497. [Google Scholar] [CrossRef]
70. Wu K, Liu G, Yu P, Ye C, Shi J, Shen Y. Prediction of hardening effect by irradiation-induced vacancy clusters with dislocation dynamics. Int J Plast. 2022;149:103160. doi:10.1016/j.ijplas.2021.103160. [Google Scholar] [CrossRef]
71. Liu G, Wu K, Yu P, Cheng X, Shi J, Ye C, et al. Atomically-informed dislocation dynamics model for prediction of void hardening in irradiated tungsten. Int J Plast. 2023;169:103727. doi:10.1016/j.ijplas.2023.103727. [Google Scholar] [CrossRef]
72. Pachaury Y, Warren G, Wharry JP, Po G, El-Azab A. Plasticity in irradiated FeCrAl nanopillars investigated using discrete dislocation dynamics. Int J Plast. 2023;167:103676. doi:10.1016/j.ijplas.2023.103676. [Google Scholar] [CrossRef]
73. Beaudoin AJ, Dawson PR, Mathur KK, Kocks UF, Korzekwa DA. Application of polycrystal plasticity to sheet forming. Comput Meth Appl Mech Eng. 1994;117(1–2):49–70. doi:10.1016/0045-7825(94)90076-0. [Google Scholar] [CrossRef]
74. Choi YS, Groeber MA, Shade PA, Turner TJ, Schuren JC, Dimiduk DM, et al. Crystal plasticity finite element method simulations for a polycrystalline Ni micro-specimen deformed in tension. Metall Mater Trans A. 2014;45(13):6352–9. doi:10.1007/s11661-014-2556-y. [Google Scholar] [CrossRef]
75. Roters F, Eisenlohr P, Hantcherli L, Tjahjanto DD, Bieler TR, Raabe D. Overview of constitutive laws, kinematics, homogenization and multiscale methods in crystal plasticity finite-element modeling: theory, experiments, applications. Acta Mater. 2010;58(4):1152–211. doi:10.1016/j.actamat.2009.10.058. [Google Scholar] [CrossRef]
76. Kardan-Halvaei M, Morovvati MR, Mollaei-Dariani B. Crystal plasticity finite element simulation and experimental investigation of the micro-upsetting process of OFHC copper. J Micromech Microeng. 2020;30(7):075005. doi:10.1088/1361-6439/ab8549. [Google Scholar] [CrossRef]
77. Wu H, Sun C. A framework of crystal plasticity finite element incorporating multiple fatigue cracking mechanisms in titanium alloys. Eng Fract Mech. 2025;327(5937):111482. doi:10.1016/j.engfracmech.2025.111482. [Google Scholar] [CrossRef]
78. Wang Z, Zhang H, Li Z, Li G, Zhang J, Zhang J, et al. Crystal plasticity finite element simulation and experiment investigation of nanoscratching of single crystalline copper. Wear. 2019;430:100–7. doi:10.1016/j.wear.2019.04.024. [Google Scholar] [CrossRef]
79. Han QN, Qiu W, He Z, Su Y, Ma X, Shi HJ. The effect of crystal orientation on fretting fatigue crack formation in Ni-based single-crystal superalloys: In-situ SEM observation and crystal plasticity finite element simulation. Tribol Int. 2018;125(2):209–19. doi:10.1016/j.triboint.2018.01.011. [Google Scholar] [CrossRef]
80. Han QN, Qiu W, Shang YB, Shi HJ. In-situ SEM observation and crystal plasticity finite element simulation of fretting fatigue crack formation in Ni-base single-crystal superalloys. Tribol Int. 2016;101:33–42. doi:10.1016/j.triboint.2016.03.025. [Google Scholar] [CrossRef]
81. Liu J, Huang M, Li Z, Zhao L, Zhu Y. Microvoid growth mechanism in FCC polycrystals and a statistical damage model. Int J Plast. 2021;137:102888. doi:10.1016/j.ijplas.2020.102888. [Google Scholar] [CrossRef]
82. Zhou X, Zan S, Zeng Y, Guo R, Wang G, Wang T, et al. Comprehensive study of plastic deformation mechanism of polycrystalline copper using crystal plasticity finite element. J Mater Res Technol. 2024;30(12):9221–36. doi:10.1016/j.jmrt.2024.06.006. [Google Scholar] [CrossRef]
83. Chandra S, Samal MK, Kapoor R, Chavan VM. Simulation of bicrystal deformation including grain boundary effects: atomistic computations and crystal plasticity finite element analysis. Comput Mater Sci. 2020;179(4):109641. doi:10.1016/j.commatsci.2020.109641. [Google Scholar] [CrossRef]
84. Yang X, Xu B, Liu E, Xiao G, Hao X, Lin J. Crystal plastic finite element model coupled with damage and mechanical properties of GH4169 polycrystalline alloy. J Mater Res Technol. 2025;38(10):3500–9. doi:10.1016/j.jmrt.2025.08.172. [Google Scholar] [CrossRef]
85. Hu L, Jiang SY, Zhang YQ, Zhu XM, Sun D. Texture evolution and inhomogeneous deformation of polycrystalline Cu based on crystal plasticity finite element method and particle swarm optimization algorithm. J Cent South Univ. 2017;24(12):2747–56. doi:10.1007/s11771-017-3688-1. [Google Scholar] [CrossRef]
86. Mao Y, Keshavarz S, Kilic MNT, Wang K, Li Y, Reid ACE, et al. A deep learning-based crystal plasticity finite element model. Scr Mater. 2025;254:116315. doi:10.1016/j.scriptamat.2024.116315. [Google Scholar] [CrossRef]
87. Dorward H, Knowles DM, Demir E, Mostafavi M, Peel MJ. Calibration and surrogate model-based sensitivity analysis of crystal plasticity finite element models. Mater Des. 2024;247(4):113409. doi:10.1016/j.matdes.2024.113409. [Google Scholar] [CrossRef]
88. Keshavarz S, Mao Y, Reid ACE, Agrawal A. Advancing material simulations: physics-informed neural networks and object-oriented crystal plasticity finite element methods. Int J Plast. 2025;185(10):104221. doi:10.1016/j.ijplas.2024.104221. [Google Scholar] [CrossRef]
89. Zhang Y, Xu P, Ding W, Jia H, Ouyang W, Cheng F. Phase-field simulation of dual-crack system hydrogen embrittlement in metallic materials. Theor Appl Fract Mech. 2024;131(22):104332. doi:10.1016/j.tafmec.2024.104332. [Google Scholar] [CrossRef]
90. Liu Z, Xu S, Ma Y, Lian H, Qu Y, Chen L. Polynomial chaos expansion-based stochastic phase field model for hydrogen-assisted cracking. Theor Appl Fract Mech. 2025;139(4):105000. doi:10.1016/j.tafmec.2025.105000. [Google Scholar] [CrossRef]
91. Si Z, Hirshikesh, Yu T, Natarajan S. An adaptive phase-field simulation for hydrogen embrittlement fracture with multi-patch isogeometric method. Comput Meth Appl Mech Eng. 2024;418(3):116539. doi:10.1016/j.cma.2023.116539. [Google Scholar] [CrossRef]
92. Liu H, Ruan R, Ma Y, Shen Y. Phase-field modelling of hydrogen-assisted fatigue cracking: theoretical framework and numerical implementation. Int J Hydrogen Energy. 2025;158:150502. doi:10.1016/j.ijhydene.2025.150502. [Google Scholar] [CrossRef]
93. Liu G, Mo H, Wang J, Shen Y. Coupled crystal plasticity finite element-phase field model with kinetics-controlled twinning mechanism for hexagonal metals. Acta Mater. 2021;202(1–2):399–416. doi:10.1016/j.actamat.2020.11.002. [Google Scholar] [CrossRef]
94. Jiang L, Liu G, Jin P, Shen Y, Wang J. The driving force for twin boundary migration in phase field model coupled to crystal plasticity finite element. Int J Plast. 2025;191:104397. doi:10.1016/j.ijplas.2025.104397. [Google Scholar] [CrossRef]
95. Mo H, Liu G, Mao Y, Shen Y, Wang J. Dual-interface model for twinning in the coupled crystal plasticity finite element-phase field method. Int J Plast. 2022;158(11):103441. doi:10.1016/j.ijplas.2022.103441. [Google Scholar] [CrossRef]
96. Wei D, Zaiser M, Feng Z, Kang G, Fan H, Zhang X. Effects of twin boundary orientation on plasticity of bicrystalline copper micropillars: a discrete dislocation dynamics simulation study. Acta Mater. 2019;176(21):289–96. doi:10.1016/j.actamat.2019.07.007. [Google Scholar] [CrossRef]
97. Lu S, Zhang X, Hu Y, Chu J, Kan Q, Kang G. Machine learning-based constitutive parameter identification for crystal plasticity models. Mech Mater. 2025;203:105263. doi:10.1016/j.mechmat.2025.105263. [Google Scholar] [CrossRef]
98. Ahmadian H, Bahrami B, Ayatollahi MR, Khosravani MR. Prediction of fracture in metallic plates based on the phase-field approach. Thin Walled Struct. 2025;213:113323. doi:10.1016/j.tws.2025.113323. [Google Scholar] [CrossRef]
99. Xiong Y, Zhao J, Zeng Q, Yuan F, Zhang X. Crack propagation behavior in metal matrix composites: a coupled nonlocal crystal plasticity and phase field modelling. J Mech Phys Solids. 2025;200:106164. doi:10.1016/j.jmps.2025.106164. [Google Scholar] [CrossRef]
100. Nairn JA, Aimene YE. Modeling multiple crack propagation in the material point method by J-integral methods accounting for other cracks intersecting the J contour. Eng Fract Mech. 2025;322(3):111143. doi:10.1016/j.engfracmech.2025.111143. [Google Scholar] [CrossRef]
101. Zhang W, Jiang W, Yu Y, Zhou F, Luo Y, Song M. Fatigue crack simulation of the 316L brazed joint using the virtual crack closure technique. Int J Press Vessels Pip. 2019;173(17):20–5. doi:10.1016/j.ijpvp.2019.04.018. [Google Scholar] [CrossRef]
102. Elguedj T, Jan Y, Combescure A, Leblé B, Barras G. X-FEM analysis of dynamic crack growth under transient loading in thick shells. Int J Impact Eng. 2018;122(8):228–50. doi:10.1016/j.ijimpeng.2018.08.013. [Google Scholar] [CrossRef]
103. Fan XJ, Qu RT, Zhang ZF. Remarkably high fracture toughness of HfNbTaTiZr refractory high-entropy alloy. J Mater Sci Technol. 2022;123:70–7. doi:10.1016/j.jmst.2022.01.017. [Google Scholar] [CrossRef]
104. Wang J, Jiang W, Li Y, Wang Q, Xu Q. Numerical assessment of cyclic J-integral △J for predicting fatigue crack growth rate. Eng Fract Mech. 2019;205(10):455–69. doi:10.1016/j.engfracmech.2018.11.031. [Google Scholar] [CrossRef]
105. Shi Z, Chen P, Wu Z, Wang Y, Li Z. Evaluation on VCCT-like methods in hyperelastic materials. Theor Appl Fract Mech. 2025;139(4):105023. doi:10.1016/j.tafmec.2025.105023. [Google Scholar] [CrossRef]
106. Chen Z, Dai Y, Liu Y. Structural fatigue crack propagation simulation and life prediction based on improved XFEM-VCCT. Eng Fract Mech. 2024;310(8):110519. doi:10.1016/j.engfracmech.2024.110519. [Google Scholar] [CrossRef]
107. Xie B, Yu T, Li R, Luo Z, Baxevanakis KP, Zhang P. Experimentally validated macro-mesoscopic simulation study on the fatigue short crack initiation and propagation in polycrystalline structure utilizing CP-XFEM. Eng Fract Mech. 2025;319(5):110995. doi:10.1016/j.engfracmech.2025.110995. [Google Scholar] [CrossRef]
108. Liang S, Zhu Y, Huang M, Li Z. Simulation on crack propagation vs. crack-tip dislocation emission by XFEM-based DDD scheme. Int J Plast. 2019;114:87–105. doi:10.1016/j.ijplas.2018.10.010. [Google Scholar] [CrossRef]
109. Shibanuma K, Suzuki Y, Kiriyama K, Suzuki K, Shirahata H. A model of cleavage crack propagation in a BCC polycrystalline solid based on the extended finite element method. Acta Mater. 2019;176:232–41. doi:10.1016/j.actamat.2019.07.013. [Google Scholar] [CrossRef]
110. Abe Y, Fukui D, Nagashima R, Nakada N, Shibanuma K. Fracture mechanics analysis of anisotropic cleavage fracture caused by transformation-induced microscopic internal stresses in lath martensite. Mater Charact. 2025;223:114905. doi:10.1016/j.matchar.2025.114905. [Google Scholar] [CrossRef]
111. Zhou H, Kinefuchi M, Takashima Y, Shibanuma K. Multiscale modelling strategy for predicting fatigue performance of welded joints. Int J Mech Sci. 2024;284:109751. doi:10.1016/j.ijmecsci.2024.109751. [Google Scholar] [CrossRef]
112. Zhou H, Suzuki Y, Kinefuchi M, Shibanuma K. Applicability of the multiscale model for predicting fatigue strength to short and long crack problems. ISIJ Int. 2022;62(10):2126–31. doi:10.2355/isijinternational.isijint-2022-205. [Google Scholar] [CrossRef]
113. Zhou H, Liu Z, Kikuchi S, Shibanuma K. Analysis of fatigue performance of austenitic stainless steels with bimodal harmonic structures based on multiscale model simulations. Mater Des. 2023;226:111657. doi:10.1016/j.matdes.2023.111657. [Google Scholar] [CrossRef]
114. Zhou H, Suzuki Y, Kinefuchi M, Schmauder S, Dogahe K, Shibanuma K. Bridging strategy between microscopic and macroscopic crack growth simulations to predict fatigue strength of steels. Int J Fatigue. 2023;168(9):107386. doi:10.1016/j.ijfatigue.2022.107386. [Google Scholar] [CrossRef]
115. Kanouté P, Boso DP, Chaboche JL, Schrefler BA. Multiscale methods for composites: a review. Arch Comput Methods Eng. 2009;16(1):31–75. doi:10.1007/s11831-008-9028-8. [Google Scholar] [CrossRef]
116. Matouš K, Geers MGD, Kouznetsova VG, Gillman A. A review of predictive nonlinear theories for multiscale modeling of heterogeneous materials. J Comput Phys. 2017;330:192–220. doi:10.1016/j.jcp.2016.10.070. [Google Scholar] [CrossRef]
117. Fish J, Shek K. Multiscale analysis of composite materials and structures. Compos Sci Technol. 2000;60(12–13):2547–56. doi:10.1016/S0266-3538(00)00048-8. [Google Scholar] [CrossRef]
118. Miehe C, Bayreuther CG. On multiscale FE analyses of heterogeneous structures: from homogenization to multigrid solvers. Numer Meth Eng. 2007;71(10):1135–80. doi:10.1002/nme.1972. [Google Scholar] [CrossRef]
119. Ghosh G, Biswas D, Bhattacharyya R. Advancements in multiscale modeling of damage in composite materials: a comprehensive review. Compos Part B Eng. 2025;307(1):112819. doi:10.1016/j.compositesb.2025.112819. [Google Scholar] [CrossRef]
120. Hu Y, Zhou G, Knezevic M, Shen Y, Wu P, Li D. Multiscale modelling with neural network-based crystal plasticity model from meso-to macroscale. Acta Mater. 2025;293:121075. doi:10.1016/j.actamat.2025.121075. [Google Scholar] [CrossRef]
121. Lee GH, Kim JH, Beom HG. Cohesive zone modeling of crack propagation in FCC single crystals via atomistic simulations. Met Mater Int. 2021;27(4):584–92. doi:10.1007/s12540-020-00693-x. [Google Scholar] [CrossRef]
122. Xie G, Wang F, Lai X, Xu Z, Zeng X. Atomistic study on crystal orientation-dependent crack propagation and resultant microstructure anisotropy in NiTi alloys. Int J Mech Sci. 2023;250(5):108320. doi:10.1016/j.ijmecsci.2023.108320. [Google Scholar] [CrossRef]
123. Elkhateeb MG, Shin YC. Molecular dynamics-based cohesive zone representation of Ti6Al4V/TiC composite interface. Mater Des. 2018;155:161–9. doi:10.1016/j.matdes.2018.05.054. [Google Scholar] [CrossRef]
124. Cai W, Sun C, Wang C, Qian L, Li Y, Fu MW. Modelling of the intergranular fracture of TWIP steels working at high temperature by using CZM-CPFE method. Int J Plast. 2022;156:103366. doi:10.1016/j.ijplas.2022.103366. [Google Scholar] [CrossRef]
125. Wang L, Shen L, Yi J, Yang X, Peng Y, Ding J, et al. Prediction model of dynamic fracture toughness of nickel-based alloys: combination of data-driven and multi-scale modelling. Eur J Mech A/solids. 2026;116(6):105892. doi:10.1016/j.euromechsol.2025.105892. [Google Scholar] [CrossRef]
126. Han F, Roters F, Raabe D. Microstructure-based multiscale modeling of large strain plastic deformation by coupling a full-field crystal plasticity-spectral solver with an implicit finite element solver. Int J Plast. 2020;125:97–117. doi:10.1016/j.ijplas.2019.09.004. [Google Scholar] [CrossRef]
127. Weng H, Bamer F, Luo C, Markert B, Yuan H. Physics-informed neural network for constitutive modeling of cyclic crystal plasticity considering deformation mechanism. Int J Mech Sci. 2025;302(5):110491. doi:10.1016/j.ijmecsci.2025.110491. [Google Scholar] [CrossRef]
Cite This Article
 Copyright © 2026 The Author(s). Published by Tech Science Press.
Copyright © 2026 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools