 Submit a Paper
Submit a Paper Propose a Special lssue
Propose a Special lssue Open Access
Open Access
ARTICLE
MiR-21/Sonic Hedgehog (SHH)/PI3K/AKT Pathway is Associated with NSCLC of Primary EGFR-TKI Resistance
The Second Department of Thoracic Oncology, Hunan Cancer Hospital/The Affiliated Cancer Hospital of Xiangya School of Medicine, Central South University, Changsha, 410013, China
* Corresponding Authors: Bolin Chen. Email: ; Lin Wu. Email:
Oncologie 2022, 24(3), 579-590. https://doi.org/10.32604/oncologie.2022.022121
Received 22 February 2022; Accepted 11 July 2022; Issue published 19 September 2022
 View Full Text
View Full Text Download PDF
Download PDFAbstract
Background: Non-small cell lung cancer (NSCLC), caused by abnormal gene drive, may have primary drug resistance after treatment with tyrosine kinase inhibitors (EGFR-TKIs). Therefore, we explore whether the primary drug-resistant NSCLC treated with EGFR-TKI is related to the miR-21/Sonic Hedgehog (SHH)/PI3K/AKT pathway. Methods: The patients from our hospital who meet the AJCC TNM staging (7th edition) stage IIIB and stage IV NSCLC were selected in this case study. Thereafter, the treatment response of EGFR-TKIs was evaluated according to the solid tumor efficacy evaluation standard (version 1.1). The patients were divided into the EGFR-TKIs primary drug resistance group (EGFR-TKIs-Primary-R) and the EGFR-TKIs sensitive group (EGFR-TKIs-Primary-S). Apoptosis level and degree of fibrosis in patients’ tumor tissues were detected by the TUNEL assay and Masson staining, respectively. The levels of miR-21 and GLI1 were measured by qRT-PCR technique. The contents of E-cadherin and Snail were detected by IF method, and the degree of PI3K/AKT phosphorylation was measured using IHC technique. Results: Compared with the EGFR-TKIs-Primary-S group, the EGFR-TKIs-Primary-R group showed lower levels of apoptosis and tumor tissue fibrosis. The levels of miR-21, GLI1, Snail, p-PI3K and p-AKT increased, while the level of E-cadherin decreased. However, the levels of total protein PI3K and AKT remained the same. Conclusion: NSCLC of primary EGFR-TKI resistance was found to be related to miR-21/SHH, the process of epithelial to mesenchymal transition (EMT), and PI3K/AKT phosphorylation. The present study provides a reference for future research in drug resistance, and paves the way to discover new therapeutic gene targets to alleviate lung cancer drug resistance.Keywords
Non-small cell lung cancer (NSCLC) is regarded as a group of diseases with abnormal gene drives in the current era. Epidermal growth factor receptor (EGFR) mutations are among the common oncogenic driver genes of NSCLC [1]. Therefore, patients diagnosed with NSCLC due to EGFR-sensitive mutations are currently remedied by tyrosine kinase inhibitors (TKIs). Due to tumor heterogeneity, 30% of the lung cancer patients triggered by EGFR-sensitive mutations have primary drug resistance to EGFR-TKIs. These drugs cannot prolong the survival rate of patients or improve clinical symptoms [2,3]. Acquired resistance of EGFR-mutant lung cancers proceeds on an average of 10 to 14 months after using EGFR TKIs [4]. The progression-free survival (PFS) of primary drug-resistant NSCLC patients is within three months [2,3] However, the underlying mechanism of primary drug resistance is still unknown. Therefore, exploring the process of primary drug resistance of EGFR-TKIs will provide us with new ideas for treating NSCLC in the clinic.
The high expression of miR-21 in NSCLC is associated with a poor response rate of EGFR-TKIs and a shorter overall survival time [5,6]. MiR-21 is considered one of the most significant miRNA molecules involved in tumorigenesis and development [7]. Numerous studies have confirmed that miR-21 not only promotes tumor growth, proliferation, anti-apoptosis and response to anti-tumor drugs, but is also overexpressed in many tumors [8]. Therefore, miR-21 is a reliable predictor of the therapeutic effect of EGFR-TKIs.
Previous literature showed that in the EGFR-TKIs drug-resistant cells, the combined application of Hedgehog (Hh) signaling pathway inhibitors and EGFR-TKIs could significantly inhibit the cell clone formation and the proliferation of EGFR-TKIs drug-resistant cells [9]. Furthermore, in the Hh signaling pathway response, SHH connected to its receptor and the SHH relative function protein released. It results in the transportation of transcription factor glioma-associated oncogene (GLI), which regulates the removal of relative genes (cell differentiation, survival, and growth). Besides, GLI 1 is the only SHH pathway activity marker [10].
Further research in the similar context proved that in glioblastoma, the SHH pathway enhanced the infiltration and migration of tumor cells through the PI3K/AKT pathway, and miR-21 could adjust necrosis of glioblastoma and cell proliferation by regulating the related pathways of SHH [11]. Moreover, miR-21 inhibited the sensitivity of EGFR-TKI in NSCLC by down-regulating PTEN, which activates the AKT and ERK pathways [11]. Therefore, it is necessary to explore whether the miR-21 has an important primary drug resistance effect in the lung cancer.
Based on the above-given facts, we reasonably and boldly speculate that the miR-21/SHH/PI3K/AKT pathway is connected to the EGFR-TKI primary drug-resistant NSCLC patients.
Patients with stage IIIB and IV NSCLC were diagnosed in our hospital by histology or cytology according to the AJCC TNM staging (7th edition) [12]. The specific diagnostic criteria are as follows:
(1) The patients who were confirmed histologically to have AJCC TNM stage IIIB and IV NSCLC and contained EGFR sensitive mutations (exon 19 deletion mutation, or 21 exon L858R mutation) were selected [13].
(2) It can be diagnosed for the first time or reoccur.
(3) There is no restriction on histological type; squamous cell carcinoma, adenocarcinoma, or other types of NSCLC were acceptable. We have done genetic testing for the above non-small cell lung cancers. For patients with EGFR-sensitizing mutations, first-line targeted therapy was performed.
(4) Patients who received their first EGFR-TKI targeted drug therapy.
Following exclusion criteria was implied:
(1) Patients with other malignant tumors at the same time.
(2) According to the judgement of the investigator, the patient suffered from other serious diseases that may have affected the follow-up and short-term survival.
(3) Patients with any other medical conditions and social/psychological problems who were judged by the researcher to be unsuitable to participate in this study.
(4) Patients who cannot accept the use of contrast-enhanced magnetic resonance imaging (MRI) or contrast-enhanced computed tomography (CT) for clinical follow-up.
We evaluated the response of each patient to EGFR-TKIs using the evaluation criteria for the efficacy of solid tumors (version 1.1) [14]. The estimate time-point of primary drug resistance relies on the time when the patient’s NSCLC progression after EGFR-TKIs treatment does not conform to Jackman’s criteria for acquired drug resistance [15].
Thereafter, we selected three patients with primary EGFR-TKIs resistance as the EGFR-TKIs-Primary-R group and then three more patients with stable disease and partial response to EGFR-TKIs as the EGFR-TKIs-Primary-S group. The clinical characteristics of the EGFR-TKIs-Primary-R group and the EGFR-TKIs-Primary-S group are presented in Table 1. The study was conducted with the informed consent of all participants and approved by the Medical Ethics Committee of Hunan Cancer Hospital (KY2021097).
2.2 Terminal Deoxynucleotidyl Transferase dUTP Nick-End Labeling (TUNEL) Assay
The degree of apoptosis in tumor tissues of patients with EGFR-TKI primary drug-resistant NSCLC was determined with the TUNEL kit (KGA704, KeyGEN, China). After the sections were sequentially sealed, permeable, biotin-blocked, and HRP labeled, DAB color development was performed on the sections. Finally, the sections were sealed with neutral gum. The microscope (BA410T, Motic, China) was employed to determine the apoptosis index.
The samples were processed by the Masson staining kit (AWI0253a/AWI0253b, Wellbio, China). After baking for 15 h, the slices were dewaxed. After we removed water from the surface of the tissue, the dye was added dropwise for 0.5 min. Then, the sample was thoroughly rinsed with the staining solution and soaked in distilled water for cleaning. The slices were soaked again with PBS solution for 10 min and stained with slurry staining solution within 5 min. For 30 s, the sample was treated with a color separation solution. The counterstain solution was treated for 7 min and then washed with ethanol. After drying, it was mounted and tested under an electron microscope (BA410T, Motic, China).
2.4 Immunofluorescence Assay (IF)
The dehydrated slices were treated in xylene for 1 h and then dehydrated with different ethanol concentrations in sequence. To restore the antigen, the slices were soaked in EDTA buffer at high temperature. After being kept at room temperature for 1 day, it was cleaned and soaked in a mixed solution (sodium borohydride solution, 75% ethanol, and Sudan black dye solution). It was blocked with BSA for 1 h. Then the sample was treated with E-cadherin primary antibody (20874-1-AP, 1:200, Proteintech, USA) and Snail primary antibody (#3879, 1:200, CST, USA), and added with the corresponding secondary antibody (SA00013-2, CoraLite488-conjugated Affinipure Goat Anti-Rabbit IgG (H + L), Proteintech, USA). Finally, the samples were stained with DAPI and observed under a fluorescence microscope (BA410T, Motic, China).
2.5 Immunohistochemistry (IHC)
After the sections were deparaffinized and dehydrated, they were soaked in 0.01 M citrate buffer at a high temperature for 20 min. After 24 h at room temperature, the slices were immersed in periodic acid for 10 min. PI3K primary antibody (ab227204, 1:100, abcam, UK), p-PI3K primary antibody (bs-5570r, 1:100, Bioss, China), AKT primary antibody (10176-2-AP, 1:100, Proteintec, USA) and p-AKT primary antibody (ab38513, 1:100, abcam, UK) were added and incubated, respectively. After that the corresponding secondary antibody was added and incubated for 30 min also. Finally, hematoxylin was dyed for 10 min, dehydrated with alcohol, treated with xylene, and thereafter mounted for observation.
2.6 Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
Trizol method was applied to extract the RNA from tissues, and an mRNA reverse transcription kit (CW2569, Beijing ComWin Biotech, China) was applied to reverse transcription cDNA. The primer sequences of miR-21, GLI1, U6, and β-actin were synthesized by Sangon Biotech (Table 2). Then a fluorescence quantitative PCR instrument (PIKOREAL96, Thermo, USA) was applied to carry out real-time monitoring of fluorescence signals and DNA amplification. U6 and β-actin were displayed as internal controls. Finally, the relative expression of genes was calculated using the 2−ΔΔCt method.

GraphPad Prism 9.0 (GraphPad Software, Inc., USA) was employed for the statistical analysis. The dataset conforms to a normal distribution and is represented as the mean ± standard deviation (X̅ ± SD). To compare the differences between two groups, Student’s T-test was used at 0.05 probability level when p-values were significant.
3.1 Elevated Expression of miR-21 and GLI1 in Tumor Tissues of NSCLC Patients with Primary EGFR-TKI Resistance
First, we measured the expression levels of miR-21 and GLI1 in tumor tissues of EGFR-TKI-resistant NSCLC patients. Data from qRT-PCR analysis (Fig. 1) showed that the contents of miR-21 and GLI1 of the EGFR-TKIs-Primary-R group were much higher than those of EGFR-TKIs-Primary-S group (p = 0.0282; p = 0.0017). Based on these findings, it was confirmed that the contents of miR-21 and GLI1 were elevated in the tumor tissues of EGFR-TKI-resistant NSCLC patients.
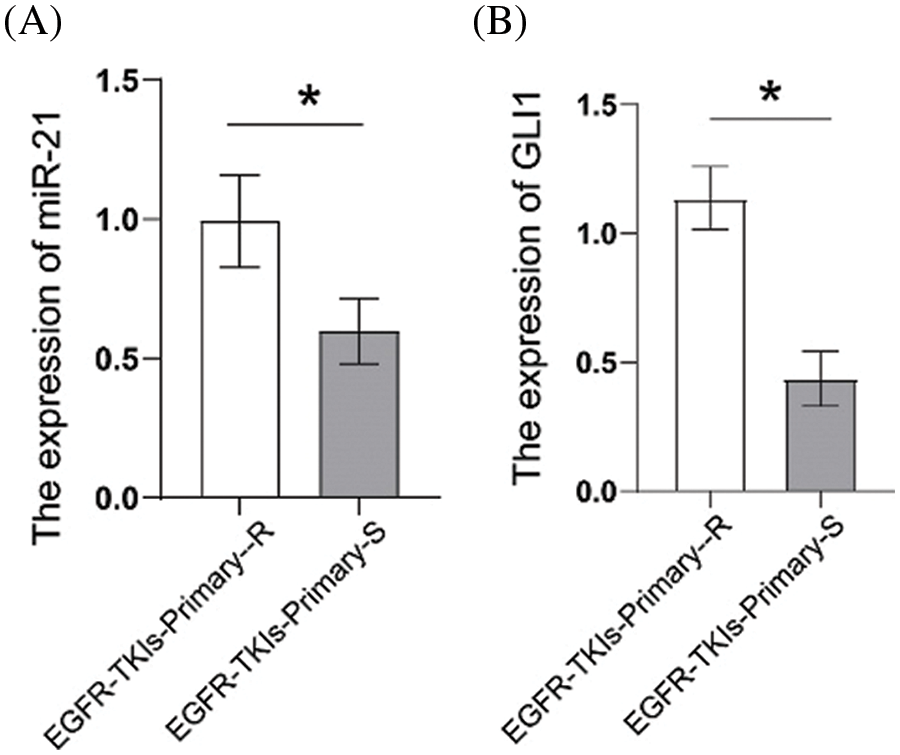
Figure 1: Elevated expression of miR-21 and GLI1 in tissues of NSCLC patients with primary EGFR-TKI resistance. (A). qRT-PCR was employed to assess the miR-21 expressions. (B). qRT-PCR was used to assess GLI1 expressions. *p < 0.05 vs. EGFR-TKIs-Primary-R
3.2 Decreased Level of Apoptosis in Tumor Tissues of NSCLC Patients with Primary EGFR-TKI Resistance
After the tumor tissues apoptosis level changes in NSCLC patients with EGFR-TKI-resistant were determined, we found that the TUNEL-positive cell number (Fig. 2) in the EGFR-TKIs-Primary-R group was much lower than the EGFR-TKIs-Primary-S group (p = 0.0006). This showed that apoptosis in the tumor tissues of EGFR-TKI-resistant NSCLC patients was reduced.
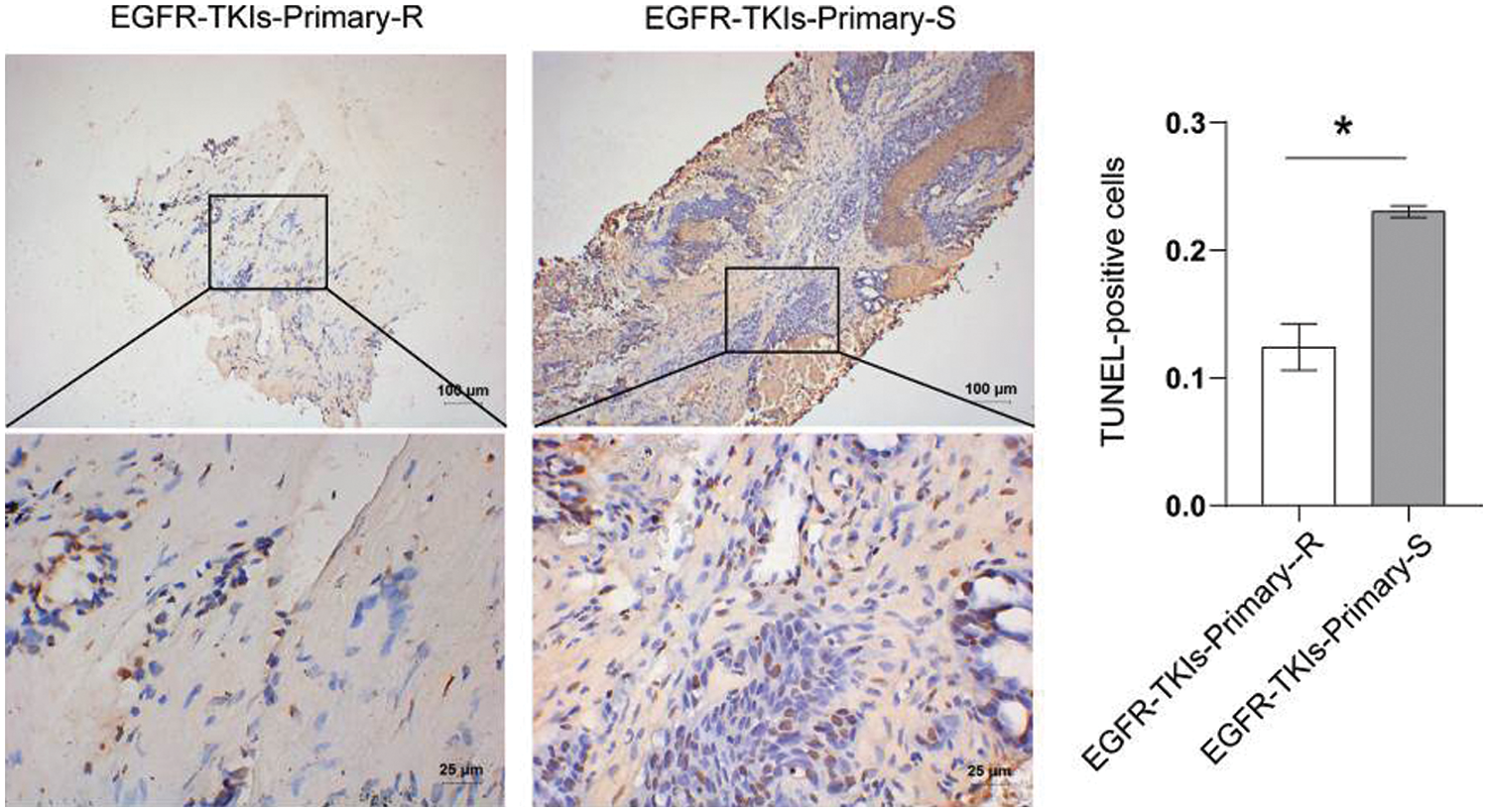
Figure 2: Decreased level of apoptosis in the tumor tissues of NSCLC patients with primary EGFR-TKI resistance. TUNEL was used to measure the level of apoptosis (TUNEL, ×400, scale bar = 25 μm; ×100, scale bar = 100 μm). *p < 0.05 vs. EGFR-TKIs-Primary-R
3.3 Decreased Degree of Fibrosis in Tumor Tissues of NSCLC Patients with Primary EGFR-TKI Resistance
Data were collected for tumor tissue analysis to estimate the degree of fibrosis changes in EGFR-TKI-resistant NSCLC patients. Fig. 3 showed that the fibrosis level of the Masson staining results in the EGFR-TKIs-Primary-R group was much lower than that of the EGFR-TKIs-Primary-S group. This indicates that the degree of fibrosis in the tumor tissues of EGFR-TKI-resistant NSCLC patients was weakened.
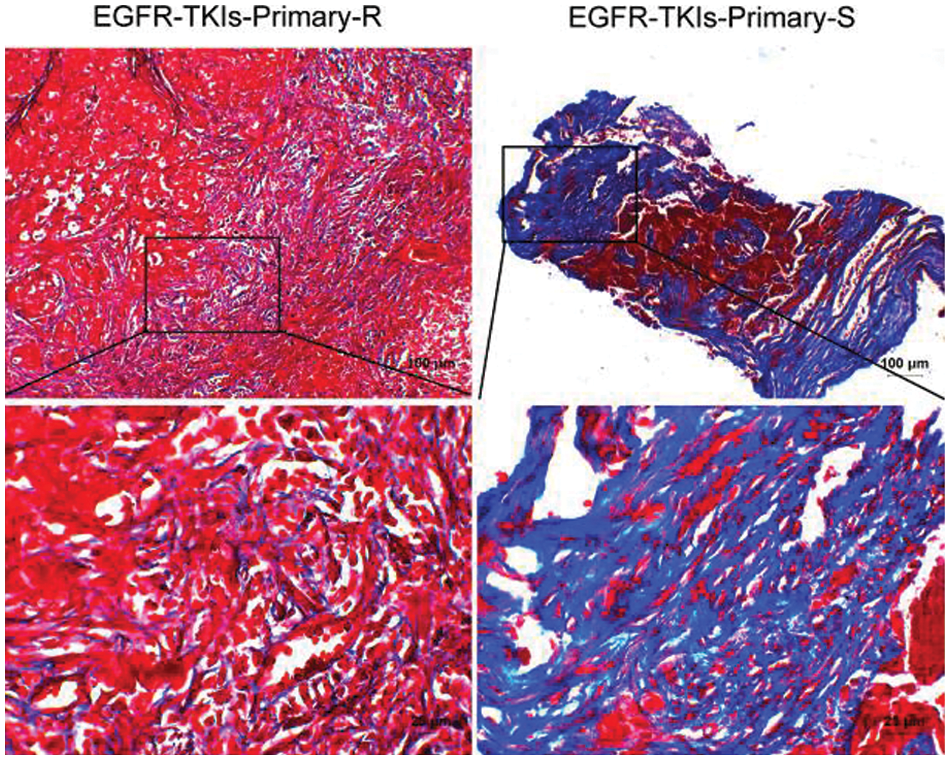
Figure 3: Decreased degree of fibrosis in the tumor tissues of NSCLC patients with primary EGFR-TKI resistance. Masson staining was applied to measure the degree of fibrosis (Masson, ×400, scale bar = 25 μm; ×100, scale bar = 100 μm)
3.4 Epithelial to Mesenchymal Transition (EMT)-Related Protein Expression in Tumor Tissues of NSCLC Patients with Primary EGFR-TKI Resistance
EMT-related proteins level changes of EGFR-TKI-resistant NSCLC patients was determined using the IF technique utilized to measure E-cadherin and Snail expression levels. Compared to the EGFR-TKIs-Primary-S group (Fig. 4), the E-cadherin content in the EGFR-TKIs-Primary-R group was much lower (p = 0.0007), whereas, the Snail content in EGFR-TKIs-Primary-R group was much higher (p = 0.0068). This demonstrates that the EMT effect of EGFR-TKI-resistant NSCLC patients was stronger.
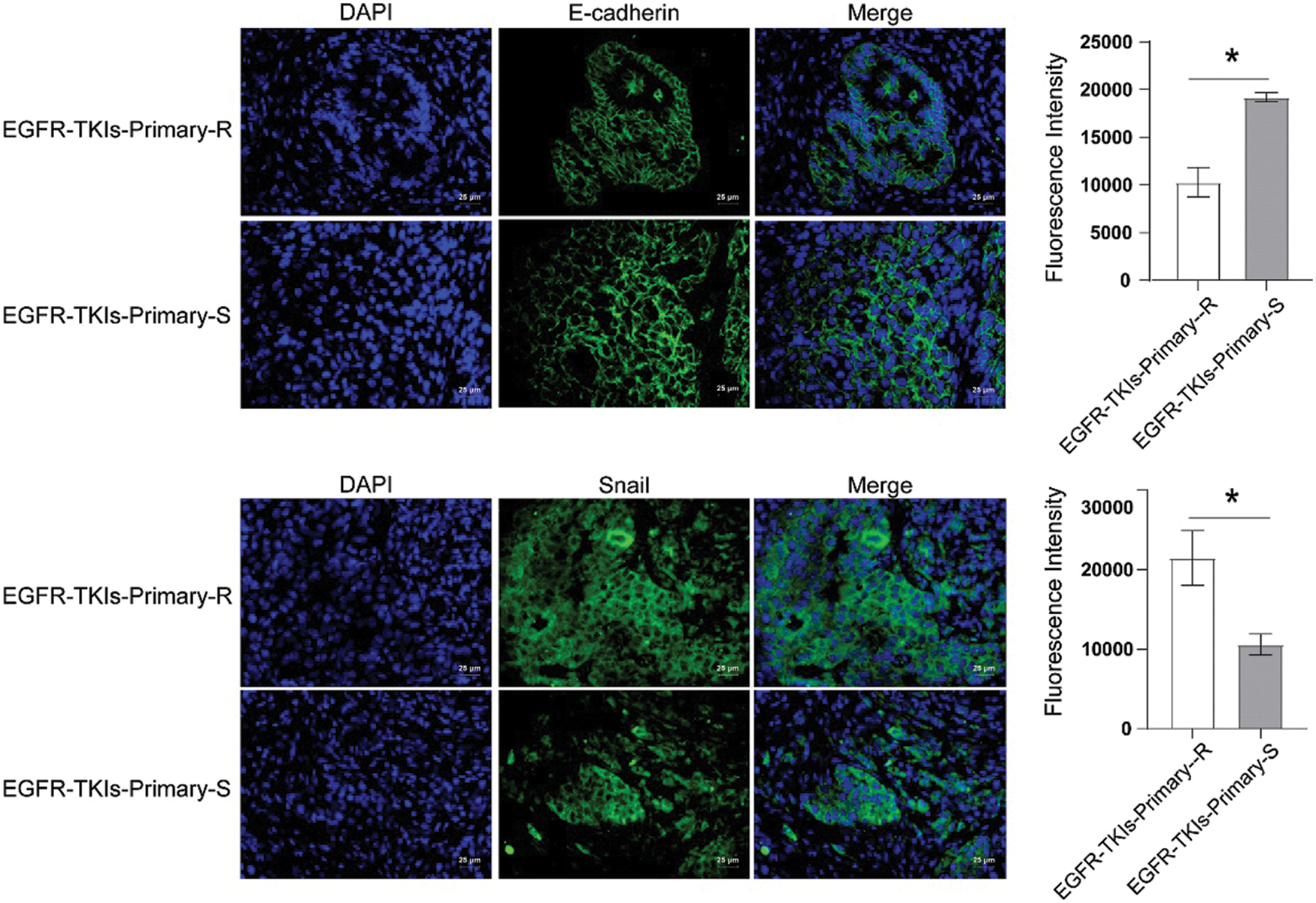
Figure 4: EMT-related protein expression in the tumor tissues of NSCLC patients with primary EGFR-TKI resistance. IF was utilized to measure the content differences of E-cadherin and Snail in groups EGFR-TKIs-Primary-R and EGFR-TKIs-Primary-S (IF, × 400, scale bar = 25 μm). *p < 0.05 vs. EGFR-TKIs-Primary-R
3.5 PI3K and its Phosphorylation Level in Tumor Tissues of NSCLC Patients with Primary EGFR-TKI Resistance
Following that, the changes in PI3K and its phosphorylation level in EGFR-TKI-resistant NSCLC patients’ tumor tissues were measured and IHC technique was applied to detect the level changes of PI3K and p-PI3K (Fig. 5). Data revealed that the content of p-PI3K in the EGFR-TKIs-Primary-R group was much higher than that of the EGFR-TKIs-Primary-S group (p = 0.0011). No significant difference was detected for the content of PI3K between the two groups. This implies that the phosphorylation level of PI3K in the tumor tissues of patients with EGFR-TKI primary drug-resistant NSCLC was increased.
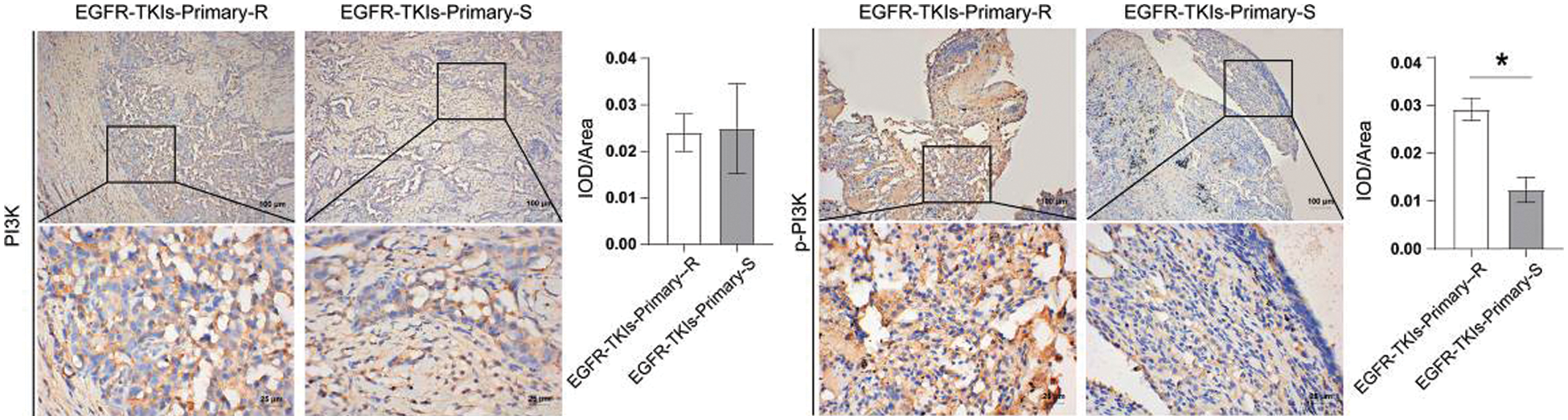
Figure 5: PI3K and its phosphorylation level in the tumor tissues of NSCLC patients with primary EGFR-TKI resistance. IHC was applied to measure the level differences of PI3K and p-PI3K in groups EGFR-TKIs-Primary-R and EGFR-TKIs-Primary-S (IHC, ×400, scale bar = 25 μm; ×100, scale bar = 100 μm).*p < 0.05 vs. EGFR-TKIs-Primary-R
3.6 AKT and Its Phosphorylation Level in the Tumor Tissues of NSCLC Patients with Primary EGFR-TKI Resistance
Finally, we assessed the changes in PI3K and its phosphorylation level in tumor tissues of EGFR-TKI-resistant NSCLC patients amd observed that the content of p-AKT in the EGFR-TKIs-Primary-R group was much higher (p = 0.0188) as ompared to the EGFR-TKIs-Primary-S group (Fig. 6). No significant differences were detected for the content of AKT between the two groups. Therefore, the phosphorylation level of AKT in the tumor tissues of patients with EGFR-TKI primary drug-resistant NSCLC was increased.
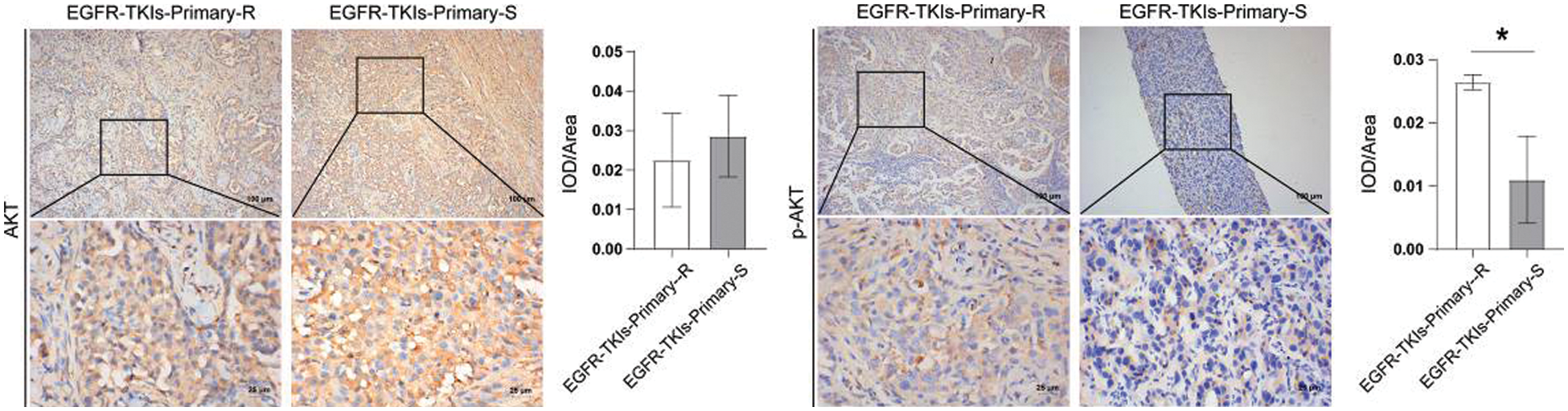
Figure 6: AKT and its phosphorylation level in the tumor tissues of NSCLC patients with primary EGFR-TKI resistance. IHC was emplyed to measure the level differences of AKT and p-AKT in groups EGFR-TKIs-Primary-R and EGFR-TKIs-Primary-S (IHC, ×400, scale bar = 25 μm; ×100, scale bar = 100 μm).*p < 0.05 vs. EGFR-TKIs-Primary-R
After careful analysis of results from present study, it can be established that EGFR-TKI primary drug-resistant NSCLC is connected to the miR-21/SHH/PI3K/AKT pathway. In addition, the expression of miR-21 and GLI1 was increased in NSCLC patients with primary EGFR-TKI resistance. Similarly, its phosphorylation of PI3K/AKT was enhanced, and the level of apoptosis and degree of tumor tissue fibrosis were weakened. Besides this, the EMT of NSCLC patients with primary EGFR-TKI resistance was stronger.
The dysregulation of specific microRNA (miRNA) may affect resistance to targeted drugs [16]. miRNA was a class of non-coding RNAs that were about 20 nucleotides in length. It worked by targeting mRNA’s 3’ untranslated region (UTR). miRNA played an important role in post-transcriptional gene regulation by inhibiting translation or influencing mRNA degradation [17]. Many studies have found that miRNA was closely related to drug resistance. miR-99a and miR-491 regulated cisplatin resistance in human gastric cancer cells by targeting CAPNS1 [18]. miR-451 was involved in resistance to paclitaxel by regulating YWHAZ in breast cancer [19]. miR-634 restored the sensitivity of drug-resistant ovarian cancer cells by targeting the RAS-MAPK pathway [20]. miR-128-3p delivered by exosomes increased the sensitivity of oxaliplatin-resistant colorectal cancer [21]. Interestingly, our studies illustrated that miR-21 is overexpressed in the PC9R cells resistant to EGFR-TKI (human lung cancer cells resistant to Gefitinib strain), and by inhibiting the expression of miR-21 could induce PC9R cell apoptosis [22]. After knocking down miR-21, the above resistance intensity was reversed [23].
In addition, GLI1 is also involved in regulating tumor cell proliferation and apoptosis [24]. GLI1 protein overexpression has been linked to a poor clinical prognosis and a lung malignant phenotype [25]. Up-regulation of GLI1 could help activate Hh signaling [26]. Inhibition of GLI1 function suppresses the Hh pathway-dependent medulloblastoma [27]. Thus, Hh pathway inhibition might be a treatment method to delay disease progression and recurrence [28]. Furthermore, an imbalance in the Hh pathway may contribute to tumor formation or accelerate tumor growth rate [28]. GLI1 activation in the Hh pathway was identified as a critical mechanism of erlotinib resistance in human NSCLC [29]. In the Hh signaling pathway response, SHH binds to its receptors and further regulates cell differentiation, survival, and growth [10]. In the meantime, EMT in cancer cells is believed to induce significant changes in the cell morphology, enhances its invasion and metastasis, and thereby contribute to the occurrence of drug resistance [30,31]. These phenotypic changes were modulated by EMT-related factors such as extracellular matrix components [30,31]. E-cadherin (intercellular adhesion complex and tumor suppressor protein) was thought to be an invasion suppressor as one of the most characteristic proteins of EMT [32]. On the contrary, overexpressed Snail could increase the metastasis ability of tumor cells [33]. After analyzing and testing the patients’ tumors, our findings were also consistent with the above cited rules. The levels of miR-21, GLI1, and Snail in the tumor tissues of patients with EGFR-TKI-resistant NSCLC were elevated, and the content of E-cadherin was decreased.
miR-21 also positively influenced the PI3K/AKT signaling pathway [22,34]. The process of overexpression of miR-21 leading to EGFR-TKI resistance in PC9R cells is also accompanied by the activation of the PI3K/AKT pathway [22]. Down-regulation of miR-21 could reduce the expression of p-AKT [23]. miR-21 inhibited PI3K/AKT signaling and switched the EMT, reducing cancer cell invasion and migration [34]. PI3K/AKT pathway inhibitors seem to help overcome malignancies [35]. Drug-resistant cells could be repaired by PI3K inhibitors [36]. PI3K/AKT activation phosphorylated several proteins, promoting tumor cell growth, proliferation, invasion, and metastasis while inhibiting cell apoptosis [37,38]. EGFR-TKIs inhibited EGFR downstream signaling pathway activity primarily through the PI3K/AKT signaling pathway [39]. After analyzing and testing the patients’ tumor, we have recorded the similar changes as cited above. The total protein content of PI3K and AKT in the tumor tissues of NSCLC patients with EGFR-TKI primary resistance remained found unchanged. The phosphorylation levels of PI3K and AKT were elevated. In general, the phosphorylation activation of the PI3K/AKT signaling pathway was involved in the process of NSCLC patient’s primary resistance to EGFR-TKI.
Limited by the sample size, we have only studied the potential pathogenic mechanism of primary drug resistance. miR-21/Sonic Hedgehog (SHH)/PI3K/AKT pathway was associated with primary EGFR-TKI resistance in NSCLC. However, these factors cannot be used as independent prognostic criteria for primary EGFR-TKI resistance. The prognostic criteria need subsequent reverse validation. We are working to collect baseline data for survival analysis and further explore independent prognostic indicators of NSCLC. Besides, we will explore the mechanism of acquired drug resistance. We hoped that our elucidation of the EGFR-TKI resistance mechanisms will provide a reference for future drug development and facilitate the advancement of more precise treatments.
Our result suggested that NSCLC of primary EGFR-TKI resistance is correlated with miR-21/SHH expression, the process of EMT, and PI3K/AKT phosphorylation. These findings have shaped our understanding of the body’s primary drug resistance in NSCLC targeted therapy. Based on present findings, future studies can also be carried out on acquired EGFR-TKI resistant NSCLC and non-adenocarcinoma of primary EGFR-TKI resistant NSCLC. Through broadening our understanding about the underlying mechanisms of EGFR TKI resistance, it is hoped that present study will provide a reference for future drug development and to facilitate the advancement of more precise and treatments.
Acknowledgement: The authors would like to thank The Hunan Cancer Hospital/The Affiliated Cancer Hospital of Xiangya School of Medicine, Central South University for their technical assistance.
Ethics Approval and Informed Consent Statement: This study was approved by The Medical Ethics Committee of Hunan Cancer Hospital (KY2021097). The study conducted with the informed consent of all participants and a written informed consent has been obtained from the patients to publish this paper.
Availability of Data and Materials: The readers can access the data used in the study from the corresponding author upon request.
Authorship: The authors confirm contribution to the paper as follows: LX: Writing-original draft; Data curation; Methodology. KL, JL, LL, FX, YX, YK, XP, QW, and JW: Data curation; Formal analysis; Visualization. BC and LW: Writing-review & editing; Project administration; Resources; Supervision. All authors have read and agreed to the published version of the manuscript.
Funding Statement: This study was supported by the Hunan Cancer Hospital Climb Plan [2020QH005], the Science and Technology Innovation Program of Hunan Province [2020SK51110], and the Scientific Research Project of Hunan Provincial Health Commission [202203102576].
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
References
1. Siegelin, M. D., Borczuk, A. C. (2014). Epidermal growth factor receptor mutations in lung adenocarcinoma. Laboratory Investigation, 94, 129–137. DOI 10.1038/labinvest.2013.147. [Google Scholar] [CrossRef]
2. Mok, T. S., Wu, Y. L., Thongprasert, S., Yang, C. H., Chu, D. T. et al. (2009). Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. The New England Journal of Medicine, 361, 947–957. DOI 10.1056/NEJMoa0810699. [Google Scholar] [CrossRef]
3. Maemondo, M., Inoue, A., Kobayashi, K., Sugawara, S., Oizumi, S. et al. (2010). Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. The New England Journal of Medicine, 362, 2380–2388. DOI 10.1056/NEJMoa0909530. [Google Scholar] [CrossRef]
4. Wu, S. G., Shih, J. Y. (2018). Management of acquired resistance to EGFR TKI-targeted therapy in advanced non-small cell lung cancer. Molecular Cancer, 17, 38. DOI 10.1186/s12943-018-0777-1. [Google Scholar] [CrossRef]
5. Shen, H., Zhu, F., Liu, J., Xu, T., Pei, D. et al. (2014). Alteration in mir-21/PTEN expression modulates gefitinib resistance in non-small cell lung cancer. PLoS One, 9, e103305. DOI 10.1371/journal.pone.0103305. [Google Scholar] [CrossRef]
6. Shen, Y., Tang, D., Yao, R., Wang, M., Wang, Y. et al. (2013). microRNA expression profiles associated with survival, disease progression, and response to gefitinib in completely resected non-small-cell lung cancer with EGFR mutation. Medical Oncology, 30, 750. DOI 10.1007/s12032-013-0750-1. [Google Scholar] [CrossRef]
7. Zhang, Z., Li, Z., Gao, C., Chen, P., Chen, J. et al. (2008). miR-21 plays a pivotal role in gastric cancer pathogenesis and progression. Laboratory Investigation, 88, 1358–1366. DOI 10.1038/labinvest.2008.94. [Google Scholar] [CrossRef]
8. Asangani, I. A., Rasheed, S. A., Nikolova, D. A., Leupold, J. H., Colburn, N. H. et al. (2008). microRNA-21 (miR-21) post-transcriptionally downregulates tumor suppressor Pdcd4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene, 27, 2128–2136. DOI 10.1038/sj.onc.1210856. [Google Scholar] [CrossRef]
9. Bai, X. Y., Zhang, X. C., Yang, S. Q., An, S. J., Chen, Z. H. et al. (2016). Blockade of hedgehog signaling synergistically increases sensitivity to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer cell lines. PLoS One, 11, e0149370. DOI 10.1371/journal.pone.0149370. [Google Scholar] [CrossRef]
10. O’Leary, K., Shia, A., Schmid, P. (2018). Epigenetic regulation of EMT in non-small cell lung cancer. Current Cancer Drug Targets, 18, 89–96. DOI 10.2174/1568009617666170203162556. [Google Scholar] [CrossRef]
11. Fu, J., Rodova, M., Nanta, R., Meeker, D., van Veldhuizen, P. J. et al. (2013). NPV-LDE-225 (Erismodegib) inhibits epithelial mesenchymal transition and self-renewal of glioblastoma initiating cells by regulating miR-21, miR-128, and miR-200. Neuro-Oncology, 15, 691–706. DOI 10.1093/neuonc/not011. [Google Scholar] [CrossRef]
12. Edge, S. B., Compton, C. C. (2010). The American joint committee on cancer: The 7th edition of the AJCC cancer staging manual and the future of TNM. Annals of Surgical Oncology, 17, 1471–1474. DOI 10.1245/s10434-010-0985-4. [Google Scholar] [CrossRef]
13. Li, W. Q., Cui, J. W. (2020). Non-small cell lung cancer patients with ex19del or exon 21 L858R mutation: Distinct mechanisms, different efficacies to treatments. Journal of Cancer Research and Clinical Oncology, 146, 2329–2338. DOI 10.1007/s00432-020-03296-6. [Google Scholar] [CrossRef]
14. Eisenhauer, E. A., Therasse, P., Bogaerts, J., Schwartz, L. H., Sargent, D. et al. (2009). New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). European Journal of Cancer, 45, 228–247. DOI 10.1016/j.ejca.2008.10.026. [Google Scholar] [CrossRef]
15. Jackman, D., Pao, W., Riely, G. J., Engelman, J. A., Kris, M. G. et al. (2010). Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. Journal of Clinical Oncology, 28, 357–360. DOI 10.1200/jco.2009.24.7049. [Google Scholar] [CrossRef]
16. Leonetti, A., Capula, M., Minari, R., Mazzaschi, G., Gregori, A. et al. (2021). Dynamic evaluation of circulating miRNA profile in EGFR-mutated NSCLC patients treated with EGFR-TKIs. Cells, 10, 1520. DOI 10.3390/cells10061520. [Google Scholar] [CrossRef]
17. Chen, P. Y., Meister, G. (2005). microRNA-Guided posttranscriptional gene regulation. Biological Chemistry, 386, 1205–1218. DOI 10.1515/bc.2005.139. [Google Scholar] [CrossRef]
18. Zhang, Y., Xu, W., Ni, P., Li, A., Zhou, J. et al. (2016). miR-99a and miR-491 regulate cisplatin resistance in human gastric cancer cells by targeting CAPNS1. International Journal of Biological Sciences, 12, 1437–1447. DOI 10.7150/ijbs.16529. [Google Scholar] [CrossRef]
19. Wang, W., Zhang, L., Wang, Y., Ding, Y., Chen, T. et al. (2017). Involvement of miR-451 in resistance to paclitaxel by regulating YWHAZ in breast cancer. Cell Death & Disease, 8, e3071. DOI 10.1038/cddis.2017.460. [Google Scholar] [CrossRef]
20. van Jaarsveld, M. T., van Kuijk, P. F., Boersma, A. W., Helleman, J., van, I. W. F. et al. (2015). miR-634 restores drug sensitivity in resistant ovarian cancer cells by targeting the Ras-MAPK pathway. Molecular Cancer, 14, 196. DOI 10.1186/s12943-015-0464-4. [Google Scholar] [CrossRef]
21. Liu, T., Zhang, X., Du, L., Wang, Y., Liu, X. et al. (2019). Exosome-transmitted miR-128-3p increase chemosensitivity of oxaliplatin-resistant colorectal cancer. Molecular Cancer, 18, 43. DOI 10.1186/s12943-019-0981-7. [Google Scholar] [CrossRef]
22. Li, B., Ren, S., Li, X., Wang, Y., Garfield, D. et al. (2014). MiR-21 overexpression is associated with acquired resistance of EGFR-TKI in non-small cell lung cancer. Lung Cancer, 83, 146–153. DOI 10.1016/j.lungcan.2013.11.003. [Google Scholar] [CrossRef]
23. Jing, C., Cao, H., Qin, X., Yu, S., Wu, J. et al. (2018). Exosome-mediated gefitinib resistance in lung cancer HCC827 cells via delivery of miR-21. Oncology Letters, 15, 9811–9817. DOI 10.3892/ol.2018.8604. [Google Scholar] [CrossRef]
24. Peralta-Arrieta, I., Trejo-Villegas, O. A., Armas-López, L., Ceja-Rangel, H. A., del Ordóñez-Luna, M. et al. (2021). Failure to EGFR-TKI-based therapy and tumoural progression are promoted by MEOX2/GLI1-mediated epigenetic regulation of EGFR in the human lung cancer. European Journal of Cancer, 160, 189–205. DOI 10.1016/j.ejca.2021.10.032. [Google Scholar] [CrossRef]
25. Herrera-Solorio, A. M., Peralta-Arrieta, I., Armas Lopez, L., Hernandez-Cigala, N., Mendoza Milla, C. et al. (2021). LncRNA SOX2-OT regulates AKT/ERK and SOX2/GLI-1 expression, hinders therapy, and worsens clinical prognosis in malignant lung diseases. Molecular Oncology, 15, 1110–1129. DOI 10.1002/1878-0261.12875. [Google Scholar] [CrossRef]
26. Ye, G., Pan, R., Zhu, L., Zhou, D. (2020). Circ_DCAF6 potentiates cell stemness and growth in breast cancer through GLI1-Hedgehog pathway. Experimental and Molecular Pathology, 116, 104492. DOI 10.1016/j.yexmp.2020.104492. [Google Scholar] [CrossRef]
27. Lu, J., Liu, L., Zheng, M., Li, X., Wu, A. et al. (2018). MEKK2 and MEKK3 suppress hedgehog pathway-dependent medulloblastoma by inhibiting GLI1 function. Oncogene, 37, 3864–3878. DOI 10.1038/s41388-018-0249-5. [Google Scholar] [CrossRef]
28. Mastrangelo, E., Milani, M. (2018). Role and inhibition of GLI1 protein in cancer. Lung Cancer, 9, 35–43. DOI 10.2147/LCTT.S124483. [Google Scholar] [CrossRef]
29. Dong, Z., Wang, Y., Ding, V., Yan, X., Lv, Y. et al. (2020). GLI1 activation is a key mechanism of erlotinib resistance in human non-small cell lung cancer. Oncology Letters, 20, 76. DOI 10.3892/ol.2020.11937. [Google Scholar] [CrossRef]
30. Singh, A., Settleman, J. (2010). EMT, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene, 29, 4741–4751. DOI 10.1038/onc.2010.215. [Google Scholar] [CrossRef]
31. Saitoh, M. (2018). Involvement of partial EMT in cancer progression. The Journal of Biochemistry, 164, 257–264. DOI 10.1093/jb/mvy047. [Google Scholar] [CrossRef]
32. Corso, G., Figueiredo, J., de Angelis, S. P., Corso, F., Girardi, A. et al. (2020). E-cadherin deregulation in breast cancer. Journal of Cellular and Molecular Medicine, 24, 5930–5936. DOI 10.1111/jcmm.15140. [Google Scholar] [CrossRef]
33. Kim, N. H., Cha, Y. H., Lee, J., Lee, S. H., Yang, J. H. et al. (2017). Snail reprograms glucose metabolism by repressing phosphofructokinase PFKP allowing cancer cell survival under metabolic stress. Nature Communications, 8, 14374. DOI 10.1038/ncomms14374. [Google Scholar] [CrossRef]
34. Yan, L. X., Liu, Y. H., Xiang, J. W., Wu, Q. N., Xu, L. B. et al. (2016). PIK3R1 targeting by miR-21 suppresses tumor cell migration and invasion by reducing PI3K/AKT signaling and reversing EMT, and predicts clinical outcome of breast cancer. International Journal of Oncology, 48, 471–484. DOI 10.3892/ijo.2015.3287. [Google Scholar] [CrossRef]
35. Papadimitrakopoulou, V. (2012). Development of PI3K/AKT/mTOR pathway inhibitors and their application in personalized therapy for non–small-cell lung cancer. Journal of Thoracic Oncology, 7, 1315–1326. DOI 10.1097/JTO.0b013e31825493eb. [Google Scholar] [CrossRef]
36. Burris, H. A. (2013). Overcoming acquired resistance to anticancer therapy: Focus on the PI3K/AKT/mTOR pathway. Cancer Chemotherapy and Pharmacology, 71, 829–842. DOI 10.1007/s00280-012-2043-3. [Google Scholar] [CrossRef]
37. Liu, R., Chen, Y., Liu, G., Li, C., Song, Y. et al. (2020). PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers. Cell Death & Disease, 11, 797. DOI 10.1038/s41419-020-02998-6. [Google Scholar] [CrossRef]
38. Song, L., Liu, S., Zhang, L., Yao, H., Gao, F. et al. (2016). MiR-21 modulates radiosensitivity of cervical cancer through inhibiting autophagy via the PTEN/Akt/HIF-1α feedback loop and the Akt-mTOR signaling pathway. Tumor Biology, 37, 12161–12168. DOI 10.1007/s13277-016-5073-3. [Google Scholar] [CrossRef]
39. Chang, F., Lee, J. T., Navolanic, P. M., Steelman, L. S., Shelton, J. G. et al. (2003). Involvement of PI3K/Akt pathway in cell cycle progression, apoptosis, and neoplastic transformation: A target for cancer chemotherapy. Leukemia, 17, 590–603. DOI 10.1038/sj.leu.2402824. [Google Scholar] [CrossRef]
Cite This Article
 Copyright © 2022 The Author(s). Published by Tech Science Press.
Copyright © 2022 The Author(s). Published by Tech Science Press.This work is licensed under a Creative Commons Attribution 4.0 International License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
 Downloads
Downloads
 Citation Tools
Citation Tools